

Abstract

Poly(ADP-ribose) polymerase-1 (PARP-1) is an enzyme involved in signaling and repair of DNA single strand breaks. PARP-1 employs NAD+ to modify substrate proteins via the attachment of poly(ADP-ribose) chains. PARP-1 is a well established target in oncology, as testified by the number of marketed drugs (e.g., Lynparza, Rubraca, Zejula, and Talzenna) used for the treatment of ovarian, breast, and prostate tumors. Efforts in investigating an uncharted region of the previously identified isoindolinone carboxamide series delivered (S)-13 (NMS-P515), a potent inhibitor of PARP-1 both in biochemical (Kd: 0.016 μM) and cellular (IC50: 0.027 μM) assays. Cocrystal structure allowed explaining NMS-P515 stereospecific inhibition of the target. After having ruled out potential loss of enantiopurity in vitro and in vivo, NMS-P515 was synthesized in an asymmetric fashion. NMS-P515 ADME profile and its antitumor activity in a mouse xenograft cancer model render the compound eligible for further optimization.
Keywords: PARP-1, inhibitor, isoindolinone, stereospecific, NMS-P515
The genetic instability of tumors arises from their deficiencies in fixing damaged DNA. These liabilities can be magnified by employing drugs that further hamper cancer DNA Damage Response (DDR) machinery.1,2 Archetype of this approach is the use of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. PARP-1, the main component of a 17-membered family of proteins named ADP-Ribosyl-Transferases Diphtheria Toxin-like (ARTDs),3,4 is a key player of the Base Excision Repair (BER) pathway involved in DNA single strand breaks (SSB) repair. PARP-1 employs NAD+ as the building block to assemble, via the release of nicotinamide, poly(ADP-ribose) chains onto several acceptor proteins (heteromodification) including PARP-1 itself (automodification).5 This post-translational event contributes in signaling the presence of DNA damage, resulting in the recruitment of proteins involved in DNA repair. Conceptually, the inhibition of PARP-1 disables BER pathway, leading to the accumulation of DNA double strand breaks (DSBs) whose repair by pathways such as the Homologous Recombination (HR) or nonhomologous end joining (NHEJ), is either more challenging or error-prone.6 The mechanism of action of such inhibitors was postulated to potentiate DNA damaging anticancer agents. The ensuing arrays of PARP-1 inhibitors, initially designed to be combined with conventional chemotherapy, finally found their way to the market (e.g., olaparib, 1; rucaparib, 2; niraparib, 3; talazoparib, 4, Figure 1) as monotherapy in those HR-deficient tumor (e.g., BRCA1/2-mutated) phenotypes that cannot withstand a DNA damage overload.7,8 To our knowledge, all the clinical and preclinical PARP-1 inhibitors under development are NAD+-competitive nicotinamide mimics, featuring a lactam ring or a primary amide conformationally locked through an intramolecular hydrogen bond with an adjacent heteroatom.9,10
Figure 1.

Marketed PARP-1 inhibitors (1–4) and isoindolinone carboxamide derivatives (5–7).
Interestingly, molecules with such an apparent architectural resemblance and similar on-target biochemical potency possess, however, striking differences in term of ARTDs selectivity profile11 and cellular and in vivo activity. A number of hypotheses (e.g., different off-target activity, trapping effects) have been invoked to account for the observed dissimilarities.12
One of the most valuable and innovative series of PARP-1 inhibitors in preclinical development is represented by the isoindolinone carboxamides chemical class. This series emerged from both an HTS campaign on our proprietary chemical collection (e.g., NMS-P118, 5, Figure 1)13 and a structure-based drug design approach performed in other laboratories (e.g., 6, Figure 1).14 Aiming at improving the binding efficiency as well as the drug-like properties of this class, we focused on the introduction of a methyl substituent15 in position 1 of the bicyclic core (see Figure 1 for numbering system). Planned compounds were considered eligible for further progression based on concurrent thresholds of biochemical activity (KD < 0.1 μM in fluorescence polarization (FP) displacement assay16 against PARP-1) and cellular inhibition of poly(ADP-ribose) synthesis (PAR assay, IC50 < 0.1 μM). Capitalizing on our acquired knowledge of the series,13 (±)-1-methyl-3-oxo-2-(piperidin-4-yl)-2,3-dihydro-1H-isoindole-4-carboxamide (7, PARP-1 KD: 0.04 μM; PAR assay IC50: 0.256 μM; Figure 1) was adopted as the starting point of our structure–activity relationship investigation. A rapid interrogation of the chemical space departing from the piperidine nitrogen atom was then performed. The planned derivatives, as exemplified by the preparation of compound (±)-13 (Scheme 1), were chemically assembled starting from 2-acetylfuran by using an intramolecular Diels–Alder as the key step.
