

Abstract
In recent years, a few successful attempts were made to repurpose the clinically approved antiarthritic gold drug, Auranofin (AF), as an anticancer agent. The present study shows that the iodido(triethylphosphine)gold(I) complex, (Et3PAuI hereafter)—an AF analogue where the thiosugar ligand is simply replaced by one iodide ligand—manifests a solution chemistry resembling that of AF and exerts similar cytotoxic and proapoptotic effects on A2780 human ovarian cancer cells in vitro. However, when evaluated in a preclinical orthotopic model of ovarian cancer, Et3PAuI produces a far superior anticancer action than AF inducing a nearly complete tumor remission. The highly promising in vivo performances here documented for Et3PAuI warrant its further evaluation as a drug candidate for ovarian cancer treatment.
Keywords: Auranofin, gold compounds, ovarian cancer, metal-based drugs, in vivo orthotopic model
Auranofin, a linear gold(I) complex bearing triethylphosphine and thioglucose tetraacetate as ligands, is an orally administered drug, in clinical use since 1985 for the treatment of rheumatoid arthritis. AF is generally reputed as a reasonably safe drug for human use, even for long chronic treatments.1,2 Beyond its clinically established antiarthritic properties, in the frame of the so-called drug repurposing strategies, AF has been extensively tested in recent years as a potential antiparasitic, antimicrobial, and anticancer agent with encouraging results.3−5 Of considerable interest are the antiproliferative properties of this compound that have been known since the early 1980s5−7 and were later assessed in the NCI 60 cancer cell line panel and in a few in vivo models.8−10 On the ground of its anticancer properties, AF in the course of the last years has entered three distinct clinical trials, two of them (ovarian and lung cancer) still ongoing and one (leukemia) closed.11−13 Results of these trials have not been disclosed yet. At the same time, considerable evidence has been gathered that a few specific biomolecular targets such as the enzyme thioredoxin reductase, the proteasome system, the NF-κB pathway and other transcription factors, are crucially involved in mediating the cellular effects of AF as well of other gold (I/III) compounds.14−18
It has been hypothesized, and then demonstrated, that the biological and pharmacological properties of AF might be kept and hopefully improved/modulated by replacing the thiosugar ligand with other ligands such as various halides. In principle, replacement of the thiosugar ligand with halide ligands may lead to an increased lipophilicity and an enhanced drug bioavailability and biodistribution. However, this modification should not affect substantially the drug’s pharmacological profile, as the invariant [Et3PAu+] moiety is assumed to be the “true pharmacophore” whereas the thiosugar ligand is believed to act mainly as a carrier ligand.18 Accordingly, the actions of iodido(triethylphosphine)gold(I) complex, Et3PAuI, and AF, have been comparatively analyzed toward ovarian cancer models both in vitro and in vivo. Et3PAuI is obtained by replacement of the thiosugar ligand with an iodide ligand according to the reported synthetic procedures18 (see Supporting Information for detailed synthesis and characterization).
The chemical structures of AF and Et3PAuI are shown in Figure 1.
Figure 1.

Chemical structures of Auranofin and iodido(triethylphosphine)gold(I).
Replacement of the thiosugar ligand with iodide results into a greatly increased lipophilic character (a logP value of 4.6 has been measured by ICP-OES in the previous work18) with respect to both Et3PAuCl and AF (this latter compound has been previously described and used experimentally for rheumatoid arthritis treatment),19 for which logP values of 1.7 and 1.6 were measured, respectively.18 In spite of that, Et3PAuI is still appreciably soluble in aqueous solutions, where it manifests a very high stability as documented by a previous systematic NMR analysis, carried out in different environments including biological media.18,20 As reported in the Supporting Information (see Figure S4), new 31P NMR results clearly demonstrate a high stability of Et3PAuI, with no changes in the structure of the molecule, even in the presence of a large excess of sodium chloride (up to 100 mM). The documented stability in physiological-like conditions makes the compound an ideal candidate for biological testing.
