

Abstract
Background
Miniature inverted-repeat transposable elements (MITEs) and long terminal repeat (LTR) retrotransposons are ubiquitous in plants genomes, and highly important in their evolution and diversity. However, their mechanisms of insertion/amplification and roles in Citrus genome’s evolution/diversity are still poorly understood.
Results
To address this knowledge gap, we developed different computational pipelines to analyze, annotate and classify MITEs and LTR retrotransposons in six different sequenced Citrus species. We identified 62,010 full-length MITEs from 110 distinguished families. We observed MITEs tend to insert in gene related regions and enriched in promoters. We found that DTM63 is possibly an active Mutator-like MITE family in the traceable past and may still be active in Citrus. The insertion of MITEs resulted in massive polymorphisms and played an important role in Citrus genome diversity and gene structure variations. In addition, 6630 complete LTR retrotransposons and 13,371 solo-LTRs were identified. Among them, 12 LTR lineages separated before the differentiation of mono- and dicotyledonous plants. We observed insertion and deletion of LTR retrotransposons was accomplished with a dynamic balance, and their half-life in Citrus was ~ 1.8 million years.
Conclusions
These findings provide insights into MITEs and LTR retrotransposons and their roles in genome diversity in different Citrus genomes.
Electronic supplementary material
The online version of this article (10.1186/s12870-019-1757-3) contains supplementary material, which is available to authorized users.
Keywords: Active transposon families, Citrus, Genome diversity, Long terminal repeat (LTR), Retrotransposons, Miniature inverted–repeat transposable elements (MITEs)
Background
Miniature inverted-repeat transposable elements (MITEs) are a type of non-autonomous DNA transposons lacking their own transposases [1]. They are widely present in eukaryotes, especially in plant genomes. MITEs have the following characteristics: (1) same as autonomous DNA transposons, MITEs possess inverted repeats flanked by small direct repeats (target site duplication, TSD) and shorter length (usually < 800 bp), (2) some MITEs may transcribe and form double strand RNAs, which may further derive into small RNAs (sRNAs) [2], and (3) MITEs can achieve a very high copy number as compared to other transposons in the host genome [3, 4].
Jiang and colleagues discovered an active MITE mPing in rice, and later they found two autonomous DNA transposons Ping and Pong through homology search using the conserved terminals of mPing. They also observed that Pong can activate the transposition of mPing and named this phenomenon “cross-mobilization” [5]. Later, Yang and colleagues confirmed cross-mobilization hypothesis experimentally and reported that MITEs can also be transposed by autonomous DNA transposons belonging to different families [6]. In addition, several studies observed that MITEs tend to be closer to genetic regions [1, 7] and can regulate gene expression [8]. Some transcripts of MITEs can form hairpin structures, which are further recognized by dicer-like (DCL) and generate small RNAs (sRNAs). This phenomenon is confirmed in Arabidopsis, rice and human [9, 10]. MITEs derived sRNAs account for about 1/4 of all the sRNAs in rice genome [2], which indicates an important regulatory role of MITEs in transcription. The insertion and elimination of MITEs can lead to presence and absence polymorphism in host genomes, which is thought to be an important aspect of host genome evolution [11].
Usually, long terminal repeat (LTR) retrotransposons start with 5′-TG-3′ and end with 5′-CA-3′. After integration, there are 4~6 bp TSD flanking their acceptor sites. Generally, autonomous LTR retrotransposons contain two genes in their internal regions: Gag and Pol. Gag encodes a structural protein of virus-like capsid, and Pol encodes a polyprotein that includes aspartic proteinase (AP), reverse transcriptase (RT), RNaseH (RH) and integrase (INT). According to the order of RT/INT and the occurrence of envelope protein (ENV), LTR retrotransposons can be further divided into Copia, Gypsy, ERV, Bel-pao and Retrovirus superfamily [12].
Previous studies indicated that there is a relative balance of LTR retrotransposons insertion and deletion in host genome. The deletion of LTR retrotransposons is mainly caused by unequal homologous recombination [13, 14] and illegitimate recombination [15]. However, intra-element unequal homologous recombination leads to the formation of solo-LTRs, which are structurally identical to the 5′ LTR or 3′ LTR ends of complete LTR retrotransposons [14] and have TSD flanks at their ends. Illegitimate recombination deletes the internal sequence of LTR retrotransposons and forms a shorter sequence of forwarding repeats at the deletion sites. Previous studies have shown that sequences deleted by illegitimate recombination are five folds higher than sequences eliminated by unequal homologous recombination in Arabidopsis [15]. LTR retrotransposons amplification was thought to be one of the main drivers that lead to the significant genome size expansion. Studies have shown that even among closely related species, significant genomic size changes can result from amplification of certain LTR retrotransposons families [16]. Hawkins et al. discovered that the specific amplification of Gorge3 LTR retrotransposons families led to significant differences in the genome size of Gossypium [17]. Besides, LTR retrotransposons tend to insert in the enhancers, repressors or promoters of downstream genes, and subsequently regulate the expression of downstream genes [18, 19]. The formation of blood orange is a good example of LTR retrotransposons based regulation of gene expression [20]. LTR retrotransposons also induce chromosome rearrangement and gene translocation [21]. In a phylogenetic study, Du et al. discovered that 5 Gypsy lineages and 6 Copia lineages had been separated before the divergence of dicot and monocot species [22]. This can be used as an annotation reference and classification marker in following studies, as well as a guide for the refinement of LTR retrotransposons sequences in annotated genomes. In Du’s study, they found that there was a positive correlation between solo-LTR formation and LTR size [22]. Later, El-Baidouri and Panaud found that there was also a stronger positive correlation between solo-LTR formation and the ratio of LTR/INTERNAL [23]. They also found that horizontal transfer of LTR retrotransposons play an important role in genome evolution [23].
Citrus is an important source of vitamins for human health and ranks at top among all the fruits. Previous studies have identified transposable elements (TEs) from published Citrus genomes [20, 24–27], but differences in the TE annotation pipelines make these results un-comparable. The mechanisms of MITEs and LTR retrotransposons accumulation and amplification, and their roles in Citrus genome evolution and diversity remain poorly understood. The publication of different Citrus genomes opens the door to compare TEs and investigate their evolution characteristics in genome diversity. In the present study, we investigated the genomes of 6 Citrus species: C. sinensis (sweet orange, genome size ~ 367 Mb, contig N50 49.89 kb), Atalantia buxifolia (Chinese box orange, genome size ~ 370 Mb, contig N50 23.89 kb), C. grandis (pummelo, genome size ~ 380 Mb, contig N50 10.62 Mb), C. ichangensis (ichang papeda, genome size ~ 391 Mb, contig N50 76.56 kb), C. medica (citron, genome size 406 Mb, contig N50 46.50 kb) and C. clementina (clementine, genome size ~ 370 Mb, contig N50 115.90 kb) [26–30].
We developed two comprehensive pipelines to annotate and analyze the MITE-related and LTR-related sequences in the above Citrus species, and then studied the amplification model of some Citrus MITE families and compared the MITE presence and absence polymorphism ratio between sweet orange and the other 5 Citrus species. We investigated the MITE relative abundance in different genomic regions and analyzed the role of MITEs in gene structure variations. MITE-derived small RNAs and their relative derived position were also investigated. Using a relatively conserved method, complete LTR retrotransposons and solo-LTRs were annotated, and we investigated the activation of different lineages and families. Relative solo-LTR abundance of different LTR retrotransposons families was also investigated. Furthermore, we studied the distribution of LTR-related sequences in sweet orange and explored the possible factors of solo-LTRs formation.