Scheme 1. Synthesis of Compound (±)-13.

Reagents and conditions: (a) 4-amino-piperidine-1-carboxylic acid tert-butyl ester, 1 M TiCl4 in DCM, TEA, DCM, reflux, then NaCNBH3, MeOH, rt, 80%; (b) maleic anhydride, toluene, reflux, 6 h, 80%; (c) 37% HCl, reflux, 70%; (d) di-tert-butyl dicarbonate, K2CO3, pyridine, MeOH, rt, 78%; (e) HOBt·NH3, EDCI·HCl, DIPEA, DMF, rt, 75%; (f) 4 M HCl in dioxane, 50 °C; (g) cyclohexanone, NaCNBH3, NaOAc, DCM/MeOH, rt, 76%.
Noteworthy, the cycloaddition delivered a single exo-diastereomer17,18 (9) with the methyl substituent opposite to the ethereal bridge. Diversification by means of a reductive amination reaction was performed at the very end of the synthesis, by employing differently substituted aromatics, heteroaromatics, aliphatic aldehydes, and ketones. This exploration allowed us to select the cyclohexyl derivative as the most compelling PARP-1 inhibitor of the series ((±)-13, PARP-1 KD < 0.03 μM16). Furthermore, compound (±)-13 is selective among the ARTDs tested (PARP-2, -3, and tankyrase-1 (TNKS-1) KD > 10 μM) and potently abrogate PAR synthesis in cells (IC50: 0.050 μM) (Table 1).
Table 1. Biochemical and Cellular Data of Compounds (±)-13 and the Separated Enantiomers.
| compound |
||||
|---|---|---|---|---|
| biochemical assay type | enzyme | (±)-13 | NMS-P515 | (R)-13 |
| FP (KD, μM) | PARP-1 | <0.03a | <0.03a | 1.32 |
| PARP-2 | >10 | >10 | ||
| PARP-3 | >10 | >10 | ||
| TNKS-1 | >10 | >10 | ||
| SPR (Kd, μM) | PARP-1 | 0.016 | 1.76 | |
| compound |
||||
|---|---|---|---|---|
| cellular assay type | cell line | (±)-13 | NMS-P515 | (R)-13 |
| PAR (IC50, μM) | HeLa | 0.050 | 0.027 | 0.162 |
Assay sensitivity limit.
The introduction of a fluorine atom meta to the primary carboxamide, which usually reinforces the cellular potency of PARP inhibitors (e.g., 2, 4, 5), was also explored within this series (Scheme 2). Thus, by subjecting previously described methyl ester (14)13 to benzylic bromination, followed by a one-pot nucleophilic displacement-ring closure sequence, intermediate isoindolinone (15) was obtained. C1 methylation using NaHMDS as the base and subsequent cyanation afforded nitrile (17), whose functional group manipulation resulted in compound (±)-18.
Scheme 2. Synthesis of Compound (±)-18.

Reagents and conditions: (a) NBS, Bz2O2, methyl pivalate, reflux then 1-cyclohexyl-piperidine-4-ylamine, TEA, acetonitrile, reflux, 40%; (b) MeI, NaHMDS 1 M in THF, rt, 90 min; (c) CuCN, DMF, reflux, 90 min, 54%; (d) 37% HCl, 100 °C then HOBt·NH3, EDCI·HCl, DIPEA, DMF, rt, 38%.
Unfortunately, fluoro-isoindolinone (±)-18 (PARP-1 KD: 0.06 μM; PAR assay IC50: 0.64 μM) proved to be less attractive than its corresponding des-fluoro analogue.