The antiproliferative properties of Et3PAuI compared to AF have been investigated in vitro toward A2780 human ovarian cancer cells. As displayed in Table 1, both compounds produce potent cytotoxic effects with IC50 values falling in the high nanomolar range. Et3PAuI is slightly more cytotoxic than AF and both result significantly more effective than cisplatin, the well-known anticancer metal compound. These results are consistent with those previously obtained in colon cancer cell lines for the same gold compounds and recently published literature18 further supporting the concept that the presence of the thiosugar ligand is not an essential requirement for the cytotoxic action and that substantial differences in the cellular uptake, despite the difference in the logP value, between Et3PAuI and AF are unlikely to occur. Remarkably, in agreement with a previous observation,18 no appreciable cytotoxic effects were detected for Et3PAuI in normal cell lines, i.e., human fibroblasts (HDF cell line) and human embryonic kidney cells (HEK293) over the concentration range 0–5000 nM.
Table 1. Inhibitory Effects of AF, Et3PAuI, and Cisplatin on A2780 Cells Following 72 h Drug Exposure As Determined by the Sulforhodamine B Assaya.
| IC50 (μM) ± SD |
|||
|---|---|---|---|
| cell lines | Et3PAuI | Auranofin | cisplatin |
| A2780 | 0.394 ± 0.174 | 0.754 ± 0.139 | 1.53 ± 0.66 |
| n | 4 | 4 | 5 |
| pb | 0.0138 | ||
Four independent experiments were performed. SD, standard deviation; n, number of independent experiments.
t test.
Then, we examined whether Et3PAuI and AF are able to induce apoptosis in A2780 cells; accordingly, we performed flow cytometry analysis of annexin V/propidium iodide-stained A2780 cells, treated with these two gold compounds, at the respective IC50 values, for 72 h. As shown in Figure S1, the percentage of apoptotic cells in AF- and Et3PAuI-treated samples is significantly increased (control vs AF, p = 0.0121; control vs Et3PAuI, p = 0.0107). In order to evaluate and quantitate apoptosis via caspase activity, we checked the activity of caspase 3 by FAM-FLICA Caspase-3 Assay Kit. The induction of apoptosis observed for AF and its iodide analogue may be related with the strong thioredoxin reductase inhibition potency. Indeed, the IC50 values for the inhibition of the enzyme are generally in nice agreement with cytotoxicity values.18
The results collected evidenced that both gold compounds induce apoptosis through a caspase 3 activation, with AF being slightly more effective than Et3PAuI (control vs AF, p = 0.0037; control vs Et3PAuI, p = 0.04, see Supporting Information).
Et3PAuI was subsequently evaluated in vivo in comparison to AF in two different ovarian cancer models, i.e., a xenograft subcutaneous and an orthotopic model. Preliminarily, drug tolerability studies were carried out to assess the in vivo safety of Et3PAuI. The single-dose tolerability was evaluated in athymic nude mice over a 3-week observation period. Et3PAuI was administered by intraperitoneal (ip) injection using two different doses (20 and 40 mg/kg). Only a single administration was performed, followed by a 3-week observation period; animals were controlled daily, and no signs of sufferings such as weight loss, mobility reduction and appetite reduction were registered. In addition, a pilot study for gold biodistribution has been carried out and a generalized higher accumulation for Et3PAuI, both in organs and in the peripheral blood, was highlighted (see Supporting Information). Based on the above observations, we can hypothesize that the detected differences in the in vivo activity of the two compounds could be due to their different lipophilic character leading to an enhanced bioavailability and more favorable pharmacokinetics for Et3PAuI.
The xenograft model consisted of 1 × 106 A2780 cells implanted subcutaneously (sc) in both flanks of the animals. The growth of the tumor masses was measured daily with caliper and the treatment started when the volume of the masses reached 60 mm3. The treatment was performed by ip injections (three times a week for 2 weeks of treatment) of AF and Et3PAuI, both 15 mg/kg. The growth of the tumor masses was monitored over 21 days and time course is shown in Figure S3A,B respectively. At the end of the experiments, mice were sacrificed and the explanted masses measured; the volume (cm3) was calculated using the ellipsoid equation.21 Remarkably, treatment with both Et3PAuI and AF was accompanied by a strong decrease of the tumor volume determined at the sacrifice (Figure 2, control: 2.96 ± 0.06 vs Et3PAuI: 0.56 ± 0.20, p < 0.01; control: 2.96 ± 0.06 vs AF: 0,79 ± 0,27, p < 0.01). The above experimental protocol did not alter the behavior, vitality, or weight of treated mice.