Results
Annotation and classification of MITEs using a MITE-hunter based pipeline
We annotated and classified MITEs using the pipeline shown in Additional file 1: Figure S1. In manual curation of MITEs seed sequences (Additional file 8: Data set S1), we found that terminal regions of many MITEs were absent (terminal absence/presence = 47:5), which suggested the necessity of boundary correction of MITE seeds calculated by MITE-Hunter [31]. All MITE-related sequences were classified into Mutator superfamily, PIF-Harbinger superfamily, and hAT superfamily, and further divided into 110 families. Each family was named as DT(A/M/H)1-n (hAT corresponds to DTA, Mutator corresponds to DTM, PIF-Harbinger corresponds to DTH, and number 1-n corresponds to specific family number). Mutator superfamily contained 82 families, hAT superfamily contained 20 families, and PIF-Harbinger superfamily contained 8 families. In total, 61,980 full-length MITEs were annotated in Citrus species, and the average length of MITE-related sequences covered ~ 3% of the total genome sequences (8~11 Mb in different genomes, Table 1). We found that 99.4% of the full-length MITEs were shorter than 800 bp, and the average length was ~ 302 bp. The average full-lengths of Mutator superfamily, PIF-harbinger superfamily and hAT superfamily were 256, 431 and 563 bp, respectively. Full-length MITEs of hAT superfamily were significantly longer than the other two super-families (Wilcox test, p-value < 0.001), and the average length of Mutator superfamily was the shortest. We also observed that the copies of top 10% full-length MITE families accounted for over 50% of the total full-length MITE copies.
Table 1.
Comparative statistics of 61,980 full-length MITEs annotated in Citrus species
| Species | Copy number | Full-length copy number | Length (Mb) |
Content (%) |
|---|---|---|---|---|
| Atalantia buxifolia | 47,856 | 10,095 | 9.10 | 2.90% |
| Citrus sinensis | 40,886 | 9529 | 8.56 | 2.81% |
| Citrus clementina | 36,950 | 9089 | 8.02 | 2.69% |
| Citrus medica | 48,872 | 12,048 | 10.79 | 2.84% |
| Citrus ichangensis | 46,421 | 10,984 | 9.70 | 2.84% |
| Citrus grandis | 39,963 | 10,235 | 8.76 | 2.51% |
One round amplification burst dominated Citrus MITEs
It is reported that MITE families in rice mainly experienced one or more round of amplification [2]. Using a similar approach in our study, we compared the pairwise divergence distributions in Citrus. We found that unimodal distribution of MITE families was dominated in Citrus, such as DTM60, DTM77 and DTH1 (Fig. 1a), whereas a few MITE families, including DTH3 and DTH6, showed a bimodal or multimodal distribution (Fig. 1b). We observed that the phylogenetic trees of MITE families with unimodal distribution showed a star shape (Fig. 1c), while those with bimodal or multimodal distributions showed two or multiple clades Fig. 1d). Taken together, MITE families with unimodal distribution mainly experienced one round amplification from a copy of the same MITE or a significant amplification of a close copy of the same family at certain time. While those with bimodal or multimodal distribution mainly experienced two or multi rounds amplification, originated from the amplification events of different members of the same MITE family at different time points.
Fig. 1.

Statistics and phylogenetic trees of MITEs. a Unimodal distribution of pairwise divergence among some representative families of full-length MITEs. b Bimodal distribution of pairwise divergence among other representative families of full-length MITEs. c Phylogenetic tree of DTM77. d Phylogenetic tree of DTH6
Ancient active MITE families and possible active MITE family in trackable past
DTM53 has over 1000 full-length copies in each of the six Citrus species, their distribution of pairwise divergence is very similar, which are all unimodal curves with mean pairwise divergence of about 0.26 (Fig. 2a). It responded to the divergence time of ~ 23 million years, which is before the divergence of Citrus and Atalantia genus. Similarly, DTM58 (mean pairwise divergence 0.45) and DTM77 (mean pairwise divergence 0.41) also experienced amplification burst before the divergence of Citrus and Atalantia genus. However, the copy numbers of full-length DTM63 in A. buxifolia, C. sinensis, C. clementina, C. ichangensis, C. medica and C. grandis are 48, 302, 201, 323, 174 and 496 respectively, and the large difference of DTM63 number indicates the recent amplification of DTM63 family. There were some very similar or identical copies in C. sinensis, C. clementina, C. ichangensis and C. grandis. The pairwise divergence of DTM63 showing a peak around the origin (Fig. 2b), indicated that DTM63 may be still an active MITE family in these four species and further research is required for confirmation. The discovered active MITEs mainly came from Tc1/mariner and PIF/Harbinger super-families, and DTM63 could be the first discovered active MITE family which belonging to Mutator superfamily. As DTM63 lacks transposase, its transposition might rely on the autonomous DNA transposons. To figure out the autonomous DNA transposons which activate the transposition of DTM63, we annotated the candidate autonomous DNA transposons and obtained 572 and 585 candidate regions in C. sinensis and C. clementine respectively. Subsequently, we scanned the candidate regions with the conserved 7 bp terminals and 9~10 bp TSD, however we had not found any Mutator-like autonomous DNA transposons which keep the same end sequence as DTM63.
Fig. 2.

Ancient active MITE family DTM53 and an active MITE family in trackable past DTM63. a Pairwise divergence among full-length MITEs of DTM53. b Pairwise divergence among full-length MITEs of DTM63
P/A polymorphism ratios between different Citrus genomes reflect genetic distance
MITE insertion and deletion inevitably cause presence or absence (P/A) polymorphism in host genomes [2]. Taken sweet orange as the reference genome, we calculated the P/A polymorphism ratio between sweet orange and the other five Citrus species (Table 2). We found that the P/A polymorphism ratio between sweet orange and the primitive Citrus variety A. buxifolia is 1.3~2.2 times that of the other four species. Therefore, MITE P/A polymorphism has strong correlation with phylogenetic relationships, as the phylogenetic relationship from the closest to farthest to sweet orange is: C. clementina (polymorphism ratio 38.38%), C. grandis (46.70%), C. ichangensis (59.89%), C. medica (65.79%) and A. buxifolia (84.49%) respectively. The above results indicate that the P/A polymorphism ratio of MITEs reflects the genetic relationship among different Citrus species.
Table 2.
