

Abstract
The pressing demand for sustainable antitumor drugs prompted us to investigate 3-chloropiperidines as potential mustard-based anticancer agents. In this study, an explorative set of variously decorated monofunctional 3-chloropiperidines (M-CePs) was efficiently synthesized through a fast and affordable route providing high yields of pure racemates and enantiomers. Consistently with their reactivity, M-CePs were demonstrated to alkylate DNA in vitro. On a panel of carcinoma cell lines, M-CePs exhibited low nanomolar cytotoxicity indexes, which showed their remarkable activity against pancreatic cancer cells and in all cases performed strikingly better than the chlorambucil control. Very interestingly, stereochemistry modulated the activity of M-CePs in unexpected ways, pointing to additional molecular mechanisms of action beyond the direct damage of genomic DNA. This encouraging combination of efficacy and sustainability suggests they are valid candidates for anticancer agent development.
Keywords: Monofunctional 3-chloropiperidines, alkylating agents, DNA cleavage, pancreatic cancer cells, chirality
For the hardly predictable mechanisms of tumor onset and the extremely variegated responses to state-of-the-art treatments, cancer represents one of the most arduous and challenging diseases of our time. Among the different anticancer drugs, DNA-alkylating agents for decades have constituted a valid therapeutic alternative. The high clinical efficacy of one of the main classes of alkylating agents, nitrogen mustards, makes them the current choice for the first-line treatment of different tumor types.1−4 However, severe side effects and incoming mechanisms of drug resistance limit the therapeutic value of such compounds, and novel effective leads are required.5,6 In this vein, we recently sought to discover new chemotherapeutics characterized by simple syntheses, an ease of ample decoration of the chemical scaffold, and a mechanism of action whose therapeutic viability had been well established by clinical practice. From the endeavor to simplify the structure of a complex natural compound with antineoplastic activity, a wide library of 3-chloropiperidines was obtained through an affordable synthetic process. In previous works, thorough investigation of the bifunctional chloropiperidine derivatives revealed efficient DNA-alkylating properties, leading to direct strand cleavage at guanine nucleotides and indirect effects on the human topoisomerase II enzyme.7−10 In this study, to further explore the chemical space of piperidine mustard-based molecules and to obtain new insights into their medicinal chemistry value, we analyzed a set of compounds bearing only a single reactive center. Our explorative series of monofunctional chloropiperidines (M-CePs) is characterized by a common 3-chloropiperidine ring with alternative substituents at the nitrogen atom, i.e., aliphatic as in 1 and 2 or aromatic as in 3 (Figure 1a). Compounds 1–3 can be easily synthesized as racemic mixtures through a fast and accessible route providing high yields and purities. In particular, heterocycles 1 and 2 were prepared as reported previously.11,12 For the synthesis of 3, dimethylpentenal was reductively aminated using benzylic amine. N-chlorination of this amine with NCS followed by iodide-catalyzed cyclization produced the desired 3-chloropiperidine (Scheme 1, I).13
Figure 1.

Chemical structures of (a) the racemic monofunctional 3-chloropiperidines and (b) the enantiomerically pure compounds.
Scheme 1. Synthesis of 3-Chloropiperidine 3 (I) and Enantiomers d-1 and l-1 (II).

Conditions: (a) benzylic amine, NaBH(OAc)3, CH2Cl2, 0 °C; (b) NCS, CH2Cl2, 0 °C; (c) catalytic Bu4NI, CHCl3, 60 °C; (d) LiAlH4, THF; (e) butyl iodide, K2CO3, THF, reflux; (f) SOCl2, pyridine, CH2Cl2, 0 °C.
In addition to the racemic heterocycles prepared by iodide-catalyzed cyclization of unsaturated N-chloroamines, we synthesized the enantiomerically pure 3-chloropiperidines D-1 and L-1 (Figure 1b) using a different procedure (Scheme 1, II). In particular, d-1 and l-1 were obtained starting from enantiomerically pure proline (d- and l-proline, respectively). At first, the amino acid was reduced to prolinol followed by N-alkylation. Next, the substitution of the hydroxyl group using thionyl chloride directly led to the 3-chloropiperidines. This reaction proceeds via a bicyclic aziridinium ion, opened through a stereoconvergent backside attack, such that the obtained 3-chloropiperidines were enantiomerically pure.