Figure 2.

In vivo anticancer activity evaluation of gold compounds in a xenograft subcutaneous (sc) model of ovarian cancer. Mean volume of tumor masses obtained at the sacrifice, after 3 weeks from injection of A2780 cells. Data are reported as the mean ± SD (*p < 0.05, **p < 0.01). Further details are reported in the Supporting Information.
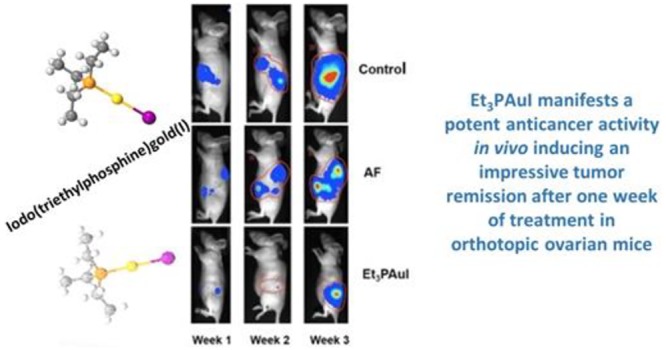
Subsequently, the in vivo anticancer activity of Et3PAuI versus AF was assessed in an orthotopic model of ovarian cancer, a model that better represents the cancer behavior and response to therapy. Thirty female athymic nude mice were injected in the ovarian bursa (5 × 105 cells each mouse) with A2780 cells transfected with the firefly luciferase gene (A2780-luc). Autopsy confirmed that ovarian cancer occurred in all animals and representative images of the procedure and of tumor masses are reported in Figure 3A. The animals were divided in three groups of treatment (10 mice each): controls, AF treated and Et3PAuI treated. The administration schedule was the following: mice received compounds (15 mg/kg) ip three times a week for 2 weeks. The rationale for such a treatment schedule relied on the schedule reported by Mirabelli et al.5 and on the above-reported biodistribution data (Supporting Information) showing that administration of 15 mg/kg gives rise to a peak peripheral blood concentration of 823.19 and 944.17 ng/mL, for AF and Et3PAuI, respectively, after 24 h. Mice were monitored daily, and the luciferase activity signal was quantified weekly (representative panels are reported in Figure 3B).
Figure 3.
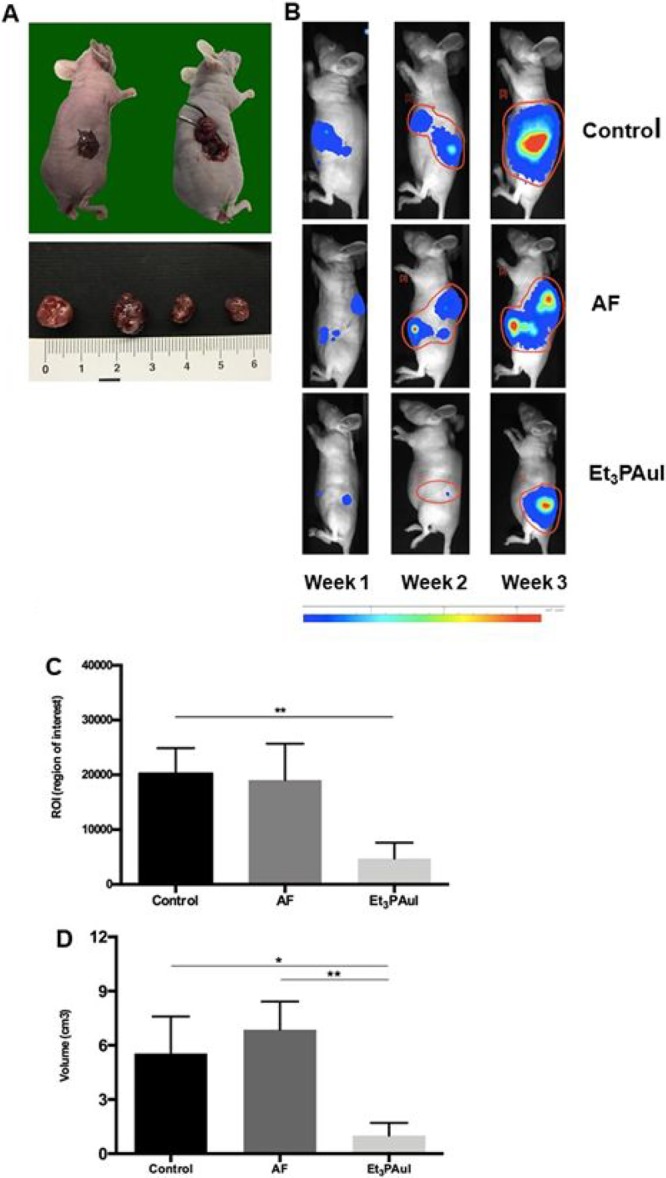
(A) Orthotopic model establishment: injection of 1 × 106 A2780-luc cells in the ovary bursa of nude mice. Representative macroscopic images of ovarian tumor masses obtained are showed in the bottom panels. (B) Representative pseudocolor BLI images tracking A2780-luc cells emission in mice, 7, 14, and 21 days after A2780-luc injection. Color bar represents light intensity levels reported as counts per minute (cpm). (C) Levels of ROI after 1 week of treatment; data are reported as mean of each group (n = 10 animals/group) ± SD (**p < 0.01). (D) Volume of tumor masses (mean ± SD) at the sacrifice after 3 weeks from injection of A2780-luc cells (control group, n = 8; AF, n = 5; Et3PAuI, n = 5). *p < 0.05, **p < 0.01. Further details are reported in the Supporting Information.
