

Abstract
JMJD3 is a member of the KDM6 subfamily and catalyzes the demethylation of lysine 27 on histone H3 (H3K27). This protein was identified as a useful tool in understanding the role of epigenetics in inflammatory conditions and in cancer as well. Guided by a virtual fragment screening approach, we identified the benzoxazole scaffold as a new hit suitable for the development of tighter JMJD3 inhibitors. Compounds were synthesized by a microwave-assisted one-pot reaction under catalyst and solvent-free conditions. Among these, compound 8 presented the highest inhibitory activity (IC50 = 1.22 ± 0.22 μM) in accordance with molecular modeling calculations. Moreover, 8 induced the cycle arrest in S-phase on A375 melanoma cells.
Keywords: Fragment-based approach, anticancer agent, one-pot microwave reaction, JMJD3 inhibitor, melanoma
JMJD3, along with tetratricopeptide repeat X-linked protein (UTX), constitutes the KDM6 subfamily, which catalyzes the demethylation of lysine 27 on histone H3 (H3K27). Both proteins share a highly homologous Jumonji C domain, endowed with Fe2+ and α-ketoglutarate,1 which recognizes the trimethylated (H3K27me3) and dimethylated (H3K27me2) lysine 27 of H3 as substrate regulating the lysine methylation state. UTX is ubiquitously expressed,2,3 and it controls the basal levels of H3K27me3 and the induction of ectoderm and mesoderm differentiation.4 JMJD3 is highly expressed in the embryonic stem cells where it regulates neuronal and epidermal differentiation; its expression is induced by inflammation5 and viral and oncogenic stimuli.6 Moreover, JMJD3 inhibits cell reprogramming,7 whereas UTX is fundamental for it.
JMJD3 has been shown to be correlated to inflammatory pathologies8,9 and neurological disorders.10,11 Different studies have also reported the key role of JMJD3 in cancers12 such as initiation and maintenance of T-cell acute lymphoblastic leukemia;13 overexpression of JMJD3 associated with the pathogenesis of Hodgkin’s lymphoma,14 metastatic prostate cancer,15 and pediatric brainstem glioma;16 promoter role in epithelial–mesenchymal transition and cell invasion.17 The reported data suggest JMDJ3 as therapeutic target both in inflammatory diseases and in cancer disorders. However, despite the great potential as therapeutic target, the specific biological function of JMJ enzymes in regulating cellular processes is still poorly understood due to the absence of selective inhibitors. Indeed, concerning JMJD3, up to date, only one JMJD3/UTX binder has been reported, GSK-J1,1 along with a series of its derivatives showing a similar or lower activity.18 Recently, we identified the quinoline-5,8 dicarboxylic acid scaffold as new potential lead compound for the development of JMJD3 inhibitors, using a fragment-based approach.19 This chemical core is endowed with an unprecedented selectivity toward the proposed biological target and a relevant inhibition potency in the micromolar range. Our successful strategy was based on in silico screening of a metal chelator fragment library (Figure S1), which was previously proposed to develop metalloprotein inhibitors.20,21 The results obtained by our investigations validated the computer-aided strategy followed, consisting in molecular docking studies against two JMJD3 conformations with catalytic charge center refined at DFT theory and integrated by molecular dynamics simulations.19 In details, we used two available X-ray structures of JMJD3 as protein models (PDB IDs: 4ASK as Model A and 2XXZ as Model B) because their structural comparison revealed different spatial rearrangements of some amino acids and of Fe2+ upon GSK-J1 binding.1
In order to further explore other chelating fragments here we report the design of new and potent binders of this attractive biological target.
From the inspection of the docked chelator library on both protein conformations (Models A and B), we selected the 2-(benzo[d]oxazol-2-yl)phenol and 2-(benzo[d]thiazol-2-yl)phenol fragments (1 and 2, Scheme 1), which were properly lodged into α-ketoglutarate cavity.
Scheme 1. Synthetic Scheme and Chemical Structures of 1, 2, and 8–11.

See experimental details in the Supporting Information.
