

Summary
Keywords: serotonin, norepinephrine, SUDEP
Objective:
Sudden unexpected death in epilepsy (SUDEP) is the leading cause of death in patients with refractory epilepsy. While the mechanisms for SUDEP are incompletely understood, seizure-induced respiratory arrest (S-IRA) has been strongly and consistently implicated. A body of evidence indicates that serotonin (5-HT), a modulator of breathing, plays a critical role in SUDEP. Because the 5-HT and norepinephrine (NE) systems interact in many biological processes and NE is known to modulate breathing and seizures, we hypothesized that NE may play a role in S-IRA and SUDEP.
Methods:
We examined the effects of pharmacological manipulation of 5-HT and NE on S-IRA and death following maximal electroshock (MES)-induced seizures in adult wild type (WT) mice, genetically 5-HT neuron deficient (Lmx1bf/f/p) mice, and chemically NE neuron deficient mice. Mice were treated with pharmacological agents targeting the serotonergic and noradrenergic systems, and subjected to seizure induction via MES while breathing was measured via whole body plethysmography.
Results:
S-IRA and death was reduced in WT mice with NE reuptake inhibitors (NRI), reboxetine and atomoxetine, selective serotonin reuptake inhibitors (SSRI), fluoxetine and citalopram, and the dual 5-HT/NE reuptake inhibitor (SNRI), duloxetine. S-IRA and death was also reduced in Lmx1bf/f/p mice with reboxetine and fluoxetine. The protective effects of the reuptake inhibitors were prevented by the α1 antagonist, prazosin. Citalopram did not reduce S-IRA and death in NE neuron deficient mice.
Significance:
These data suggest that 5-HT and NE critically interact in modulation of breathing following a seizure and potentially inform preventive strategies for SUDEP.
Introduction
Epilepsy is a heterogeneous group of neurological disorders marked by recurrent unprovoked seizures1. One in 26 Americans will develop epilepsy sometime in their life2. One-third of epilepsy patients will not become seizure free after treatment with two or more antiepileptic drugs (AED), and will be diagnosed with refractory epilepsy3. These patients are at high risk for sudden unexpected death in epilepsy (SUDEP), the leading cause of death in patients with refractory epilepsy. SUDEP is second only to stroke in terms of years of potential life lost due to a neurological condition4, and thus is a major public health problem.
Despite its societal cost, the etiology of SUDEP has yet to be elucidated; however, considerable evidence implicates seizure-induced respiratory arrest (S-IRA) in SUDEP5–8. While seizure-related cardiac dysregulation has also been strongly implicated and surely plays an important role, in recorded cases of SUDEP occurring in epilepsy monitoring units, terminal respiratory arrest precedes terminal asystole9. Furthermore, cardiac effects have been shown to occur secondarily to hypoxemia, or hypoxia10–12.
Due to its role in modulation of breathing, arousal, and seizures, serotonin (5-hydroxytrypamine; 5-HT) has been implicated in the etiology of SUDEP. Patients with refractory epilepsy have a decreased risk for ictal hypoxemia if treated with selective serotonin reuptake inhibitors (SSRI)13. The SSRI, fluoxetine, prevents S-IRA and death in DBA/2 mice following audiogenic seizures6. The SSRIs fluoxetine, sertraline, fluvoxamine, and paroxetine prevent S-IRA and death in DBA/1 mice following audiogenic seizures14–16. Similarly, the SSRI citalopram prevents S-IRA in WT mice following maximal electroshock (MES)-induced seizures, but not in 5-HT neuron deficient mice (Lmx1bf/f/p)8;17. Similar protective effects are afforded by 5-HT2A receptor agonists, and the protective effects of these agonists and citalopram are prevented with 5-HT2A receptor antagonists8. Similarly, the 5-HT2 antagonist cyproheptadine increases S-IRA in DBA/2 mice6.
While there is ample evidence in support of a role for 5-HT in S-IRA, other neurotransmitters modulate breathing, arousal, and seizures, and could play a role in post-ictal respiratory control and SUDEP. One of these is norepinephrine (NE)18. In two studies using DBA/1 mice, it was demonstrated that both the SSRIs fluvoxamine19 and fluoxetine20, which have considerable activity at α1 noradrenergic receptors, and the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine were more effective than the more 5-HT selective SSRI, paroxetine, in preventing S-IRA and subsequent death15. Recent evidence demonstrates that the norepinephrine reuptake inhibitor (NRI), atomoxetine, can also reduce susceptibility to S-IRA and death in DBA/1 mice21,22.
Since NE can modulate seizures23 and breathing18, is released by NE-producing locus coeruleus (LC) neurons following a seizure24, and is modulated in the LC by 5-HT25;26, we hypothesized that the NE and 5-HT systems interact in mediating S-IRA. In this study we tested this hypothesis by pharmacologically manipulating the NE and 5-HT systems in Lmx1bf/f/p mice, their phenotypically WT littermates (Lmx1bf/f), and C57BL/6 mice with or without toxin-mediated destruction of NE neurons in the LC, while monitoring seizure severity, respiratory activity, and survival following MES-induced seizures.
Methods
Ethical approval
All procedures and protocols were approved by the Institutional Animal Care and Use Committee at the University of Iowa, Carver College of Medicine in accordance with AAALAC guideline. Animals that did not succumb during experiments were euthanized with overdose of ketamine (100 mg/kg) and xylazine (10 mg/kg). Effort was made to minimize discomfort and use a minimum number of animals.
Experimental animals
Adult (2–4 mos) male C57BL/6, Lmx1bf/f, and Lmx1bf/f/p mice from our colony were used in these studies. C57BL/6 mice were originally obtained from Jackson Laboratories (Bar Harbor, ME). Lmx1bf/f and Lmx1bf/f/p mice were originally obtained from Zhoufeng Chen at Washington University, St. Louis, MO17 and subsequently bred in our facilities. Mice were housed in cages in a 12 h light/12 h dark schedule with food and water available ad libitum. Breeding and genotyping of Lmx1bf/f and Lmx1bf/f/p mice has been described previously27;28. Briefly, Lmx1bf/f mice carry two floxed Lmx1b alleles but are phenotypically normal, while Lmx1bf/f/p mice carry two floxed Lmx1b alleles and are hemizygous for ePet1-Cre and thus have a nearly complete elimination of 5-HT neurons in the central nervous system8;17;28.
Surgery
All surgeries were performed with aseptic technique under isoflurane anesthesia (1–5% inh. induction; 0.5–2% inh. maintenance). A subset of animals was implanted with EEG/EMG headmounts (8206; Pinnacle Technologies, Inc.; Lawrence, KS) to verify seizures and correlate with breathing cessation8. Four holes were made in the skull with a 23 ga. needle 1 mm anterior to bregma and 1 mm posterior to lambda, ± 2 mm from midline. The headmount was fastened to the skull with stainless steel machine screws (000–120, 0.1 in. anterior, 0.125 in. posterior; Small Parts; Miami Lakes, FL) inserted into the holes. Electrical connectivity was enhanced with silver epoxy (M.G. Chemicals Ltd.; Ontario, Canada). EMG leads were secured in nuchal muscles ±2 mm from midline. Subcutaneous ECG electrodes (Plastics One; Roanoke, VA) were implanted over the left chest wall and within the right axilla as described previously8. Another subset of mice was implanted with guide cannulae (Plastics One) into the right lateral ventricle (AP, −0.3 mm; ML, +1.0 mm, DV, −1.8 mm from bregma) for acute phenylephrine or vehicle application. Cannula placement was verified post-hoc via Nissl stain. Mice received meloxicam (2 mg/kg, i.p.) pre-operatively and once daily for at least two days post-operatively, and were closely monitored for at least five days after the surgery. Sutures were removed seven days post-operatively.