Statistics of MITE P/A polymorphism ratio between sweet orange and the other five Citrus species
| Feature | Atalantia buxifolia | Citrus clementina | Citrus medica | Citrus ichangensis | Citrus grandis |
|---|---|---|---|---|---|
| Common loci | 7571 | 9233 | 9727 | 9324 | 9745 |
| Absence in C. sinensis | 3268 | 1787 | 3982 | 3081 | 2442 |
| Absence in other genomes | 3129 | 1757 | 2417 | 2503 | 2109 |
| Commonly present collinear region loci | 1174 | 5689 | 3328 | 3740 | 5194 |
| Polymorphic ratio (%) | 84.49 | 38.38 | 65.79 | 59.89 | 46.70 |
MITEs preferentially inserted in gene flanking regions and play important role in genome diversity
To study whether MITEs favorably inserted in the gene related regions, we calculated the distribution of MITEs insertion in gene regions (from transcription start sites (TSS) to transcription termination sites (TTS)), upstream and downstream of gene regions in sweet orange, pummelo and clementine. We observed that the MITEs insertion distribution patterns were very similar in the three Citrus species, and different peaks were observed within 1 kb of upstream and downstream gene regions respectively (Fig. 3a), indicating that MITEs are preferentially inserted in gene flanking regions. Then we analyzed the distribution of MITEs in different genomic regions, including 5′ and 3′ untranslated regions (UTRs), introns, promoters (defined as 1 kb upstream of TSS) and intergenic regions. Considering that most of the genomic regions are intergenic regions, we calculated the relative abundance of MITEs, and observed that the relative density of MITEs was the most abundant in promoter regions, and the least abundant in gene regions (5′ and 3′ UTRs, introns, Fig. 3b). Therefore, MITEs preferentially inserted in gene flanking regions especially in promoters, indicating a cis-regulatory role of MITEs for their downstream genes [32, 33].
Fig. 3.

MITEs preferentially inserted in gene flanking regions and play important roles in genome diversity. a The distribution of MITEs inserted in upstream and downstream of gene regions in three Citrus species. b The relative abundance of MITEs in different genomic regions. c The stable and unstable MITEs in 6 Citrus species. d A MITE uniquely inserted into 5′ UTR of C. grandis genome. e A MITE uniquely inserted into intron of C. grandis genome. f A MITE uniquely inserted into C. grandis genome, and a gene is annotated in the insertion region. (d-f: Gray shading represents collinear regions. Blue shading represents collinear MITE regions. Orange large rectangles, small rectangles and lines respectively represent CDS, UTR and intron. Blue rectangles and inverted triangle respectively represent MITE and MITE absence loci)
We compared the copies of different MITE families to investigate their distribution pattern and role in Citrus genome evolution, the abundance of different MITE families in six Citrus genomes was shown in Additional file 3: Figure S3. There were two types of MITEs sequence distribution pattern. Stable genomic distribution pattern indicated MITE families were similar with almost same copy number in different Citrus species, such as DTM53, DTM58 and DTM77. These MITE families experienced amplification burst before the speciation of Citrus and Atalantia with very few recent MITE insertions or deletions. However, unstable genomic distribution of MITEs families showed obvious distribution bias across different species. For example, the ratios of DTM63 in C. grandis, C. ichangensis and C. sinensis were much higher than C. medica and A. buxifolia (Fig. 3c), owning to recent transpositions. DTM62 had more copies in A. buxifolia than the other 5 Citrus species, and DTM51 had more copies in C. media than the others. Comparing with stable MITE families, unstable MITE families contributed more divergence to Citrus and might play an important role in the diversity of Citrus genome.
We defined MITEs present in all species as conserved MITEs and the other as non-conserved. The proportion of conserved MITEs inserted into promotor (18.13%) and 5′/3′ UTR regions (1.72%) was lower than non-conserved MITEs (25.76% in promotors and 4.31% in UTRs), which indicated a strong selection effect on MITEs insertion in these regions (Additional file 5: Table S1). Interestingly, newly-inserted MITEs tended to be abundant in promotor regions (26.08% in C. clementina, 27.17% in C. sinensis and 26.63% in C. grandis) comparing to the ancient inserted MITEs (23.97% in C. clementina, 25.21% in C. sinensis and 23.69% in C. grandis) (Additional file 6: Table S2). Figure 3d showed a MITE uniquely inserted into 5′ UTR of C. grandis, Fig. 3e showed an example of MITE inserted into intron of C. grandis and the MITE insertion in intron did not alter the gene structure, and Fig. 3f showed a MITE inserted into C. grandis and a gene was annotated in the insertion region, while there were no genes annotated in the collinear region of C. clementina and C. sinensis genomes respectively. The above examples indicate that MITEs play important roles in genome structure variations in Citrus genomes.
MITE-derived sRNAs were predominantly derived from MITEs terminals
We collected a total of 14,664,233 unique sRNA tags from previous studies, among them 7,258,262 tags could be aligned to the sweet orange reference genome, and 935,213 tags could be aligned to MITE-related sequences. While filtering out the unmapped tags, MITE-derived sRNAs accounted for 12.9% of the total amount. By looking into the length distribution of MITE-derived sRNAs, we observed that they are predominately 24 bp (Fig. 4a) and derived from different positions of MITEs (Fig. 4b). In comparison with the relative positions from where sRNAs were derived, we found that their distribution in sweet orange is different from the rice [2]. There were only two peaks at both ends with a valley in the middle, indicating that the middle of MITEs derived less sRNAs than the other region in sweet orange, whereas there was another peak in the middle in rice [2].
Fig. 4.
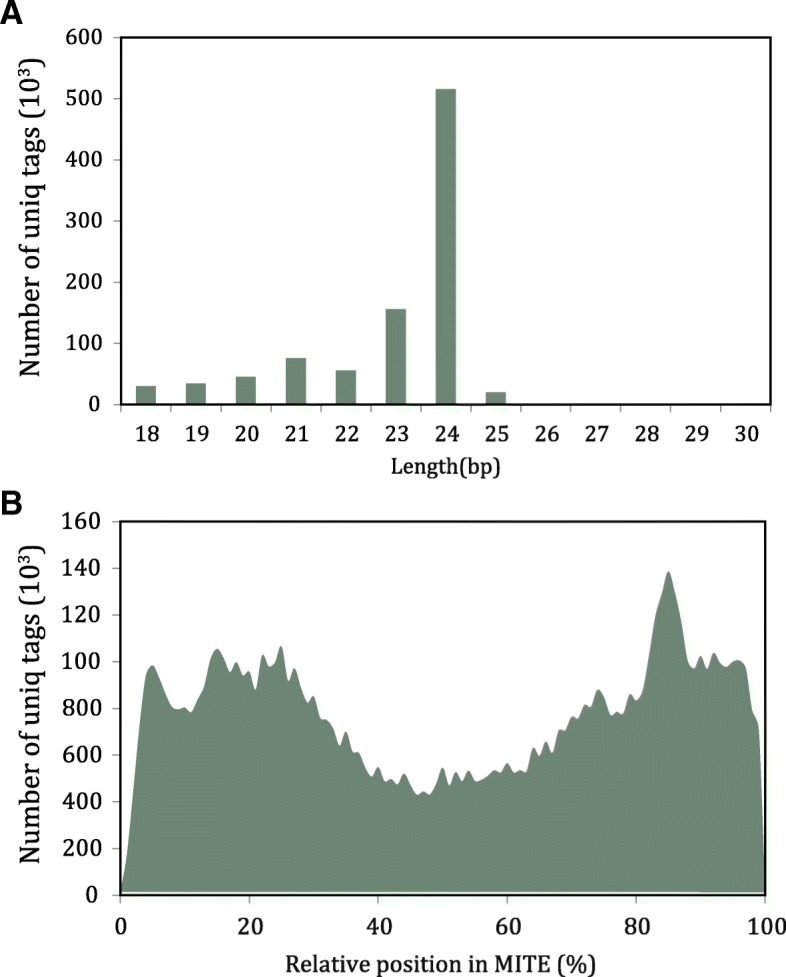
Distribution of MITE-derived small RNAs. a Length distribution of MITE-derived small RNAs. b The relative position distribution of MITE-derived small RNAs
LTR retrotransposons annotation, classification and characterization
Although Wicker and colleagues proposed the “80–80-80” rule for TEs family classification [34], it is still controversial in different LTR studies. In a recent study, it is suggested that use of another cutoff (60% identity and 70% coverage) is more appropriate for the Uwum family in Zea mays and RLC_Gmr6/18 family in soybean [23]. Considering that both the complete LTR retrotransposons and solo-LTRs have an intact LTR, a cutoff of 75% identity was chosen to classify all LTR retrotransposons into different Citrus families. Totally, we obtained 13,371 solo-LTRs and 6670 complete LTR retrotransposons from 340 families (Table 3; Additional file 9: Data set S2). The number of solo-LTRs was roughly equivalent in the 6 species except for Atalantia buxifolia and C. sinensis, whereas complete LTR retrotransposons varies from 392 to 1904. Considering that the completeness of C. grandis is much better than other genomes, its assembly quality for complete LTR retrotransposon regions would be much better than the other five genomes. In addition, the strategy we used might miss some low-copy LTR retrotransposons families of A. buxifolia. In this way, C. grandis would be a more appropriate choice for further investigation. There were two distinct peaks in the length distribution of complete LTR retrotransposons (Fig. 5a), one peak was around ~ 5.4 kb and the other ~ 12.4 kb, which corresponded to the peaks of Copia and Gypsy super-families, respectively. The average length of complete LTR retrotransposons in Copia superfamily was ~ 6.5 kb, and the average length of complete LTR retrotransposons in Gypsy superfamily was ~ 9.3 kb. Consistent with previous studies, Gypsy LTR retrotransposons were significantly longer than Copia LTR retrotransposons (Wilcox test p-value < 0.001) [22, 23, 26, 35, 36]. The ratio of Gypsy-like elements to Copia-like elements was 1.5 in C. grandis, which is slightly higher than in soybean (1.4) [22], but much lower than in rice (4.9) [35] and sorghum (3.7) [36]. In addition, the ratio of solo-LTR to complete LTR retrotransposons was 1.4, which is higher than that in maize, soybean and rice [23].