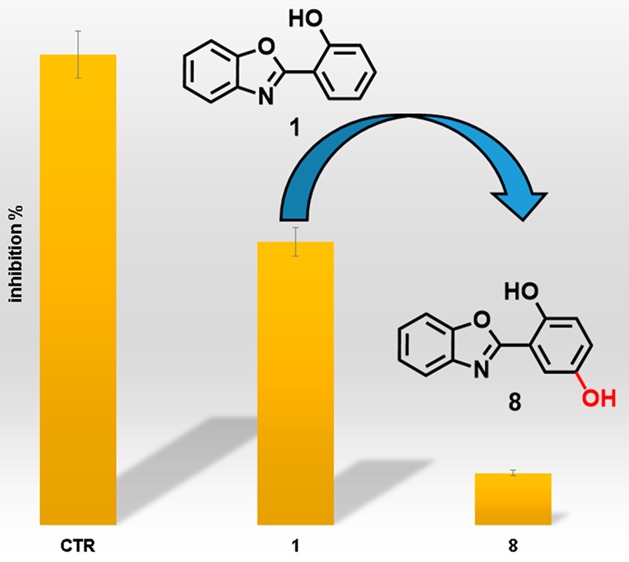
The selected fragments do not present any carboxylic group, despite the previously selected scaffold B11 (Figure S1).19 Their binding mode, especially with Model B, suggests that they could fill the cavity devoted to host the methylated lysine (Figure S2). Thus, we evaluated the ability of 1 and 2 to inhibit JMJD3 by AlphaScreen protocol (Figure 1).
Figure 1.

Effect of compounds 1, 2, 8–11 (25 μM), and GSK-J1 (10 μM, reference compound) on JMJD3 activity. Data are given as the means ± SEM, n = 3.
The experiments highlighted that 1 inhibits the enzymatic activity of 40%, whereas 2 was inactive (Figure 1). Hence, we selected 1 as starting scaffold for the synthetic modification in order to improve the affinity toward JMJD3. The docked pose of 1 suggested a modification of phenol moiety to increase the interaction network with α-ketoglutarate pocket of JMJD3, with surrounding residues: T1387, K1381, N1400, W1410, V1472 (Figure S2). Thus, we screened on both protein conformations (Models A and B) a small library of 52 chemical-diverse substituents endowed with H-bond acceptor/donors (Figure S3), considering the synthetic feasibility and commercial availability of reactives. Our analysis was based on the docked energy and visual inspection. In particular, in our growing fragment strategy, we paid attention on the influence of the inserted substituents to preserve the original docked pose of parent scaffold and the favorable interactions with α-ketoglutarate cavity, as well as maintaining the bidentate coordination manner with Fe2+ ion.1,19,22,23 The docking outcomes of all generated compounds (8–59, Scheme 1, Table S1, and Figure S3) lead to a focused collection of benzoxazole derivatives (8–11, Scheme 1), selected for the synthesis and useful to provide preliminary information for structure–activity relationship.
The results obtained on Model A revealed that 8–11 fill equivalent spaces. Compounds 8–11 (Figures 2 and S4–S6) coordinate in bidentate manner the Fe2+ ion as observed by cocrystallized GSK-J1 (Figure 2C).1 The benzoxazole portion of 8–11 makes π–π interactions with the side chain of H1390, as observed for GSK-J1, and is H-bonded to Y1379. A π–π interaction is also observed for the phenol moiety of 8–10 and the methoxybenzodioxole of 11 with Y1381. The hydroxyl group of phenolic ring of 8–10 is further H-bonded to H1470. Actually, 8–11 differ for the interaction given by the different substituents of the R aromatic ring (Scheme 1). Indeed, the hydroxyl group at C-4 of phenol ring of 8 establishes H-bond with K1381, T1387, and N1400 (Figure 2A). The carboxylic groups of 9 gives a salt bridge with K1381 and hydrogen bonds with T1387 and N1400 (Figure S4). The fluorine at C-4 of 10 points toward the K1381 (Figure S5).24 Finally, the methoxybenzodioxole of 11 interacts with K1381 and T1387 (Figure S6).