Physicochemical and ADME profiling of (±)-13 demonstrated that the compound is moderately soluble (140 μM) at neutral pH, it is highly permeable with an excellent metabolic stability in vitro, and it has a high plasma protein binding (see Supporting Information, Table S1). Compound (±)-13 modestly inhibited two P450 cytochrome isoforms (CYP-2D6 IC50: 0.60 μM; CYP-2B6 IC50: 4.90 μM) out of eight tested. The ensuing evaluation of IV/OS pharmacokinetic parameters in mice further corroborated the drug-like properties of (±)-13, as it is slowly cleared from the body, with high exposure and complete oral bioavailability (Table 2).
Table 2. Preliminary Pharmacokinetic Parameters of Compounds (±)-13 and NMS-P515.
| PK dataa iv |
PK
dataa per os |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | doseb (mg/kg) | Cmax (μM) | AUC∞ (μM·h) | CL (mL/min/kg) | Vss (L/kg) | t1/2 (h) | dosec (mg/kg) | Cmax (μM) | AUC∞ (μM·h) | t1/2 (h) | Fd (%) |
| (±)-13 | 5 | 2.5 ± 0.3 | 6.3 ± 0.0 | 32.6 ± 0.8 | 3.6 ± 0.4 | 1.8 ± 0.1 | 10 | 2.5 ± 0.4 | 13.5 ± 1.72 | 3.1 ± 0.7 | 100 |
| NMS-P515 | 10 | 4.9 ± 0.6 | 8.2 ± 0.8 | 53.4 ± 8.0 | 6.8 ± 1.0 | 2.1 ± 0.4 | 10 | 1.4 ± 0.2 | 4.9 ± 0.1 | 2.6 ± 0.9 | 78.3 |
Harlan Nu/Nu Mice; n = 3 animals per study.
Dosed iv (intravenous administration): 50% PEG 400 in glucosate for (±)-13 and in situ hydrochloride salt; 1% Tween 80 in dextrose for NMS-P515.
Dosed per os (oral administration): 0.5% methocel.
Bioavailability.
Following the initial preclinical profiling of compound (±)-13, we next scrutinized the contribution of each separated enantiomer to the overall bioactivity picture of the racemate. Chiral HPLC on a preparative scale allowed to separate up to 2 g of (±)-13 with 93% recovery and ≥99.5:0.5 enantiomeric ratio for each enantiomer. X-ray cocrystal structures of (S)-13 and (R)-13 with chicken PARP-1 catalytic domain (both solved at 2.1 Å; Figure 2) permitted the unambiguous attribution of their absolute stereochemistry.
Figure 2.
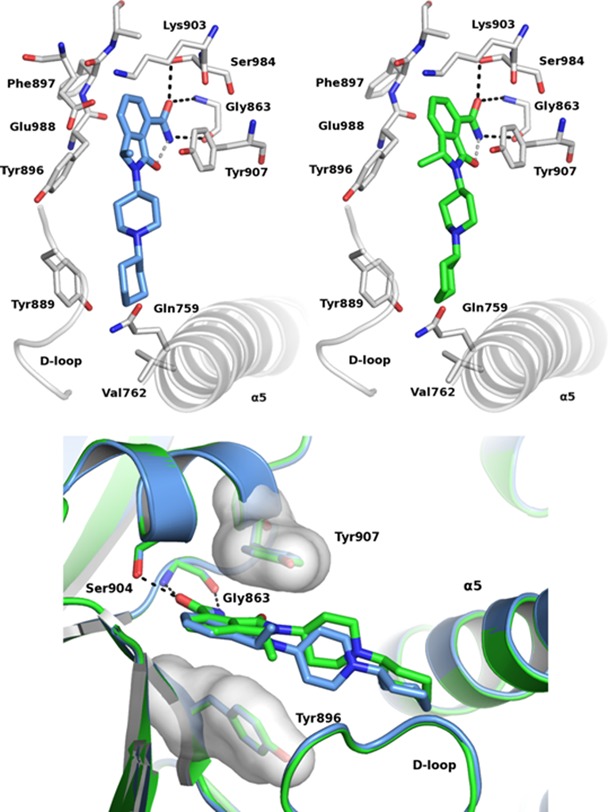
Cocrystal structure of (S)-13 (NMS-P515, blue, top left) and (R)-13 (green, top right) with chicken PARP-1 CD and their overlapping (bottom).