Following the workflow adopted for the bifunctional analogues, M-CePs were investigated to assess their alkylating activity.8−10 Along with compounds 1–3, we tested enantiomers d-1 and l-1 to explore the effect of stereogenic centers on the reactivity of M-CePs. As a widely used nitrogen mustard compound, chlorambucil was also tested as a reference drug for comparative purposes. The DNA cleavage assay evaluated the ability of compounds to induce changes in supercoiled plasmid (SC) electrophoretic mobility: the alkylation of DNA followed by single- or double-strand breakage leads to rearrangements in the topology of the plasmid, generating the open circular (OC) and/or linearized (L) forms with distinct mobilities in an agarose gel. In our initial assay, supercoiled plasmid pBR322 was incubated with the M-CePs for 2 h at 37 °C. As shown in Figure 2a, the test compounds showed a limited reactivity, a different outcome when considering the marked potency exhibited in the same assays by selective members of the bifunctional series.8−10 Under these conditions, the chlorambucil control was shown to be inactive at all concentrations, while only compounds 2 and 3 at the highest concentration (50 μM) caused the conversion of supercoiled DNA into the OC form. Compound 1 and its pure enantiomers were inactive, thus indicating that the double methylation of the piperidine ring prompts the reactivity of M-CePs: the inductive effect of methyl groups might stabilize the aziridinium ion, thus accelerating its formation and the subsequent nucleophilic attack. In addition, the two methyl groups induce an angle contraction (Thorpe–Ingold effect), leading to a faster aziridinium ion formation.14 These results are consistent with the relative percentage of aziridinium ion and hydroxyl-substituted species detected by electrospray ionization mass spectrometry (ESI-MS) upon incubation of a water solution of 1 and 2 for 2 h at 37 °C in the absence of the nucleic acid (Figure S1).
Figure 2.

DNA cleavage activity of 3-chloropiperidines after incubation for (a) 2 h and (b) 18 h. The supercoiled pBR322 plasmid (120 ng) was incubated with increasing concentrations (5 and 50 μM) of the compounds (1, 2, 3, d-1, l-1, and chlorambucil) at 37 °C for 2 and 18 h in BPE buffer. Cleavage of supercoiled DNA (SC) into its open circular (OC) and linearized (L) forms was analyzed by 1% agarose electrophoresis in TAE buffer. DNA was stained with GelRed. C, supercoiled plasmid control; Chl, chlorambucil.
In light of the limited cleavage observed upon a short incubation with the DNA substrate (Figure 2a), the time-dependent reactivity of M-CePs was further investigated by prolonging the incubation to 18 h. Under these conditions, all racemates were shown to efficiently react with DNA causing the nicking and linearization of the substrate at 5 μM and its fragmentation at 50 μM, once more performing better than the control chlorambucil (Figure 2b). Though the racemate 1 showed activity, the ring methylation was confirmed to enhance potency as in compound 2 and, with a different nitrogen substituent, also in compound 3. With regard to d-1 and l-1, we report here for the first time marked differences in reactivity between pure enantiomers of this class of nitrogen mustard derivatives. As shown in Figure 2b, the R configuration of l-1 strikingly favors plasmid cleavage over d-1 bearing the S conformation in position 3. While the former enantiomer is more active than the racemate 1, d-1 was shown to be inactive even after prolonged incubation.