Figure 2.
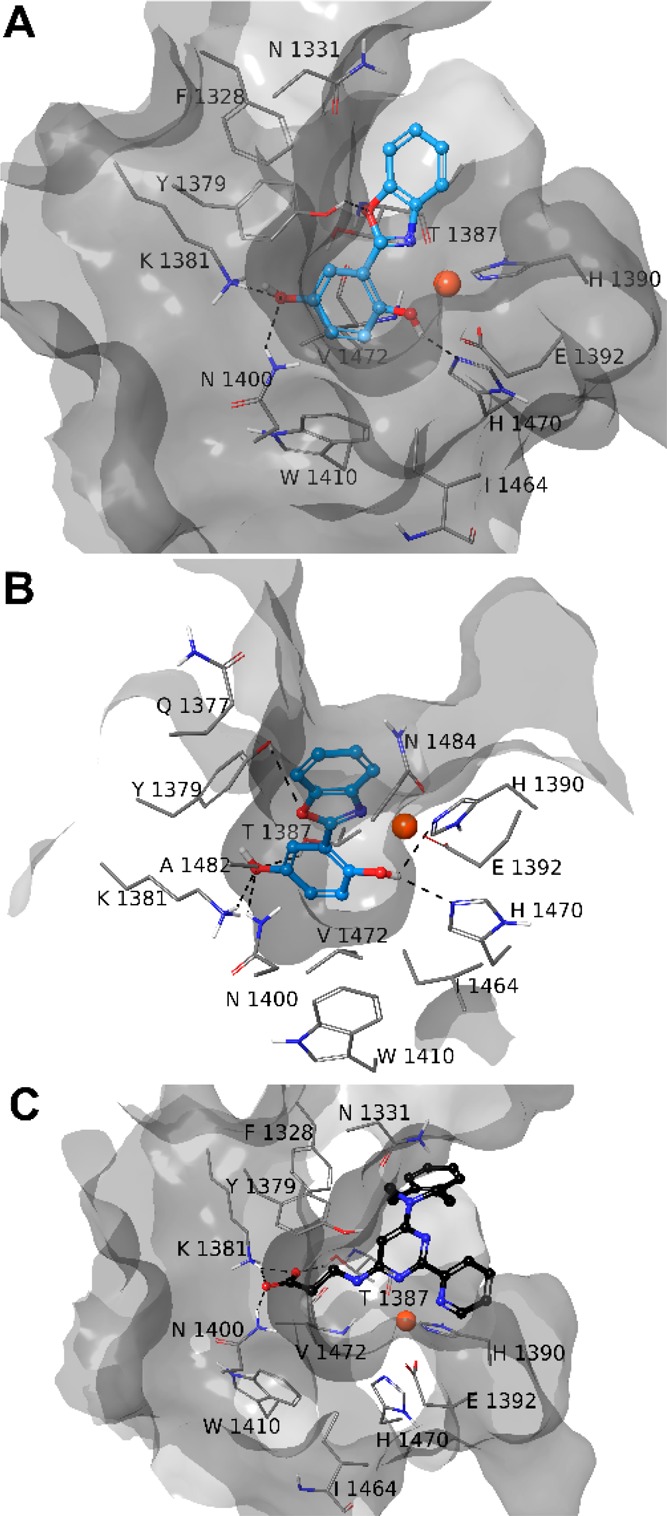
Three-dimensional model of the interactions of 8-Model A (A), 8-Model B (B), and GSK-J1-Model A (C) (PDB ID 4ASK). JMJD3 is represented by molecular surface and tube, 8 (cyan) and GSK-J1 (black) by sticks and balls. The atom color codes are C (8), cyan; C (GSK-J1), black; C (JMJD3), gray; polar H, white; N, dark blue; O, red. The dashed black lines indicate the H-bonds between ligand and protein.
