

Abstract

A series of isatin–dihydropyrazole hybrids have been synthesized in order to assess their potential as anticancer agents. In particular, 12 compounds were evaluated for their antiproliferative activity toward A549, IGR39, U87, MDA-MB-231, MCF-7, BT474, BxPC-3, SKOV-3, and H1299 cell lines, and human foreskin fibroblasts. Four compounds exhibited interesting antiproliferative activity and were further examined to determine their EC50 values toward a panel of selected tumor cell lines. The best compounds were then investigated for their induced mechanism of cell death. Preliminary structure–activity relationship indicates that the presence of a substituent such as a chlorine atom or a methyl moiety in position 5 of the isatin nucleus is beneficial for the antitumor activity. EMAC4001 proved the most promising compound within the studied series with EC50 values ranging from 0.01 to 0.38 μM.
Keywords: Anticancer agents, isatin-dihydropyrazole hybrids, apoptosis inducers
Several anticancer chemotherapeutic agents act by causing cell death either by directly inhibiting the synthesis of DNA or by interfering with its function. Unfortunately, they are generally not specific for tumor cells and are therefore associated with high toxicity. Not surprisingly, nowadays, the focus of the scientific community is oriented toward the development of new target-directed, more specific cytotoxic agents. Such agents must be able to inhibit or modulate identified molecular targets that are involved in the control of cancer cells, such as signal transduction, apoptosis, transcription regulation, matrix invasion, and angiogenesis. Our research group is currently involved in several projects regarding the design and synthesis of anticancer agents directed toward several targets such as human carbonic anhydrases (hCA),1−5 DNA G-quadruplex, and hCA/COX2 dual inhibitors.6−9 However, it is commonly recognized that cancer is a complex multifactorial disease and, therefore, cannot be treated with a single drug therapy. Accordingly, new agents, combining diverse pharmacophores in a single hybrid molecule, might represent a goal for the treatment of cancer and indeed a big effort has been put into the identification of anticancer multitarget hybrid agents.10−18 In this respect, isatin is commonly recognized as a privileged scaffold in drug design.2,19−24 Moreover, it is a highly represented structural motif in kinase inhibitor anticancer drugs (Figure 1).25−28 The structure–activity relationships of the isatin based multikinase inhibitor nintedanib, as well as its drug development to phase III clinical trial, has been recently reported.28 More in detail the relevant role of the isatin nitrogen and of the carbonyl group in positions 1 and 2, as H-bond donor and acceptor network, was outlined. However, with respect to the structurally similar, multikinase inhibitor sunitinib, the isatin nucleus was, in this case, decorated by a methoxycarbonyl group in position 6, instead of a fluorine atom in position 5. On this basis, we have synthesized a new series of dihydropyrazole isatin dihydrothiazole hybrids EMAC4000, 4001, 4003, 4005, 4007, 4008, 4011, 4012, 4014, 4015, 4018, and 4019 to evaluate their activity toward diverse cancer cell lines. Analogous compounds have been previously reported, and the most relevant structural features that are essential or beneficial for the activity have been outlined.23
Figure 1.

Structurally related multikinase inhibitor sunitinib (VEGFR, PDGFR, KIT, RET), nintedanib (VEGFR, EGFR, PDGFR), and EMAC4000, 4001, 4003, 4005, 4007, 4008, 4011, 4012, 4014, 4015, 4018, and 4019 derivatives.
Prompted by these observations, we aimed to further investigate the effect of both the introduction of diverse substituents on the isatin nucleus and of the replacement of the naphthalen-2-yl group with a thiazol-2-yl ring on the biological activity. EMAC compounds were synthesized slightly modifying previously reported methods (Scheme 1).2,29
Scheme 1. Synthetic Pathway to Compounds EMAC4000, 4001, 4003, 4005, 4007, 4008, 4011, 4012, 4014, 4015, 4018, and 4019.

Reagents and conditions: (i) 2-acetylnaphtalene or 2-acetylthiophene, ethanol, NaOH 10% water solution, 0 °C; (ii) thiosemicarbazide, ethanol, KOH 5%, reflux; (iii) ethyl bromoacetate, R-isatin, dry sodium acetate, acetic acid, reflux.
