

Abstract
The role of water in protein-ligand binding has been an intensely studied topic in recent years; however, how ligand protonation state change perturbs water has not been considered. Here we show that water dynamics and interactions can be controlled by the protonation state of ligand using continuous constant pH molecular dynamics simulations of two closely related model systems, β-secretase 1 and 2 (BACE1 and BACE2), in complex with a small-molecule inhibitor. Simulations revealed that, upon binding, the inhibitor pyrimidine ring remains deprotonated in BACE1 but becomes protonated in BACE2. Pyrimidine protonation results in water displacement, rigidification of the binding pocket, and shift in the ligand binding mode from water-mediated to direct hydrogen bonding. These findings not only support but also rationalize the most recent structure-selectivity data in BACE1 drug design. Binding-induced protonation state changes arc likely common; our work offers a glimpse at how modeling protein-ligand binding while allowing ligand titration can further advance the understanding of water and structure-based drug design.
Graphical Abstract
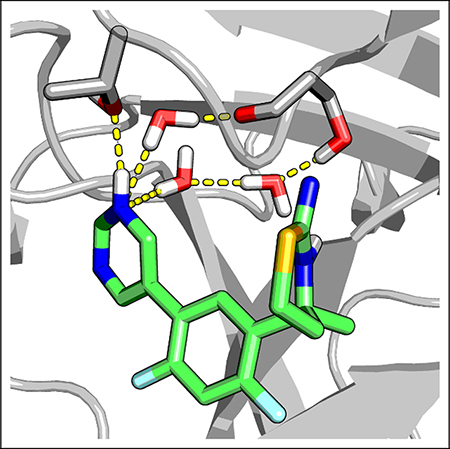
Over the past decade, experiments, theories, and simulations have refined our view of water. Hydrophobic effects, once thought to be exclusively driven by a change in the entropy of water, can also have an entropy origin,1,2 and ordered water molecules at the protein-ligand binding site may increase or decrease the binding free energy. 3,4 In recent years, binding-induced protonation state changes of proteins have been discussed; 5–9 however, a similar topic for small-molecule ligands has not gained much attention. Furthermore, how a change in the ligand protonation state can impact the water and their involvement in protein-ligand binding has not been investigated. Here we use continuous constant pH molecular dynamics (CpHMD) simulations to illustrate how a binding-induced change in the ligand protonation state perturbs water, protein dynamics, and hydrogen bonding.
As model systems, we consider, β-secretase 1 (BACE1) and its paralog β-secretase 2 (BACE2). BACE1 cleaves the amyloid precursor protein (APP) at the extracellular site to generate the β-amyloid peptide; as such it has been intensely pursued as a pharmaceutical target for Alzheimer’s disease. In vitro experiments showed that the APP cleavage activity of BACE1 occurs in the pH range 3.5–5.5.13,14 BACE2 has been shown to cleave APP with a similar bell-shaped pH profile;14–16 however, its biological functions are not fully understood, BACE2 is highly expressed in pigment cells, and deletion of the gene in mice and zebrafish results in a lack of pigmentation,17 Thus, BACE2 is considered an off-target in BACE1 drug design.18
BACE1 and BACE2 share similar sequences (71% similarity) and structures (Fig. 1a). The active site contains a dyad of aspartic acids, Asp32 (Asp48 in BACE2) and Asp228 (Asp241 in BACE2), which respectively act as the catalytic acid and base in the peptide cleavage reaction. All small-molecule BACE1 inhibitors contain an aspartyl binding motif19 (ABM, the endo-and exo-cyclic amino groups shown in Fig. 1b), which forms hydrogen bonds with the aspartyl dyad. Lying over the catalytic dyad is an 11-residue β-hairpin loop, commonly known as the flap, which is an important structural feature in BACE-1 related proteases (Fig. 1a). The flap of BACE1 has been shown, both experimentally and computationally, to undergo pH-dependent motion relative to the active site.13‘20‘21 Located next to the catalytic dyad and on the opposite side from the active site is the S3 subpocket, another conserved feature among BACE1-related proteases (Fig. 1b). Some highly potent BACE1 inhibitors reach into the S3 subpocket to form direct interactions, for example, the Merck compound MK8698.22 Analysis of crystal structures suggested that direct interactions with the S3 subpocket release four coordinated waters and cause the pocket to narrow. 18,23 Displacement of ordered waters to the bulk increases entropy and therefore can improve binding affinity.8,18 However, a recent study by Johannson et al, suggested that the resulting improved affinity comes at a cost of lowering selectivity over BACE2.18 Based on the analysis of BACE1 cocrystal structures and activity data, they found that compounds that reach into the S3 subpoeket and displace water show little or no selectivity over BACE2, while those preserving the binding-site water show greatly improved selectivity.18 Thus, the S3 subpoeket water appears to be a key player in tuning the balance between potency and selectivity.