The docked poses of 8–11 into Model B showed a similar pattern of interactions established by the common structural portion (Figures 2 and S4–S6). Indeed, the Fe2+ ion is coordinated in a bidentate manner by the nitrogen and the hydroxyl group of 8–10, whereas the oxygen of the five-membered ring is H-bonded to Y1379. The OH group is also H-bonded to H1470. The compound 11 structurally differs from its congeners also for a methoxy group instead of a hydroxyl group, which is able to coordinate the Fe2+. The phenol ring of 8–10 and the methoxybenzodioxole of 11 establish π–π interactions with Y1379. The substituents, introduced to get 8–11, basically give the same pattern of interactions observed into Model A.
We further investigated the stability of 8–11–JMJD3 complexes by molecular dynamics simulations (50 ns, 310 K, see experimental details in the Supporting Information).19,25,26 The trajectory analysis revealed that 8 makes different contacts with protein residues, and it keeps most of all interactions observed from the docked pose during the entire simulation (>50%) with both protein models (Figure 3B). The backbone-positional RMSD of Model A bound to 8 shows a better stability during molecular dynamics simulations with respect to the reference structure, with a comparable behavior on both protein models (Figure 3A). The atom relative orientation of 8 is kept during the simulation with Models A and B as highlighted by the RMSD of heavy atoms (Figure 3A, right plot). The trajectory of 9-Model A showed oscillations (Figure S7), and fluctuations are observed for 9 atom relative orientation. Indeed, at the end of simulation, 9 moved away from α-ketoglutarate pocket of Model A. Lower oscillations are observed with Model B (Figure S7) with respect to the simulation with Model A. The 10-Model A trajectory is rather constant, whereas for 10-Model B a larger deviation from the initial position is observed especially in the first 5 ns of the simulation (Figure S8). Very few contacts with the surrounding amino acids are observed during the simulation for 10 with Models A and B. Fluctuations of the trajectory are found for 11 with both Models (Figure S9). In particular, 11 keeps the bidentate coordination of the Fe2+ with Model A for the whole trajectory, whereas with Model B, just for 75% of the simulation time the nitrogen is involved in coordination of the metal ion (Figure S9). Compound 11 gives H-bonds with K1381, T1387, and N1440 for ≤10% of simulation time (Figure S9). Collectively, the integrated analysis of docking results and molecular dynamics on both models suggested a higher activity of 8 compared to 9–11 as experimentally confirmed by AlphaScreen analysis.
Figure 3.
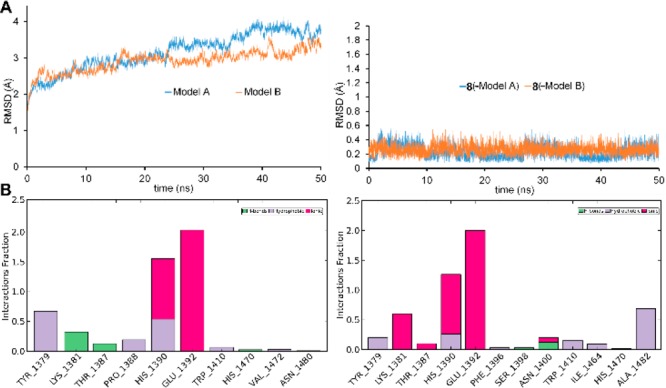
(A) Heavy atom-positional RMSD (Å) as function of simulation time (ns): on the left Models A (blue line) and Model B plot; on the right 8 plot. (B) Protein–ligand contact histograms during the simulation of 8-Model A (left) and 8-Model B (right).
For the synthesis of compounds 8–11, classical approaches to benzoxazole derivatives involve cyclocondensation of carboxylic acid derivatives such as esters and acyl chlorides with o-aminophenols through initial nucleophilic attack at an activated carbonyl group by the aniline moiety or through oxidative cyclocondensation with aldehydes.27 However, these methodologies suffer from several limitations in the preparation of the starting materials and the use of oxidizing agents, which are not compatible with the presence of para-hydroxyl groups in the molecule. Hence, we opted for a microwave-assisted coupling between the carboxylic acid and 2-hydroxyaminophenol under catalyst and solvent-free conditions (Scheme 1).28
The reaction mixture was irradiated in a microwave oven. After several attempts to determine the optimum reaction conditions, we obtained the best result setting the reaction temperature at 250 °C for 2 min. Following this procedure, the desired products were obtained in good yields, after extraction and purification by semipreparative RP-HPLC chromatography on C18 column (see experimental details in Supporting Information). The structures of the synthesized compounds 8–11 were fully characterized by MS and NMR spectra (Figures S11–S22).
