

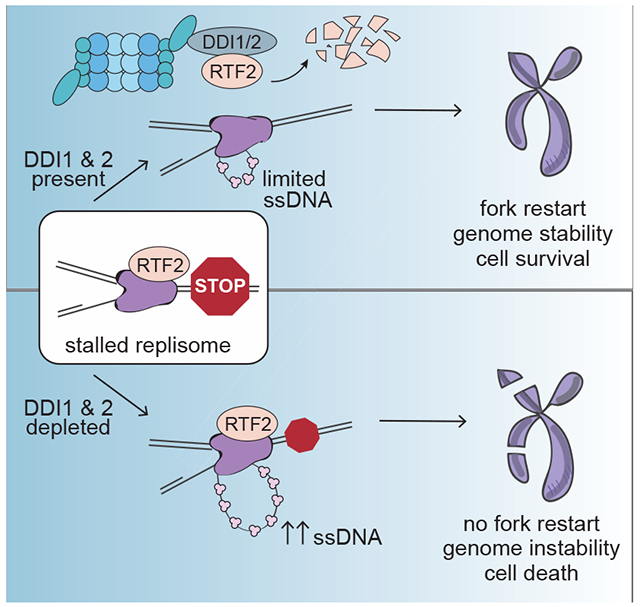
Summary
The protection and efficient restart of stalled replication forks is critical for the maintenance of genome integrity. Here we identify a regulatory pathway that promotes stalled forks recovery from replication stress. We show that the mammalian replisome component C20orf43/RTF2 (homologous to S. pombe Rtf2) must be removed for fork restart to be optimal. We further show that the proteasomal shuttle proteins DDI1 and DDI2 are required for RTF2 removal from stalled forks. Persistence of RTF2 at stalled forks results in fork restart defects, hyperactivation of the DNA damage signal, accumulation of ssDNA, sensitivity to replication drugs, and chromosome instability. These results establish that RTF2 removal is a key determinant for the ability of cells to manage replication stress and maintain genome integrity.
Keywords: DNA replication, replication stress, ubiquitin proteasome system, DDI1, DDI2, iPOND, C20orf43, RTF2, hydroxyurea, aphidicolin
eTOC
Recovery of normal DNA replication after encountering obstacles to replication fork progression is a critical aspect of protecting the genome from damage. Kottemann et al. identify RTF2, a replisome component that must be removed by the proteasome shuttles DDI1 and DDI2 to allow an effective response to replication stress
Graphical Abstract
Introduction
In each cell cycle, the replication machinery encounters replication fork barriers. They include DNA lesions that impede fork progression, repetitive, secondary structure-forming sequences (rDNAs, centromeres, telomeres), and collisions between the replisome and transcriptional machinery (reviewed in Dalgaard JZ, 2011; Hamperl and Cimprich, 2016; Zeman and Cimprich, 2014). Changes in replication conditions (usage of replication of origins, replication speed, dNTP pools), such as those caused by oncogenic transformation, also cause replication fork dysfunction (reviewed in Macheret and Halazonetis, 2015; Hills and Diffley, 2014). In recent years, the term “replication stress” was coined to encompass the diverse ways that the proper progression of the replisome may be impeded.
Response to replication stress is an essential aspect of DNA damage response (DDR) in cells, and the consequences of inadequate response result in genome instability and contributes to human disease (Hills and Diffley, 2014; Kim et al., 2016; Lambert and Carr, 2013; Lee et al., 2007; Macheret and Halazonetis, 2015; Willis et al., 2014; Zhang et al., 2009). To counter replication stress, cells use a host of factors that promote replication fork stability during normal elongation and that respond specifically to stalled replication forks. The fork protection complex, comprised of TIMELESS (TIM) and TIPIN in human cells (Tof1 and Scm3 in S. cerevisiae and Swi1 and Swi3 in S. pombe) travels with the elongating replisome. It stabilizes forks by coupling polymerase and helicase activity during unperturbed replication, and is required for establishing checkpoint signaling following fork stalling (Chou and Elledge, 2006; Errico et al., 2007; Gotter et al., 2007; Unsal-Kacmaz et al., 2005; Yoshizawa-Sugata and Masai, 2007).