Maximal electroshock seizure induction and analysis
Seizures were induced via MES (50 mA, 200 msec, 60 Hz, sine wave), delivered via saline moistened ear clip electrodes connected to a rodent shocker (Hugo Sachs Elektronik; Grünstraße, Germany). Mice were video monitored for post-hoc analysis of seizure phenotype. Seizure severity was determined as described previously8 by the extension flexion (E/F) ratio, which is the ratio of the time the hindlimbs were extended ≥ 90˚ to the time they were flexed < 90˚. Seizures were assessed by EEG recording in a subset of mice.
Assessment of breathing via whole body plethysmography
Animals were acclimated to the recording apparatus for at least one hour a day for two days. Compressed room air (21% O2 / 79% N2; Praxair; Cedar Rapids, IA) flowed through the chamber at 0.40–0.45 L/min. Flow was balanced with a flow meter (Cole-Parmer; Vernon Hills, IL) and vacuum regulator (McMaster-Carr; Elmhurst, IL). An ultra-low volume pressure transducer (Honeywell, Morris Plains, NJ) on the recording chamber detected pressure waves associated with breathing. The transducer signal was amplified (100X) and band-pass filtered (0.3–30 Hz) with a data conditioning amplifier (Brownlee Precision 440; NeuroPhase; Palo Alto, CA). Signals were digitized (NI-USB-6008; National Instruments; Austin, TX) and passed to a computer running custom MATLAB software (Mathworks; Natick, MA), as described previously8;27.
Respiratory data (100 s before and 30 min after treatments; 100 s before and 180 s after seizures) were analyzed in 10 s epochs for respiratory rate (RR), tidal volume (VT), and minute ventilation (VE). A mechanical ventilator (MiniVent 845; Hugo Sachs Elektronik) was used to calibrate the transducer signal. VT was calculated using a barometric method29 of transforming on the basis of breath amplitude, barometric pressure (www.wunderground.com), flow rate (Cole-Parmer), mouse temperature (rectal thermocouple; Physitemp), and air temperature (Physitemp Instruments, Inc; Clifton, NJ). In these studies S-IRA was always associated with death. Thus, S-IRA was defined as an apnea which occurred as a result of the seizure and persisted until the death of the animal.
EEG/EMG/ECG data collection and analysis
In the subset of mice instrumented for recording, EEG, EMG, and ECG signals were collected using the same custom software as described previously8;27;30. EEG signals were filtered (0.5 and 200 Hz) and amplified (50,000X). EMG signals were filtered (10 and 200 Hz) and amplified (50,000X). ECG signals were filtered (0.3 and 300 Hz) and amplified (20,000X). Data were sampled at 1000 Hz.
Histology
After all experiments were completed, all animals were euthanized, intracardially perfused, and brains were extracted and cryoprotected as described previously31. Frozen brains were cryostat sectioned (30 μm; Leica, Buffalo Grove, IL), and placed into free floating wells or mounted onto slides. Sections were washed (0.0125% Tween 20; 15 min), incubated in citrate buffer (10 mM sodium citrate; 0.05% Tween 20; 60° C; 20 min), placed into blocking solution (10% goat serum; 0.3% Triton-X; 60 min), incubated overnight with 5% goat serum/PBS and primary antibody [Aves Labs; chicken anti-tyrosine hydroxylase (TH), 1:2000], and then incubated in flurophore-conjugated secondary antibody (goat anti-chicken, 1:500) in 0.3% Triton-X in PBS. Mounted sections were observed with an epifluorescent microscope (Zeiss). Stereological cell counting of TH+ cells was performed using the optical fractionator approach and unbiased counting rules with 3-dimensional 200 × 150 × 10 μm counting frames using a 40× objective lens (Stereoinvestigator; MBF Biosciences; Williston, VT). TH+ cells were counted in three sections containing bilateral LC for each animal and compared to DSP-4 treated and vehicle treated mice. Tissue from i.c.v. cannulated animals was Nissl stained (0.1% cresyl violet) using standard procedures in the lab31. Only mice in which the cannula was verified in the lateral ventricle were included in analysis.
Drugs
Ketamine and xylazine were obtained from Midwest Veterinary Supply (Lakeville, MN). Meloxicam was obtained from Norbrook Laboratories (Overland Park, KS). Atomoxetine hydrochloride (1044469) and fluoxetine hydrochloride (F132) were obtained from Sigma-Aldrich (St. Louis, MO). Citalopram hydrobromide (1427), reboxetine mesylate (1982), N-(2-Chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride (DSP-4,2958), prazosin hydrochloride (0623), (S)-duloxetine hydrochloride (4798), propranolol hydrochloride (0624), and (R)-(−)-phenylephrine hydrochloride (2838) were obtained from Tocris Biosciences (Minneapolis, MN). Fluoxetine, citalopram, prazosin, and reboxetine were dissolved in 1% DMSO. DSP-4, phenylephrine, atomoxetine, propranolol, and duloxetine were dissolved in saline (0.9% NaCl).
Results
Systemic administration of NRIs prevented S-IRA and mortality
To determine whether noradrenergic stimulation could affect S-IRA and death, mice were subjected to seizure induction via MES 30 minutes following systemic application (i.p.) of vehicle or the NRIs reboxetine (0.1, 0.3, 1, 3, 10, or 30 mg/kg) or atomoxetine (10 mg/kg). Reboxetine and atomoxetine reduced S-IRA compared to vehicle (Figure 1A-D). Neither reboxetine at any tested dose nor atomoxetine had a significant effect on VT, VE, or RR compared to vehicle (Table 1). Reboxetine 30 mg/kg significantly reduced seizure severity (Table 2). Atomoxetine and lower doses of reboxetine had no significant effect on seizure severity (Table 2).
Figure 1. Norepinephrine reuptake inhibitors and an α1 agonist prevent S-IRA and death following MES induced seizures in C57BL/6 mice.
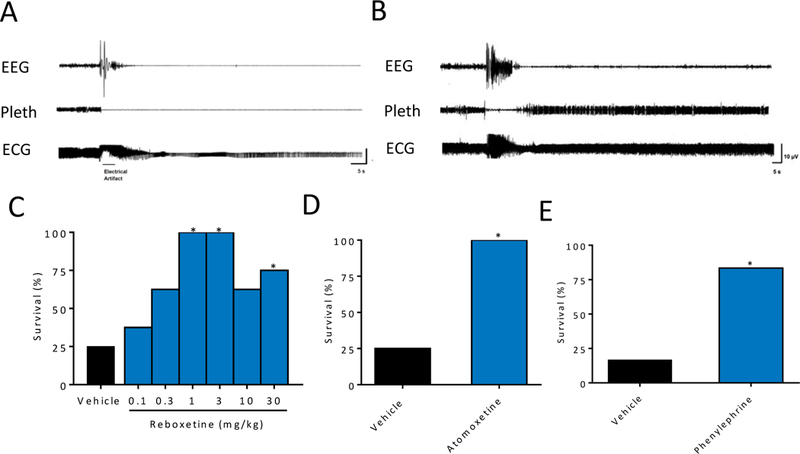
A,B) 140-s raw EEG, breathing (Pleth), and ECG traces demonstrating death from a seizure following vehicle treatment (A) and survival from a seizure following reboxetine treatment (B). C-E) Percent survival from MES-induced seizures following systemic (i.p.) application of A) reboxetine (RBX; 0, 0.1, 0.3, 1, 3, 10, and 30 mg/kg i.p., p = 1.00 for 0.1 mg/kg, p = 0.31 for 0.3 mg/kg, p = 0.007 for 1 mg/kg and 3 mg/kg; p = 0.3 for 10 mg/kg, p = 0.04 for 30 mg/kg) and B) atomoxetine (AXM; 10 mg/kg, p = 0.007), and C) following i.c.v. application of phenylephrine (1 μM, 1 μl, p = 0.03 ) or vehicle. n = 6–8 per group, as indicated in Table 1. *, p < 0.05 compared to vehicle.
Table 1.