Table 3.
Comparative statistics of 13,371 solo-LTRs and 6670 complete LTR retrotransposons
| Species | Complete LTR | Solo-LTR | LTR content(%) |
|---|---|---|---|
| Atalantia buxifolia | 392 | 1417 | 20.66 |
| Citrus sinensis | 487 | 1946 | 24.81 |
| Citrus clementina | 1742 | 2498 | 29.31 |
| Citrus medica | 1205 | 2389 | 32.18 |
| Citrus ichangensis | 940 | 2416 | 23.87 |
| Citrus grandis | 1904 | 2705 | 28.32 |
Fig. 5.

Length distribution and phylogenetic trees of LTRs. a Length distribution of complete LTRs (green whiskers and black boxes represent average length distribution, and white circles represent median). b Copia-like superfamily RT domain phylogenetic tree. c Gypsy-like superfamily RT domain phylogenetic tree. Citrus also keeps six Gypsy lineages (reference LTR retrotransposons are shown as italic with circles on branches, others are LTR families in Citrus)
By calculating the distribution of LTR retrotransposons in sweet orange genome, we found that different from the relatively uniform genomic distribution of MITEs, the distribution of LTR retrotransposons was quite heterogeneous. There were significant peaks in centromere-proximal regions along different chromosomes, which is consistent with the previous studies of other species [37]. A possible explanation for this phenomenon is that, centromere-proximal regions are recombination-suppressed, which leads to the suppression of unequal homology recombination and illegitimate recombination, therefore, LTR retrotransposons in centromere-proximal regions are accumulated.
Twelve conserved LTR retrotransposon lineages are present in Citrus
In the previous studies, the Copia-like superfamily was grouped into 6 lineages, Angela, Ale, Bianca, Ivana, Maximus and TAR [12, 38, 39], while Gypsy-like superfamily was further grouped into another 6 lineages, Tekay, Galadriel, CRM, Reina, Athila and Tat [40]. Among these, Tekay, Galadriel, CRM and Reina were Chromovirus clades. These 12 lineages were separated before the divergence of dicot and monocot species. We retrieved the sequences of RT domains from public data and the LTR retrotransposons identified in this study, including 91 Gypsy-like RT and 174 Copia-like RT sequences. Then we constructed the phylogenetic trees of RT domain (Fig. 5b, c). From the phylogenetic trees, it was observed that all the 12 lineages were identified in Citrus. In addition, the branches of the corresponding families were classified according to the topological structure of the phylogenetic tree.
To investigate the activation and status of these 12 lineages, we counted the family and element numbers of each lineage (Table 4). In Gypsy-like superfamily, Tat had the most families (33), and Athila contained the maximum number of complete LTR retrotransposons (1754). However, in Copia-like superfamily, Bianca had the most families (86) and complete LTR retrotransposons (1071), while Angela had the least families (5) and TAR contained the least complete LTR retrotransposons (96).
Table 4.
Statistics of LTR retrotransposons in 12 conserved citrus lineages
| Lineages | Number of families | Number of Complete LTRs | Average Complete LTR length (bp) | Number of solo-LTRs | Average solo-LTR length (bp) |
|---|---|---|---|---|---|
| Ale | 51 | 637 | 5259 | 219 | 267 |
| Ivana | 10 | 155 | 5574 | 132 | 343 |
| Maximus | 13 | 439 | 10,070 | 1542 | 1319 |
| TAR | 10 | 96 | 6208 | 178 | 750 |
| Angela | 5 | 277 | 8689 | 1313 | 1537 |
| Bianca | 86 | 1071 | 5832 | 961 | 585 |
| Reina | 25 | 309 | 4712 | 357 | 394 |
| Tekay | 2 | 26 | 9068 | 326 | 2187 |
| Galadriel | 5 | 193 | 5416 | 317 | 473 |
| CRM | 2 | 109 | 8054 | 605 | 940 |
| Athila | 23 | 1754 | 11,324 | 4435 | 1302 |
| Tat | 33 | 732 | 10,211 | 1180 | 838 |
We further calculated the average length of complete LTR retrotransposons and solo-LTRs for each lineage. Complete LTR retrotransposons of different Copia and Gypsy lineages showed significant length difference (Kruskal-Wallis rank sum test, p-value < 0.001), and solo-LTRs also showed significant length difference (Kruskal-Wallis rank sum test, p-value < 0.001). The Maximus lineage had an average length of ~ 10 kb, which is the longest in complete Copia LTR retrotransposons. Compared to a previous study, the average length of the Copia lineages in Citrus was roughly equivalent, although Bianca has more families and complete LTR retrotransposon members in Citrus than in rice, Arabidopsis and Triticeae [12].