Activation of the ATR kinase is central to replication stress response and is triggered by the accumulation of RPA-coated single stranded DNA (ssDNA) at stalled forks (Bass et al., 2016; Byun et al., 2005; Cortez et al., 2001; Costanzo et al., 2003; Haahr et al., 2016; Zou and Elledge, 2003). ATR promotes checkpoint signaling to prevent the firing of new origins into a refractory replication environment by activating the checkpoint kinase CHK1, and directly facilitates the recruitment of repair proteins to the fork by phosphorylating scaffolding substrates like γH2AX (Branzei and Foiani, 2009; Dungrawala et al., 2015; Friedel et al., 2009).
Recent data from the Cortez laboratory have shown that the replisome is stable in the presence of replicative stress even when the ATR is inhibited (Dungrawala et al., 2015). The slow dissociation of the replisome proteins is associated with the slow replication termination of forks under stress, rather than active removal of the replisome proteins from the stalled forks. The replisome stability is not dependent on ATR activity, consistent with previous yeast data (De Piccoli et al., 2012). In the absence of ATR signaling, however; stalled forks are converted into double strand breaks (DSBs) during S phase, a progressive process known as fork collapse (Dungrawala et al., 2015; Toledo et al., 2013). DSB can also manifest in mitosis in response to under-replicated DNA in a manner dependent on structure-specific nucleases (Minocherhomji et al., 2015).
Another major pathway of fork protection is the shielding of stalled replication forks from inappropriate nuclease activity. In particular, the BRCA2 and RAD51 proteins prevent the MRE11 nuclease from degrading the nascent strand during replication stress (Schlacher et al., 2011; Schlacher et al., 2012). They have been shown to work through stabilization of replication forks reversed by SNF2-family remodelers including SMARCAL1, ZRANB3, and HLTF (Kolinjivadi et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017; Vujanovic et al., 2017)
In this study, we identify the proteasomal shuttle proteins DDI1 and DDI2 as participants in the cellular response to DNA replication stress necessary for the maintenance of genomic stability. The yeast proteasome shuttle proteins, Rad23, Ddi1 and Dsk2, function by virtue of modular ubiquitin-like (UBL) and ubiquitin-associated (UBA) domains that allow them to interact with both the proteasome and ubiquitinated substrates (Elsasser et al., 2002; Kang et al., 2006). Proteasomal shuttle proteins have been previously linked to aspects of the DNA damage response and DNA replication. Rad23 and the human orthologs RAD23A and RAD23B have been implicated in nucleotide excision repair (NER) (reviewed in (Dantuma et al., 2009). Furthermore, the proteasomal shuttle protein VCP functions in origin licensing and replication termination during normal S phase and in the repair of UV damage and DSBs where it removes Ku70/80 after NHEJ (Acs et al., 2011; He et al., 2014; van den Boom et al., 2016) , reviewed in (Franz et al., 2016).
Using conditions of DDI1 and 2 proteasomal shuttle depletion in human cells, we identify removal of a replisome component Replication Termination Factor 2 (RTF2), also known as C20orf43, as a key step during proper response to replication stress. Without RTF2’s removal, the cells cannot complete replication and suffer genome instability.
Results
DDI1 and DDI2 associate with the proteasome and replication fork proteins.
The human homologs of S. cerevisiae DNA damage-inducible 1 (Ddi1), DDI1 and DDI2, are understudied proteins containing a conserved N-terminal ubiquitin-like (UBL) domain and a C-terminal RVP domain (Figure 1A). The UBL domain facilitates binding both to ubiquitin receptors and ubiquitin, fulfilling the bifunctional requirement for shuttle proteins (Nowicka et al., 2015). The Drosophila Ddi1 homolog rngo has also been shown to bind both ubiquitin and the proteasome (Gomez et al., 2011; Morawe et al., 2011). DDI1 and DDI2 exhibit 35% identity to S. cerevisiae Ddi1, and 72% identity to each other at the protein level.
Figure 1: Proteasome shuttles DDI1 and DDI2 function in replication stress response.