Effects of drugs on breathing.
| Mean Frequency, breath/min (SD) | Mean Tidal Volume, ml/g (SD) | Mean Ventilation, ml/min/g (SD) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre-Injection | Post-Injection | p-value | Pre-Injection | Post-Injection | p-value | Pre-Injection | Post-injection | p-value | |
| Vehicle, n = 8 |
185.45 (27.89) | 193.85 (17.69) | 0.484 | 20.67 (4.22) | 21.91 (3.25) | 0.521 | 3.78 (0.67) | 4.24 (0.72) | 0.210 |
| Atomoxetine 10 mg/kg, n = 8 |
170.63 (29.51) | 182.59 (27.98) | 0.419 | 21.50 (3.83) | 20.68 (2.94) | 0.641 | 3.72 (1.21) | 3.83 (1.07) | 0.852 |
| Reboxetine 0.1 mg/kg, n = 8 |
187.37 (32.69) | 173.63 (32.26) | 0.412 | 20.65 (5.90) | 21.40 (3.91) | 0.771 | 3.79 (0.92) | 3.74 (1.15) | 0.930 |
| Reboxetine 0.3 mg/kg, n = 8 |
165.70 (39.17) | 166.49 (43.05) | 0.970 | 22.44 (4.63) | 19.96 (3.10) | 0.230 | 3.75 (1.28) | 3.37 (1.17) | 0.545 |
| Reboxetine 1 mg/kg, n = 8 |
172.75 (42.63) | 188.89 (34.24) | 0.418 | 21.39 (2.91) | 23.59 (4.97) | 0.299 | 3.68 (0.94) | 4.53 (1.01) | 0.207 |
| Reboxetine 3 mg/kg, n = 8 |
178.84 (26.40) | 177.51 (28.02) | 0.924 | 21.68 (4.27) | 21.78 (2.21) | 0.956 | 3.85 (0.80) | 3.84 (0.53) | 0.968 |
| Reboxetine 10 mg/kg, n = 8 |
180.89 (36.28) | 178.06 (38.25) | 0.882 | 21.29 (3.22) | 21.18 (3.70) | 0.952 | 3.89 (1.05) | 3.75 (0.88) | 0.772 |
| Reboxetine 30 mg/kg, n = 8 |
163.61 (32.19) | 182.51 (27.24) | 0.226 | 22.38 (4.60) | 23.40 (5.19) | 0.685 | 3.73 (1.38) | 4.22 (0.97) | 0.425 |
|
i.c.v. Vehicle, n = 7 |
172.53 (32.36) | 168.11 (24.03) | 0.778 | 20.35 (3.36) | 20.76 (4.02) | 0.839 | 3.54 (1.04) | 3.43 (0.48) | 0.804 |
|
i.c.v. Phenylephrine 1 μM, n = 7 |
181.33 (41.98) | 167.02 (34.84) | 0.501 | 21.87 (4.85) | 23.04 (3.83) | 0.626 | 3.89 (0.85) | 3.86 (1.20) | 0.958 |
| Fluoxetine 10 mg/kg n = 8 |
184.46 (23.92) | 173.96 (36.14) | 0.566 | 22.57 (5.24) | 21.90 (2.94) | 0.791 | 4.16 (1.10) | 3.77 (0.74) | 0.492 |
| Citalopram 20 mg/kg, n = 6 |
177.47 (20.72) | 188.76 (24.40) | 0.408 | 22.42 (5.61) | 20.58 (3.07) | 0.498 | 3.93 (0.90) | 3.88 (0.77) | 0.920 |
| Duloxetine 10 mg/kg, n = 8 |
198.91 (30.87) | 214.42 (20.56) | 0.257 | 23.63 (5.13) | 24.13 (3.94) | 0.830 | 4.68 (1.24) | 5.18# (0.99) | 0.388 |
| Propranolol 10 mg/kg, n = 8 |
174.74 (28.99) | 218.62 (50.66) | 0.052 | 19.75 (3.49) | 21.00 (5.18) | 0.580 | 3.41 (0.20) | 4.45* (1.04) | 0.015 |
| Prazosin 0.3 mg/kg, n = 7 |
163.03 (43.28) | 169.49 (39.69) | 0.776 | 18.33 (4.77) | 19.49 (1.41) | 0.534 | 2.96 (0.93) | 3.33# (0.88) | 0.459 |
| Prazosin 1.0 mg/kg, n = 7 |
191.36 (36.95) | 149.39*# (19.61) | 0.021 | 19.48 (1.56) | 19.23 (2.21) | 0.811 | 3.72 (0.72) | 2.87*# (0.56) | 0.030 |
| Prazosin 3.0 mg/kg, n = 7 |
201.87 (26.74) | 166.74*# (25.41) | 0.027 | 20 .56 (2.92) | 19.01 (3.36) | 0.375 | 4.16 (0.83) | 2.74*# (0.69) | 0.005 |
| Prazosin 10 mg/kg, n = 7 |
188.81 (19.50) | 152.64*# (36.01) | 0.038 | 22.50 (2.48) | 17.50*# (3.69) | 0.012 | 4.24 (0.52) | 2.69*# (0.94) | 0.003 |
| Prazosin + Atomoxetine 10 mg/kg, n = 8 |
219.81 (37.00) | 160.9* (16.89) | 0.001 | 23.09 (5.08) | 16.45* (5.31) | 0.023 | 5.04 (0.44) | 5.67*# (0.58) | 0.028 |
| Prazosin + Reboxetine 30 mg/kg, n = 8 |
192.15 (22.33) | 155.27*# (32.71) | 0.046 | 18.92 (3.07) | 16.99*# (3.59) | 0.031 | 3.64 (0.72) | 2.64*# (0.74) | 0.041 |
| Prazosin + Reboxetine 3 mg/kg, n = 7 |
179.88 (22.36) | 148.20*# (30.81) | 0.048 | 20.29 (3.31) | 14.73*# (3.10) | 0.007 | 3.65 (0.77) | 2.55*# (0.79) | 0.022 |
| Prazosin 10 mg/kg + Fluoxetine 10 mg/kg, n = 8 |
184.82 (21.32) | 160.01# (25.32) | 0.096 | 20.21 (1.00) | 16.91*# (2.11) | 0.004 | 3.74 (0.53) | 2.71*# (0.58) | 0.009 |
| Prazosin 10 mg/kg + Citalopram 20 mg/kg, n = 8 |
187.48 (14.42) | 163.06# (23.01) | 0.052 | 20.46 (3.24) | 18.42 (4.16) | 0.360 | 3.82 (0.45) | 2.98*# (0.63) | 0.032 |
| Prazosin 10 mg/kg + Duloxetine 10 mg/kg, n = 8 |
210.38 (41.63) | 168.27* (21.44) | 0.023 | 21.75 (4.27) | 17.89 (3.49) | 0.068 | 4.58 (0.14) | 3.14* (0.73) | <0.001 |
| Prazosin 10 mg/kg + Propranolol 10 mg/kg, n = 8 |
228.63 (32.54) | 162.47* (33.52) | 0.001 | 22.88 (5.30) | 16.55* (4.14) | 0.019 | 5.18 (0.89) | 2.71*# (0.92) | <0.001 |
| DSP-4 Vehicle, n = 8 |
177.81 (26.18) | 177.83 (32.35) | 0.999 | 22.18 (3.03) | 22.44 (5.42) | 0.906 | 3.92 (0.68) | 3.93 (0.95) | 0.983 |
| DSP-4 Citalopram 20 mg/mg, n = 7 |
179.20 (42.80) | 175.34 (24.84) | 0.840 | 21.77 (4.29) | 21.38 (2.89) | 0.845 | 3.98 (1.54) | 3.76 (0.80) | 0.745 |
Pre- and post-injection breathing changes across measures of frequency, tidal volume, and ventilation for all drug groups expressed as mean (standard deviation). Post-injection breathing for drug combinations was taken after injection of the second drug. P-values reflect post-injection breathing compared to pre-injection breathing of same drug. Unless otherwise noted, all drugs were injected i.p. n = 6–8 per group as indicated.