Briefly, an ethanol solution of 2-acetylnaphthalene (for the synthesis of EMAC4000, 4001, 4003, 4005, 4007, 4008) or 2-acetylthiophene (for the synthesis of EMAC4011, 4012, 4014, 4015, 4018, 4019) was reacted at 0 °C with an equimolar amount of 4-methoxybenzaldehyde in the presence of 1.2 equiv of sodium hydroxide 10% water solution. The obtained solids were crystallized from ethanol. The obtained diarylpropenones were reacted with thiosemicarbazide in refluxing ethanol by adding a freshly prepared KOH 5% ethanol solution. The formation of the dihydrothiazole ring and the condensation of the substituted isatin was accomplished in a single three component step. The 3,5-diaryldihydropyrazole, ethyl bromoacetate, and the appropriate isatin derivative were refluxed in acetic acid in the presence anhydrous sodium acetate to give the desired products EMAC4000, 4001, 4003, 4005, 4007, 4008, 4011, 4012, 4014, 4015, 4018, and 4019.
All compounds were characterized by means of analytical and spectroscopic methods (SI, Tables S1 and S2, and Figures S2–S36) and then evaluated for their ability to inhibit tumor cell growth. First, the activity of the new derivatives was evaluated for antiproliferative activity in the MTT assay at a fixed concentration of 10 μM toward a panel of nine human cancer cell lines, namely A549 (lung carcinoma), IGR39 (melanoma), U87 (glioblastoma), MDA-MB-231 (triple-negative breast cancer), MCF-7 (breast adenocarcinoma), BT474 (invasive ductal carcinoma), H1299 (non-small-cell lung carcinoma), SKOV-3 (ovarian cancer), and BxPC-3 (pancreatic adenocarcinoma) cell lines, and human foreskin fibroblasts. When tested toward cancer cell lines, some of the compounds exhibited antiproliferative activity (Table 1). In particular, compounds bearing a 2-naphthyl substituent in position 3 of the dihydropyrazole ring were generally more active than their corresponding 2-thiophenyl analogues. Although with some differences, compounds EMAC4001, EMAC4007, and EMAC4008 were found to be the most active toward the entire cell panel.
Table 1. Antiproliferative Activity of Compounds EMAC4000, 4001, 4003, 4005, 4007, 4008, 4011, 4012, 4014, 4015, 4018, and 4019 at 10 μM concentration.


Interestingly, EMAC4012 and EMAC4019 were the most active within the 2-thiophenyl series when tested on IGR39 and U87 and IGR39, respectively, with antiproliferative activity comparable to that of the 2-naphthyl analogues. Nevertheless, it should be noted that these derivatives are the analogues of the two most potent compounds of the 2-naphthyl series EMAC4001 and EMAC4008, indicating that the presence of the 5-chloro or of its isostere 5-CH3 substituent is optimal for the antiproliferative activity within this class of compounds. Prompted by these encouraging results, we measured the EC50 values of the most active compounds of the 2-naphthyl series, EMAC4001, EMAC4007, and EMAC4008, and of the best performing derivative within the 2-thiophenyl series, EMAC4012, on a panel of selected cancer cell lines (Table 2).
Table 2. EC50 Values of EMAC4001, EMAC4007, EMAC4008, and EMAC4012 toward a Panel of Selected Tumor Cells.


All compounds exhibited EC50 values in the low micromolar to high nanomolar range. EMAC4001 was the most potent within all the tested compounds with EC50 values ranging from 0.01 μM against H1299 to 0.38 μM against U87 cells (Table 2). The substitution of the 2-naphthyl moiety with the 2-thiophenyl group in the position 3 of the dihydropyrazole ring in EMAC4012 led to an evident decrease of the potency and to EC50 values of 2.97 μM and 5.76 μM toward IGR39 and U87 cell lines, respectively. Interestingly, when tested on A549, IGR39, and U87, EMAC4008 exhibited the highest activity compared with compounds EMAC4001, EMAC4007, and EMAC4012, with EC50 values of 0.18 μM, 0.14 μM, and 0.23 μM, respectively. On the basis of these results, it can be observed that the 5-Cl-isatin is generally the most efficient, but at least in some cases, its isosteric replacement with the 5-CH3-isatin is well tolerated or even more advantageous. Furthermore, to better characterize the biological behavior of these derivatives, we investigated the mechanism of cellular death when the cells are treated with half of the EC50 concentration of compounds EMAC4001 and EMAC4008. Results are presented in Figure 2, and the percentage of apoptotic and necrotic cells is reported in Figure 3.
