

Abstract

Among human carbonic anhydrase (CA) inhibitors, the α,γ-diketocarboxylic acids and esters are still poorly investigated. Here, we report the first compounds of this class (1–6) acting as potent inhibitors at low nanomolar level against the cancer-related human CA IX and XII, and 2–3 magnitude orders selective toward the cytosolic isoforms hCA I and II. At enzymatic level, the α,γ-diketoacids 1–3 were more effective inhibitors compared to the corresponding ethyl esters 4–6. The phenyl- and α-naphthyl-containing compounds (1, 3, 4, and 6) behaved as dual hCA IX/XII inhibitors, while the β-naphthyl analogues (2 and 5) exhibited hCA IX-selective inhibition. In MG63 and HOS osteosarcoma (OS) cell lines, the ethyl esters 5 and 6 displayed dose-dependent reduction of viability and proliferation after 72 h treatment, with 6 being more potent than 5 likely for its dual hCA IX/XII inhibition.
Keywords: Metalloenzyme, carbonic anhydrase, carboxylic acid, selective inhibition, antitumor
Carbonic anhydrase (CA) inhibitors (CAIs) possess a range of pharmacologic applications for the treatment and prevention of diseases connected with enhanced secretion of electrolytes, pH regulation, metabolic modulation, etc.1−4 In fact, many CAIs are used as diuretics, antiglaucoma agents, antiepileptics, antiobesity, and antitumor drugs.1−4 In addition to primary sulfonamides and their isosters (sulfamates and sulfamides), several other chemotypes with CA inhibitory properties and diverse mechanisms of CA inhibition were reported in the last years.5,6 Indeed, sulfonamides and their congeners, as well as dithio-/monothiocarbamates, xanthates, hydroxamates, and some carboxylates coordinate the zinc ion from the enzyme active site potently inhibiting the CA catalytic activity.5,6 Compounds that inhibit CAs by anchoring to the zinc-coordinated water molecule are the phenols, polyamines, sulfocoumarins, some carboxylic acid esters, and the thioxocoumarins,7,8 whereas the coumarins show a distinct CA inhibition mechanism, acting as prodrug inhibitors, and after hydrolysis to the corresponding 2-hydroxycinnamic acids, occlude the entrance to the active site.9,10 The fourth inhibition mechanism, out of the active site binding, was also reported for a carboxylic acid derivative.11 Thus, from the above data it is possible to observe that the carboxylates and their derivatives may interact with these enzymes by at least four different inhibition mechanisms, making them of great interest for the design of inhibitors with diverse profiles for the many CA isoforms present in vertebrates (15 α-class isoforms) or other organisms (with seven distinct CA genetic families present in organisms all over the tree of life).5,6 Indeed, several drug design studies of carboxylic acids as CAIs were reported in the last years based on aromatic/heterocyclic scaffolds, with several such derivatives also being characterized by X-ray crystallographic techniques.11−14 In some cases, highly isoform-selective inhibitors were detected,12−14 as well as compounds with enhanced inhibitory activity against pathogenic versus human enzymes.14
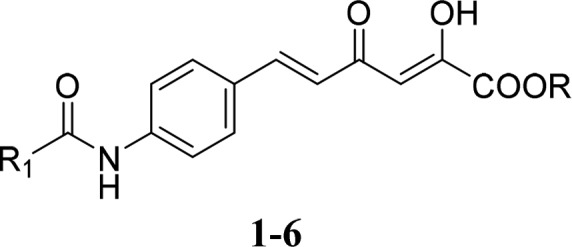
With the only exception of the carboxylic acid derivatives reported by Sechi et al., which showed inhibitory activity in the low micromolar/submicromolar range against a panel of human and fungal CAs,14 the α,γ-diketo acid scaffold has been poorly investigated for the interaction with CAs so far. Therefore, we designed a few 6-aromatically substituted-2,4-dioxo-5-hexenoic acids and their ethyl esters 1–6 (Figure 1) as potential CAIs, studied their inhibition mechanism by using kinetic and computational techniques, and evaluated their effects in two human osteosarcoma (OS) cell lines, MG63 and HOS, highly expressing the cancer-related CA isoforms IX and XII.
Figure 1.

New α,γ-diketocarboxylic acids and ethyl esters 1–6 as CAIs (R = H, ethyl; R1 = phenyl, α- or β-naphthyl).