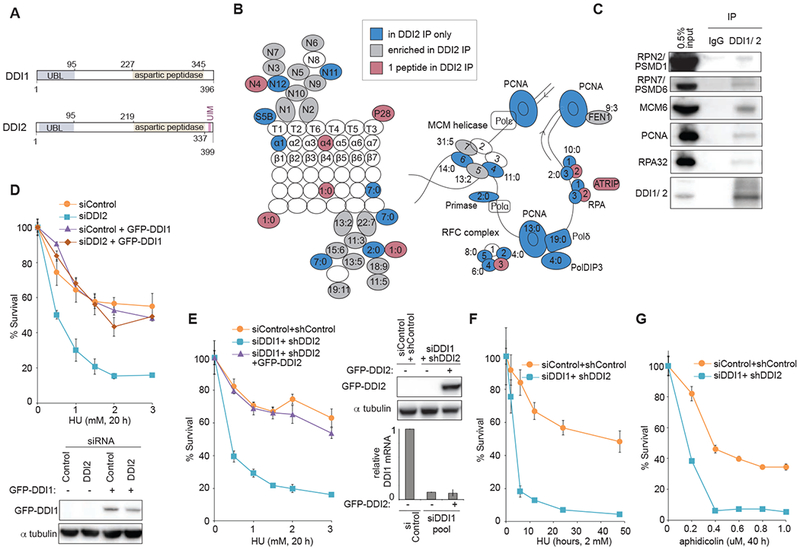
(A) Schematic of human DDI1 and DDI2 proteins highlighting their domain structure. Yeast Ddil contains both the UBL and UBA domains typical of shuttle proteins. Human DDI1/2 lack the UBA domain but feature an atypical UBL domain that can bind both ubiquitin and proteasomal ubiquitin receptors. A C-terminal ubiquitin interacting motif (UIM) is specific to human DDI2 (Nowicka et al., 2015; Siva et al., 2016; Trempe et al., 2016). A retroviral aspartyl protease (RVP) domain is also unique to DDI1/2 shuttle proteins (Sirkis et al., 2006). (B) Schematic of the proteasome (RPN, RPT, α, and β subunits) and DNA replication proteins identified to interact with DDI2 after crosslinking with DTSSP. Labeled in blue are proteins identified in the pulldown of GFP-tagged DDI2 but not GFP-only control. In grey are proteins enriched at least twofold in the GFP-DDI2 pulldown compared to GFP control IP. In purple are proteins identified with only one peptide in the GFP-DDI2 pulldown. Ratios in the figure indicate the number of peptides identified in the GFP-DDI2 pulldown to number of peptides in the GFP only control pulldown. (C) Validation of a subset of identified interactions by western blot following IP of endogenous DDI1/2 from cell lysates in the presence of the protein crosslinker DTSSP. All pulldowns for western blot were done in the presence of benzonase. (D) Graphs showing survival of U2OS cells transiently transfected with a pool of DDI2 siRNAs and cross-complemented by stable expression of GFP-DDI1 construct, and a western blot showing GFP-DDI1 expression in these cells. Cells were treated with the indicated doses of HU for 20 h, washed, released, allowed to grow for 7 days, and counted. (E) Graphs showing survival of U2OS cells transiently transfected with a pool of siRNAs against DDI1 or control and stably expressing shDDI2 #1 or control shRNA, that have been complemented with GFP-tagged DDI2, a western blot showing levels of GFP-DDI2, and a graph of relative DDI1 mRNA. Cells were treated as in D. (F) Graphs showing survival of U2OS cells transiently transfected with a pool of siRNAs against DDI1 and stably expressing shDDI2 #1. Cells were treated with 2 mM HU for the indicated time, washed, released, allowed to grow for 7 days, and counted. (G) Graphs showing survival of U2OS cells transiently transfected with a pool of siRNAs against DDI1 or control and stably expressing shDDI2 #1 or control shRNA. Cells were treated with the indicated dose of aphidicolin for 40 h, washed, released, allowed to grow for 7 days, and counted. Error bars represent SEM for 3 replicates. See also Figure S1
To identify DDI1/2 binding targets, we performed an unbiased proteomic screen for factors that preferentially associate with a GFP-tagged DDI2. Consistent with data reported in yeast, the first major network identified among DDI-interacting factors was the ubiqutin-proteasome system (UPS). DDI2 co-purified members of the non-ATPase subunit of the 19S regulatory cap (Figure 1B), most prominently RPN1, which functions with RPN2 as the major proteasomal ubiquitin receptor for UBL domains (Gomez et al., 2011). Unexpectedly, DDI2 also co-purified with numerous replication factors, including 4 out of 6 members of the MCM replicative helicase, all 3 members of the ssDNA-binding RPA heterotrimer, 4 out of 5 members of the RFC sliding clamp loader complex along with PCNA itself, and DNA polymerase delta, among others (Figure 1B). We validated a subset of these interactions by performing an IP of either GFP-DDI2 (Figure S1A) or with an antibody that recognizes both endogenous DDIs (Figure 1C). Taken together, these data suggest that DDI1 and DDI2 might function at the interface between the UPS and the replisome.