The synthesized compounds 8–11 were investigated for inhibition of JMJD3 (at 25 μM) by means of AlphaScreen (Figure 1). The experimental tests revealed that 8 exhibited the highest inhibitory activity (90%). Compounds 9 and 10 also moderately decreased the enzyme activity by 27% and 20%, respectively. The analogue 11 did not show any inhibitory activity. It is noteworthy that these experimental data are in good qualitative agreement with the theoretical outcomes, highlighting the structural features of modified phenolic portion as responsible for the binding affinity toward the macromolecule. Compound 8 showed an IC50 value of 1.22 ± 0.22 μM against JMJD3 by AlphaScreen assay. We also evaluated the inhibition activity of 8 against UTX, the highly structural related isoform of JMJD3. Unlike GSK-J1, 8 showed an interesting selectivity (about 21-fold, Table 1) toward JMJD3. Then, we calculated the ligand efficiency (LE) of 8. This parameter, taking into account the ligand size and potency, is useful to evaluate the quality of a disclosed hit.29,30 Indeed, a LE > 0.3 kcal/mol is usually considered promising for a fragment hit. Since for 8, we obtained a LE = 0.48 kcal/mol, this fragment was confirmed to be a good structural seed to develop new JMJD3 inhibitors. On a side note, the LE value of 8 is even better than our proposed quinoline-5,8 dicarboxylic acid scaffold, previously identified by the same fragment-based strategy.19
Table 1. IC50 ± SEM and LE Value of 8.
| IC50 (μM) |
|||
|---|---|---|---|
| JMJD3 | UTX | LEa (kcal/mol) | |
| 8 | 1.2 ± 0.2 | 25.7 ± 2.3 | 0.48 |
Calculated at 25 °C by using the equation: LE = (1.37/HA) × pIC50, where HA is number of non-hydrogen atoms.
Recently, the JMJD3 promoting role in progression and metastasis of melanoma has been reported.31 These studies also suggest a contrasting role of UTX and JMJD3 in melanoma, with UTX working as tumor suppressor. Thus, we decide to investigate the ability of 8 to affect cell cycle progression and/or induced cell death of A375 cells. First, we evaluated the cell permeability of 8 with the well-validated parallel artificial membrane permeability assay (PAMPA) technique, obtaining an acceptable apparent permeability value (Papp) of 9.12 × 10–7 ± 1.67 × 10–8 cm/s. Then, the A375 melanoma cells were incubated with compound 8 at different concentrations (1–5–10–20–40–80–100 μM). After 24 h the cells were collected, incubated with iced ethanol 70% for 2 h, and stained with propidium iodide for flow cytometer analysis. Compound 8 showed to affect melanoma cell proliferation in a concentration-dependent manner, with a significant effect at 20 μM. The higher effect was reached at 100 μM by arresting cell cycle in S phase (Figure 4 and Table S2). Moreover, we obtained the IC50 (24.7 ± 1.1 μM) of 8 through the FACS titration, to avoid interference in MTT assays being a colored compound. These results are in agreement with the previous studies showing that JMJD3 can also gather at DNA double-strand break sites (DSBs) to facilitate the expression of specific genes, such as Ku80, which is a key mediator protein in NHEJ pathway (nonhomologous end-joining).32 In particular, Ku proteins are essential for the S-phase cycle transition and Ku80–/– cells present a stronger S checkpoint response33 after a DNA damage.
Figure 4.