The general synthetic pathway followed for the preparation of derivatives 1–6 is depicted in the Scheme 1. The (E)-4-(4-nitrophenyl)but-3-en-2-one 7, prepared as reported in the literature,15 was dissolved in ethanol and treated with a solution of stannous chloride dihydrate in concentrated hydrochloric acid at 0 °C to obtain the reduced product (E)-4-(4-aminophenyl)but-3-en-2-one 8, which was then subjected to acylation reaction with the appropriate commercial aroyl chloride in the presence of triethylamine in dry DCM at 0 °C in order to afford the intermediates 9–11. The compounds 4–6, obtained by treating 9–11 with diethyl oxalate in the presence of sodium ethoxide in dry THF at room temperature under nitrogen atmosphere, were finally hydrolyzed to the corresponding α,γ-diketocarboxylic acids 1–3 with 0.7 M lithium hydroxide in a mixture of THF and methanol.
Scheme 1. Synthetic Route to Compounds 1–6.

Reagents and conditions: (a) 2.8 M SnCl2 in 37% HCl, EtOH, 0 °C, 24 h; (b) appropriate aroyl chloride, Et3N, dry DCM, 0 °C, 2–24 h; (c) diethyl oxalate, EtONa, dry THF, N2 atmosphere, rt, 3–5 h; (d) 0.7 M LiOH, THF/MeOH, rt, 6–8 h.
Full experimental details regarding the synthesis and physicochemical characterization of final (1–6) and intermediate (8–11) compounds are outlined in Supporting Information.
The CA inhibition profile of compounds 1–6 was evaluated by applying a stopped flow CO2 hydrase assay16 against four physiologically significant CA isoforms (I, II, IX, and XII), in comparison with acetazolamide (AAZ) as reference drug.
The following SAR can be gathered from the inhibition data shown in Table 1: (i) The cytosolic hCA I and II are the least inhibited isoforms by α,γ-diketocarboxylic acids 1–3 and their ethyl esters 4–6. Most derivatives act as low micromolar inhibitors, with inhibition constant (KI) values spanning between 2.08 and 9.4 μM. Compounds 1, 3, and 6 do not inhibit hCA I below 10 μM, and 4 exhibits the same behavior against hCA II.
Table 1. Inhibitory Potencies of Derivatives 1–6 versus hCA I, II, IX, and XII.
|
KI (nM)a |
||||||
|---|---|---|---|---|---|---|
| cpd | R | R1 | hCA I | hCA II | hCA IX | hCA XII |
| 1 | H | Ph | >10000 | 5034 | 12.5 | 7.5 |
| 2 | H | β-naph | 8981 | 2084 | 11.3 | 767.6 |
| 3 | H | α-Naph | >10000 | 2580 | 12.8 | 8.7 |
| 4 | ethyl | Ph | 7331 | >10000 | 90.2 | 34.6 |
| 5 | ethyl | β-naph | 6178 | 9443 | 12.6 | 249.1 |
| 6 | ethyl | α-naph | >10000 | 8654 | 37.9 | 8.9 |
| AAZ | 250 | 12 | 25 | 5.7 | ||
Mean from three different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values).
Carboxylic acids and mostly esters are known to be relatively less effective CA inhibitors than sulfonamides, with most derivatives of these classes of compounds acting in the micromolar range.11−14 The present results against the cytosolic isoforms confirm these evidences. No significant difference stood out between acids 1–3 and esters 4–6 in the inhibition of hCA I. Conversely, hCA II was more than 2-fold better inhibited by compounds 1–3 than their ethyl esters. Switching the phenyl group to α- or β-naphthyl moiety does not affect the inhibitory properties against hCA I and II within the two subsets of molecules. (ii) The tumor-associated hCA IX and XII are potently inhibited by both subsets of derivatives. Compounds 1–6 inhibited hCA IX in the low nanomolar range, exhibiting KI values spanning between 11.3 and 90.2 nM. The nature of the outer aromatic ring does not affect the hCA IX inhibitory properties of acid derivatives 1–3, being their KI values not significantly diverse (11.3–12.8 nM). Solely the β-naphthyl ethyl ester 5 exhibits analog KI (12.6 nM) to carboxylic acids 1–3, whereas the switching of the aromatic moiety to the phenyl (4) and α-naphthyl (6) ones worsens the inhibitory activity by three- (37.9 nM) or 7-fold (90.2 nM), respectively. (iii) With the exception of the β-naphthyl derivatives 2 and 5 (KI values = 767.6 and 249.1 nM, respectively), the hCA XII is potently inhibited by the other screened derivatives that show comparable inhibitory properties in the low nanomolar range (7.5–34.6 nM) independently from the nature of the zinc binding group (ZBG). Anyway, it should be noted that the phenyl ester derivative 4 is 4-fold less effective than its carboxylic acid cognate 1. (iv) A clear selectivity of action against hCA IX and XII over the cytosolic hCA I and II stands out from the data in Table 1 for most of the reported derivatives, with β-naphthyl derivatives 2 and 5 that can be considered hCA IX-selective inhibitors.