The plasmid cleavage observed in the electrophoretic analysis is typically connected to DNA alkylation, which causes destabilization of the nucleobases, formation of abasic sites, and phosphodiester bond hydrolysis, finally resulting in single- or double-strand breaks.15,16 To prove such a mechanism of action (MOA), we further investigated the reactivity of M-CePs toward DNA by ESI-MS. The rather inactive compound 1 was tested in comparison to the most active analogue 2 (Figure 1). The double-stranded oligodeoxynucleotide substrate (dsDNA) replicates the sequence used in our previous studies.7 Experiments were performed as a function of concentration and time, conforming to the conditions employed in gel electrophoresis experiments, i.e., incubating dsDNA with either 5 or 50 μM compound, followed by incubation at 37 °C for 2 or 18 h. In accordance with the DNA cleavage activity seen on the plasmid, we observed evident DNA modifications induced by the test compounds at the larger dose. Figure 3 shows representative ESI-MS spectra of the reaction mixtures resulting from the incubation of 50 μM compound 1 or compound 2 with the nucleic acid substrate after 2 and 18 h. Only the region containing the 6– charge state is shown for the sake of clarity. By comparing the alkylating activity of 1 and 2, we observe a substantial difference in the reactivity of the two analogues toward the DNA substrate. After incubation for 2 h in the presence of derivative 1 (Figure 3a), we recognized exclusively the signal corresponding to the unreacted DNA substrate (labeled [dsDNA-6H]6–), whereas additional peaks corresponding to 3-chloropiperidine–DNA adducts were detected in the presence of compound 2 (Figure 3b). By matching experimental masses with values calculated from the dsDNA sequence and the structure of 2, we were able to identify each peak. Additional products were also observed with masses that were 133.04 units lower than that of the unreacted substrate, which were consistent with the formal loss of a G nucleobase to leave an abasic site produced by hydrolytic attack (Figure 3b). Table S1 summarizes experimental and calculated masses and provides the assignment of the various species detected in Figure 3. The results revealed that the initial DNA alkylation was directed toward guanine residues, which represent the alkylation site preferred by nitrogen mustards as well as by the bifunctional chloropiperidine derivatives.7,9 The presence in the dsDNA substrate of several sites susceptible to attack by M-CePs (i.e., guanine residues) explains the identification of multiple adducts, while the fast nature of the depurination process can account for the simultaneous detection of species containing alkylation products and abasic sites. The same type of reactions was monitored for ≤18 h to assess the effects of incubation time on product distribution. Longer incubation intervals promoted the formation of adducts and of abasic lesions by hydrolysis of alkylated guanine nucleobases in the presence of either compound 1 or 2 (panel c or d, respectively, of Figure 3). Compound 2 produced more abundant DNA damage compared to analogue 1, confirming that the presence of the methyl groups in the 3-chloropiperidine ring positively affects the reactivity of M-CePs toward DNA.
Figure 3.

ESI-MS spectra of the reaction mixture obtained by incubating the duplex dsDNA with compounds 1 and 2 both at 50 μM for 2 h (a and b, respectively) and 18 h (c and d, respectively) at 37 °C. The analyses were performed in 150 mM ammonium acetate. Lower-intensity signals near free/bound species consist of sodium and ammonium adducts. For the sake of clarity, the spectra show only the region containing the 6– charge state of the dsDNA. dsDNA, double bonds (=); adduct, red angles; base hydrolysis, winged V’s.
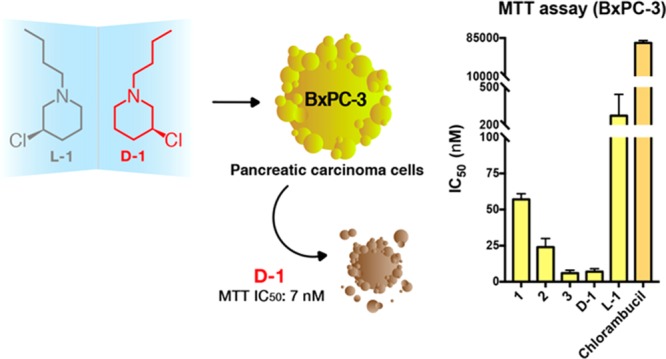
As a step forward in the evaluation of the anticancer potential of our agents, we investigated the activity of M-CePs on colon (HCT-15), ovarian (2008), and pancreatic (BxPC-3) carcinoma cells. Table 1 reports the calculated IC50 values of test compounds for the three cell lines after treatment for 72 h.
Table 1. MTT IC50 Values of Monofunctional 3-Chloropiperidines for HCT-15, 2008, and BxPC-3 Cell Lines.
| MTT
assay IC50 value (nM)a |
|||
|---|---|---|---|
| HCT-15 | 2008 | BxPC-3 | |
| 1 | 474 ± 110 | 329 ± 40 | 57 ± 4 |
| 2 | 254 ± 10 | 174 ± 10 | 24 ± 6 |
| 3 | 300 ± 81 | 107 ± 10 | 6 ± 2 |
| d-1 (S) | 181 ± 80 | 102 ± 30 | 7 ± 2 |
| l-1 (R) | 5640 ± 3103 | 4600 ± 1490 | 274 ± 170 |
| Chlb | 49700 ± 3310 | 12500 ± 2050 | 75300 ± 5110 |
MTT IC50 values with the associated standard deviations of test compounds on HCT-15, 2008, and BxPC-3 cancer cell lines after treatment for 72 h. IC50 values were calculated by a four-parameter logistic model (P < 0.05).