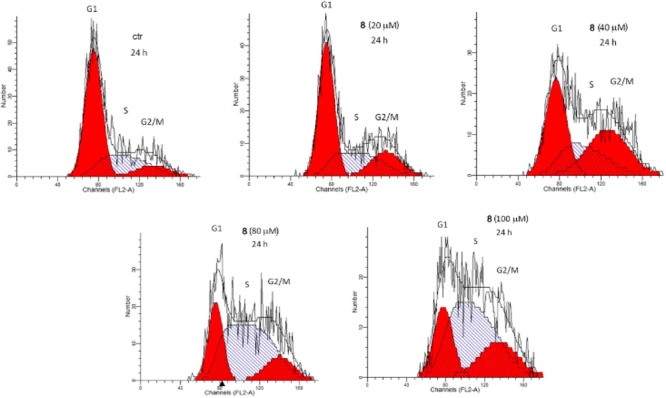
Flow cytometer analysis of representative cell cycle of A375, treated with 8 (n = 3).
A375 melanoma cell lines were incubated by 100 μM 8 for 4 and 24 h and lysed for SDS-PAGE for a Western blot assay and the nitrocellulose membrane was blotted with an anti-H3K27me2 antibody. After 24 h of treatment with 8, a slight decrease of the H3K27me2 level was observed (Figure S10). The absence of a clear result is probably due to the low inhibitor activity of 8 against UTX, which continues exerting the demethylase activity.
In conclusion, by a fragment-based approach we have identified a novel promising hit to develop new JMJD3 inhibitors. Indeed, the identified benzoxazole derivative 8 inhibits the protein in the low micromolar range and is able to induce cell cycle arrest of A375 cancer cells. Furthermore, the low molecular weight and the remarkable LE value of 8 promote this chemical core for the hit to lead optimization step for the development of new benzoxazole-based JMJD3 inhibitors for melanoma treatment.
Acknowledgments
We thank Dr. Eduardo M. Sommella (Department of Pharmacy, University of Salerno, Italy) for HR mass spectra.
Glossary
ABBREVIATIONS
- JMJD3
Jumonji domain-containing protein 3
- UTX
ubiquitously transcribed X chromosome tetratricopeptide repeat protein
- H3K27
lysine 27 on histone H3
- H3K27me3
trimethylated lysine 27 on H3
- H3K27me2
dimethylated lysine 27 on H3
- NHEJ
nonhomologous end-joining
- DFT
density functional theory
- RMSD
root-mean-square deviation
- MW
microwave
- LE
ligand efficiency
- DSBs
DNA double-strand break sites
- NMR
nuclear magnetic resonance
- MS
mass spectroscopy
- SEM
standard error of the mean
- FACS
fluorescence assisted cell sorting
- PAMPA
parallel artificial membrane permeability assay
- Papp
apparent permeability
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00589.
Computational details, synthetic procedures, AlphaScreen method, characterization data, NMR and mass spectra, cell assay, PAMPA test, Western blot (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) and Fondazione Cariplo (grant TRIDEO Id. 17099 to S.D.M.).
The authors declare no competing financial interest.
Supplementary Material
References
- Kruidenier L.; Chung C. W.; Cheng Z.; Liddle J.; Che K.; Joberty G.; Bantscheff M.; Bountra C.; Bridges A.; Diallo H.; Eberhard D.; Hutchinson S.; Jones E.; Katso R.; Leveridge M.; Mander P. K.; Mosley J.; Ramirez-Molina C.; Rowland P.; Schofield C. J.; Sheppard R. J.; Smith J. E.; Swales C.; Tanner R.; Thomas P.; Tumber A.; Drewes G.; Oppermann U.; Patel D. J.; Lee K.; Wilson D. M. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Martini M. L.; Jin J. Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 2018, 118, 989–1068. 10.1021/acs.chemrev.6b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A. Epigenetic drug discovery: a success story for cofactor interference. Philos. Trans. R. Soc., B 2018, 373, 20170069. 10.1098/rstb.2017.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales Torres C.; Laugesen A.; Helin K. Utx is required for proper induction of ectoderm and mesoderm during differentiation of embryonic stem cells. PLoS One 2013, 8, e60020 10.1371/journal.pone.0060020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F.; Totaro M. G.; Prosperini E.; Notarbartolo S.; Testa G.; Natoli G. The histone H3 lysine-27 demethylase jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Barradas M.; Anderton E.; Acosta J. C.; Li S.; Banito A.; Rodriguez-Niedenfuhr M.; Maertens G.; Banck M.; Zhou M. M.; Walsh M. J.; Peters G.; Gil J. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; Li Q.; Ayers S.; Gu Y.; Shi Z.; Zhu Q.; Chen Y.; Wang H. Y.; Wang R. F. Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell 2013, 152, 1037–1050. 10.1016/j.cell.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F.; Narang V.; Yap Z. H.; Tusi B. K.; Burgold T.; Austenaa L.; Bucci G.; Caganova M.; Notarbartolo S.; Casola S.; Testa G.; Sung W. K.; Wei C. L.; Natoli G. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 2009, 28, 3341–3352. 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F.; Totaro M. G.; Prosperini E.; Notarbartolo S.; Testa G.; Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Przanowski P.; Dabrowski M.; Ellert-Miklaszewska A.; Kloss M.; Mieczkowski J.; Kaza B.; Ronowicz A.; Hu F.; Piotrowski A.; Kettenmann H.; Komorowski J.; Kaminska B. The signal transducers Stat1 and Stat3 and their novel target JMJD3 drive the expression of inflammatory genes in microglia. J. Mol. Med. (Heidelberg, Ger.) 2014, 92 (3), 239–254. 10.1007/s00109-013-1090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Li T.; Li J.; Yang J.; Liu H.; Zhang X. J.; Le W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014, 21, 369–380. 10.1038/cdd.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barradas M.; Anderton E.; Acosta J. C.; Li S.; Banito A.; Rodriguez-Niedenfuhr M.; Maertens G.; Banck M.; Zhou M. M.; Walsh M. J.; Peters G.; Gil J. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P.; Tsirigos A.; Welstead G. G.; Trimarchi T.; Bakogianni S.; Xu L.; Loizou E.; Holmfeldt L.; Strikoudis A.; King B.; Mullenders J.; Becksfort J.; Nedjic J.; Paietta E.; Tallman M. S.; Rowe J. M.; Tonon G.; Satoh T.; Kruidenier L.; Prinjha R.; Akira S.; Van Vlierberghe P.; Ferrando A. A.; Jaenisch R.; Mullighan C. G.; Aifantis I. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. 10.1038/nature13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderton J. A.; Bose S.; Vockerodt M.; Vrzalikova K.; Wei W.; Kuo M.; Helin K.; Christensen J.; Rowe M.; Murray P. G.; Woodman C. B. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 2011, 30, 2037–2043. 10.1038/onc.2010.579. [DOI] [PubMed] [Google Scholar]
- Xiang Y.; Zhu Z.; Han G.; Lin H.; Xu L.; Chen C. D. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007, 17, 850–857. 10.1038/cr.2007.83. [DOI] [PubMed] [Google Scholar]
- Hashizume R.; Andor N.; Ihara Y.; Lerner R.; Gan H.; Chen X.; Fang D.; Huang X.; Tom M. W.; Ngo V.; Solomon D.; Mueller S.; Paris P. L.; Zhang Z.; Petritsch C.; Gupta N.; Waldman T. A.; James C. D. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadoss S.; Chen X.; Wang C.-Y. Histone demethylase KDM6B promotes epithelial-mesenchymaltransition. J. Biol. Chem. 2012, 287, 44508–44517. 10.1074/jbc.M112.424903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Wang X.; Chen L.; Huang M.; Tang W.; Zuo J.; Liu Y.-C.; Shi Z.; Liu R.; Jingkang S.; Xiong B. Design and discovery of new pyrimidine coupled nitrogen aromatic rings as chelating groups of JMJD3 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 721–725. 10.1016/j.bmcl.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Giordano A.; del Gaudio F.; Johansson C.; Riccio R.; Oppermann U.; Di Micco S. Virtual fragment screening identification of a novel quinoline-5,8-dicarboxylic acid derivative as selective JMJD3 inhibitor. ChemMedChem 2018, 13, 1160–1164. 