A few LTR retrotransposons families were active in trackable past and play an import role in Citrus genome diversity
Generally, LTR retrotransposons constantly inserted and eliminated in a long-term cycle and maintain the host genome size in a dynamic balance. Through computing the insertion time of LTR retrotransposons, we obtained the LTR retrotransposons insertion time curve (Fig. 6a) and found that the LTR retrotransposons insertion time followed an exponential distribution and their half-life in Citrus was ~ 1.8 million years. Meanwhile, we noticed that only a few LTR retrotransposons families were active in trackable past, which were consistent with the previous studies [23]. In C. grandis, only eight families, i.e., RLG1, RLG2, RLG3, RLG4, RLG5, RLC7, RLG9 and RLG12 contained more than 30 complete LTR retrotransposons, and the member of RLG1 (476) were larger than the total members (322) of the other seven families. In addition, LTR retrotransposon families with the most copy numbers of complete LTR retrotransposons (such as RLG1, RLG2, RLG3) were usually active recently (Fig. 6b), which indicated that ancient LTR retrotransposons were rapidly removed from the genome by unequal homology recombination and illegitimate recombination. Although copies of complete LTR retrotransposons were highly dependent on the genome integrity, however the copies of solo-LTRs showed less dependency (Table 4). Thus, we compared the copies of different LTR retrotransposons families to investigate whether solo-LTRs played any roles in Citrus genome diversity (Fig. 6c). Some families (such as RLC2, RLG5 and RLG16) have similar number of copies among the 6 Citrus genomes, indicating these LTR retrotransposons were less active and divergent among different Citrus species, we called them stable LTR families. In contrast, some unstable LTR families showed distinct composition among 6 Citrus species. For example, 136 solo-LTRs were found belonging to RLG25 in A. buxifolia, but in each of the other 5 Citrus genomes there were less than 10 copies. Above finding suggested that RLG25 might be more active in A. buxifolia and more copies of solo-LTRs had been accumulated through unequal recombination than the other 5 Citrus species.
Fig. 6.

Insertion time and formation factor of LTR. a LTR insertion time distribution. b Insertion time distribution of three LTR families. c Relative content of solo-LTRs in six Citrus genomes. Some LTR families keep relative stable like RLC2 and RLG5, while other LTR families shows unstable and may become more active in one specie like RLG25. d Solo-LTR formation factor. The ratio of LTR-length/Internal-length shows clear positive correlation with the ratio of Solo copy/Complete copy
Insertion time might contribute to solo-LTRs formation
To avoid miss annotation of solo-LTRs which mainly formed by the internal unbalanced homologous recombination of complete LTR retrotransposons and truncated LTR retrotransposons, we treated a region as a solo-LTR only if there were two 4~6 bp exact TSD flanks around. Different from previous studies, only complete LTR retrotransposons were adopted, because LTR retrotransposons without TSD may be the consequence of assembly error, boundary annotation error and inter-element unequal recombination which was shown to be rare in Arabidopsis [15]. We took the value of S/C (solo-LTR/complete LTR) to evaluate the relationship between solo-LTR formation and some relative factors (Fig. 6d). Our result revealed a significant correlation between S/C and LTR retrotransposons insertion time (Spearman’s rank correlation r = 0.455, p-value < 0.01), which showed disagreement with the result in soybean [22]. Similar significant correlation was detected between S/C and LTR size (Spearman’s rank correlation r = 0.627, p-value < 0.01). We confirmed that there was a strong correlation between S/C and the ratio of LTR/INTERNAL (Spearman’s rank correlation r = 0.691, p-value < 0.01), which agrees with El-Baidouri’s study [23]. Taken together, we identified that solo-LTRs formation was related with several factors, and longer LTR will favor more stable pair of 5′ and 3′ LTR if they are not too distant, and that the longer the insertion time of LTR retrotransposons, the higher possibility of the unequal recombination will be.
Discussion
Annotation and comparison of MITEs and LTR retrotransposons from multiple genomes provides a new insight into Citrus evolution and genome diversity
Although a few studies have revealed the important roles of MITEs and LTR retrotransposons in Citrus [20, 24, 27], their pivotal role in Citrus evolution and diversity has long been overlooked. This is the first study focusing on MITEs and LTR retrotransposons annotation and comparison in Citrus to investigate their role especially in genome diversity and evolution. Annotation of MITEs and LTR retrotransposons provides a useful resource for researchers who are interested in Citrus MITEs and LTR retrotransposons.
The insertion of MITEs in the Citrus genome leads to massive polymorphism, where the inter-genus polymorphism ratio of MITE Insertion Polymorphism (MIP) is much higher than intra-genus, which reflects the genetic relationships among different species. Comparison of MITEs and LTR retrotransposons reveals that some MITEs and LTR retrotransposons are relatively stable in Citrus genomes, which shows little composition divergence among Citrus genomes, such as DTM53 and RLC2. In contrast, some MITEs and LTR retrotransposons are quite unstable, such as DTM63 and RLG25, they greatly reshape the Citrus genomes and some MITEs even play a role in gene structure variations. It remains to be seen that whether other plants show similar MIP among different species and share the same MITEs and LTR retrotransposons stable and unstable composition patterns.
MITEs and LTR retrotransposons: similarities and difference
MITEs and LTR retrotransposons are the most important types of transposons and highly characterized in plant genomes. MITEs account for 2.51 to 2.90%, while LTR retrotransposons account for 20.66 to 29.31% in 6 different Citrus species. Generally, the average length of LTR retrotransposons is much longer than that of MITEs and account for the most majority part of genome. Both MITEs and LTR retrotransposons are dominated by a few families of full-length (complete) copies. However, the distribution of the two types of transposable elements is quite different, LTR retrotransposons related sequences are enriched in centromere-proximal regions, whereas MITE-related sequences are distributed relatively even across the chromosomes (Fig. 7a-d). Both MITEs and LTR retrotransposons can insert into promoter regions to regulate downstream gene expression as revealed by some previous studies [20, 27].
Fig. 7.

LTR-RTs and MITEs distribution on some representative chromosomes (window size = 1 Mb; step size = 100 kb). a LTR retrotransposons and MITEs distribution on chromosome 1. b LTR retrotransposons and MITEs distribution on chromosome 4. c LTR retrotransposons and MITEs distribution on chromosome 7. d LTR retrotransposons and MITEs distribution on chromosome 9
DTM63 was active in the trackable past and plays a role in gene-structure variation
The large difference of DTM63 copy number in 6 Citrus species identified in this study as well as DTM63 new insertions in citrus bud mutant discovered by Ke et al. [41] indicates that DTM63 is possibly be an active Mutator-like MITE family in the trackable past and may still be active in Citrus. The average length of DTM63 is ~ 150 bp. We have found some manually confirmed cases of DTM63 insertion in promoter regions, which indicate DTM63 potential role in gene structure variations and may regulate downstream gene expression (Additional file 4: Figure S4; homologous regions listed in Additional file 7: Table S3). In our previous study we reported that MITE insertion is involved in emergence of nucleus polyembryony in Citrus [27], which also highlight the pivotal regulatory role of MITEs in promotor regions. In further studies, whether DTM63 insertion change downstream gene expression and even cause phenotype mutations in Citrus will be investigated.
The number of complete LTR retrotransposons depends highly on genome integrity
We notice that the number of complete LTR retrotransposons highly depends on the genome integrity. For example, C. grandis was sequenced by Pacbio long reads (contig N50, 10.62 Mb) and C. clementina was sequenced using Sanger sequencing method (contig N50, 115.9 kb), while the other 4 genomes were sequenced and assembled using next-generation sequencing (NGS) approach (all their contig N50 < 80 kb). Therefore, the number of identified complete LTR retrotransposons in C. grandis is the highest, followed by C. clementina, while the other 4 genomes have less complete LTR retrotransposons. As the 5’LTR and 3′ LTR of complete LTR retrotransposons, especially newly inserted LTR retrotransposons should be identical, but in this case, NGS assembly software tend to disrupt the 5’LTR and 3’LTR into different contigs, while long reads spanning the 5′ /3’LTR with flanking sequence usually overcome this biasness. The length of MITEs is relatively short (usually < 800 bp), therefore the number of full-length MITEs is basically unaffected by genome integrity.