, p < 0.05 compared to pre-injection values.
, p < 0.05 compared to vehicle.
Table 2.
Effects of drugs on seizure severity.
| Extension-Flexion Ratio (SD) | ||
|---|---|---|
| Mean | p-value | |
| Vehicle, n = 8 | 14.46 (5.81) | - |
| Atomoxetine 10 mg/kg, n = 8 | 11.34 (1.14) | 0.158 |
| Reboxetine 0.1 mg/kg, n = 8 | 11.36 (3.81) | 0.228 |
| Reboxetine 0.3 mg/kg, n = 8 | 10.17 (2.24) | 0.072 |
| Reboxetine 1 mg/kg, n = 8 | 10.63 (2.31) | 0.105 |
| Reboxetine 3 mg/kg, n = 8 | 11.13 (4.67) | 0.227 |
| Reboxetine 10 mg/kg, n = 8 | 8.80 (5.87) | 0.073 |
| Reboxetine 30 mg/kg, n = 8 | 7.11* (5.03) | 0.017 |
| i.c.v. Vehicle, n = 7 | 14.65 (8.80) | 0.985 |
| i.c.v. Phenylephrine 1 mM, n = 7 | 10.30 (2.93) | 0.136 |
| Fluoxetine 10 mg/kg, n = 8 | 15.64 (3.19) | 0.622 |
| Citalopram 20 mg/kg, n = 6 | 14.61 (3.94) | 0.958 |
| Duloxetine 10 mg/kg, n = 8 | 14.27 (2.15) | 0.936 |
| Propranolol 10 mg/kg, n = 8 | 15.90 (1.51) | 0.509 |
| Prazosin 0.3 mg/kg, n = 7 | 17.43 (4.23) | 0.694 |
| Prazosin 1 mg/kg, n = 7 | 4.38* (3.58) | 0.002 |
| Prazosin 3 mg/kg, n = 7 | 12.58 (7.34) | 0.598 |
| Prazosin 10 mg/kg, n = 7 | 2.81* (1.06) | <0.001 |
| Prazosin 10 mg/kg + Atomoxetine 10 mg/kg, n = 8 | 4.41* (1.71) | <0.001 |
| Prazosin 10 mg/kg + Reboxetine 3 mg/kg, n = 8 | 6.85* (5.93) | 0.026 |
| Prazosin 10 mg/kg + Reboxetine 30 mg/kg, n = 8 | 5.25* (1.03) | 0.003 |
| Prazosin 10 mg/kg + Fluoxetine 10 mg/kg, n = 8 | 6.91* (0.78) | 0.003 |
| Prazosin 10 mg/kg + Citalopram 20 mg/kg, n = 8 | 4.41* (2.01) | <0.001 |
| Prazosin 10 mg/kg + Duloxetine 10 mg/kg, n = 8 | 4.83* (1.64) | <0.001 |
| Prazosin 10 mg/kg + Propranolol 10 mg/kg, n = 8 | 7.30* (1.66) | 0.005 |
| DSP-4 Vehicle, n = 8 | 15.76 (2.43) | 0.569 |
| DSP-4 Citalopram 20 mg/kg, n = 7 | 14.61 (3.94) | 0.955 |
Extension-flexion ratio of seizures after drug administration for all groups. Values are expressed as mean (standard deviation) with p-values provided that reflect the drug group compared to extension-flexion ratio of vehicle group. Unless otherwise noted, all drugs were injected i.p. n = 6–8 per group as indicated.
, p < 0.05 compared to vehicle.
Intracerebroventricular application of the α1 agonist, phenylephrine, reduced S-IRA and mortality in WT mice
Systemic activation of the NE system with NRIs effectively reduced S-IRA in WT mice. Excitatory effects of NE on breathing have been shown to occur through an α1 receptor mediated mechanism18;32;33. To determine whether α1 receptor activation could prevent S-IRA and mortality, the α1 agonist phenylephrine (1 μM, 1 μl) was applied i.c.v. 60 min prior to seizure induction via MES. Phenylephrine does not cross the blood brain barrier and has been delivered in a similar manner to cause central nervous system effects34. Phenylephrine applied in this manner prevented S-IRA and death compared to vehicle (Figure 1E). I.c.v. application of phenylephrine did not significantly affect breathing (Table 1) or seizure severity (Table 2).
Systemic blockade of α1 receptors prevented the protective effects of NRIs on S-IRA and mortality in WT mice
To determine whether the protective effects of reboxetine were exerted through activation of the α1 receptors, mice underwent seizure induction via MES following pre-treatment with one of the following: (1) two doses of vehicle; (2) the α1 receptor antagonist prazosin (10 mg/kg) followed by vehicle; (3) prazosin (10 mg/kg) followed by reboxetine (3.0 mg/kg); or (4) prazosin (10 mg/kg) followed by atomoxetine (10 mg/kg); i.p. injections, 30 min apart. The protective effects of reboxetine and atomoxetine were prevented by pre-treatment with prazosin (Figure 2A). All groups that received prazosin also exhibited less severe seizures compared to vehicle (Table 2). As expected18, breathing was significantly reduced by the administration of prazosin with vehicle, prazosin and reboxetine, prazosin and atomoxetine (Table 1).
Figure 2. The α1 antagonist, prazosin, prevents the protective effects of reboxetine, citalopram, and fluoxetine on S-IRA and death following MES induced seizures in C57BL/6 mice.
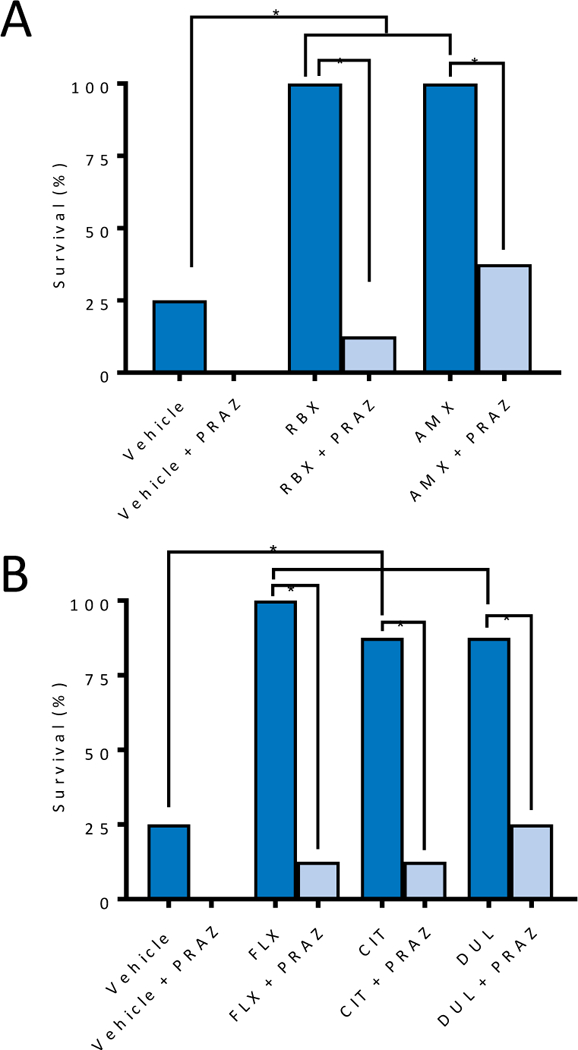
A) Percentage of C57BL/6J mice surviving prazosin (PRAZ, 10 mg/kg, p = 0.47 compared to vehicle), reboxetine (3 mg/kg), alone (p = 0.007 compared to vehicle) or together (p = 0.001 compared to reboxetine alone), and atomoxetine (10 mg/kg), alone (p = 0.007 compared to vehicle) or together (p = 0.04 compared to atomoxetine alone) in C57BL/6J mice compared to control. RBX alone redrawn form Figure 1C. B) Percentage of C57BL/6J mice surviving MES-induced seizure when α1 antagonist prazosin (PRAZ, 10 mg/kg) was co-administered with either vehicle, fluoxetine (FLX, 10 mg/kg, p = 0.001), citalopram (CITAL, 20 mg/kg, p = 0.01), duloxetine (DUL, 10 mg/kg, p = 0.04), or when FLX, CITAL, or DUL were given with vehicle (FLX, p = 0.007; CITAL, p = 0.04; and DUL, p = 0.04). n = 6–8 per group, as indicated in Table 1. *, p < 0.05 between groups.