Chlorambucil.
All of the tested compounds exhibited a very promising cytotoxic effect on the selected panel of cancer cells, especially when seen in comparison to the effect of the chlorambucil control. IC50 values of all of the racemates are in the range of 100–400 nM in the case of colorectal and ovarian tumor cells (2008 and HCT-15 tumor cell lines), at least 2 orders of magnitude lower than that of the reference drug (IC50 for chlorambucil with HCT-15, 49.7 μM; IC50 for chlorambucil with 2008, 12.5 μM). Very interestingly, the cytotoxicity of 1–3 toward the pancreatic carcinoma cell line tested was particularly striking, exhibiting IC50 values in the range 6–60 nM (BxPC-3 cell line). This tropism becomes particularly relevant in light of the scarce susceptibility of pancreatic tumor cells to chlorambucil (IC50 for chlorambucil with BxPC-3, 75.3 μM). As expected from the reactivity observed toward DNA in vitro, 2 was more cytotoxic than 1, suggesting that methyl substituents in the 3-chloropiperidine ring enhance the reactivity and consequent cytotoxicity of the former analogue. The introduction of an N-benzyl substituent further stimulates the cytotoxicity of compound 3 especially on BxPC-3 cells.
The analysis of the cytotoxicity of enantiomers d-1 and l-1 in the three cancer cell lines was revealing. As shown in Table 1, d-1 was the most active compound, again with a clear tropism for pancreatic cancer cells, while its enantiomer, l-1, was less cytotoxic, with an eudismic ratio of ∼40 in the case of the BxPC-3 cells. It has to be noted, however, that although it is the least active M-CeP, l-1 is more active than chlorambucil in ovarian and colon cancer cells and ≤2 orders of magnitude more potent than the control in the pancreatic cancer cell line. These results are particularly relevant considering that pancreatic cancer represents one of the most insidious tumor types frequently associated with a poor prognosis for the affected patients.
In addition to the low nanomolar indexes of cytotoxicity, it is interesting to note what these findings reveal when considered alongside the evidence of DNA alkylation and cleavage discussed above. Unexpectedly, d-1 turned out to be the most cytotoxic compound of the entire series, although it was inactive in the DNA cleavage assay. On the contrary, its mirror image, l-1, found to efficiently nick and fragment the plasmid in vitro, happened to be much less cytotoxic.
To gain more insights into the contribution of lipophilicity of the different M-CePs to their cytotoxicity, we performed the non-cell-based parallel artificial membrane permeation assay (PAMPA).17 The PAMPA simulates the passive diffusion of analytes through biological membranes by employing a 96-well sandwich device endowed with a filter pretreated with a specific lipid mixture. Compound solutions are added to a donor well, while the artificial membrane at the bottom of the well allows permeation toward the acceptor well according to the compounds’ physicochemical properties. Ultraviolet–visible (UV–vis) analysis is employed for a fast quantification of the analyte in each compartment. Prior to our PAMPA analysis, we adjusted the typical assay conditions taking into consideration the reactivity of M-CePs and their fast interconversion into different species, namely, the unreacted (U), aziridinium ion (N+), and hydroxylated (OH) species. None of these three species has an adequate UV–vis signal, so we resorted to UPLC coupled to ESI-MS for their separation and quantification. Moreover, we decreased the assay incubation time to minimize the occurrence of water-reacted species. As shown in Figure S2, after incubation for 1 h the test compounds were mostly present as unreacted species in the assay buffer. We then proceeded with PAMPA analysis. The aqueous compartments consisted of PBS (pH 7.4), and the membrane was pretreated with 1% l-α-phosphatidylcholine in n-dodecane. Results reported in Figure S3 show the compartment distribution for all M-CePs. The racemic mixtures 2 and 3 (predicted logP values of 3.43 and 3.92, respectively) were faster than 1 (predicted logP of 2.49) in leaving the donor compartment after incubation for 1 h, which is consistent with their increased lipophilicity. However, compounds 2 and 3 were barely detected in the acceptor compartment, suggesting their entrapment in the phospholipidic layer. Longer incubation times allowed us to exclude their irreversible interaction with membrane components (data not shown). Interestingly, the compartment distribution profiles of the two enantiomers after 1 h reveal that l-1 is appreciably faster to diffuse than d-1, suggesting a different pattern of interaction with the chiral l-α-phosphatidylcholine membrane component. The chirality of the artificial membrane used in the PAMPA assay may selectively favor or hinder the passive permeation of chiral compounds. Because d-1 is the most cytotoxic but least permeable enantiomer, PAMPA analysis of the chiral compounds suggests the involvement of active mechanisms of uptake into cells.