The in silico assessment of the binding mode of compounds 2 and 5 into the hCA II and IX highlights that 2-hydroxy-4-oxohexa-2,5-dienoic acids, and their ethyl esters are able to occupy the catalytic region of the binding pockets by coordinating the zinc ion and interacting with residues nearby. Docking studies followed by a refinement in a VSGB solvent model show that the coordination occurs between the metal ion and the deprotonated carboxylic group of 2 or the oxo-group of the ethyl ester of 5 (Figure 2). The molecular architecture of the two active sites thoroughly affects the binding modes. The hCA II/IX Phe131/Val130 mutation modulates the H-bonds network that the 2-hydroxy-4-oxohexa-2,5-dien portions can form within the pockets. The presence of several H-bonds effectively reinforces the carboxylate coordination to the metal ion of 2 and 5 in hCA IX (Figure 2A,B). The carboxy or carbethoxy moieties of 2 and 5, respectively, accept two H-bonds from the backbone NH of Thr200 and Thr201. The side chain OH group of Thr201 is involved in an interesting pattern of H-bonds with the 2-hydroxy-4-oxohexa-2,5-diene portions of the ligands under investigation, acting both as donor and acceptor group. In particular, the OH group acts as both H-bond donor toward the 2-hydroxy and to the 4-oxo moieties of 2, while it participates to a three center H-bond involving the analogue deprotonated groups in 5. The ethyl moiety of 5 accommodates into the pocket lined by Val121, Val142, and Trp210. The naphthamidophenyl fragment of both molecules orients toward the hydrophobic half of hCA IX active site, with van der Waals interactions taking place with Val130, Asp131, and Arg129.
Figure 2.

Docking of 2 (A) and 5 (B) into hCA IX. Docking of 2 into hCA II (C).
The above-mentioned hCA II/IX Phe131/Val130 mutation makes the hCA II binding site less roomy if compared to that one of hCA IX preventing, de facto, the positioning of the ligand as described for hCA IX. Nonetheless, the carboxylates maintain the zinc-coordination and a H-bond with the backbone NH Thr200. The 2-hydroxy-4-oxohexa-2,5-diene portions lack the proper H-bond distances with Thr201 because of the rotation undergone by the ligands to accommodate the naphthamidophenyl core toward His64, Ans62, and Asn67 (Figure 1C). These evidence support the observed CA IX/II selective inhibition profiles. In fact, the binding mode of 2 within the hCA II active site prevents the coordination bond stabilization, which enhances the hCA IX inhibition efficacy of the compounds more than two-orders of magnitude if compared to that toward hCA II.
Chemotypes endowed with a selectivity ratio spanning between 2 and 3 orders of magnitude for hCA IX and XII over both I and II have a great potential as starting points for the design of novel CAIs as antitumor agents devoid of undesired side effect related to promiscuous activity. Since in a previous paper we have demonstrated that there is a strong rationale for the use of CA IX inhibitors in human OS models,17 we tested the β-naphthyl derivatives 2 and 5 together with the α-naphthyl ethyl ester 6 (0–100 μM, 72 h) in two different OS cell lines (MG63 and HOS) that highly express CA IX and/or XII (see Figure S1 in Supporting Information). Likely for the reduced cell permeability due to its acidic nature, 2 did not show any inhibitory effect on OS cell growth, while the two ethyl esters 5 and 6 affected MG63 and HOS cell viability in a dose-dependent way (Figure 3). In particular, 5 decreased by 50% the viability of both the tested cell lines at 50 μM, while 6 at 25 μM arrested the viability of MG63 and HOS cells at 16.4 and 19.7%, respectively, thus being the most potent of the two compounds.