Whole genome comparison reveals the role of LTR retrotransposons in genome diversity
Our whole genome LTRs annotation and comparison give insights into the role of different LTR retrotransposons families in genomes diversity. Some LTR retrotransposons are less activated with few new solo-LTR formation in recent years and contributed less diversity among different Citrus genomes, while some families are highly divergent among different Citrus genomes and may play a part in Citrus genome diversity. As we highlighted above, the number of complete LTR retrotransposons highly depends on genome integrity, further studies on more complete Citrus genomes will be needed to validate whether complete LTR retrotransposons shows same patterns of solo-LTRs. Although an insertion of Copia-like retrotransposon upstream of Ruby (a MYB transcriptional activator) was identified and controlled the expression of Ruby [20], which further reveals the regulatory role of LTR insertions for downstream genes. It remains to be seen whether other LTRs insertions show similar effect and even impact the phenotype of Citrus or not.
Conclusions
In this study, we focused on MITEs and LTR retrotransposons in 6 Citrus species and found that MITE-related sequences accounted for about 3% of each genome, while LTR retrotransposons accounted for about 21.2% to ~ 33.1%. One-round amplification dominates the amplification mode of MITE families with high-copy numbers. Among these, DTM63 was possibly the first discovered active Mutator-like MITE family in Citrus. 12.9% of the small RNAs in sweet orange were derived from MITE-related sequences, indicating an important regulatory role of MITEs. The insertion of MITEs in the Citrus genome led much polymorphism, and the inter-genus ratio of MIP is much higher than intra-genus, which reflects the genetic relationships. Moreover, by comparing the LTR retrotransposons content on chromosomes, we found that LTRs predominantly enrich in centromere-proximal region, and Solo LTRs formation has a positive relation with LTR insertion time, LTR size and the ratio of LTR/INTERNAL. The insertion and elimination of LTR-RTs accomplished by a dynamic balance in Citrus genomes, and the half-life of LTR-RTs is longer than Arabidopsis, Rice and Medicago truncatula. These findings provide insights into MITEs and LTR retrotransposons and their roles in genome diversity in different Citrus species genomes.
Methods
Genomic sequences
C. sinensis, A. buxifolia, C. grandis, C. ichangensis, and C. medica were sequenced by us [27] and available at C. sinensis annotation project database (http://citrus.hzau.edu.cn/orange/). While, the genome of C. clementine was downloaded from Citrus Genome Database (https://www.citrusgenomedb.org/).
MITE annotation, manual curation and classification
MITE-Hunter [31] (default parameters) was used to search all MITE candidates from the selected genomes followed by UCLUST, identity 80% was used to cluster the candidates. We picked at least 1 representative from each cluster to perform manual correction for TSD and terminal inverted repeats (TIR). We removed the false-positive sequences which did not have TSD and TIR features, and then removed the sequences with length greater than 800 bp to keepF41 in line with the previous studies. Since some of the sequences have changed, we perform the second clustering (family) with the same parameters. MITEs superfamily classification was referred to a previous study [42]. Finally, we manually picked one MITE with complete TIR and TSD as representative from each family to construct Citrus MITE database. RepeatMasker 4.0.2 (parameters “-pa 6 -s -nolow –xsmall -excln”) [43] was used to annotate MITE-related sequences for all 6 selected genomes, and an in-house Perl script was written to retrieve all MITE-related sequences (Additional file 1: Figure S1).
MITE amplification mode and time
MITE-related sequences which cover over 90% of the representatives were treated as full-length MITEs. MUSCLE [44] was used to align the full-length MITEs which belong to the same family, and then MEGA6 [45] was used to construct phylogenetic tree with neighbor-joining method. According to the Jukes-Cantor method [46], we wrote a Perl script to calculate the pairwise divergence of each MITE family. Using kiwifruit’s average substitution rate of 2.81 × 10− 9 substitutions per synonymous site per year in coding region [47] and 2 times the rate in non-coding region (r = 5.62 × 10− 9), and formula T = k/2r [13], we estimated the amplification time of several MITE families with high-copy number.
Putative Mutator-liker DDE regions in C. sinensis and C. clementina were obtained with TARGeT (e value<=0.01) pipeline [48]. Flanking sequences with 10 kb upstream and downstream of the putative region were retrieved. A Perl script was written to find the conserved terminal sequences of 5′-GGACTTG-3′, 5′-CAGGTCC-3′ (allowing 1 mismatch in the terminals), and 9~10 bp TSD in all candidates.
P/A polymorphism of MITEs
Flanking sequences with 1 kb upstream and downstream of the full-length MITEs were retrieved from all six genomes. First, the flanking sequences of MITE from the other five genomes were aligned to sweet orange genome using BLAST (e value< 10− 50). Then the flanking sequences of sweet orange were aligned to the other five genomes. If the pair of flanking sequences was the best hits and anchored to the same scaffold/chromosome, same strand, and the distance between the two anchored sites was less than 1 kb, the MITE loci were thought to be allelic. If there was a MITE-related sequence of the same MITE family in the above target allelic loci, the MITE insertion was thought to be present in both genomes, otherwise the MITE insertion was thought to be specific for the query genomes and absent from the target genome. A common MITE locus index was created based on the pairwise relation between C. sinensis and the other five Citrus species to analyze its distribution and role across all the six Citrus genomes. We defined the first 1 kb sequence region of the gene transcription start site as the promoter region. Bedtools [49] v2–2.26.0 was used to identify the insertion/presence of MITEs in different genome regions (5’UTR, 3’UTR, intron, intergenic, promoter). DREME tool of the MEME Suite (http://meme-suite.org/tools/dreme) was used for MOTIF analysis. Several in-house PERL scripts were written to measure relative abundance density of MITEs in Citrus genomes.
MITE-derived small RNAs
In order to prevent deviation of relative position statistics caused by incomplete MITE, the length of all the MITE’s sequences with the difference of less than 1% of the length of the seed sequence was selected from each family. BOWTIE2 [50] allowing for 1 base mismatch was used to compare small RNAs came from previous studies to the selected MITE’s sequences. Small RNAs are believed to be derived from the MITE if they significantly aligned against MITE.
LTR retrotransposons annotation and classification
tRNAScan-SE [51] was used to predict the tRNAs of 6 species with default parameter, and then all tRNAs were combined after filtering pseudogenes and redundant sequences to make a Citrus tRNA database. The tRNA data was used to predict the PBS of LTR retrotransposons with LTR_FINDER [52] by using developed tRNA database. LTR_FINDER parameter “-w 2” was used to predict LTR retrotransposons. Similarly, as performed in a previous study [53], clustering and filtering was performed to reduce false positives, only candidates with 5’TG, 5’CA, PBS, PPT, 3’CA and 3’TG were considered in the further analyses. The annotation work flow for LTR is shown step by step in Additional file 2: Figure S2.
LTR retrotransposons phylogenetic tree
Three frame translation of INTERNAL sequences was employed, and hmmsearch (e value ≤10− 6) was used to search the profile of Gypsy-like RT (PF00078) and Copia-like RT (PF07727). RT domain amino acid sequences longer than 100 aa were retrieved from the translation. We chose one typical RT sequence from each family, and premature stop codons were eliminated from the sequences. Combined with the representatives we collected from the previous studies, Muscle was used to align the RT sequences. MEGA6 [45] (default parameters) was used to construct phylogenetic tree with neighbor-joining method.