Thus, based on chiral HPLC retention time (see Supporting Information, Figure S1), the first eluting peak was demonstrated to be the (S)-13 enantiomer (followed by the (R)-13 peak). As soon as the enantiomers were tested, it became clear that (S)-13 (NMS-P515) solely contributes to the overall biochemical potency and the cellular activity (PARP-1 KD < 0.03 μM;16 PAR assay IC50: 0.027 μM) showed by (±)-13 (Table 1). To further substantiate FP displacement data, both (S)-13 and (R)-13 were also tested against PARP-1 catalytic domain using surface plasmon resonance (SPR) binding assay (Table 1).16 As this technique does not carry any sensitivity limit, evidence of the different behavior between enantiomers emerged even more strikingly ((S)-13, PARP-1 Kd: 0.016 μM; (R)-13, PARP-1 Kd: 1.76 μM). Same differences were also appreciated at the cellular level (Table 1 and Figure 3A). Similarly to (±)-13, also NMS-P515 was selective against PARP-2, -3, and TNKS-1 with a recorded KD > 10 μM on each enzyme (Table 1). The identification of NMS-P515 as a stereoselective PARP-1 inhibitor comes along with the effort in rationalizing its superior activity compared to (R)-13. Having successfully gathered PARP-1 cocrystal data for both enantiomers, a structure-based hypothesis was formulated. As expected,13,14 the isoindolinone core of both NMS-P515 and (R)-13 is anchored to the nicotinamide pocket of PARP-1 by the classical hydrogen-bond triad with Gly863 and Ser904 and by a π–π interaction with the phenyl ring of Tyr907. The 1-cyclohexyl-4-piperidinyl substituent, which departs from the bicyclic scaffold, extends outside the nicotinamide site and fills a cleft defined by the D-loop and the helix 5 of the helical bundle domain. However, NMS-P515 and (R)-13 overall orientations are slightly different (Figure 2). NMS-P515 C1 methyl nicely adapts to the binding site. Differently, the C1 methyl in (R)-13 points toward Tyr896, forcing the whole molecule to rotate about 10° outward from the pocket to avoid any steric clash. The net result is a poorer binding site fit for (R)-13 compared to NMS-P515.
Figure 3.
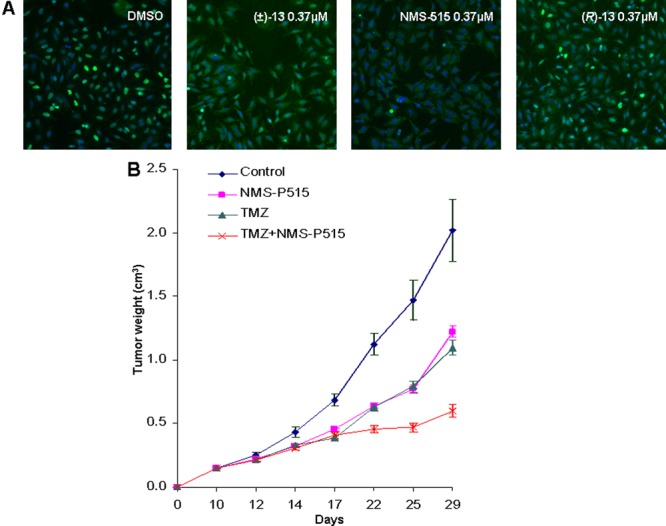
(A) ArrayScan Images of HeLa cells treated with dimethyl sulfoxide (DMSO, control), and same concentration (0.37 μM) of (±)-13 (NMS-P515) and (R)-13 followed by hydrogen peroxide-induced PAR formation (blue, Hoechst (DNA); green, antibody anti-PAR). (B) Tumor growth inhibition of Capan-1 pancreatic cancer xenograft model (control, blue line; NMS-P515, purple line; Temozolomide, green line; NMS-P515 + Temozolomide, red line).