The incongruence between the relative potencies of DNA cleavage and the actual cytotoxicity of the pure enantiomers, although pointing to the value of M-CePs as potential anticancer compounds, partially questions our starting hypothesis about the “intended” MOA of these reactive molecules. To clarify whether the unexpected outcome may depend on concerted activities, M-CePs were further evaluated for their capability to inhibit the DNA decatenation of human topoisomerase IIα (hTopoIIα), a well-known therapeutic target overexpressed in tumors. This enzyme is a fundamental regulator of the cellular dynamics involved in the maintenance of genome integrity and in the modulation of chromatin accessibility during replication and transcription.18−21 The rationale for this hypothesis comes from our previous work that proved that DNA lesions induced by bifunctional analogues impaired the decatenating activity of the enzyme.7 However, the topoisomerase II decatenation assay performed by incubating the substrate DNA with all of the new M-CePs for 2 and 18 h at 37 °C showed in all cases very poor activity: only compounds 2 and 3 partially inhibited the enzyme at the highest concentration (50 μM) and longer times (data not shown). These results rule out the hypothesis that hTopoIIα represents an extra target of inhibition by M-CePs, strengthening their differences with the bifunctional analogues and leaving open the possibility of additional mechanisms being responsible for the cytotoxicity of these promising chemotherapeutics. As highlighted by preliminary ESI-MS experiments with RNA substrates (Figure S4), macromolecular targets besides DNA might be intercepted by M-CePs in cells and their future investigation could delineate more exhaustively the MOA of the new derivatives. On the other hand, diversified DNA repair mechanisms are likely to account for the enhanced susceptibility of tested tumor cells to M-CeP alkylators and their preferential tropism toward the BxPC-3 line. In fact, imbalances in the base excision repair (BER) of N-alkyl lesions and in other mechanisms of recombinational repair represent consolidated factors that affect genome integrity that contribute to the cellular response to DNA alkylation damage.22 In this vein, we plan to analyze the repair patterns elicited in cancer cells exposed to M-CePs to thoroughly dissect the basis of their enhanced antiproliferative effect.
In conclusion, this study led to the discovery of a promising set of monofunctional 3-chloropiperidines successfully obtained through an efficient and affordable synthetic path. All compounds were shown to be highly active on carcinoma cells and particularly cytotoxic against the pancreatic BxPC-3 tumor cell line. Interestingly, the direct damage observed on isolated DNA is not sufficient to explain their nanomolar cytotoxicity, especially when considering the enantiomeric couple. A key determinant of cell sensitivity to alkylating agents is the carrier-mediated cell uptake, typically involved in mechanisms of resistance to nitrogen mustards.23 In this sense, differences in the cellular accumulation of the analyzed enantiomers might exist and need to be elucidated as a possible reason for their unexpected cytotoxicity profile. Finally, given the precious information derived from the analysis of the enantiomeric couple of d-1 and l-1, we plan to synthesize and test the pure enantiomers of all compounds to expand our library of monofunctional 3-chloropiperidines, to gain complete structure–activity relationships, and to identify leads with improved antitumor properties against different models of pancreatic cancers.
Experimental Section
Chemistry
All solvents were purified by distillation prior to use and in case of anhydrous solvents dried and stored under a nitrogen atmosphere. Commercially available reagents were used as supplied if not stated otherwise. Syntheses using anhydrous solvents were carried out under Schlenk conditions. For purification by flash chromatography, silica gel 60 (Merck) was used. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance II 400 spectrometer (1H at 400 MHz; 13C at 100 MHz) in deuterated solvents. Chemical shifts were determined by reference to the residual solvent signals. High-resolution ESI mass spectra were recorded in methanol using an ESImicroTOF spectrometer (Bruker Daltonics). The adopted synthetic procedures provided good yields of highly pure compounds (at least 95% purity of the final products as judged by NMR and in one case elemental analysis). Detailed synthetic procedures are reported in the Supporting Information.