Systemic blockade of α1 receptors prevented the protective effects of SSRIs and SNRI on S-IRA and mortality in WT mice
We previously found that systemic application of the SSRI, citalopram, could prevent S-IRA following seizure induced by MES and postulated that this might represent a way to augment the respiratory system to reduce the effects of seizures on breathing and survival8. To determine whether this type of “breathing support” could also be affected by α1 receptor blockade, mice underwent seizure induction via MES after pre-treatment with one of the following: (1) two doses of vehicle; (2) prazosin (0.3, 1.0, 3.0, and 10 mg/kg) followed by vehicle; (3) vehicle followed by prazosin (10 mg/kg); (4) prazosin (10 mg/kg) followed by fluoxetine (10 mg/kg); 5) vehicle followed by citalopram (20 mg/kg); 6) vehicle followed by duloxetine (10 mg/kg); 7) prazosin (10 mg/kg) followed by duloxetine (10 mg/kg); or 8) prazosin (10 mg/kg) followed by citalopram (20 mg/kg); i.p. injections 30 min apart. Prazosin at 0.3, 1.0, 3.0, and 10 mg/kg had no significant effect on S-IRA compared to vehicle. Fluoxetine, citalopram, and duloxetine alone prevented S-IRA and improved survival (Figure 2B). Prazosin 1.0 mg/kg significantly reduced seizure severity (Table 2). Prazosin 10 mg/kg prevented the protective effects of fluoxetine, citalopram, and duloxetine (Figure 2B). Prazosin in combination with vehicle, fluoxetine, citalopram, or duloxetine significantly reduced seizure severity (Table 2). Prazosin at 1.0, 3.0, and 10 mg/kg significantly reduced breathing (Table 1). Prazosin 10 mg/kg reduced breathing when co-administered with fluoxetine, citalopram, and duloxetine (Table 1).
Systemic α1 noradrenergic receptor blockade prevented the protective effects of fluoxetine and reboxetine on S-IRA and mortality in Lmx1bf/f/p mice
We previously found that citalopram could prevent S-IRA following MES in Lmx1bf/f mice and that this effect was lost in Lmx1bf/f/p mice that do not have 5-HT neurons in the CNS8. To determine whether this effect would hold true for other SSRIs, we tested fluoxetine (10 mg/kg) in Lmx1bf/f and Lmx1bf/f/p mice. Fluoxetine, prevented S-IRA following MES induced seizures in Lmx1bf/f mice (Figure 3A); however, somewhat surprisingly, fluoxetine also prevented S-IRA in Lmx1bf/f/p mice (Figure 3A). Given that these mice do not have 5-HT transporters17, we hypothesized that fluoxetine might be exerting its effect in Lmx1bf/f/p mice through another mechanism, such as α1 receptor activation.
Figure 3. α1 blockade prevented protective effects of fluoxetine and reboxetine on S-IRA in Lmx1bf/f/p mice.
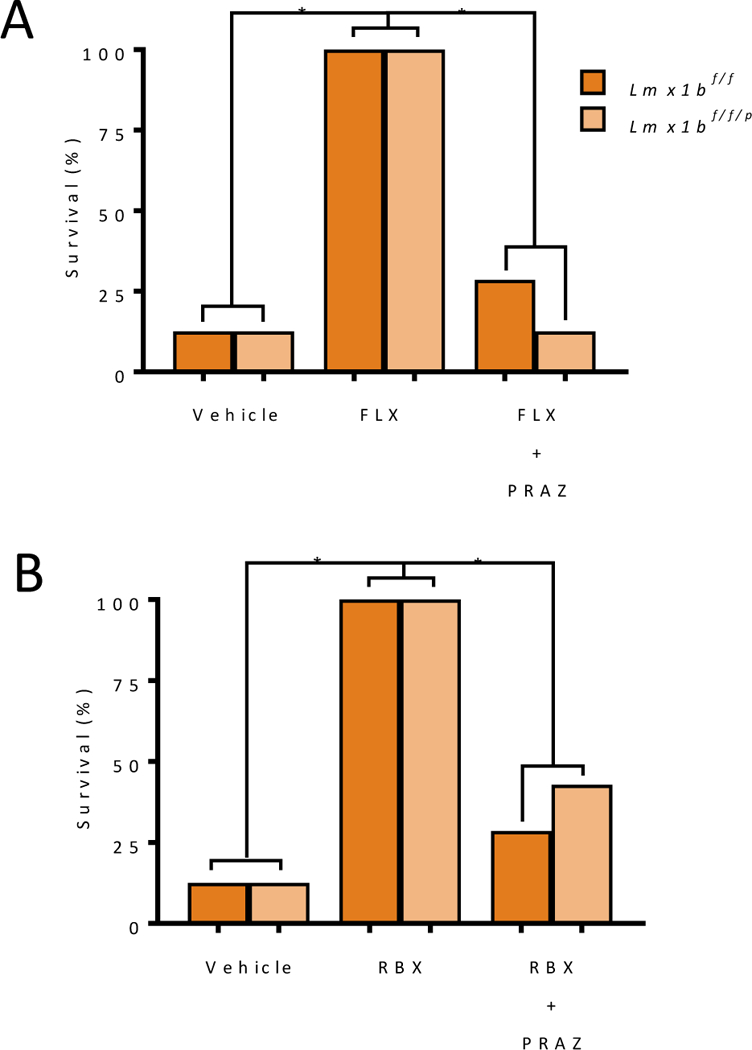
A) Percentage of Lmx1bf/f/p mice surviving MES-induced seizure when administered fluoxetine i.p. (FLX, 10 mg/kg, p = 0.001 for both Lmx1bf/f and Lmx1bf/f/p compared to vehicle) or fluoxetine and prazosin compared to vehicle and Lmx1bf/f controls (p = 0.007 compared to Lmx1bf/f fluoxetine alone and p = 0.001 compared to Lmx1bf/f/p fluoxetine alone). B) Percentage of Lmx1bf/f/p mice surviving MES-induced seizure when administered reboxetine (RBX, 3 mg/kg, p = 0.001 for both Lmx1bf/f and Lmx1bf/f/p compared to vehicle) alone or administered reboxetine with prazosin (PRAZ, 10 mg/kg) compared to vehicle and Lmx1bf/f controls (p = 0.007 for Lmx1bf/f and p = 0.03 for Lmx1bf/f/p. n = 7–8. *, p < 0.05.
To test this, reboxetine (3 mg/kg, i.p.) was given to Lmx1bf/f and Lmx1bf/f/p mice 30 min prior to seizure induction. Reboxetine prevented S-IRA in both genotypes (Figure 3B). α1 blockade prevented the protective effects of NRI and SSRI application on S-IRA (Figure 2). To determine whether the reduction in S-IRA and death seen in Lmx1bf/f/p mice treated with fluoxetine or reboxetine was also due to α1 receptor activation, Lmx1bf/f/p mice were given prazosin (10 mg/kg) followed 30 min later by fluoxetine (10 mg/kg) or reboxetine (3 mg/kg) and were then subjected seizures induction 30 min later. Prazosin prevented the protective effects of fluoxetine and reboxetine on S-IRA and death in Lmx1bf/f and Lmx1bf/f/p mice (Figure 3).
Systemic application of citalopram did not reduce S-IRA and mortality in NE neuron depleted mice.