As the preclinical progress of enantiopure synthetic materials brings about a higher cost of goods compared to the corresponding racemate, a number of experiments aimed at judging the maintenance of the stereochemical integrity of NMS-P515 in different conditions was planned. Thus, exposure of NMS-P515 to a wide pH range (1.5, 7.4, and 10) at room temperature for up to 24 h did not result in any erosion of its enantiopurity. Similarly, NMS-P515 stability in human plasma was evaluated after incubation at 37 °C for up to 24 h, again with no apparent detection of (R)-13. Finally, to investigate enantiomer integrity in vivo, mice were treated with a single dose (100 mg/kg, OS) of NMS-P515, and blood samples were withdrawn after 45 min (corresponding to nearly the Tmax value of (±)-13 as measured in the PK experiments). Also in this case, chiral HPLC analysis showed complete preservation of NMS-P515 enantiopurity.
Detailed physicochemical and ADME profiling of NMS-P515 are reported in Supporting Information Table S1. NMS-P515 is more soluble at neutral pH (>225 μM) than its corresponding racemate, its plasma protein binding is slightly lower, and it does not inhibit any of the P450 cytochrome isoforms against which it was tested (CYPs IC50 > 31 μM). The NMS-P515 pharmacokinetic profile mostly overlaps with the one of (±)-13, however with some noteworthy differences (Table 2). Thus, NMS-P515 clearance and volume of distribution were nearly twice the one of the racemate. On the contrary, the exposure of NMS-P515 resulted lower than that of (±)-13, with an oral bioavailability of 78%. NMS-P515’s further progression as a lead compound was, however, challenged by the lack of an efficient and scalable asymmetric synthesis, which is mandatory to overcome the limitations of a costly and time-consuming chiral HPLC separation. After a number of unfruitful attempts,19 the engagement of the previously described20 (S)-1-furan-2-yl-ethylamine in a reductive amination reaction with 1-cyclohexyl-piperidin-4-one was key for the proper installation of the desired stereocenter (Scheme 3). The resulting (S)-19 proved to be optically pure (>99.5/0.5 enantiomeric ratio by chiral HPLC, using ad hoc prepared (±)-19 as standard). Diels–Alder reaction, followed by functional group manipulation, resulted in the asymmetric synthesis of (S)-13 (NMS-P515), with a 21% overall yield from (S)-1-furan-2-yl-ethylamine and a 95.5/4.5 enantiomeric ratio.
Scheme 3. Asymmetric Synthesis of NMS-P515.

Reagents and conditions: (a) 1-cyclohexyl-piperidin-4-one, 1 M TiCl4, DCM, then NaCNBH3, MeOH, 52%; (b) maleic anhydride, toluene, 110 °C; (c) 37% HCl, 100 °C; (d) HOBt·NH3, EDCI·HCl, Et3N, THF, 40%.
The partial erosion of the stereochemical integrity during the synthesis of (S)-13 was ascribed to the harsh reaction conditions involved in the dehydration–aromatization step. The batch of NMS-P515 produced through asymmetric synthesis recapitulates the biochemical and cellular activity of (S)-13 arising from chiral HPLC separation (data not shown). NMS-P515 was then subjected to in vivo efficacy studies, both as monotherapy and in combination, on subcutaneously implanted Capan-1 pancreatic (BRCA2-mutated) mouse xenografts. Thus, administration of NMS-P515 for 12 days (once a day, oral dose of 80 mg/kg21) as a methocel suspension clearly reduced the tumor growth (maximal tumor growth inhibition observed, 48%; maximum body weight loss, 6%; Figure 3B). An even more pronounced effect (maximal tumor growth inhibition, 79%; maximum body weight loss, 17%; Figure 3B), was observed when animals were treated with NMS-P515 (12 days, once a day oral dose of 80 mg/kg) in combination with Temozolomide, a DNA damaging drug (5 days starting from day 3, once a day IV dose of 62.5 mg/kg). For comparison, the same dose of Temozolomide, administered as single agent, resulted in maximal tumor growth inhibition of 46%, with a maximum body weight loss of 8% (Figure 3B).
In summary, mindful exploration of the C1-methylated isoindolinone, a privileged scaffold within the realm of preclinical PARP-1 inhibitors, delivered NMS-P515. This compound potently and selectively inhibits the target enzyme, with a clear mechanism of action in cells. After a thorough assessment of the preservation of its stereochemical integrity, both in vitro and in vivo, NMS-P515 was deeply profiled in terms of physicochemical and pharmacokinetic properties. The ensuing (scalable) asymmetric synthesis of NMS-P515 overcomes the limitations associated with preparative chiral HPLC separation. NMS-P515’s lead-like performance was reinforced by preliminary antitumor efficacy studies in vivo that render this PARP-1 stereospecific inhibitor eligible for further optimization.