Figure 2.
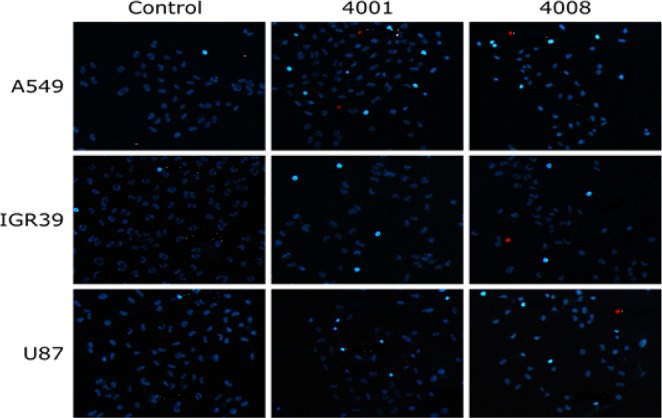
Visualization of apoptotic (bright blue) and necrotic (red) cells after treatment with 1/2 EC50 of EMAC4001 and EMAC4008.
Figure 3.

(a) Percentage of necrotic cells after treatment with 1/2 EC50 of EMAC4001 and EMAC4008. (b) Percentage of apoptotic cells after treatment with 1/2 EC50 of EMAC4001 and EMAC4008.
Results were more than encouraging. In all three considered cell lines, the percentage of apoptotic cells ranges between 13.5% and 27%. Conversely, when the number of necrotic cells is considered, EMAC4001 or EMAC4008 induced necrosis of less than 1% of the cell population. Results show that tested compounds induce cell death mostly through apoptosis. Overall these results indicate that a specific mechanism, such as the inhibition of a signaling pathway, might be the target of EMAC derivatives.
Although further studies are needed to clarify and identify the exact mechanism of action of such derivatives, our data indicate that the hybridization of 5-chloroisatin with 3,5-diaryldihydropyrazoles by the interposition of a dihydrothiazole spacer is a promising approach to the identification of anticancer agents. With this information in our hand, we are encouraged to further investigate these scaffolds in order to optimize their activity and pharmacokinetic properties.
Acknowledgments
The authors acknowledge the “Ufficio Valorizzazione dei Risultati della Ricerca” of Sardegna Ricerche Technological Park, Pula (CA), Italy. The authors also thank the COST action CA15135 (Multitarget Paradigm for Innovative Ligand Identification in the Drug Discovery Process MuTaLig) for support.
Glossary
Abbreviations
- hCA
human carbonic anhydrase
- COX2
cyclooxygenase2
- VEGFR
vascular endothelial growth factor
- PDGFR
platelet-derived growth factor receptor
- KIT
Mast/stem cell growth factor receptor
- RET
proto-oncogene tyrosine-protein kinase receptor
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00596.