Bioassays
Chemical Reagents
Dilutions of M-CePs were freshly prepared from a 10 mM DMSO stock in Milli-Q water and were instantly reacted with the DNA or RNA substrate to avoid the typical quenching effects of the aqueous environment. Agarose Low EEO was purchased from Eppendorf. The other chemical reagents, including salts and solvents, were purchased from Sigma-Aldrich.
DNA Cleavage Assay
Compounds were evaluated for their ability to induce cleavages on supercoiled plasmid pBR322 via the electrophoretic cleavage assay. Experiments were performed by exploiting previously reported materials and protocols at the specified incubation time.8−10
Mass Spectrometric Analysis
The oligonucleotides (dsDNA and ssRNA) used for mass spectrometry analysis were purchased from Integrated DNA Technologies (IDT) and stored as reported in previous works.7 Samples containing a final nucleic acid substrate concentration of 2 μM were added with the selected compound at a final concentration of either 5 or 50 μM in BPE buffer at 37 °C for 2 or 18 h, as indicated in the text. All samples were analyzed by direct infusion ESI on Thermo Fisher Scientific LTQ-Orbitrap Velos mass spectrometer as previously reported for the bifunctional derivatives.7
Decatenation Assay
The inhibition of the activity of human topoisomerase IIα by M-CePs was evaluated through the kinetoplast DNA (kDNA) decatenation assay using a kit provided by Inspiralis Ltd. and following the protocol reported previously.7,24,25
Cytotoxicity Assays
Colon (HCT-15) and pancreatic (BxPC-3) carcinoma cell lines were obtained from American Type Culture Collection. The human ovarian 2008 cancer cell line was kindly provided by G. Marverti (Department of Biomedical Science, University of Modenaand Reggio Emilia, Modena, Italy). Cell lines were maintained in logarithmic phase at 37 °C in a 5% carbon dioxide atmosphere using RPMI-1640 medium (Euroclone) containing 10% fetal calf serum (Euroclone), antibiotics (50 units/mL penicillin and 50 μg/mL streptomycin), and 2 mM l-glutamine. The MTT assay was performed following previously reported protocols.26,27 Test compounds were dissolved in DMSO, and a calculated amount of the drug solution was added to the cell growth medium to a final solvent concentration of 0.5%, which had no detectable effect on cell killing.
PAMPA Assay
The parallel artificial membrane permeation assay (PAMPA) was performed as suggested by Kansy et al., with minor modifications.17 Plates were purchased from Sigma-Aldrich as well as the components of the lipid mixture, which consisted of 1% l-α-phosphatidylcholine in n-dodecane. Compound dilutions prepared in PBS (pH 7.4) at a concentration of 500 μM from 10 mM DMSO stocks constituted the solution applied to donor wells. PBS containing 5% DMSO was employed as receiver buffer added to acceptor wells. Upon incubation for 1 h at room temperature, aliquots from the initial solution, donor, and acceptor compartments were analyzed by UPLC-MS in the presence of 20 μM caffeine as an internal standard to permit relative quantification of the compound. Areas under the peak corresponding to the masses of interest were calculated with MassLynx software (Waters). The reported compartment distribution for test compounds considered the sum of unreacted and reacted species detected in the donor, acceptor, and membrane compartments (the last one indirectly determined as the difference between the relative amount of compound in the initial solution and in the aqueous compartments). UPLC-MS analysis was performed in positive ion mode using a Xevo G2-XS Qtof instrument (Waters), coupled to an Acquity UPLC BEH C18 2.1 mm × 50 mm, 130 Å column (Waters). The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The column temperature was set at 25 °C, and the injection volume was 10 μL. Samples coming from PAMPA plates in PBS and 5% DMSO were diluted 1:10 in the UPLC mobile phase prior to being loaded. The applied gradient program (0.2 mL/min flow rate) was as follows: 2 to 98% B from 1 to 11 min, 98% B from 11 to 14 min, 98 to 2% B from 14 to 14.5 min, and 2% B from 14.5 to 18.5 min. ESI-MS analysis was performed at a desolvation temperature of 450 °C and using a capillary voltage of 3000 V.