DDI1/2 are required for cellular survival following replication stress
To determine whether DDI1/2 function during replication stress response, we evaluated survival of cells depleted of DDI1 or DDI2 to stress-causing drugs. Depleting DDI1 or DDI2 in U2OS or DDI1 in BJ cells led to substantially decreased survival after hydroxyurea (HU) treatment compared to controls (Figure S1B-F), indicating a deficiency in managing replication stress. Co-depletion of DDI1 together with DDI2 resulted in further sensitization to HU (Figure S1D). Complementation with GFP-DDI1 rescued the cellular sensitivity caused by DDI2 depletion and vice versa (Figure 1D-E), suggesting that they function together or redundantly during stress response. Therefore, the majority of experiments that follow were performed in cells with a dual-depletion of DDI1 and DDI2 (DDI1/2).
Next, we addressed if DDI1/2 depleted cells respond differently to a specific types of replication stress. A time course of HU exposure revealed increasing relative sensitivity of the DDI1/2 depleted cells compared to control with longer exposures to HU (Figure 1F). The DDI1/2 depleted cells also exhibited sensitivity to aphidicolin treatment (Figure 1G), an inhibitor of the replicative polymerase (Ohashi et al., 1978), to gemcitabine (Figure S1G), a chain-terminator (Huang et al., 1991), and more modest sensitivity to a panoply of DNA damaging agents that cause transient fork stalling followed by recruitment of dedicated DDR pathway components (Figure S1H-J). These results collectively demonstrate that DDI1/2 are critical factors during replication stress.
DDI1/2 promote removal of RTF2 and other proteins from replication forks
Based upon DDI1/2 interaction with the proteasome and their suggested role as a proteasome shuttle, we hypothesized that DDI1/2 are required for the turnover of a specific factor or factors at replication forks, whose removal is critical for proper replication stress response. To identify these factors, we performed isolation of proteins on nascent DNA (iPOND) (Sirbu et al., 2013) to enrich for replisome factors, coupled with stable isotope labeling with amino acids in cell culture (SILAC)-based quantitative mass spectrometry to compare protein occupancy at the fork between DDI1/2 knockdown and control cells during replication stress (Figure 2A). Within the identified replication and repair factors, ten proteins emerged as enriched two-fold or greater in the DDI1/2 knockdown cells (Figure 2B). We validated a subset of these enrichment results by western blot of iPOND or whole cell lysates (Figure 2C and S2A).
Figure 2: DDI1/2 mediate the removal of RTF2 from replication forks.