Experimental procedures and compound characterization (PDF)
Author Contributions
∇ R.M. and V.P. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Melis C.; Distinto S.; Bianco G.; Meleddu R.; Cottiglia F.; Fois B.; Taverna D.; Angius R.; Alcaro S.; Ortuso F.; Gaspari M.; Angeli A.; Del Prete S.; Capasso C.; Supuran C. T.; Maccioni E. Targeting Tumor Associated Carbonic Anhydrases IXAnd XII: Highly Isozyme Selective Coumarin and Psoralen Inhibitors. ACS Med. Chem. Lett. 2018, 9, 725–729. 10.1021/acsmedchemlett.8b00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis C.; Meleddu R.; Angeli A.; Distinto S.; Bianco G.; Capasso C.; Cottiglia F.; Angius R.; Supuran C. T.; Maccioni E. Isatin: A Privileged Scaffold for the Design of Carbonic Anhydrase Inhibitors. J. Enzyme Inhib. Med. Chem. 2017, 32, 68–73. 10.1080/14756366.2016.1235042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco G.; Meleddu R.; Distinto S.; Cottiglia F.; Gaspari M.; Melis C.; Corona A.; Angius R.; Angeli A.; Taverna D.; Alcaro S.; Leitans J.; Kazaks A.; Tars K.; Supuran C. T.; Maccioni E. N-Acylbenzenesulfonamide Dihydro-1,3,4-Oxadiazole Hybrids: Seeking Selectivity Toward Carbonic Anhydrase Isoforms. ACS Med. Chem. Lett. 2017, 8, 792–796. 10.1021/acsmedchemlett.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meleddu R.; Maccioni E.; Distinto S.; Bianco G.; Melis C.; Alcaro S.; Cottiglia F.; Ceruso M.; Supuran C. T. New 4-[(3-cyclohexyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzene-1-Sulfonamides, Synthesis And Inhibitory Activity Toward Carbonic Anhydrase I, II, IX, XII. Bioorg. Med. Chem. Lett. 2015, 25, 3281–3284. 10.1016/j.bmcl.2015.05.076. [DOI] [PubMed] [Google Scholar]
- Meleddu R.; Distinto S.; Cottiglia F.; Angius R.; Gaspari M.; Taverna D.; Melis C.; Angeli A.; Bianco G.; Deplano S.; Fois B.; Del Prete S.; Capasso C.; Alcaro S.; Ortuso F.; Yanez M.; Supuran C. T.; Maccioni E. Tuning the Dual Inhibition of Carbonic Anhydrase and Cyclooxygenase by Dihydrothiazole Benzensulfonamides. ACS Med. Chem. Lett. 2018, 9, 1045–1050. 10.1021/acsmedchemlett.8b00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca R.; Moraca F.; Costa G.; Nadai M.; Scalabrin M.; Talarico C.; Distinto S.; Maccioni E.; Ortuso F.; Artese A.; Alcaro S.; Richter S. N. Identification of G-Quadruplex DNA/RNA Binders: Structure-Based Virtual Screening and Biophysical Characterization. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 1329–1340. 10.1016/j.bbagen.2016.12.023. [DOI] [PubMed] [Google Scholar]
- Rocca R.; Costa G.; Artese A.; Parrotta L.; Ortuso F.; Maccioni E.; Pinato O.; Greco M. L.; Sissi C.; Alcaro S.; Distinto S.; Moraca F. Hit Identification of a Novel Dual Binder for h-telo/c-myc G-Quadruplex by a Combination of Pharmacophore Structure-Based Virtual Screening and Docking Refinement. ChemMedChem 2016, 11, 1721–1733. 10.1002/cmdc.201600053. [DOI] [PubMed] [Google Scholar]
- Rocca R.; Moraca F.; Costa G.; Alcaro S.; Distinto S.; Maccioni E.; Ortuso F.; Artese A.; Parrotta L. Structure-Based Virtual Screening of Novel Natural Alkaloid Derivatives as Potential Binders of H-Telo and C-Myc DNA G-Quadruplex Conformations. Molecules 2015, 20, 206–223. 10.3390/molecules20010206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaro S.; Musetti C.; Distinto S.; Casatti M.; Zagotto G.; Artese A.; Parrotta L.; Moraca F.; Costa G.; Ortuso F.; MacCioni E.; Sissi C. Identification and Characterization of New DNA G-Quadruplex Binders Selected by a Combination of Ligand and Structure-Based Virtual Screening Approaches. J. Med. Chem. 2013, 56, 843–855. 10.1021/jm3013486. [DOI] [PubMed] [Google Scholar]