Seven noradrenergic neuron populations (A1-A7) have been identified in the CNS35. Several have been implicated in breathing regulation18. Among these, neurons from A6 (locus coeruleus; LC), have facilitory effects on respiratory networks18;36. Given the previously identified role of LC on breathing18, seizures23, and 5-HT signaling modulation37, the LC was investigated here as opposed to other catecholaminergic nuclei. The above findings which demonstrate that prazosin could prevent the protective effects of SSRI on S-IRA suggest that these effects may be dependent on NE. To begin to examine contributions of NE from LC neurons we employed the DSP-4 neurotoxin, a small molecule toxin that, through ill-defined mechanisms, targets the noradrenergic transporter and selectively destroys terminals originating form LC, and in some cases cell bodies in the LC38. In order to determine an optimal treatment strategy for reducing NE cell bodies in the LC, groups of C57BL/6 mice received either one injection of saline, or one (50 or 75 mg/kg) or two (50 mg/kg; seven days apart) injections of DSP-4. One week after the last injection mice were intracardially perfused and the brain tissue was processed for histology. DSP-4 dose dependently reduced the number of TH+ cells in the LC, with the double-injection strategy having the greatest effect (Figure 4A-C). We decided to use this treatment strategy to determine whether LC neuron loss could alter the effect of SSRI on S-IRA.
Figure 4. Citalopram does not prevent S-IRA and death in mice with selective depletion of noradrenergic neurons in the LC.
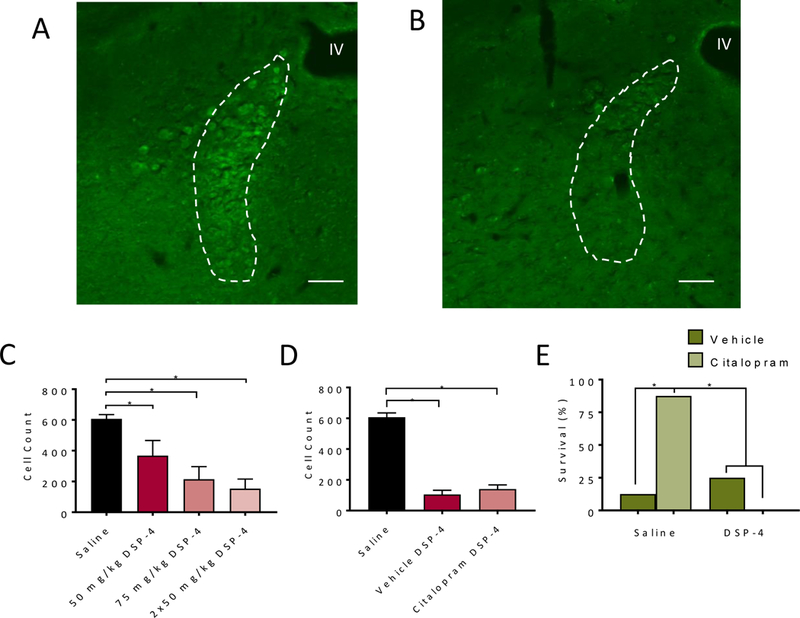
A,B) Representative micrographs from coronal sections of C57BL/6 mice treated with saline (A) or two doses DSP-4 (50 mg/kg) and immunostained with an antibody against tyrosine hydroxylase in locus coeruleus. IV, fourth ventricle. Scale bars, 100 μm. C) TH+ cell counts in animals administered vehicle or varying doses of DSP-4 (one dose of 50 mg/kg (p = 0.003) or 75 mg/kg (p < 0.001), or 2 doses of 50 mg/kg (p < 0.001); n = 3 per group). D) TH+ cell counts in animals which underwent maximal electroshock with citalopram 20 mg/kg (p < 0.001) or vehicle pre-treatment (p < 0.001, p = 0.072 between vehicle and citalopram groups) one week after the second of two 50 mg/kg DSP-4 doses (n = 7–8 per group), as indicated on Table 1. *, p < 0.05. E) Percentage of saline and DSP-4 treated mice surviving seizures induced by MES following treatment with vehicle (p = 0.02 vehicle with DSP-4 compared to citalopram without DSP-4) or citalopram (20 mg/kg, p = 0.001 citalopram with DSP-4 compared to citalopram without DSP-4). n = 8 per group. *, p < 0.05.
Two additional groups were treated with two doses of saline or DSP-4 (50 mg/kg) seven days apart as above. One week after the second injection, animals were subjected to seizure induction via MES 30 minutes following pre-treatment with either vehicle or citalopram (20 mg/kg). Citalopram was chosen for these experiments since it is a relatively more 5-HT selective SSRI. DSP-4 treatment had no effect on S-IRA in vehicle treated animals (Figure 4E); however, DSP-4 treatment prevented the protective effects of citalopram on S-IRA (Figure 4E). Following these studies, animals were euthanized and the extent of NE cell loss in the LC was evaluated. Again, there was similar reduction of TH+ cell bodies in the LC of mice that received saline or citalopram prior to MES (Figure 4D). There was no difference in the degree of cell loss between the saline or citalopram groups (Figure 4D). There was no effect of saline pre-treatment or citalopram pre-treatment (Table 1) on breathing or seizure severity (Table 2) in DSP-4 treated animals.
Administration of non-selective β-antagonist reduced S-IRA in C57Bl/6 mice while co-administration with α1 antagonist reversed this effect
To examine whether β-receptors might play a role in S-IRA, the β-receptor antagonist propranolol (10 mg/kg) was administered i.p. 30 minutes before MES-induced seizure. Propranolol significantly reduced S-IRA and death compared to vehicle (n = 8, p = 0.041). Pre-treatment with prazosin 10 mg/kg reversed this effect compared to propranolol alone (n = 8, p = 0.041). Administration of propranolol increased ventilation compared to pre-injection (Table 1) and did not affect seizure-severity (Table 2).
Discussion
A preponderance of evidence implicates 5-HT in S-IRA and SUDEP 39;40. Recent data have implicated NE in S-IRA21;22. Here we demonstrate that 5-HT and NE may act as part of a circuit that regulates breathing following a seizure. Both NE and 5-HT are modulated by seizures41–43, are involved in breathing regulation18;44, and have anti-epileptic properties41;45. α1 adrenergic receptor density is decreased in the epileptogenic foci of patients with intractable partial epilepsy46. Despite the antiepileptic properties of NE and 5-HT, drugs employed in this study that modulate these neurotransmitter systems did not significantly alter MES seizure severity.
Recently, the NRI atomoxetine was shown to reduce S-IRA and death in the DBA/1 audiogenic seizure mouse model21;22. Here we found that atomoxetine, as well as another NRI, reboxetine, reduced S-IRA and death following seizures induced in a different seizure model, MES. In this study we further showed that the protective effect of NE is mediated through an α1 receptor mechanism. α1 receptors are expressed in key respiratory nodes and α1 receptor activation positively modulates breathing18. Intriguingly, we found that α1 receptor blockade could also prevent the protective effects of SSRI, suggesting that NE may act downstream of 5-HT in regulating breathing following a seizure. While, this is not the first study to implicate NE in seizure-related breathing changes, these results meaningfully extend the literature, particularly as it pertains to interactions between the 5-HT and NE systems. It is clear from the existing literature that serotonergic upregulation is protective against seizure-induced death in a variety of animal models; however, the current study suggests that the protective effect of serotonergic upregulation may be predicated on normally functioning noradrenergic neurotransmission.
Similar to what was reported for atomoxetine21, the maximal effect of reboxetine in protecting against S-IRA following MES-induced seizures was seen at mid-range doses and diminished at higher doses. For atomoxetine, the authors attributed the effect to increased activation of α2 receptors at higher doses which inhibited norepinephrine release21. Reboxetine also exhibits α2 receptor binding at higher doses47. Furthermore, reboxetine has been previously shown to act on α2 adrenoceptors on NE neurons in the LC to dose-dependently decrease neuronal firing48.