Acknowledgments
The authors thank Dr. M. Bellini for technical assistance in performing UPLC-MS analysis.
Glossary
Abbreviations
- M-CePs
monofunctional 3-chloropiperidines
- NCS
N-chlorosuccinimide
- THF
tetrahydrofuran
- SC
supercoiled plasmid
- OC
open circular plasmid form
- L
linearized plasmid form
- ESI-MS
electrospray ionization mass spectrometry
- MOA
mechanism of action
- dsDNA
double-stranded DNA
- G
guanine nucleobase
- IC50
50% inhibitory concentration
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- SD
standard deviation
- PAMPA
parallel artificial membrane permeation assay
- UPLC
ultra-performance liquid chromatography
- hTopoIIα
human topoisomerase IIα
- BER
base excision repair
- DMSO
dimethyl sulfoxide
- ssRNA
single-stranded RNA
- kDNA
kinetoplast DNA.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00580.
Relative percentages of the reaction species detected by ESI-MS upon incubation of 1 and 2 for 2 h at 37 °C (Figure S1), names, descriptions, and experimental and calculated masses for species detected in the ESI-MS spectra reported in Figure 3 (Table S1), relative percentages of the reaction species detected by UPLC-MS upon incubation of test compounds for 1 h at room temperature (RT) in PBS buffer (Figure S2), PAMPA assay compartment distribution of M-CePs 1, 2, 3, d-1, l-1, and chlorambucil (Chl) upon incubation for 1 h at room temperature (RT) (Figure S3), ESI-MS spectra of the reaction mixture obtained by incubating the ssRNA with 50 μM compound 2 for 18 h at 37 °C (Figure S4), and detailed synthetic procedures (PDF)
Author Present Address
∥ F.K.: University Hospital Bonn, Institute for Experimental Immunology, Sigmund-Freud-Str. 25, 53127 D-Bonn, Germany.
A.S. has received funding from the European Union’s Horizon 2020 Research and Innovation programme under Marie Skłodowska-Curie Grant Agreement 751931.
The authors declare no competing financial interest.
Supplementary Material
References
- Avendaño C.; Menendez J. C.. Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Elsevier Science: Amsterdam, 2015. [Google Scholar]
- Hurley L. H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2 (3), 188–200. 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- Rajski S. R.; Williams R. M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98 (8), 2723–2796. 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- Siddik Z. H.Mechanisms of Action of Cancer Chemotherapeutic Agents: DNA-Interactive Alkylating Agents and Antitumour Platinum-Based Drugs. In The Cancer Handbook, 1st ed.; Alison M. R., Ed.; John Wiley and Sons Ltd.: Hoboken, NJ, 2002. [Google Scholar]
- Noll D. M.; Mason T. M.; Miller P. S. Formation and repair of interstrand cross-links in DNA. Chem. Rev. 2006, 106 (2), 277–301. 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasci L.; Xu Z. Y.; Bello V.; Aloyz R. The role of DNA repair in nitrogen mustard drug resistance. Anti-Cancer Drugs 2002, 13 (3), 211–220. 10.1097/00001813-200203000-00002. [DOI] [PubMed] [Google Scholar]
- Sosic A.; Zuravka I.; Schmitt N. K.; Miola A.; Gottlich R.; Fabris D.; Gatto B. Direct and Topoisomerase II Mediated DNA Damage by Bis-3-chloropiperidines: The Importance of Being an Earnest G. ChemMedChem 2017, 12 (17), 1471–1479. 10.1002/cmdc.201700368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuravka I.; Roesmann R.; Sosic A.; Gottlich R.; Gatto B. Bis-3-chloropiperidines containing bridging lysine linkers: Influence of side chain structure on DNA alkylating activity. Bioorg. Med. Chem. 2015, 23 (6), 1241–1250. 10.1016/j.bmc.2015.01.050. [DOI] [PubMed] [Google Scholar]
- Zuravka I.; Roesmann R.; Sosic A.; Wende W.; Pingoud A.; Gatto B.; Gottlich R. Synthesis and DNA cleavage activity of Bis-3-chloropiperidines as alkylating agents. ChemMedChem 2014, 9 (9), 2178–2185. 10.1002/cmdc.201400034. [DOI] [PubMed] [Google Scholar]