Figure 1: Structures of BACE1 and BACE2 and their binding interactions with the Eli Lilly inhibitor LY2811376.

a) Cartoon representations of BACE1 (PDB ID 1SGZ10) and BACE2 (PDB ID 3ZKQ11). The flap is shown in red; the catalytic dyad and the S3 pocket residues are rendered as orange and green surfaces, respectively. b) A two-dimensional view of the Eli Lilly compound LY2811376 associated with BACE1 (PDB ID 4YBI12). Residues in the S1, S1′, S2 and S3 subpockets are indicated. The hydrogen bonding interactions with the dyad carboxylate groups are shown. The titratable pyrimidinyl nitrogen is circled.
Intrigued by the above finding, we set out to understand the moderate selectivity of a small-molecule BACE1 inhibitor LY2811376 developed by Eli Lilly,12 using CpHMD simulations with pH repliea-exehange. By simultaneously evolving conformational dynamics and protonation states at a specified pH and allowing different pH replicas to swap configurations in a Monte-Carlo fashion (see a recent review24), pH-EEX CpHMD is ideally suited for studying BACE1 and related systems that display pH-dependent dynamics and functions.13,20 Using this method, we previously characterized the pH-dependent interaction of LY2811376 with the catalytic dyad in BACE1.8,9 Here we examine the dynamics of waters and interactions in the S3 subpoeket of both BACE1 and BACE2 in complex with LY2811376, We found that the inhibitor displaces a larger number of waters, causes the S3 subpoeket to narrow in BACE2, The effect on water and protein dynamics is related to the binding-induced protonation of the inhibitor pyrimidine ring, which allows the formation of a direct hydrogen bond with the protein. These data illustrate how a change in the ligand protonation state can perturb binding-site water and modulate protein-ligand binding.
CpHMD simulations were conducted for apo BACE2 and holo BACE2 in complex with LY2811376. A pH replica-exchange sampling protocol was used, which included 20/24 pH replicas in the pH range 1.3–8/−1–8 for the apo/holo protein. The aggregate simulation time for each system was about 600 ns (see Supporting Information for convergence analysis). The trajectories for the apo and holo BACE1 were taken from our previous simulations. 8 LY2811376 binds BACE1 and BACE2 in similar modes. The ABM occupies the shallow S1′ subpoeket and forms two hydrogen bonds with the catalytic aspartates (Fig. 1b), while the pyrimidine ring occupies the S3 subpoeket, and is flanked by the two loop regions Ser10–Glyl3 and Gly230-Thr232 (Fig. 1b). Simulations of the apo proteins showed that the S3 subpocket is filled with five water molecules in both BACE1 and BACE2 (Fig. 2a), consistent with the similar pocket widths in apo BACE1 and BACE2 (Fig. 2d, dashed lines). One water molecule was mobile and exchanged with the bulk; however, four water molecules remained in similar locations throughout the simulations (Fig. 2b). These water molecules were captured by crystal structures10–12,26 and have been noted as important in several experimental studies.18,19,26
Figure 2: Water dynamics in the apo and holo forms of BACE1 and BACE2.

a) Number of water molecules in the S3 subpocket as a function of pH for apo BACE1 (blue) and BACE2 (red). The subpocket is defined by a 5-Å sphere, centered at the heavy-atom center of mass of loop residues Ser10-Gly13 and Gly230-Thr232, which flank the pyrimidinyl ring (see b). b) Zoomed-in view of the S3 subpocket with the inhibitor overlayed onto the apo BACE2 structure. Red spheres indicate crystal water molecules; those within the defined S3 subpocket are numbered. An isosurface of water density at 0.65 amu/Å3 (light cyan) was created with the VolMap plugin of VMD 25 based on the apo BACE2 simulation. c) Number of displaced water molecules in the S3 subpocket upon ligand binding to BACE1 and BACE2. d) Probability distribution of the S3 subpocket width of BACE1 and BACE2. The pocket width is defined as the distance between the heavy-atom centers of mass of the two aforementioned loop regions. Dashed lines represent the data from the apo simulations. Data from pH 4.9 were used. Distributions for other pH conditions are similar.