To assess the role of NE neurons from LC in preventing S-IRA, we pre-treated WT mice with DSP-4, resulting in ~80% loss of NE neurons in LC. In our previous study, citalopram prevented S-IRA in WT mice8. Here we found that citalopram was ineffective at reducing S-IRA in mice subjected to LC NE neuron destruction, suggesting that NE neuron activity may be necessary to prevent S-IRA.
The non-selective β-antagonist propranolol significantly reduced S-IRA and death compared to vehicle. The 10 mg/kg dose was used to maximize the drug’s effects during S-IRA. However, it has been reported that propranolol can potentiate activity of NE at the α receptors through norepinephrine reuptake inhibition49. Given that α1 blockade with prazosin was sufficient to prevent the effect of propranolol, we believe that this effect was a result of the reuptake inhibition described, although a specific role for β-receptors cannot be ruled out and future experiments examining a role of β-receptors are warranted.
With the exception of phenylephrine, all drugs were applied systemically. The systemic nature of the drug application does not reveal the specific site of receptor activation. As there are α1 receptors on respiratory nuclei in the brainstem, this might be the site of action18. While there are α1 receptors on 5-HT neurons, that NRIs were effective in reducing S-IRA and death in Lmx1bf/f/p mice and that this effect was similarly prevented with prazosin, suggests that α1 receptors are normally activated somewhere other than on 5-HT neurons.
Prazosin reduced seizure severity in all groups receiving it. Despite reducing seizure severity, prazosin still prevented the protective effects of NRI, SNRI, and SSRI. This anti-seizure property of prazosin has been described previously, but had been attributed to a melatonin-2 receptor mechanism23. Perhaps melatonin receptor mechanisms should also be probed in S-IRA. Prazosin also decreased breathing as expected. Thus, the fact that prazosin prevented the protective effects of the reuptake inhibitors, could be attributed to the significant pre-ictal reduction in breathing and not necessarily a specific effect of α1 blockade.
Here we found that decreasing NE neurons in the LC with the neurotoxin DSP-4 prevented the protective effect of citalopram on S-IRA and death. This suggests that noradrenergic input is important in regulating breathing after a seizure. One explanation is that 5-HT neurons modulate breathing after a seizure through NE neurons in the LC. Thus, eliminating the NE neurons in the LC disallows 5-HT to protect against S-IRA. However, 5-HT usually exerts inhibitory effects on NE neurons in the LC50. Another possible explanation is that the 5-HT and NE systems interact at a final common effector site, such as the respiratory nuclei, which express both α1 and 5-HT receptors18. In this sense, enhancing extracellular concentrations of NE or 5-HT is sufficient to protect breathing following a seizure, and may suggest that other pharmacological way of enhancing breathing would also be effective.
The MES model is not a model of epilepsy, per se, but rather is a model of acute seizure induced in a seizure naïve brain. Given that most witnessed SUDEP cases occur following post-ictal respiratory arrest, it is reasonable to try to model SUDEP in animals by eliciting post-ictal respiratory arrest. A limitation to the MES model is that each animal undergoes only a single seizure induction. In the DBA/1 and DBA/2 mouse models of audiogenic seizures, in which S-IRA and death are prevented with mechanical ventilation and mice can undergo serial testing and serve at their own controls6;39; however, an advantage to the MES model is that it is simple to employ and seizure inductions are immediate.
In this study, prazosin, an exogenous antagonist, was used to examine a role for the α1 noradrenergic receptor in seizure-induced death. However, prazosin may have off-target effects both systemically and in the central nervous system. Convergent evidence provided by optogenetic and transgenic manipulations or blood brain barrier impermeable drugs delivered i.c.v. will be critical to fully characterizing this effect.
Epilepsy, especially refractory epilepsy, is highly prevalent. SUDEP is an entirely too common consequence of refractory epilepsy. While pathophysiological mechanisms for SUDEP are poorly understood, it is clear that seizure related respiratory dysregulation plays a large role. Understanding these mechanisms and relevant neurotransmitter systems involved is vital to development of prophylactic strategies for patients at highest risk for SUDEP. 5-HT mechanisms have been implicated, including by our group, in seizure-induced respiratory arrest and death. Here we corroborate recent data implicating NE in the modulation of post-ictal respiration and provide additional mechanistic insights. As we continue to learn more about who is at greatest risk for SUDEP, studies like this one will help to inform pharmacologic prophylactic strategies, and may even inform choice of anti-epileptic therapy by taking into account mechanisms of action and off-target effects. Perhaps pharmacologic prophylaxis will involve combination therapy targeting serotonergic and noradrenergic systems.
Statistical Analysis
All statistical analyses were completed using Graphpad Prism 7 (GraphPad Software Inc., La Jolla, CA) or Microsoft Excel (Redmond, WA). Two-tailed Fisher’s Exact Tests were used to compare survival between groups. Unpaired two-tailed student’s t-tests were employed to compare E/F ratio and pre/post-injection respiratory parameters. Comparisons were made between the effects of the different experimental manipulations on survival, breathing, seizure severity, and post-ictal respiratory recovery. Analyses were corrected for multiple comparisons. Unless otherwise specified, data is expressed as mean ± standard deviation. The sample sizes of groups were determined using power calculations based on the observed effect sizes seen in pilot data in the lab. Significance threshold was set at p < 0.05 for all conditions.
Key points summary.
SUDEP is the leading cause of death in patients with refractory epilepsy and thus is a major public health problem.
A large body of evidence implicates serotonin in SUDEP; however, other neurotransmitters such as norepinephrine may play a role.
Seizure-induced respiratory arrest was prevented by NRIs, SNRIs, SSRIs; α1 noradrenergic blockade prevented these protective effects.
The results suggest that norepinephrine may act as an intermediary in serotonergic modulation of breathing following a seizure.
Manipulation of noradrenergic pathways with or without manipulation of serotonergic pathways may be helpful for preventing SUDEP.
Acknowledgments
This work was supported by NIH/NINDS K08 NS069667 and R01 NS095842, a Targeted Research Initiative in Morbidity and Mortality award from the Epilepsy Foundation, and the Beth Levitt Tross Professorship in Epilepsy Research to G.F.B., NIH/NINDS T32 NS007421 to K.G.D. and B.S.P., the Post-Comprehensive Exam Fellowship from the University of Iowa Graduate College to B.S.P., and Iowa Center for Research by Undergraduates and Iowa Neurosciences Institute Summer Scholars Fellowships to S.W.K.
Footnotes
Disclosure of conflicts of interest
The authors have no conflict of interest to disclose.