Both gold compounds effectively reduced the tumor burden (Figure 3B); notably, Et3PAuI is already very effective in antitumor activity after few doses (i.e., 1 week of treatment) leading to an almost complete tumor remission, as evidenced by the drastic reduction of the bioluminescent signal (ROI levels) compared to the control group (control: 20260 ± 4631 vs Et3PAuI: 4729 ± 2881; p < 0,01) (see graph in Figure 3C).
The tumor masses of the sacrificed animals after 3 weeks of treatment were collected and measured with caliper; the volume (cm3) was calculated using the ellipsoid equation even for this model.21 Most important, the tumor masses of the animals treated with Et3PAuI showed a very strong reduction of the volume compared both to the control group (control vs Et3PAuIp < 0.05) and to the AF group (AF: 6.86 ± 1.57 vs Et3PAuI: 1.00 ± 0.70, p < 0.01) (Figure 3D). This latter evidence points out that Et3PAuI is a more effective anticancer drug compared with AF in our animal model.
In conclusion, we have shown here that a specific structural modification of the repurposed gold(I) drug AF, now under consideration in clinical trials for cancer treatment, i.e., the substitution of the thiosugar ligand with iodide, affords a distinct gold(I) complex Et3PAuI, whose anticancer profile in vitro roughly resembles that of AF. This confirms that, even in the present ovarian cancer model, the thiosugar ligand is not mechanistically indispensable and that the [Et3PAu]+ moiety is likely to be the true pharmacophore. Very interestingly, in our in vivo models, Et3PAuI, has turned out to be far more effective than AF in contrasting tumor growth. Particularly impressive are the results and the differences between AF and Et3PAuI observed in the orthotopic model of ovarian cancer. Et3PAuI is found to induce a very large tumor regression after only 1 week of treatment, as pointed out by the decrease of the bioluminescent signal. A conspicuous shrinkage of the tumor is still evident after 3 weeks. The enhanced anticancer activity observed in this orthotopic model might well arise from the far greater lipophilicity of Et3PAuI, which might lead to a more favorable pharmacokinetics and biodistribution. Notably, the improved in vivo anticancer activity of Et3PAuI over AF does not bring about any increased systemic toxicity, making Et3PAuI a promising new drug candidate. In view of its far superior efficacy in animal models, Et3PAuI merits a more extensive preclinical testing as it might be a valuable alternative to AF in cancer clinical trials.