- Hassan A. H. E.; Choi E.; Yoon Y. M.; Lee K. W.; Yoo S. Y.; Cho M. C.; Yang J. S.; Kim H. I.; Hong J. Y.; Shin J.-S.; Chung K.-S.; Lee J.-H.; Lee K.-T.; Lee Y. S. Natural Products Hybrids: 3,5,4’-Trimethoxystilbene-5,6,7-Trimethoxyflavone Chimeric Analogs as Potential Cytotoxic Agents Against Diverse Human Cancer Cells. Eur. J. Med. Chem. 2019, 161, 559–580. 10.1016/j.ejmech.2018.10.062. [DOI] [PubMed] [Google Scholar]
- Abbas S. H.; Abd El-Hafeez A. A.; Shoman M. E.; Montano M. M.; Hassan H. A. New Quinoline/Chalcone Hybrids as Anti-Cancer Agents: Design, Synthesis, and Evaluations of Cytotoxicity and PI3K Inhibitory Activity. Bioorg. Chem. 2019, 82, 360–377. 10.1016/j.bioorg.2018.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrenti V.; Pittala V.; Romeo G.; Amata E.; Dichiara M.; Marrazzo A.; Turnaturi R.; Prezzavento O.; Barbagallo I.; Vanella L.; Rescifina A.; Floresta G.; Tibullo D.; Di Raimondo F.; Intagliata S.; Salerno L. Targeting Heme Oxygenase-1 With Hybrid Compounds to Overcome Imatinib Resistance in Chronic Myeloid Leukemia Cell Lines. Eur. J. Med. Chem. 2018, 158, 937–950. 10.1016/j.ejmech.2018.09.048. [DOI] [PubMed] [Google Scholar]
- Palermo A. F.; Diennet M.; El Ezzy M.; Williams B. M.; Cotnoir-White D.; Mader S.; Gleason J. L. Incorporation of Histone Deacetylase Inhibitory Activity into the Core of Tamoxifen - A New Hybrid Design Paradigm. Bioorg. Med. Chem. 2018, 26, 4428–4440. 10.1016/j.bmc.2018.07.026. [DOI] [PubMed] [Google Scholar]
- Ning W.; Hu Z.; Tang C.; Yang L.; Zhang S.; Dong C.; Huang J.; Zhou H.-B. Novel Hybrid Conjugates with Dual Suppression of Estrogenic and Inflammatory Activities Display Significantly Improved Potency Against Breast Cancer. J. Med. Chem. 2018, 61, 8155–8173. 10.1021/acs.jmedchem.8b00224. [DOI] [PubMed] [Google Scholar]
- Maestro A.; Martin-Encinas E.; Alonso C.; Martinez de Marigorta E.; Rubiales G.; Vicario J.; Palacios F. Synthesis of Novel Antiproliferative Hybrid Bis-(3-Indolyl)Methane Phosphonate Derivatives. Eur. J. Med. Chem. 2018, 158, 874–883. 10.1016/j.ejmech.2018.09.011. [DOI] [PubMed] [Google Scholar]
- Froehlich T.; Kiss A.; Woelfling J.; Mernyak E.; Kulmany A. E.; Minorics R.; Zupko I.; Leidenberger M.; Friedrich O.; Kappes B.; Hahn F.; Marschall M.; Schneider G.; Tsogoeva S. B. Synthesis of Artemisinin-Estrogen Hybrids Highly Active Against Hcmv, P. Falciparum, and Cervical and Breast Cancer. ACS Med. Chem. Lett. 2018, 9, 1128–1133. 10.1021/acsmedchemlett.8b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavo F.; Pucci S.; Fasoli F.; Lammi C.; Moretti M.; Mucchietto V.; Lattuada D.; Viani P.; De Palma C.; Budriesi R.; Corradini I.; Dowell C.; Mcintosh J. M.; Clementi F.; Bolchi C.; Gotti C.; Pallavicini M. Potent Anti-Glioblastoma Agents by Hybridizing the Onium-Alkyloxy-Stilbene Based Structures of an Alpha7-, alpha9-nAChR Antagonist and of a Pro-Oxidant Mitocan. J. Med. Chem. 2018, 61, 10531. 10.1021/acs.jmedchem.8b01052. [DOI] [PubMed] [Google Scholar]
- Kucuksayan E.; Ozben T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. 10.2174/1568026616666160927155515. [DOI] [PubMed] [Google Scholar]