Water 1, which was present in both the apo and holo simulations, forms hydrogen bonds with Val31 and Tyr14 in BACE1 and corresponding residues in BACE2. Water 3 and 4 were displaced in the simulations of the holo proteins. Water 2 is most interesting. It remains in holo BACE1 and bridges the hydrogen bonds between the inhibitor’s pyrimidine nitrogen and Ser229 (Fig. 3a); however, it is displaced in holo BACE2. To quantify the water dynamics upon ligand binding, we calculated the total number of displaced water in the S3 subpoeket as a function of pH. In the enzyme active pH range (3.5–5.5), nearly two water molecules are released in BACE1, while one or two more are released in BACE2 (Fig. 2c). This data is consistent with the qualitative picture (Fig. 2b), which shows that while water 3 and 4 are displaced in both proteins, water 2 is displaced only in BACE2. Interestingly, the difference in the water displacement is correlated with the change in the pocket width, defined as the center-of-mass distance between the aforementioned two loop regions. The probability distribution of the pocket width remains unchanged when the inhibitor binds BACE1; however it becomes narrower and the peak shifts to a smaller value for BACE2 binding (Fig. 2d). This data indicates that, upon binding, the S3 subpoeket in BACE1 remains open and flexible, while it narrows and rigidities in BACE2.
Figure 3: LY2811376 forms water-mediated or direct hydrogen bonds in the S3 subpockets of BACE1 and BACE2.

a) Representations of the ligand forming the water-mediated hydrogen bond with Ser229 (ligand shown in green) and the direct hydrogen bond with Thr232 (ligand shown in gray). b) Probabilities of forming the water-mediated hydrogen bonds in BACE1 (blue) and BACE2 (red) as functions of pH. c) Probabilities of deprotonation for the pyrimidinyl nitrogen in solution (green), BACE1 and BACE2 as functions of pH. The pKa’s in solution, BACE1, and BACE2 are 3.7, 4.2, and 6.4, respectively. d) Probabilities of forming the direct hydrogen bond in BACE1 and BACE2 as functions of pH.
Johansson et al. observed that a significant difference between the selective and nonse-lective compounds is that the former do not perturb the S3 water in BACE1, keeping the pocket in an open state,18 However, no observation was made regarding BACE2, due to the lack of cocrystal structures. Considering that LY2811376 is ten fold selective for BACE1 over BACE2, our data is consistent with the above observation; however, our data further suggests that the mechanism for selectivity is the increased perturbation of the S3 water in BACE2 compared to BACE1.
Several past studies of BACE1 co-crystal structures noted that inhibitors that occupy the S3 subpoeket form a water-mediated hydrogen bond with a serine.18,19,26 Consistent with the crystal structures, our simulations showed that the pyrimidinyl nitrogen can accept hydrogen bonds from two nearby water molecules, which are in turn hydrogen bonded with either the sidechain hydroxyl or the backbone carbonyl of Ser229 in the S3 subpocket of BACE1 (Fig. 3a). Importantly, the water-bridged hydrogen bonds only appear above pH ~3.5, and the occupancy increases to a maximum at pH ~5.5, coinciding with the active pH range of the enzyme (Fig. 3b, blue). In this pH range, the pyrimidinyl nitrogen becomes increasingly deprotonated with pH (Fig. 3c, blue). Surprisingly, below the active pH range, the protonated pyrimidinyl nitrogen can donate a hydrogen bond to the hydroxyl oxygen of Thr232, which is near Ser229 (Fig. 3d, blue). We now turn to BACE2. In the active pH range, there is no water-mediated hydrogen bond between the pyrimidine and BACE2 (Fig. 3b, red), as the pyrimidinyl nitrogen is fully protonated (Fig. 3c, red) and forms a direct hydrogen bond with the threonine equivalent to Thr232 in BACE1 (Fig. 3d, red).