The distribution of LTR retrotransposons on chromosomes
Using the above LTR annotation results and taking the sweet orange as reference example, chromosomes were split into 1 Mb windows, and the windows < 1 Mb for each chromosome were not taken into account in further analyses. The distribution of the LTR on the chromosome is counted.
LTR insertion time
As in the transposition process, the newly generated LTR retrotransposon will only take one of the LTRs as template. In this way, the 5’LTR and 3’LTR of the newly generated LTR retrotransposon will be identical. During evolutionary time, 5’LTR and 3’LTR may accumulate spontaneous mutations. LTR insertion time (T) formula T = k/2r, using a substitution rate of r = 5.62 × 10− 9 substitutions per site per year for calculations. MUSCLE was used to align the 5′-LTR and 3′-LTR of each complete LTR retrotransposon and the distance between LTRs was corrected by the Jukes-Cantor method [46].
In addition, we inferred the insertion time of LTR in an exponential distribution. By using R language curve fitting nonlinear least squares (NLS) function (fitting equation y = a ∗ 2b ∗ x), initial value a = 270, b = − 0.5 and the exponential function, we explained the number and time of LTR insertion. Furthermore, for the LTR family with higher copy number in the Citrus species, the insertion time of each member was calculated separately, and then the family member amplification time was mapped.
Solo-LTR formation
C. grandis was sequenced with the PacBio sequencing technology, so we chose C. grandis as the representative for solo-LTR formation analysis. Families with less than 10 complete LTR retrotransposons were not included in this study, as mis-annotation and omission of some family members are unavoidable, and families with more complete LTR retrotransposons would be more robust. As solo-LTRs only have one LTR, the methods used for complete LTR retrotransposons are not feasible for solo-LTRs studies. Therefore, we used the average insertion time of complete LTR retrotransposons of each family to represent the insertion time of solo-LTRs. The average INTERNAL sequence length was also calculated. The calculation of Spearman’s relationship between S/C and LTR insertion time, LTR size and LTR/INTERNAL were done using R software.
Additional files
Figure S1. The pipeline of MITE classification and annotation. (DOCX 151 kb)
Figure S2. The pipeline of LTR retrotransposons classification and annotation. (DOCX 475 kb)
Figure S3. Relative abundance of MITE families in Citrus species. (DOCX 1605 kb)
Figure S4. Manually confirmed DTM63 insertion sites. (A) Manually confirmed case of C. grandis. (B). Manually confirmed case of C. sinensis. (C) Manually confirmed case of C. clementina. (D) Manually confirmed case of C. ichngensis. Sequences in blue represent TSD and the “N” in red represent a copy full-length DTM63. The corresponding homologous regions were listed in Additional file 7: Table S3. (DOCX 584 kb)
Table S1. Information of conserved and non-conserved MITEs insertion between different genomic regions (DOCX 12 kb)
Table S2. Information of newly inserted MITEs in different genomic regions of three Citrus species. (DOCX 13 kb)
Table S3. Information of DTM63 insertion sites. (DOCX 14 kb)
Data Set S1. The seed file of MITEs. (FA 43 kb)
Data Set S2. The seed file of LTR retrotransposons. (FA 2229 kb)
Acknowledgements
Authors would like to thank the Senior Editors and Associate Editors of the journal as well as the anonymous reviewers who have generously given up valuable time to review our work and given us useful suggestions for improvements.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFD1000101), National Natural Science Foundation of China (31871269, 31571351), and the Fundamental Research Funds for the Central Universities (2662017PY043). Authors are very thankful to these organizations.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its additional files].
Abbreviations
- LTR
Long terminal repeat
- MITEs
Miniature inverted–repeat transposable elements
- RE
Reverse transcriptase
- sRNAs
Small RNAs
- TSD
Target site duplication
- TSS
Transcription start sites
- TTS
Transcription termination sites
Authors’ contributions
YL and MTQ contributed to the conception and design of the analysis, acquisition of data, analysis of the data, performing the experiments, and writing of the original draft of the manuscript. JWF, YD and SW contributed to the bioinformatics analysis of LTR retrotransposons and MITEs. GW, LK and QX analyzed the genomic data and edited/reviewed the manuscript. LLC acquired the funding, contributed to the conception and design of the analysis and acquisition of data, supervise and analyze the data, and improve the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bureau TE, Ronald PC, Wessler SR. A computer-based systematic survey reveals the predominance of small inverted-repeat elements in wild-type rice genes. Proc Natl Acad Sci U S A. 1996;93:8524–8529. doi: 10.1073/pnas.93.16.8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu C, Chen J, Zhang Y, Hu Q, Su W, Kuang H. Miniature inverted-repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa. Mol Biol Evol. 2012;29:1005–1017. doi: 10.1093/molbev/msr282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feschotte C, Jiang N, Wessler SR. Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002;3:329–341. doi: 10.1038/nrg793. [DOI] [PubMed] [Google Scholar]
- 4.Smit A, Riggs AD. Tiggers and DNA transposon fossils in the human genome. Proc Natl Acad Sci U S A. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR. An active DNA transposon family in rice. Nature. 2003;421:163–167. doi: 10.1038/nature01214. [DOI] [PubMed] [Google Scholar]
- 6.Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR. Tuned for transposition: molecular determinants underlying the hyperactivity of a stowaway MITE. Science. 2009;325:1391–1394. doi: 10.1126/science.1175688. [DOI] [PubMed] [Google Scholar]
- 7.Oki N, Yano K, Okumoto Y, Tsukiyama T, Teraishi M, Tanisaka T. A genome-wide view of miniature inverted-repeat transposable elements (MITEs) in rice, Oryza sativa ssp. japonica. Genes Genetic Syst. 2008;83:321–329. doi: 10.1266/ggs.83.321. [DOI] [PubMed] [Google Scholar]
- 8.Zerjal T, Joets J, Alix K, Grandbastien MA, Tenaillon MI. Contrasting evolutionary patterns and target specificities among three tourist-like MITE families in the maize genome. Plant Mol Biol. 2009;71:99–114. doi: 10.1007/s11103-009-9511-0. [DOI] [PubMed] [Google Scholar]
- 9.Piriyapongsa J, Mariño-Ramírez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176:1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piriyapongsa J, Jordan IK. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA. 2008;14:814–821. doi: 10.1261/rna.916708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampath P, Murukarthick J, Izzah NK, et al. Genome-wide comparative analysis of 20 miniature inverted-repeat transposable element families in Brassica rapa and B. oleracea. PLoS One. 2014;9:e94499. doi: 10.1371/journal.pone.0094499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wicker T, Keller B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007;17:1072–1081. doi: 10.1101/gr.6214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101:12404–12410. doi: 10.1073/pnas.0403715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitte C, Panaud O. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol Biol Evol. 2003;20:528–540. doi: 10.1093/molbev/msg055. [DOI] [PubMed] [Google Scholar]