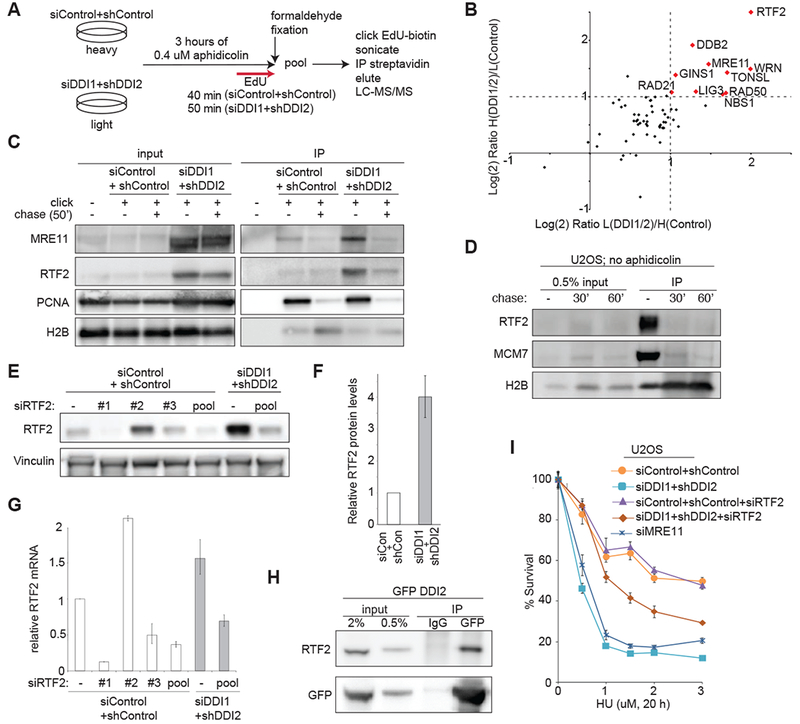
(A) Schematic of SILAC/iPOND experiment comparing replication fork occupancy during aphidicolin treatment in control vs. DDI1/2 depleted cells. The experiment was performed twice, with light/heavy label swap. (B) Graph of the Log(2) ratio of a curated list of DDI-target candidates among replication and repair proteins identified by iPOND in DDI1/2 co-depleted/control cells in two label swap experiments. Proteins reaching the enrichment threshold of Log(2)=1/−1 are named and shown in red. Proteins were quantified using the average area. (C) Validation of mass spectrometry results by iPOND and western blot. Cells were treated with 0.4 μM aphidicolin for 3 h then labeled with EdU for 50′ in the presence of aphidicolin. For chase, cells were not treated with aphidicolin and were labeled with EdU for 10′ then with thymidine for 50′. (D) Western blot showing iPOND analysis of H2B, MCM7, and RTF2 during unperturbed replication. U2OS cells were labeled with EdU for 10′ followed by 0′, 30′, or 60′ of thymidine chase. (E) Validation of RTF2 stabilization in whole cell extract. Control or DDI1/2 co-depleted cells were treated with the indicated siRNAs against RTF2 or control. Error bars represent SEM n=at least 3. (F) Quantification of relative protein levels of RTF2 in control or DDI1/2 depleted cells. (G) Quantification of relative mRNA levels of RTF2 in control, RTF2, or DDI1/2 depleted cells. (H) Coimmunoprecipitation of GFP-DDI2 and RTF2. Stably expressing GFP-DDI2 U2OS cells treated with 2 μM MG132 were subject to immunoprecipitation by anti-GFP or an IgG control, using an IP buffer containing 5 mg/ml NEM, and immunoblotted for RTF2. (I) Graph showing survival of U2OS cells treated with the indicated doses of HU. Cells were depleted of MRE11, control, or DDI1/2 with and without knockdown of RTF2. Error bars represent SEM n=3. See also Figure S2
DDB2, also known as XPE, a nucleotide excision repair protein, was stabilized in the DDI1/2 depleted cells, but we were not able to detect DDB2 reliably at the replication fork by iPOND, consistent with published iPOND data. (Figure S2A, data not shown) (Dungrawala et al., 2015; Ruthemann et al., 2016; Tang et al., 2000). It is therefore likely that DDB2 is a bona fide target of DDI1/2 but is unlikely to be the culprit at the replisome. The members of the MRN complex - MRE11, RAD50, and NBS1 - as well as WRN helicase were also highly enriched at the fork by iPOND. We validated that MRE11 is enriched at the fork by western blot (Figure 2C).
The top enriched protein identified by iPOND-SILAC was RTF2, also known as C20orf43. RTF2 is a previously unstudied human homolog of the S. pombe Replication Termination Factor 2 (Rtf2) which is required to maintain replication fork stalling at a prescribed Replication Termination Site (RTS) to preserve unidirectional replication of the mating type locus (Inagawa et al., 2009). Data from fission yeast suggest that Rtf2 interacts with the replisome and may constrain restart and repair factors although the exact mechanism of Rtf2 function at the RTS is still unknown. Based on its function in S. pombe, RTF2 emerged as a particularly promising candidate since we were searching for a factor that impedes fork progression and repair when it is inappropriately enriched at the stalled replication fork.