10.1002/cmdc.201800198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A.; Johnson S. L.; Jacobsen J. A.; Miller M. T.; Chen L. H.; Pellecchia M.; Cohen S. M. Chelator Fragment Libraries for Targeting Metalloproteinases. ChemMedChem 2010, 5, 195–199. 10.1002/cmdc.200900516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen J. A.; Fullagar J. L.; Miller M. T.; Cohen S. M. Identifying chelators for metalloprotein inhibitors using a fragment-based approach. J. Med. Chem. 2011, 54, 591–602. 10.1021/jm101266s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco S.; Terracciano S.; Cantone V.; Fischer K.; Koeberle A.; Foglia A.; Riccio R.; Werz O.; Bruno I.; Bifulco G. Discovery of new potent molecular entities able to inhibit mPGES-1. Eur. J. Med. Chem. 2018, 143, 1419–1427. 10.1016/j.ejmech.2017.10.039. [DOI] [PubMed] [Google Scholar]
- Botta A.; Sirignano E.; Popolo A.; Saturnino C.; Terracciano S.; Foglia A.; Sinicropi M. S.; Longo P.; Di Micco S. Identification of Lead Compounds as Inhibitors of STAT3: Design, Synthesis and Bioactivity. Mol. Inf. 2015, 34, 689–697. 10.1002/minf.201500043. [DOI] [PubMed] [Google Scholar]
- Giordano A.; Forte G.; Massimo L.; Riccio R.; Bifulco G.; Di Micco S. Discovery of new erbB4 inhibitors: repositioning an orphan chemical library by inverse virtual screening. Eur. J. Med. Chem. 2018, 152, 253–263. 10.1016/j.ejmech.2018.04.018. [DOI] [PubMed] [Google Scholar]
- Di Micco S.; Renga B.; Carino A.; D’Auria M. V.; Zampella A.; Riccio R.; Fiorucci S.; Bifulco G. Structural insights into Estrogen Related Receptor-b modulation:4-Methylenesterols from Theonella swinhoei sponge as the first example of marine natural antagonists. Steroids 2014, 80, 51–63. 10.1016/j.steroids.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Chini M. G.; Malafronte N.; Vaccaro M. C.; Gualtieri M. J.; Vassallo A.; Vasaturo M.; Castellano S.; Milite C.; Leone A.; Bifulco G.; De Tommasi N.; Dal Piaz F. Identification of limonol derivatives as heat shock protein 90 (hsp90) inhibitors through a multidisciplinary approach. Chem. - Eur. J. 2016, 22, 13236–13250. 10.1002/chem.201602242. [DOI] [PubMed] [Google Scholar]
- Rajasekhar S.; Maiti B.; Chanda K. A Decade update on benzoxazoles, a privileged scaffold in synthetic organic chemistry. Synlett 2017, 28, 521–541. 10.1055/s-0036-1588671. [DOI] [Google Scholar]
- Kumar R.; Selvam C.; Kaur G.; Chakraborti A. K. microwave-assisted direct synthesis of 2-substituted benzoxazoles from carboxylic acids under catalyst and solvent-free conditions. Synlett 2005, 9, 1401–1404. 10.1055/s-2005-868509. [DOI] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: A useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Park W.-Y.; Hong B.-J.; Lee J.; Choi C.; Kim M.-Y. H3K27 Demethylase JMJD3 employs the NF-kB and BMP signaling pathways to modulate the tumor microenvironment and promote melanoma progression and metastasis. Cancer Res. 2016, 76, 161–170. 10.1158/0008-5472.CAN-15-0536. [DOI] [PubMed] [Google Scholar]
- Williams K.; Christensen J.; Rappsilber J.; Nielsen A. L.; Johansen J. V.; Helin K. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS One 2014, 9, e96545 10.1371/journal.pone.0096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. Y.; Wang X.; Wang H.; Chen D. J.; Li G. C.; Iliakis G.; Wang Y. Ku affects the ATM-dependent S phase checkpoint following ionizing radiation. Oncogene 2002, 21 (41), 6377–6381. 10.1038/sj.onc.1205782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.