- 15.Devos KM, Brown JK, Bennetzen JL. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002;12:1075–1079. doi: 10.1101/gr.132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennetzen JL. Mechanisms and rates of genome expansion and contraction in flowering plants. Genetica. 2002;115:29–36. doi: 10.1023/A:1016015913350. [DOI] [PubMed] [Google Scholar]
- 17.Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16:1252–1261. doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kashkush K, Feldman M, Levy AA. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genetics. 2003;33:102–106. doi: 10.1038/ng1063. [DOI] [PubMed] [Google Scholar]
- 19.Schramke V, Allshire R. Hairpin RNAs and retrotransposon LTRs effect RNAi and chromatin-based gene silencing. Science. 2003;301:1069–1074. doi: 10.1126/science.1086870. [DOI] [PubMed] [Google Scholar]
- 20.Butelli E, Licciardello C, Zhang Y, et al. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell. 2012;24:1242–1255. doi: 10.1105/tpc.111.095232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao H, Jiang N, Schaffner E, Stockinger EJ, van der Knaap E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science. 2008;319:1527–1530. doi: 10.1126/science.1153040. [DOI] [PubMed] [Google Scholar]
- 22.Du J, Tian Z, Hans CS, et al. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 2010;63:584–598. doi: 10.1111/j.1365-313X.2010.04263.x. [DOI] [PubMed] [Google Scholar]
- 23.El Baidouri M, Panaud O. Comparative genomic paleontology across plant kingdom reveals the dynamics of TE-driven genome evolution. Genome Biol Evol. 2013;5:954–965. doi: 10.1093/gbe/evt025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernet GP, Asíns MJ. Identification and genomic distribution of gypsy like retrotransposons in Citrus and Poncirus. Theor Appl Genet. 2003;108:121–130. doi: 10.1007/s00122-003-1382-1. [DOI] [PubMed] [Google Scholar]
- 25.Biswas MK, Chai L, Mayer C, Xu Q, Guo W, Deng X. Exploiting BAC-end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in Citrus. Mol Biol Rep. 2012;39:5373–5386. doi: 10.1007/s11033-011-1338-5. [DOI] [PubMed] [Google Scholar]
- 26.Xu Q, Chen LL, Ruan X, et al. The draft genome of sweet orange (Citrus sinensis) Nat Genet. 2013;45:59–66. doi: 10.1038/ng.2472. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Xu Y, Zhang S, et al. Genomic analyses of primitive, wild and cultivated Citrus provide insights into asexual reproduction. Nat Genet. 2017;49:765–772. doi: 10.1038/ng.3839. [DOI] [PubMed] [Google Scholar]
- 28.Wu GA, Prochnik S, Jenkins J, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during Citrus domestication. Nat Biotechnol. 2014;32:656–662. doi: 10.1038/nbt.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terol J, Naranjo MA, Ollitrault P, Talon M. Development of genomic resources for Citrus clementina: characterization of three deep-coverage BAC libraries and analysis of 46,000 BAC end sequences. BMC Genomics. 2008;9:423. doi: 10.1186/1471-2164-9-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu GA, Terol J, Ibanez V, et al. Genomics of the origin and evolution of Citrus. Nature. 2018;554:311–316. doi: 10.1038/nature25447. [DOI] [PubMed] [Google Scholar]
- 31.Han Y, Wessler SR. MITE-hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010;38:e199. doi: 10.1093/nar/gkq862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang G, Lee YH, Jiang Y, Shi X, Kertbundit S, Hall TC. A twoedged role for the transposable element Kiddo in the rice ubiquitin2 promoter. Plant Cell. 2005;17:1559–1568. doi: 10.1105/tpc.104.030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naito K, Zhang F, Tsukiyama T, et al. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature. 2009;461:1130–1134. doi: 10.1038/nature08479. [DOI] [PubMed] [Google Scholar]
- 34.Wicker T, Sabot F, Hua-Van A, et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 35.Tian Z, Rizzon C, Du J, et al. Do genetic recombination and gene density shape the pattern of DNA elimination in rice long terminal repeat retrotransposons? Genome Res. 2009;19:2221–2230. doi: 10.1101/gr.083899.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paterson AH, Bowers JE, Bruggmann R, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 37.Du J, Tian Z, Bowen NJ, Schmutz J, Shoemaker RC, Ma J. Bifurcation and enhancement of autonomous-nonautonomous retrotransposon partnership through LTR swapping in soybean. Plant Cell. 2010;22:48–61. doi: 10.1105/tpc.109.068775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Liu JS. LTR retrotransposon landscape in Medicago truncatula: more rapid removal than in rice. BMC Genomics. 2008;9:382. doi: 10.1186/1471-2164-9-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mascagni F, Giordani T, Ceccarelli M, Cavallini A, Natali L. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genomics. 2017;18:634. [DOI] [PMC free article] [PubMed]
- 40.Domingues DS, Cruz GM, Metcalfe CJ, et al. Analysis of plant LTR-retrotransposons at the fine-scale family level reveals individual molecular patterns. BMC Genomics. 2012;13:1. doi: 10.1186/1471-2164-13-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ke L, Yu H, Xu H, Xie Z, Deng X, Xu Q. Identification the Citrus bud mutants by transposon display technology. Acta Horticulturae Sinica. 2017;44:1207–16.
- 42.Han Y, Qin S, Wessler SR. Comparison of class 2 transposable elements at superfamily resolution reveals conserved and distinct features in cereal grass genomes. BMC Genomics. 2013;14:1. doi: 10.1186/1471-2164-14-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhi D, Raphael BJ, Price AL, Tang H, Pevzner PA. Identifying repeat domains in large genomes. Genome Biol. 2006;7:R7. doi: 10.1186/gb-2006-7-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jukes TH, Cantor CR. Evolution of protein molecules. Mammalian Protein Metabolism. 1969;3:132. [Google Scholar]
- 47.Shi T, Huang H, Barker MS. Ancient genome duplications during the evolution of kiwifruit (Actinidia) and related Ericales. Ann Botany. 2010;106:497–504. doi: 10.1093/aob/mcq129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han Y, Burnette JM, Wessler SR. TARGeT: a web-based pipeline for retrieving and characterizing gene and transposable element families from genomic sequences. Nucleic Acids Res. 2009;37:e78. doi: 10.1093/nar/gkp295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–W268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steinbiss S, Willhoeft U, Gremme G, Kurtz S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009;37:7002–7013. doi: 10.1093/nar/gkp759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The pipeline of MITE classification and annotation. (DOCX 151 kb)
Figure S2. The pipeline of LTR retrotransposons classification and annotation. (DOCX 475 kb)
Figure S3. Relative abundance of MITE families in Citrus species. (DOCX 1605 kb)
Figure S4. Manually confirmed DTM63 insertion sites. (A) Manually confirmed case of C. grandis. (B). Manually confirmed case of C. sinensis. (C) Manually confirmed case of C. clementina. (D) Manually confirmed case of C. ichngensis. Sequences in blue represent TSD and the “N” in red represent a copy full-length DTM63. The corresponding homologous regions were listed in Additional file 7: Table S3. (DOCX 584 kb)
Table S1. Information of conserved and non-conserved MITEs insertion between different genomic regions (DOCX 12 kb)
Table S2. Information of newly inserted MITEs in different genomic regions of three Citrus species. (DOCX 13 kb)
Table S3. Information of DTM63 insertion sites. (DOCX 14 kb)
Data Set S1. The seed file of MITEs. (FA 43 kb)
Data Set S2. The seed file of LTR retrotransposons. (FA 2229 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article [and its additional files].