Acknowledgments
We gratefully acknowledge the technical skill and support of Rita Perego (Protein Production), Emiliana Corti and Rosita Lupi (Assay Development), Francesco Sola and Ivan N. Fraietta (Cellular Biology), Barbara Forte and Mikhail Y. Krasavin (Medicinal Chemistry), and Fabio Zuccotto (Molecular Modeling).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00569.
Synthetic procedures and analytical characterization for all new compounds described. Experimental details for analytical and preparative enantiomers separation, evaluation of NMS-P515 stereochemical erosion in vitro and in vivo, X-ray crystallography, ADME in vitro data, and biochemical and cellular assays. Detailed description of preliminary pharmacokinetic and in vivo efficacy studies (PDF)
Accession Codes
Reported structures have been deposited to the Protein Data Bank (PDB) with accession codes: 618M and 618T.
Author Present Address
† (V.D.) Bracco Imaging S.p.A., via Caduti di Marcinelle, 13-20134 Milano, Italy.
Author Present Address
‡ (M.G.) Helsinn Healthcare SA, via Pian Scairolo 9-6912 Pazzallo-Lugano, Switzerland.
Author Present Address
§ (F.R.-S.) Quantitative Pharmacology and Drug Disposition (QPD), NBE Bioanalytics Unit MERCK Bioindustry Park I-10010 Colleretto Giacosa (TO), Italy.
Author Present Address
∥ (A.S.) Fresenius Kabi Business Unit API, piazza Maestri del Lavoro, 7-20063 Cernusco sul Naviglio, Milano, Italy.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
All procedures adopted for housing and handling of animals were in strict compliance with Italian and European guidelines for Laboratory Animal Welfare and the protocols were approved by IRB.
The authors declare no competing financial interest.
Supplementary Material
References
- Hengel S. R.; Spies M. A.; Spies M. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chem. Biol. 2017, 24, 1101–1119. 10.1016/j.chembiol.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A.; Yan Y.; Gerson S. L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018, 66–67, 24–29. 10.1016/j.dnarep.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O.; Hassa P. O.; Lüscher B.; Schüler H.; Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Di Girolamo M.; Fabrizio G. The ADP-Ribosyl-Transferases Diphtheria Toxin-Like (ARTDs) Family: An Overview. Challenges 2018, 9, 24. 10.3390/challe9010024. [DOI] [Google Scholar]
- Bürkle A.; Virág L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 2013, 34, 1046–1065. 10.1016/j.mam.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Lord C. J.; Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth A.; Lord C. J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors?. Nat. Rev. Clin. Oncol. 2018, 15, 564–576. 10.1038/s41571-018-0055-6. [DOI] [PubMed] [Google Scholar]
- Ringley J. T.; Moore D. C.; Patel J.; Rose M. S. Poly (ADP-ribose) polymerase inhibitors in the management of ovarian cancer: A drug class review. P&T 2018, 43, 549–556. [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-Q.; Wang P.-Y.; Wang Y.-T.; Yang G.-F.; Zhang A.; Miao Z.-H. An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. J. Med. Chem. 2016, 59, 9575–9598. 10.1021/acs.jmedchem.6b00055. [DOI] [PubMed] [Google Scholar]