- Javid M. T.; Rahim F.; Taha M.; Nawaz M.; Wadood A.; Ali M.; Mosaddik A.; Shah S. A. A.; Farooq R. K. Synthesis, SAR Elucidations and Molecular Docking Study of Newly Designed Isatin Based Oxadiazole Analogs as Potent Inhibitors of Thymidine Phosphorylase. Bioorg. Chem. 2018, 79, 323–333. 10.1016/j.bioorg.2018.05.011. [DOI] [PubMed] [Google Scholar]
- Teng Y.-O.; Zhao H.-Y.; Wang J.; Liu H.; Gao M.-L.; Zhou Y.; Han K.-L.; Fan Z.-C.; Zhang Y.-M.; Sun H.; Yu P. Synthesis and Anti-Cancer Activity Evaluation of 5-(2-Carboxyethenyl)-Isatin Derivatives. Eur. J. Med. Chem. 2016, 112, 145–156. 10.1016/j.ejmech.2015.12.050. [DOI] [PubMed] [Google Scholar]
- Rana S.; Blowers E. C.; Tebbe C.; Contreras J. I.; Radhakrishnan P.; Kizhake S.; Zhou T.; Rajule R. N.; Arnst J. L.; Munkarah A. R.; Rattan R.; Natarajan A. Isatin Derived Spirocyclic Analogues with α-Methylene-γ-butyrolactone as Anticancer Agents: A Structure–Activity Relationship Study. J. Med. Chem. 2016, 59, 5121–5127. 10.1021/acs.jmedchem.6b00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Zhao H.-Y.; Han K.-L.; Yang Y.; Song B.-B.; Guo Q.-N.; Fan Z.-C.; Zhang Y.-M.; Teng Y.-O.; Yu P. 5-(2-Carboxyethenyl) Isatin Derivative Induces G2/M Cell Cycle Arrest and Apoptosis in Human Leukemia K562 Cells. Biochem. Biophys. Res. Commun. 2014, 450, 1650–1655. 10.1016/j.bbrc.2014.07.053. [DOI] [PubMed] [Google Scholar]
- Havrylyuk D.; Zimenkovsky B.; Vasylenko O.; Gzella A.; Lesyk R. Synthesis of New 4-Thiazolidinone-, Pyrazoline-, and Isatin-Based Conjugates with Promising Antitumor Activity. J. Med. Chem. 2012, 55, 8630–8641. 10.1021/jm300789g. [DOI] [PubMed] [Google Scholar]
- Lin H.-H.; Wu W.-Y.; Cao S.-L.; Liao J.; Ma L.; Gao M.; Li Z.-F.; Xu X. Synthesis and Antiproliferative Evaluation of Piperazine-1-Carbothiohydrazide Derivatives of Indolin-2-One. Bioorg. Med. Chem. Lett. 2013, 23, 3304–3307. 10.1016/j.bmcl.2013.03.099. [DOI] [PubMed] [Google Scholar]
- Dweedar H. E.; Mahrous H.; Ibrahim H. S.; Abdel-Aziz H. A. Analogue-Based Design, Synthesis and Biological Evaluation of 3-Substituted-(methylenehydrazono)indolin-2-ones as Anticancer Agents. Eur. J. Med. Chem. 2014, 78, 275–280. 10.1016/j.ejmech.2014.03.058. [DOI] [PubMed] [Google Scholar]
- Cao J.; Gao H.; Bemis G.; Salituro F.; Ledeboer M.; Harrington E.; Wilke S.; Taslimi P.; Pazhanisamy S.; Xie X.; Jacobs M.; Green J. Structure-Based Design and Parallel Synthesis of N-Benzyl Isatin Oximes as JNK3MAP Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2891–2895. 10.1016/j.bmcl.2009.03.043. [DOI] [PubMed] [Google Scholar]
- Theou-Anton N.; Faivre S.; Dreyer C.; Raymond E. Benefit-Risk Assessment of Sunitinib in Gastrointestinal Stromal Tumours and Renal Cancer. Drug Saf. 2009, 32, 717–34. 10.2165/00002018-200932090-00003. [DOI] [PubMed] [Google Scholar]
- Roth G. J.; Binder R.; Colbatzky F.; Dallinger C.; Schlenker-Herceg R.; Hilberg F.; Wollin S.-L.; Kaiser R. Nintedanib: From Discovery to the Clinic. J. Med. Chem. 2015, 58, 1053–1063. 10.1021/jm501562a. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Maccioni E.; Secci D.; Bolasco A.; Chimenti P.; Granese A.; Befani O.; Turini P.; Alcaro S.; Ortuso F.; Cirilli R.; La Torre F.; Cardia M. C.; Distinto S. Synthesis, Molecular Modeling Studies, and Selective Inhibitory Activity against Monoamine Oxidase of 1-Thiocarbamoyl-3,5-diaryl-4,5-dihydro-(1H)- pyrazole Derivatives. J. Med. Chem. 2005, 48, 7113–7122. 10.1021/jm040903t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.