- Zuravka I.; Sosic A.; Gatto B.; Gottlich R. Synthesis and evaluation of a bis-3-chloropiperidine derivative incorporating an anthraquinone pharmacophore. Bioorg. Med. Chem. Lett. 2015, 25 (20), 4606–4609. 10.1016/j.bmcl.2015.08.042. [DOI] [PubMed] [Google Scholar]
- Noack M.; Göttlich R. Iodide-Catalysed Cyclization of Unsaturated N-Chloroamines: A New Way to Synthesise 3-Chloropiperidines. Eur. J. Org. Chem. 2002, 2002 (18), 3171–3178. 10.1002/1099-0690(200209)2002:18<3171::AID-EJOC3171>3.0.CO;2-L. [DOI] [Google Scholar]
- Göttlich R. Copper(I)-Catalyzed Intramolecular Addition of N-Chloroamines to Double Bonds under Aprotic Conditions. Towards a Stereoselective Catalytic Radical Reaction. Synthesis 2000, 2000 (11), 1561–1564. 10.1055/s-2000-7605. [DOI] [Google Scholar]
- Göttlich R.; Noack M. Samarium(II)-iodide catalysed addition of N-chloroamines to double bonds, an iodide-catalysed reaction. Tetrahedron Lett. 2001, 42 (44), 7771–7774. 10.1016/S0040-4039(01)01620-3. [DOI] [Google Scholar]
- Bachrach S. M. The gem-dimethyl effect revisited. J. Org. Chem. 2008, 73 (6), 2466–2468. 10.1021/jo702665r. [DOI] [PubMed] [Google Scholar]
- Gates K. S. An overview of chemical processes that damage cellular DNA: spontaneous hydrolysis, alkylation, and reactions with radicals. Chem. Res. Toxicol. 2009, 22 (11), 1747–1760. 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates K. S.; Nooner T.; Dutta S. Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem. Res. Toxicol. 2004, 17 (7), 839–856. 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- Kansy M.; Senner F.; Gubernator K. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998, 41 (7), 1007–10. 10.1021/jm970530e. [DOI] [PubMed] [Google Scholar]
- Brill S. J.; DiNardo S.; Voelkel-Meiman K.; Sternglanz R. DNA topoisomerase activity is required as a swivel for DNA replication and for ribosomal RNA transcription. NCI Monographs 1987, 4, 11–15. [PubMed] [Google Scholar]
- Nitiss J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9 (5), 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M. O.; Larsen M. K.; Barthelmes H. U.; Hock R.; Andersen C. L.; Kjeldsen E.; Knudsen B. R.; Westergaard O.; Boege F.; Mielke C. Dynamics of human DNA topoisomerases IIalpha and IIbeta in living cells. J. Cell Biol. 2002, 157 (1), 31–44. 10.1083/jcb.200112023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- Fu D.; Calvo J. A.; Samson L. D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12 (2), 104–120. 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscow J. A.; Swanson C. A.; Cowan K. H. Decreased melphalan accumulation in a human breast cancer cell line selected for resistance to melphalan. Br. J. Cancer 1993, 68 (4), 732–737. 10.1038/bjc.1993.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane A.; Sullivan D. M. DNA topoisomerase II-catalyzed DNA decatenation. Methods Mol. Biol. 2000, 95, 13–23. 10.1385/1-59259-057-8:13. [DOI] [PubMed] [Google Scholar]
- Furlanetto V.; Zagotto G.; Pasquale R.; Moro S.; Gatto B. Ellagic acid and polyhydroxylated urolithins are potent catalytic inhibitors of human topoisomerase II: an in vitro study. J. Agric. Food Chem. 2012, 60 (36), 9162–9170. 10.1021/jf302600q. [DOI] [PubMed] [Google Scholar]
- Montagner D.; Gandin V.; Marzano C.; Erxleben A. DNA damage and induction of apoptosis in pancreatic cancer cells by a new dinuclear bis(triazacyclonane) copper complex. J. Inorg. Biochem. 2015, 145, 101–107. 10.1016/j.jinorgbio.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Alley M. C.; Scudiero D. A.; Monks A.; Hursey M. L.; Czerwinski M. J.; Fine D. L.; Abbott B. J.; Mayo J. G.; Shoemaker R. H.; Boyd M. R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48 (3), 589–601. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.