Acknowledgments
This study was supported by contributions of Associazione Italiana per la Ricerca sul Cancro, Milan (AIRC-IG16049) and Istituto Toscano Tumori (Messori- ITT-2015) to L.Me. T.M. thanks AIRC-FIRC (Fondazione Italiana per la Ricerca sul Cancro, Milan) 3-years Fellowship for Italy (Project Code: 18044) and University of Pisa (PRA_2017_25). CIRCMSB and ente CRF are gratefully acknowledged. Animal experiments have been approved and authorized in accordance with the Italian law, reference code: 113/2016-PR 03/2016.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00007.
Synthesis and purity of Et3PAuI, logP determinations, apoptosis plots; cell death and caspase activity; cell growth inhibition studies (sulforhodamine B assay); tolerability studies; biodistribution studies; in vivo anticancer activity evaluation xenograft subcutaneous models plots; NMR stability (PDF)
Author Contributions
∇ These authors contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Bombardier C.; Ware J.; Russell I. J.; Larson M.; Chalmers A.; Read J. L. Auranofin therapy and quality of life in patients with rheumatoid arthritis. Results of a multicenter trial. Am. J. Med. 1986, 81, 565–578. [DOI] [PubMed] [Google Scholar]
- Shaw C. F. Gold-Based Therapeutic Agents. Chem. Rev. 1999, 99, 2589–2600. 10.1021/cr980431o. [DOI] [PubMed] [Google Scholar]
- Cassetta M. I.; Marzo T.; Fallani S.; Novelli A.; Messori L. Drug repositioning: auranofin as a prospective antimicrobial agent for the treatment of severe staphylococcal infections. BioMetals 2014, 27, 787–791. 10.1007/s10534-014-9743-6. [DOI] [PubMed] [Google Scholar]
- Capparelli E. V.; Bricker-Ford R.; Rogers M. J.; McKerrow J. H.; Reed S. L. Phase I Clinical Trial Results of Auranofin, a Novel Antiparasitic Agent. Antimicrob. Agents Chemother. 2017, 61, e01947. 10.1128/AAC.01947-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabelli C. K.; Johnson R. K.; Sung C. M.; Faucette L.; Muirhead K.; Crooke S. T. Evaluation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin, a coordinated gold compound, in murine tumor models. Cancer Res. 1985, 45, 32–39. [PubMed] [Google Scholar]
- Simon T. M.; Kunishima D. H.; Vibert G. J.; Lorber A. Cellular antiproliferative action exerted by auranofin. J. Rheumatol. Suppl. 1979, 5, 91–97. [PubMed] [Google Scholar]
- Mirabell C. K.; Johnson R. K.; Hill D. T.; Faucette L. F.; Girard G. R.; Kuo G. Y.; Sung C. M.; Crooke S. T. Correlation of the in vitro cytotoxic and in vivo antitumor activities of gold(I) coordination complexes. J. Med. Chem. 1986, 29, 218–223. 10.1021/jm00152a009. [DOI] [PubMed] [Google Scholar]
- Hou G.-X.; Pan-Pan L.; Shengyi Z.; Mengqi Y.; Jianwei L.; Jing Y.; Yumin H.; Wen-Qi J.; Shijun W.; Peng H. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. 2018, 9, 89. 10.1038/s41419-017-0159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios Perez M. V.; Roife D.; Dai B. B.; Kang Y.; Li X.; Pratt M.; Fleming J. B. Auranofin to prevent progression of pancreatic ductal adenocarcinoma. J. Clin. Oncol. 2016, 34, 236. 10.1200/jco.2016.34.4_suppl.236. [DOI] [Google Scholar]
- Roh J. L.; Jang H.; Kim E. H.; Shin D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer, Antioxid. Antioxid. Redox Signaling 2017, 27, 106–114. 10.1089/ars.2016.6841. [DOI] [PubMed] [Google Scholar]
- Auranofin in Treating Patients With Recurrent Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. https://clinicaltrials.gov/ct2/show/NCT01747798.