Figure 3.

Cell viability assay. The treatment of OS cells with the two ethyl esters 5 and 6 for 72 h reduced cell viability in a dose-dependent way, as revealed by the indirect acid phosphatase viability assay. Treatment with 2 did not produce any cytotoxicity on OS cells.
In order to confirm this viability indirect test, the effectiveness of 5 and 6 was then evaluated by erythrosine B dye vital staining in both MG63 and HOS OS cell lines. Both compounds displayed strong antiproliferative effects in the low micromolar range after 72 h treatment (Figure 4), with 6 [IC50 values = 14.7 μM (MG63) and 15.6 μM (HOS)] being more potent than 5 [IC50 values = 45.8 μM (MG63) and 19.2 μM (HOS)]. When tested in the same cell lines to determine their putative effect on apoptosis induction through detection of caspase 3 and 7 activity, neither 6 (at 25 μM) or 5 (at 50 μM) induced any pro-apoptotic activity (see Figure S2 in Supporting Information). Thus, the decreased number of cells after compounds 5 and 6 treatment could be a consequence of a cell cycle arrest.
Figure 4.

Cell proliferation assay. The percentage of cell growth with respect to untreated cells is significantly lower (p < 0.05) after the treatment with compound 5 or 6 for all the tested doses.
The evidence that the dual CA IX/XII inhibitor α-naphthyl ester 6 resulted more effective than the CA IX-selective β-naphthyl regioisomer 5 in inhibiting the proliferation of both the OS cell lines suggests the importance of the simultaneous inhibition of both the hCA IX and XII isoforms, at least in this context.
In summary, we have reported herein a new series of α,γ-diketocarboxylic acids and ethyl esters (1–6) potent at nanomolar level and selective toward the human CA isoforms IX and XII, highly expressed in tumor cells, and scarcely potent against the cytosolic CAs I and II. This unique inhibitory profile displayed by 1–6 is different from previously reported α,γ-diketoacids and esters (see compounds 1–3, 6a,b in ref (14)), which showed unselective, low micromolar inhibition against hCA I, II, IX, and XII, with only one compound (6a in ref (14)) potent against hCA IX and XII at submicromolar level.14
In general, among the new α,γ-diketoacids the carboxylic acids 1–3 displayed similar or higher potency than the corresponding ethyl esters 4–6 against the hCA IX and XII, and the phenyl- and α-naphthyl-substituted compounds of the two series (1, 3, 4, and 6) behaved as dual hCA IX/XII inhibitors, while the β-naphthyl analogues (2 and 5) exhibited hCA IX-selective inhibition. Molecular modeling studies performed on 2 and 5 docked into hCA II and hCA IX highlighted their mechanism of action, due to coordination between the enzyme zinc ion and the carboxylate group of 2 or the carbonyl group of 5. In comparison with the binding mode within the hCA II active site, the better accommodation (more interactions and H-bonds) of the ligands into the hCA IX binding site accounted for their higher selectivity toward hCA IX. When tested in MG63 and HOS OS cells to determine their effects on cell viability, 2 was totally inactive likely due to its acidic nature (low cell permeability), while 5 and 6 displayed dose-dependent reduction of viability after 72 h treatment. As antiproliferative agents, the α-naphthyl ester 6 was more potent than the β-naphthyl analogue 5, probably because 6 was a dual hCA IX/XII inhibitor, while 5 exerted selective hCA IX inhibition.
Acknowledgments
This work was supported by funds from PRIN 2015 (prot.20152TE5PK) (to A.M.), AIRC IG 2016 (no. 19162) (to A.M.), and The Italian Ministry of the Health, Financial Support for Scientific Research (“5 per 1000 2015” (to N.B.).
Glossary
ABBREVIATIONS
- AAZ
acetazolamide
- CA
carbonic anhydrase
- CAI
carbonic anhydrase inhibitors
- OS
osteosarcoma
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00023.