- Kirby I. T.; Cohen M. S. Small-molecule inhibitors of PARPs: From tools for investigating ADP-Ribosylation to therapeutics. Curr. Top. Microbiol. Immunol. 2018, 420, 211. 10.1007/82_2018_137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A.-G.; Ekblad T.; Karlberg T.; Löw M.; Pinto A. F.; Trésaugues L.; Moche M.; Cohen M. S.; Schüler H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; O’Connor M. J.; de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanism of action. Sci. Transl. Med. 2016, 8, 362ps17. 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- Papeo G.; Posteri H.; Borghi D.; Busel A. A.; Caprera F.; Casale E.; Ciomei M.; Cirla A.; Corti E.; D’Anello M.; Fasolini M.; Forte B.; Galvani A.; Isacchi A.; Khvat A.; Krasavin M. Y.; Lupi R.; Orsini P.; Perego R.; Pesenti E.; Pezzetta D.; Rainoldi S.; Riccardi-Sirtori F.; Scolaro A.; Sola F.; Zuccotto F.; Felder E. R.; Donati D.; Montagnoli A. Discovery of 2-[1-(4,4-difluorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carboxamide (NMS-P118): a Potent, Orally Available and Highly Selective PARP-1 Inhibitor for Cancer Therapy. J. Med. Chem. 2015, 58, 6875–6898. 10.1021/acs.jmedchem.5b00680. [DOI] [PubMed] [Google Scholar]
- Gandhi V. B.; Luo Y.; Liu X.; Shi Y.; Klinghofer V.; Johnson E. F.; Park C.; Giranda V. L.; Penning T. D.; Zhu G.-D. Discovery and SAR of substituted 3-oxoisoindoline-4-carboxamides as potent inhibitors of poly(ADP-ribose) polymerase (PARP) for the treatment of cancer. Bioorg. Med. Chem. Lett. 2010, 20, 1023–1026. 10.1016/j.bmcl.2009.12.042. [DOI] [PubMed] [Google Scholar]
- Sun S.; Fu J. Methyl-Containing Pharmaceuticals: Methylation in Drug Design. Bioorg. Med. Chem. Lett. 2018, 28, 3283–3289. 10.1016/j.bmcl.2018.09.016. [DOI] [PubMed] [Google Scholar]
- Papeo G.; Avanzi N.; Bettoni S.; Leone A.; Paolucci M.; Perego R.; Quartieri F.; Riccardi-Sirtori F.; Thieffine S.; Montagnoli A.; Lupi R. Insights into PARP inhibitors selectivity using fluorescence polarization and surface plasmon resonance binding assays. J. Biomol. Screening 2014, 19, 1212–1219. 10.1177/1087057114538319. [DOI] [PubMed] [Google Scholar]
- Zubkov F. I.; Boltukhina E. V.; Turchin K. F.; Borisov R. S.; Varlamov A. V. New synthetic approach to substituted isoindolo[2,1-a]quinoline carboxylic acids via intramolecular Diels-Alder reaction of 4-(N-furyl-2)-4-arylaminobutenes-1 with maleic anhydride. Tetrahedron 2005, 61, 4099–4113. 10.1016/j.tet.2005.02.017. [DOI] [Google Scholar]
- Boltukhina E. V.; Zubkov F. I.; Nikitina E. V.; Varlamov A. V. Novel approach to isoindolo[2,1-a]quinolines: synthesis of 1- and 3-halo-substituted 11-oxo-5,6,6a,11-tetrahydroisoindolo[2,1-a]quinoline-10-carboxylic acids. Synthesis 2005, 1859–1875. 10.1055/s-2005-869948. [DOI] [Google Scholar]
- Di Mola A.; Palombi L.; Massa A.. An overview of asymmetric synthesis of 3-substituted isoindolinones. In Targets in Heterocyclic Systems, 18th ed.; Attanasi O. A., Noto R., Spinelli D., Eds.; Italian Society of Chemistry: Rome, Italy, 2014; pp 113–140. [Google Scholar]
- Alvaro G.; Martelli G.; Savoia D.; Zoffoli A. Synthesis of (S)- and (R)-1-(2-furyl)alkylamines and (S)- and (R)-α-amino acids through the addition of organometallic reagents to imines derived from (S)-valinol. Synthesis 1998, 1773–1777. 10.1055/s-1998-2227. [DOI] [Google Scholar]
- Such an aggressive BRCA-2 mutated pancreatic tumor model requires high doses of PARP-1 inhibitors (approaching the maximum tolerated one) to show efficacy. For (±)-13, maximum tolerated dose in this model proved to be 100 mg/kg (data not shown). As enantiomer NMS-P515 is the actual responsible for the target inhibition observed with (±)-13, a theoretical efficacious dose of 50 mg/kg can thus be envisioned. However, being that NMS-P515’s oral bioavailability and exposure level are lower than its racemate (see Table 2), an intermediate dose (80 mg/kg) was selected for efficacy studies.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.