The pyrimidine ring has a solution (model) pKa of 3.7; as such it is deprotonated at the pH (4.5) of peak enzyme activity,13 which is also the typical pH used in the BACE1 binding assays,18,27 However, our simulations revealed that in association with BACE1 the pyrimidine ring has a similar pKa (4.2) as the solution value, but in association with BACE2, the pKa is shifted up bv 2.7 units to 6.4 (Fig. 3c), Thus, at pH 4.5, the inhibitor remains at least partially deprotonated upon binding BACE1 but becomes protonated upon binding BACE2. The difference in protonation state is reflected in the different mechanism of hydrogen bonding, i.e. water-mediated in BACE1 versus direct hydrogen bonding in BACE2. Johansson et al, suggested that the formation of water-mediated hydrogen bonds in BACE1 is important for selectivity,18 but why it leads to selectivity against BACE2 was not explained. Our data demonstrate that a selective compound may become protonated in BACE2 and lose the ability to accept water-bridged hydrogen bonds in the S3 subpoeket. Thus, the loss of water-mediated interactions in BACE2 may be a key contributor to selectivity.
The effect of the binding-induced ligand protonation state change on water network has not been investigated until now. Our data revealed that protonation of the pyrimidine ring, an important and frequently used building block in small-molecule drug design, increases the number of displaced water, rigidities the binding pocket, and shifts the ligand binding mode from water-mediated to direct hydrogen bonding. While a more detailed study is warranted in the future, the current work offers a glimpse at how modeling protein-ligand binding coupled to ligand titration can further advance the understanding of water and structure-based drug design.
Methods and Protocols.
The initial structure of apo BACE2 was taken from the PDB data bank (PDB ID: 3ZKQ11). A few missing residues (remote from the ligand binding site) were added using SWISS-MODEL.28 Since BACE1 and BACE2 structures are highly similar, following our previous work,9 the inhibitor LY2811376 was manually docked to BACE2 by aligning the crystal structure of apo BACE2 with the crystal structure of BACE1 in complex with the same inhibitor (PDB ID: 4YBI12). The docking pose was validated by monitoring the replica-exchange trajectories. Missing hydrogens were added, assuming doubly protonated histidines and otherwise standard protonation states for all sidechains, using the HBUILD facility in CHAEMM.29 An in-house CHAEMM script was used to add dummy hydrogens to the sidechains of Asp and Glu in preparation for the CpHMD titration. The structures were then solvated in truncated octahedron water boxes with at least 10 Å between the protein and the box edges. Water molecules within 2.6 Å from the protein heavy atoms were removed. The total number of atoms in the solvated systems was about 55,000. The inclusion of explicit ions is not required for system neutralization in the hybrid-solvent CpHMD simulations, as the fluctuating system net charge cannot be compensated by a fixed number of ions. In lieu of explicit counterions, the particle-mesh Ewald calculation includes a neutralizing plasma to maintain charge neutrality. To account for salt effects on the pica pKa’s, an ionic strength of 0.05 M was used in the generalized Born calculations for the hybrid-solvent CpHMD, consistent with our previous work on BACE1 and related systems.8,9
All simulations were performed using the CHAEMM package (version e36a6).29 The hybrid-solvent CpHMD was invoked through the PHMD module. 30 The pH replica-exchange protocol was invoked through the EEPSTE module.30 The proteins were represented by the CHAEMM22/CMAP all-atom force field,31,32 and water was represented by the CHAEMM modified TIP3P model.29 The parameters for LY2811376 were optimized previously by us. 8 The systems underwent energy minimization, 120 ps heating, and 280 ps equilibration with hybrid-solvent CpHMD turned on and pH set to 7. In the production run, the pH replica-exchange protocol30 was additionally used (see below for more details). All settings in the CpHMD simulations were program defaults and can be found in our earlier work30 and CHAEMM documentation (http://www.eharmm.org). The SHAKE algorithm33 was applied to bonds involving hydrogen atoms. Simulations were run under an NPT ensemble at a temperature of 300 K maintained by the Nose-Hoover thermostat and a pressure of 1 atm maintained by the Langevin piston pressure-coupling algorithm (see CHAEMM documentation, http://www.charmm.org) For the van der Waals calculations a switching function of 10 to 12 Å was used. A real-space cutoff of 12 Å and a sixth-order interpolation with approximately 1-Å grid were applied in the electrostatics calculations with the smooth particle-mesh Ewald method.34
In the CpHMD simulations, all Asp, Glu, and His sidechains were allowed to titrate, while Lys and Arg were kept in the protonated states. The latter can be justified, as the model pKa’s are outside of the simulation pH range and structural analysis ruled out possible pKa shifts to the simulation pH range. The simulation of apo BACE2 utilized 20 pH replicas in the pH range 1.3–8, with a spacing of 0.3 pH units from pH 1.3 to 5.5 and 0.5 pH units from pH 5.5 to 8; the total simulation time was 600 ns (30 ns per replica). The holo BACE2 simulation utilized 24 pH replicas in the pH range −1–8 using increments of 0.3/0.5 pH units; the total simulation time was 744 ns (31 ns per replica). The exchange rates were on average 44% and 46% for the apo and holo simulations, respectively. Except for the pKa calculations, which utilized the entire trajectories, the initial 10 ns from each replica was discarded in the analysis.