Experimental procedures for the synthesis and physicochemical characterization of all new compounds; material and methods for CA inhibition assays, in silico studies, cell based assays (PDF)
Author Contributions
A.N., D.R., A.Mai, F.P., N.B., and C.T.S. assisted manuscript preparation, data interpretation, and project overview. A.L., D.T., and D.R., compound synthesis. A.N., P.G., and C.T.S., in vitro profiling for hCAs inhibition and molecular modeling. F.P., A.M., and N.B., biological assays on OS cell lines. D.R., A.Mai, and C.T.S. conceived the study and followed the whole project. All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Supuran C. T. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin. Ther. Pat. 2018, 28, 709–712. 10.1080/13543776.2018.1523897. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Supuran C. T. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin. Ther. Pat. 2018, 28, 729–740. 10.1080/13543776.2018.1508453. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin. Ther. Pat. 2018, 28, 713–721. 10.1080/13543776.2018.1519023. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. 10.1042/BCJ20160115. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. How many carbonic anhydrase inhibition mechanisms exist?. J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. 10.3109/14756366.2015.1122001. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discovery 2017, 12, 61–88. 10.1080/17460441.2017.1253677. [DOI] [PubMed] [Google Scholar]
- Lopez M.; Vu H.; Wang C. K.; Wolf M. G.; Groenhof G.; Innocenti A.; Supuran C. T.; Poulsen S. A. Promiscuity of carbonic anhydrase II. Unexpected ester hydrolysis of carbohydrate-based sulfamate inhibitors. J. Am. Chem. Soc. 2011, 133, 18452–18462. 10.1021/ja207855c. [DOI] [PubMed] [Google Scholar]
- Maresca A.; Temperini C.; Vu H.; Pham N. B.; Poulsen S. A.; Scozzafava A.; Quinn R. J.; Supuran C. T. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. 10.1021/ja809683v. [DOI] [PubMed] [Google Scholar]
- Davis R. A.; Vullo D.; Maresca A.; Supuran C. T.; Poulsen S. A. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1539–1543. 10.1016/j.bmc.2012.07.021. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio K.; Carradori S.; Monti S. M.; Buonanno M.; Secci D.; Vullo D.; Supuran C. T.; De Simone G. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem. Commun. (Cambridge, U. K.) 2015, 51, 302–305. 10.1039/C4CC07320G. [DOI] [PubMed] [Google Scholar]
- Langella E.; D’Ambrosio K.; D’Ascenzio M.; Carradori S.; Monti S. M.; Supuran C. T.; De Simone G. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of Carbonic Anhydrases. Chem. - Eur. J. 2016, 22, 97–100. 10.1002/chem.201503748. [DOI] [PubMed] [Google Scholar]
- Cadoni R.; Pala N.; Lomelino C.; Mahon B. P.; McKenna R.; Dallocchio R.; Dessì A.; Carcelli M.; Rogolino D.; Sanna V.; Rassu M.; Iaccarino C.; Vullo D.; Supuran C. T.; Sechi M. Exploring Heteroaryl-pyrazole carboxylic acids as human Carbonic Anhydrase XII inhibitors. ACS Med. Chem. Lett. 2017, 8, 941–946. 10.1021/acsmedchemlett.7b00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi M.; Innocenti A.; Pala N.; Rogolino D.; Carcelli M.; Scozzafava A.; Supuran C. T. Inhibition of α-class cytosolic human carbonic anhydrases I, II, IX and XII, and β-class fungal enzymes by carboxylic acids and their derivatives: new isoform-I selective nanomolar inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5801–5806. 10.1016/j.bmcl.2012.07.094. [DOI] [PubMed] [Google Scholar]
- Shokat K.; Uno T.; Schultz P. G. mechanistic studies of an antibody-catalyzed elimination-reaction. J. Am. Chem. Soc. 1994, 116, 2261–2270. 10.1021/ja00085a004. [DOI] [Google Scholar]
- Khalifah R. G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [PubMed] [Google Scholar]
- Perut F.; Carta F.; Bonuccelli G.; Grisendi G.; Di Pompo G.; Avnet S.; Sbrana F. V.; Hosogi S.; Dominici M.; Kusuzaki K.; Supuran C. T.; Baldini N. Carbonic anhydrase IX inhibition is an effective strategy for osteosarcoma treatment. Expert Opin. Ther. Targets 2015, 19, 1593–1605. 10.1517/14728222.2016.1086339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.