Additional information
Competing interests
None of the authors has any competing interests to disclose.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Fisher RS. The New Classification of Seizures by the International League Against Epilepsy 2017. Curr Neurol Neurosci Rep 2017;17:48. [DOI] [PubMed] [Google Scholar]
- 2.Hesdorffer DC, Logroscino G, Benn EK et al. Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology 2011;76:23–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Z, Brodie MJ, Liew D et al. Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol 2018;75:279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia 2014;55:1479–85. [DOI] [PubMed] [Google Scholar]
- 5.Nashef L, Garner S, Sander JW et al. Circumstances of death in sudden death in epilepsy: interviews of bereaved relatives. J Neurol Neurosurg Psychiatry 1998;64:349–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia 2006;47:21–6. [DOI] [PubMed] [Google Scholar]
- 7.Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain 2008;131:3239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchanan GF, Murray NM, Hajek MA et al. Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol 2014;592:4395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryvlin P, Nashef L, Lhatoo SD et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol 2013;12:966–77. [DOI] [PubMed] [Google Scholar]
- 10.Seyal M, Pascual F, Lee CY et al. Seizure-related cardiac repolarization abnormalities are associated with ictal hypoxemia. Epilepsia 2011;52:2105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park KJ, Sharma G, Kennedy JD et al. Potentially high-risk cardiac arrhythmias with focal to bilateral tonic-clonic seizures and generalized tonic-clonic seizures are associated with the duration of periictal hypoxemia. Epilepsia 2017;58:2164–71. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y, Bravo E, Thirnbeck CK et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest 2018;128:1141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bateman LM, Li CS, Lin TC et al. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia 2010;51:2211–4. [DOI] [PubMed] [Google Scholar]
- 14.Faingold CL, Randall M. Effects of age, sex, and sertraline administration on seizure-induced respiratory arrest in the DBA/1 mouse model of sudden unexpected death in epilepsy (SUDEP). Epilepsy Behav 2013;28:78–82. [DOI] [PubMed] [Google Scholar]
- 15.Faingold CL, Kommajosyula SP, Long X et al. Serotonin and sudden death: differential effects of serotonergic drugs on seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav 2014;37:198–203. [DOI] [PubMed] [Google Scholar]
- 16.Zeng C, Long X, Cotten JF et al. Fluoxetine prevents respiratory arrest without enhancing ventilation in DBA/1 mice. Epilepsy Behav 2015;45:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao ZQ, Scott M, Chiechio S et al. Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci 2006;26:12781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viemari JC, Tryba AK. Bioaminergic neuromodulation of respiratory rhythm in vitro. Respir Physiol Neurobiol 2009;168:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shachar D, Klein E, Tabak A et al. Effect of single and repeated administration of fluvoxamine on noradrenaline release in rat brain. Eur J Pharmacol 1997;332:237–43. [DOI] [PubMed] [Google Scholar]
- 20.Fuller RW, Wong DT, Robertson DW. Fluoxetine, a selective inhibitor of serotonin uptake. Med Res Rev 1991;11:17–34. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Zhao H, Feng HJ. Atomoxetine, a norepinephrine reuptake inhibitor, reduces seizure-induced respiratory arrest. Epilepsy Behav 2017;73:6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao H, Cotten JF, Long X et al. The effect of atomoxetine, a selective norepinephrine reuptake inhibitor, on respiratory arrest and cardiorespiratory function in the DBA/1 mouse model of SUDEP. Epilepsy Res 2017;137:139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray M, Mediratta PK, Reeta K et al. Receptor mechanisms involved in the anticonvulsant effect of melatonin in maximal electroshock seizures. Methods Find Exp Clin Pharmacol 2004;26:177–81. [DOI] [PubMed] [Google Scholar]
- 24.Meierkord H, Shorvon S, Lightman SL. Plasma concentrations of prolactin, noradrenaline, vasopressin and oxytocin during and after a prolonged epileptic seizure. Acta Neurol Scand 1994;90:73–7. [DOI] [PubMed] [Google Scholar]
- 25.Cedarbaum JM, Aghajanian GK. Afferent projections to the rat locus coeruleus as determined by a retrograde tracing technique. J Comp Neurol 1978;178:1–16. [DOI] [PubMed] [Google Scholar]
- 26.Kaehler ST, Singewald N, Philippu A. Dependence of serotonin release in the locus coeruleus on dorsal raphe neuronal activity. Naunyn Schmiedebergs Arch Pharmacol 1999;359:386–93. [DOI] [PubMed] [Google Scholar]
- 27.Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci USA 2010;107:16354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purnell BS, Hajek MA, Buchanan GF. Time-of-day influences on respiratory sequelae following maximal electroshock-induced seizures in mice. J Neurophysiol 2017;118:2592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drorbaugh JE,FENN WO A barometric method for measuring ventilation in newborn infants. Pediatrics 1955;16:81–7. [PubMed] [Google Scholar]
- 30.Hajek MA, Buchanan GF. Influence of vigilance state on physiologic consequences of seizures and seizure-induced death in mice. J Neurophysiol 2016;115:2286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith HR, Leibold NK, Rappoport DA et al. Dorsal raphe serotonin neurons mediate CO2-induced arousal from sleep. J Neurosci 2018;38:1915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viemari JC, Ramirez JM. Norepinephrine differentially modulates different types of respiratory pacemaker and nonpacemaker neurons. J Neurophysiol 2006;95:2070–82. [DOI] [PubMed] [Google Scholar]
- 33.Al-Zubaidy ZA, Erickson RL, Greer JJ. Serotonergic and noradrenergic effects on respiratory neural discharge in the medullary slice preparation of neonatal rats. Pflugers Arch 1996;431:942–9. [DOI] [PubMed] [Google Scholar]
- 34.Zhang ZH, Felder RB. Hypothalamic corticotrophin-releasing factor and norepinephrine mediate sympathetic and cardiovascular responses to acute intracarotid injection of tumour necrosis factor-alpha in the rat. J Neuroendocrinol 2008;20:978–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aston-Jones G, Shipley MT, Chouvet G et al. Afferent regulation of locus coeruleus neurons: anatomy, physiology and pharmacology. Prog Brain Res 1991;88:47–75. [DOI] [PubMed] [Google Scholar]
- 36.Hilaire G, Viemari JC, Coulon P et al. Modulation of the respiratory rhythm generator by the pontine noradrenergic A5 and A6 groups in rodents. Respir Physiol Neurobiol 2004;143:187–97. [DOI] [PubMed] [Google Scholar]
- 37.Cedarbaum JM, Aghajanian GK. Activation of locus coeruleus neurons by peripheral stimuli: modulation by a collateral inhibitory mechanism. Life Sci 1978;23:1383–92. [DOI] [PubMed] [Google Scholar]
- 38.Hormigo S, Horta JA Junior, Gomez-Nieto R et al. The selective neurotoxin DSP-4 impairs the noradrenergic projections from the locus coeruleus to the inferior colliculus in rats. Front Neural Circuits 2012;6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng HJ, Faingold CL. Abnormalities of serotonergic neurotransmission in animal models of SUDEP. Epilepsy Behav 2017;71:174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richerson GB, Buchanan GF. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia 2011;52 Suppl 1:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bagdy G, Kecskemeti V, Riba P et al. Serotonin and epilepsy. J Neurochem 2007;100:857–73. [DOI] [PubMed] [Google Scholar]
- 42.Jobe PC. Neurotransmitters and epilepsy: an overview. Fed Proc 1984;43:2503–4. [PubMed] [Google Scholar]
- 43.Jobe PC, Laird HE, Ko KH et al. Abnormalities in monoamine levels in the central nervous system of the genetically epilepsy-prone rat. Epilepsia 1982;23:359–66. [DOI] [PubMed] [Google Scholar]
- 44.Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci 2004;5:449–61. [DOI] [PubMed] [Google Scholar]
- 45.Mishra PK, Kahle EH, Bettendorf AF et al. Anticonvulsant effects of intracerebroventricularly administered norepinephrine are potentiated in the presence of monoamine oxidase inhibition in severe seizure genetically epilepsy-prone rats (GEPR-9s). Life Sci 1993;52:1435–41. [DOI] [PubMed] [Google Scholar]
- 46.Briere R, Sherwin AL, Robitaille Y et al. Alpha-1 adrenoceptors are decreased in human epileptic foci. Ann Neurol 1986;19:26–30. [DOI] [PubMed] [Google Scholar]
- 47.Invernizzi RW, Parini S, Sacchetti G et al. Chronic treatment with reboxetine by osmotic pumps facilitates its effect on extracellular noradrenaline and may desensitize alpha(2)-adrenoceptors in the prefrontal cortex. Br J Pharmacol 2001;132:183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grandoso L, Pineda J, Ugedo L. Comparative study of the effects of desipramine and reboxetine on locus coeruleus neurons in rat brain slices. Neuropharmacology 2004;46:815–23. [DOI] [PubMed] [Google Scholar]
- 49.Tuross N, Patrick RL. Effects of propranolol on catecholamine synthesis and uptake in the central nervous system of the rat. J Pharmacol Exp Ther 1986;237:739–45. [PubMed] [Google Scholar]
- 50.Dremencov E, el MM, Blier P. Noradrenergic augmentation of escitalopram response by risperidone: electrophysiologic studies in the rat brain. Biol Psychiatry 2007;61:671–8. [DOI] [PubMed] [Google Scholar]