Supplementary Material
Acknowledgement
The authors thank the National Science Foundation (CBET1435957) and National Institutes of Health (GM098818) for funding.
Footnotes
Supporting Information Available
Supporting Information contains supplementary figures.
References
- (1).Biela A; Nasief NN; Betz M; Heine A; Hangauer D; Klebe G Dissecting the Hydrophobic Effect on the Molecular Level: The Role of Water, Enthalpy, and Entropy in Ligand Binding to Thermolysin. Angew. Chem. Int. Ed 2013, 52, 1822–1828. [DOI] [PubMed] [Google Scholar]
- (2).Setny P; Baron R; McCammon JA How Can Hydrophobic Association Be Enthalpy Driven? J. Chem. Theory Comput 2010, 6, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Breiten B; Lockett MR; Sherman W; Fujita S; Al-Sayah M; Lange H; Bowers CM; Heroux A; Krilov G; Whitesides GM Water Networks Contribute to Entahlpy/Entropy Compensation in Protein-Ligand Binding. J. Am. Chem. Soc 2013, 135, 15579–15584. [DOI] [PubMed] [Google Scholar]
- (4).Fox JM; Kang K; Sastry M; Sherman W; Sankaran B; Zwart PH; Whitesides GM Water-Restructuring Mutations Can Reverse the Thermodynamic Signiture of Ligand Binding to Human Carbonic Anhydrase. Angew. Chem. Int. Ed 2017, 56, 3833–3837. [DOI] [PubMed] [Google Scholar]
- (5).Wallace JA; Shen JK Unraveling a trap-and-trigger mechanism in the pH-sensitive self-assembly of spider silk proteins. J. Phys. Chem. Lett 2012, 3, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Onufriev AV; Alexov E Protonation and pK changes in protein-ligand binding. Q. Rev. Biophys 2013, 46, 181–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Petukh M; Ste S; Alexov E The Role of Protonation States in Ligand-Receptor Recognition and Binding. Curr. Pharm. Des 2013, 19, 4182–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ellis CR; Tsai C-C; Hou X; Shen J Constant pH Molecular Dynamics Reveals pH-Modulated Binding of Two Small-Molecule BACE1 Inhibitors. J. Phys. Chem. Lett 2016, 7, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Harris RC; Tsai C-C; Ellis CR; Shen J Proton-Coupled Conformational Al-lostery Modulates the Inhibitor Selectivity for β-Secretase. J. Phys. Chem. Lett 2017, 8, 4832–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hong L; Tang J Flap position of free memapsin 2 (β-secretase), a model for flap opening in aspartic protease catalysis. Biochemistry 2004, 43, 4689–4695. [DOI] [PubMed] [Google Scholar]
- (11).Banner DW; Gsell B; Benz J; Bertschinger J; Burger D; Brack S; Cuppuleri S; Debulpaep M; Gast A; Grabulovski D et al. Mapping the conformational space accessible to BACE2 using surface mutants and cocrystals with Fab fragments, Fynomers and Xaperones. Acta Crystallogr. D 2013, 69, 1124–1137. [DOI] [PubMed] [Google Scholar]
- (12).May PC; Dean RA; Lowe SL; Martenyi F; Sheehan SM; Boggs LN; Monk SA; Mathes BM; Mergott DJ; Watson BM et al. Robust Central Reduction of Amyloid-β in Humans with an Orally Available, Non-Peptidic β-Secretase Inhibitor. J. Neurosci 2011, 31, 16507–16516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shimizu H; Tosaki A; Kaneko K; Hisano T; Sakurai T; Nukina N Crystal structure of an active form of BACE1, an enzyme responsible for amyloid β protein production. Mol. Cell. Biol 2008, 28, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grüninger-Leitch F; Schlatter D; Küng E; Nelböck P; Döbeli H Substrate and inhibitor profile of BACE (β-secretase) and comparison with other mammalian aspartic proteases. J. Biol. Chem 2002, 277, 4687–4693. [DOI] [PubMed] [Google Scholar]