- Sirolimus and Auranofin in Treating Patients With Advanced or Recurrent Non-Small Cell Lung Cancer or Small Cell Lung Cancer. https://clinicaltrials.gov/ct2/show/NCT01737502.
- Phase I and II Study of Auranofin in Chronic Lymphocytic Leukemia (CLL). https://clinicaltrials.gov/ct2/show/NCT01419691.
- Scalcon V.; Bindoli A.; Rigobello M. P. Significance of the mitochondrial thioredoxin reductase in cancer cells: An update on role, targets and inhibitors. Free Radical Biol. Med. 2018, 127, 62–79. 10.1016/j.freeradbiomed.2018.03.043. [DOI] [PubMed] [Google Scholar]
- Magherini F.; Fiaschi T.; Valocchia E.; Becatti M.; Pratesi A.; Marzo T.; Massai L.; Gabbiani C.; Landini I.; Nobili S.; Mini E.; Messori L.; Modesti A.; Gamberi T. Antiproliferative effects of two gold(I)-N-heterocyclic carbene complexes in A2780 human ovarian cancer cells: a comparative proteomic study. Oncotarget 2018, 9, 28042–28068. 10.18632/oncotarget.25556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratesi A.; Gabbiani C.; Michelucci E.; Ginanneschi M.; Papini A. M.; Rubbiani R.; Ott I.; Messori L. Insights on the mechanism of thioredoxin reductase inhibition by Gold N-heterocyclic carbene compounds using the synthetic linear Selenocysteine containing C-terminal peptide hTrxR(488–499): An ESI-MS investigation. J. Inorg. Biochem. 2014, 136, 161–169. 10.1016/j.jinorgbio.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Micale N.; Schirmeister T.; Ettari R.; Cinellu M. A.; Maiore L.; Serratrice M.; Gabbiani C.; Massai L.; Messori L. Selected cytotoxic gold compounds cause significant inhibition of 20S proteasome catalytic activities. J. Inorg. Biochem. 2014, 11, 79–82. 10.1016/j.jinorgbio.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Marzo T.; Cirri D.; Gabbiani C.; Gamberi T.; Magherini F.; Pratesi A.; Guerri A.; Biver T.; Binacchi F.; Stefanini M.; Arcangeli A.; Messori L. Auranofin, Et3PAuCl, and Et3PAuI Are Highly Cytotoxic on Colorectal Cancer Cells: A Chemical and Biological Study. ACS Med. Chem. Lett. 2017, 8, 997–1001. 10.1021/acsmedchemlett.7b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton B. M.; Mc Gusty E.; Walz D. T.; Di Martino M. J. Oral gold. Antiarthritic properties of alkylphosphinegold coordination complexes. J. Med. Chem. 1972, 15, 1095–1098. 10.1021/jm00281a001. [DOI] [PubMed] [Google Scholar]
- Marzo T.; Cirri D.; Pollini S.; Prato M.; Fallani S.; Cassetta M. I.; Novelli A.; Rossolini G. M.; Messori L. Auranofin and its Analogues Show Potent Antimicrobial Activity Covering Multiresistant Pathogens: Structure-Activity Relationships. ChemMedChem 2018, 13, 2448–2454. 10.1002/cmdc.201800498. [DOI] [PubMed] [Google Scholar]
- Crociani O.; Zanieri F.; Pillozzi S.; Lastraioli E.; Stefanini M.; Fiore A.; Fortunato A.; D’Amico M.; Masselli M.; De Lorenzo E.; Gasparoli L.; Chiu M.; Bussolati O.; Becchetti A.; Arcangeli A. hERG1 channels modulate integrin signaling to trigger angiogenesis and tumor progression in colorectal cancer. Sci. Rep. 2013, 3, 3308. 10.1038/srep03308. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.