- (15).Abdul-Hay SO; Sahara T; McBride M; Kang D; Leissring MA Identification of BACE2 as an avid β-amyloid-degrading protease. Mol. Neurodegener 2012, 7, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Andrau D; Dumanchin-Njock C; Ayral E; Vizzavona J; Farzan M; Boisbrun M; Fulcrand P; Hernandez J-F; Martinez J; Lefranc-Jullien S et al. BACE1- and BACE2-expressing human cells characterization of β-amyloid precursor protein-derived catabolites, design of a novel fluorimetric assay, and identification of new in vitro in-hibitors. J. Biol. Chem 2003, 278, 25859–25866. [DOI] [PubMed] [Google Scholar]
- (17).Vassar R; Kuhn P-H; Haass C; Kennedy ME; Rajendran L; Wong PC; Lichtenthaler SF Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem 2014, 130, 4–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Johansson P; Kaspersson K; Gurrell IK; Bäck E; Eketjäll S; Scott CW; Cebers G; Thorne P; McKenzie MJ; Beaton H et al. Toward β-Secretase-1 Inhibitors with Improved Isoform Selectivity. J. Med. Chem 2018, 61, 3491–3502. [DOI] [PubMed] [Google Scholar]
- (19).Yuan J; Venkatraman S; Zheng Y; McKeever BM; Dillard LW; Singh SB Structure-Based Design of β-Site APP Cleaving Enzyme 1 (BACE1) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem 2013, 56, 4156–4180. [DOI] [PubMed] [Google Scholar]
- (20).Ellis CR; Shen J pH-Dependent Population Shift Regulates BACE1 Activity and Inhibition. J. Am. Chem. Soc 2015, 137, 9543–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kim MO; Blachly PG; McCammon JA Conformational Dynamics and Binding Free Energies of Inhibitors of BACE-1: From the Perspective of Protonation Equilibria. PLoS Comput. Biol 2015, 11, e1004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Stamford AW; Scott JD; Li SW; Babu S; Tadesse D; Hunter R; Wu Y; Misiaszek J; Cumming JN; Gilbert EJ et al. Discovery of an Orally Available, Brain Penetrant BACE1 Inhibitor That Affords Robust CNS A β Reduction. ACS Med. Chem. Lett 2012, 3, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Geschwindner S; Ulander J; Johansson P Ligand Binding Thermodynamics in Drug Discovery: Still a Hot Tip? J. Med. Chem 2015, 58, 6321–6335. [DOI] [PubMed] [Google Scholar]
- (24).Chen W; Morrow BH; Shi C; Shen JK Recent development and application of constant pH molecular dynamics. Mol. Simul 2014, 40, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cohen J; Arkhipov A; Braun R; Schulten K Imaging the Migration Pathways for O2, CO, NO, and Xe Inside Myoglobin. Biophys. J 2006, 91, 1844–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Malamas MS; Erdei J; Gunawan I; Turner J; Hu Y; Wagner E; Fan K; Chopra R; Olland A; Bard J et al. Design and Sythesis of 5,5’-Disubstituted Amino-hydantoins as Potent and Selective Human β-Secretase (BACE1) Inhibitors. J. Med. Chem 2010, 53, 1146–1158. [DOI] [PubMed] [Google Scholar]
- (27).Butler CR; Brodney MA; Beck EM; Barreiro G; Nolan CE; Pan F; Vajdos F; Parris K; Varghese AH; Helal CJ et al. Discovery of a Series of E cient, Centrally E cacious BACE1 Inhibitors through Structure-Based Drug Design. J. Med. Chem 2015, 58, 2678–2702. [DOI] [PubMed] [Google Scholar]
- (28).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TA; Rempfer C; Bordoli L et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartles C; Boresch S et al. CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wallace JA; Shen JK Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange. J. Chem. Theory Comput 2011, 7, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).MacKerell AD Jr.; Bashford D; Bellott M; Dunbrack RL Jr.; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (32).MacKerell AD Jr.; Feig M; Brooks CL III Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc 2004, 126, 698–699. [DOI] [PubMed] [Google Scholar]
- (33).Ryckaert JP; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (34).Essmann U; Perera L; Berkowitz ML; Darden T; Hsing L; Pedersen LG A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.