Consistent with previous iPOND (Dungrawala et al., 2015) and nascent chromatin capture experiments (Alabert et al., 2014), human RTF2 associates with nascent DNA even during unperturbed replication (Figure 2D). To determine if RTF2 is a target of DDI1/2, we assessed the relative protein and mRNA levels of RTF2 in control and DDI1/2 knockdown cells with and without RTF2 depletion. RTF2 was highly stabilized at the protein level by DDI1/2 knockdown, with levels approaching a four-fold increase, consistent with the differences seen by iPOND (Figure 2E-F), and without commensurate changes in mRNA levels (Figure 2G). In U2OS and HeLa cells, DDI1 and DDI2 knockdown both exhibited increases in RTF2 levels (Figure S2B and C). In BJ cells, only DDI1 knockdown had the stabilization effect, correlating increased RTF2 levels with the HU sensitivity (Figure S2D, S1E-F)). DDI1/2 knockdown extended RTF2 half-life during cycloheximide treatment (Figure S2E-F), consistent with posttranslational stabilization. The proteasome inhibitor MG132 also stabilized RTF2 during cycloheximide treatment in U2OS and BJ cells (Figure S2F and G), establishing that proteasome-mediated degradation is the central contributor to its stability.
RTF2 and GFP-DDI2 co-immunoprecipitate (Figure 2H), which supports a model in which DDI1/2 delivers RTF2 to the proteasome for degradation. However, the regulation of the interaction remains to be established. To determine if DDI1/2 travels with RTF2 and the replication fork, we performed iPOND analysis on cells expressing GFP-DDI2. While DDI2 is present on nascent chromatin, it does not appear to be significantly enriched at the fork (Figure S2H), indicating that it is probably not stably associated with the traveling replisome.
If RTF2 is the key factor at the stressed replication fork in DDI1/2 depleted cells, one would expect that removing excess RTF2 would ameliorate the observed replication stress sensitivity. To test this hypothesis, we depleted RTF2 down to the approximate level of control cells. siRNA treatment against RTF2 in control cells has no effect on cellular survival after HU treatment; however, depletion of RTF2 in the DDI1/2 knockdown cells resulted in a significant increase in resistance to HU (Figure 2I and Figure S2I). Almost complete depletion of RTF2 in the DDI1/2 knockdown cells showed a more modest rescue, possibly due to poor growth of RTF2 depleted cells (Figure S2J). Failure to restore survival to the level of control may indicate that turnover of other factors, perhaps the MRN complex, is relevant to the phenotype, or it could be a consequence of relative DDI1/2 and RTF2 levels in individual cells and at individual forks. Knockdown of MRE11 alone yielded HU sensitivity similar to DDI1/2 knockdown (Figure 2I), making a rescue experiment not feasible. It remains to be determined, if the MRN complex contributes to the phenotypes seen in the DDI1/2 knockdown cells.
Removal of RTF2 from replisome promotes cell cycle progression after replication stress
To determine when and how DDI1/2 knockdown cells fail following exposure to HU, we analyzed their cell cycle. We first asked if DDI knockdown cells were able to resume replication on a global level following fork stalling and cell cycle synchronization with HU. Control and DDI1/2 depleted cells were able to re-enter active S-phase en masse following release from 20 h 2 mM HU treatment, with equivalent BrdU incorporation (Figure 3A). DDI1/2 knockdown and control cells at first proceeded through the cell cycle with similar kinetics; however, by 14-16 hours the cells exhibited a late S-phase delay and a defect in G1 entry (Figure 3A). Apoptosis was elevated in DDI1/2 depleted cells compared to control, with a striking increase in cleaved-caspase-3 staining around 16 hours post-release (Figure 3B, S3A), which corresponds to late-S/G2, the timepoint at which cell cycle profile began to reveal aberrancies. Nearly half of apoptotic cells during the 16-hour apoptotic surge were positive for EdU and cleaved caspase 3 (Figure S3B), indicating that they were undergoing apoptosis during or soon after DNA replication. Importantly, depletion of RTF2, resulted in a striking reduction in cleaved caspase-3 positive cells following HU compared to DDI1/2 depletion alone (Figure 3B). These data suggest that while the bulk of the genome can be successfully replicated after HU treatment in the DDI1/2 depleted cells, these cells, due to the persistence of RTF2 at the replication fork under stress, experience conditions that prevent full replication and cell cycle progression resulting in cell death.
Figure 3: Stabilization of RTF2 by DDI1/2 depletion causes impaired recovery from replication stress.
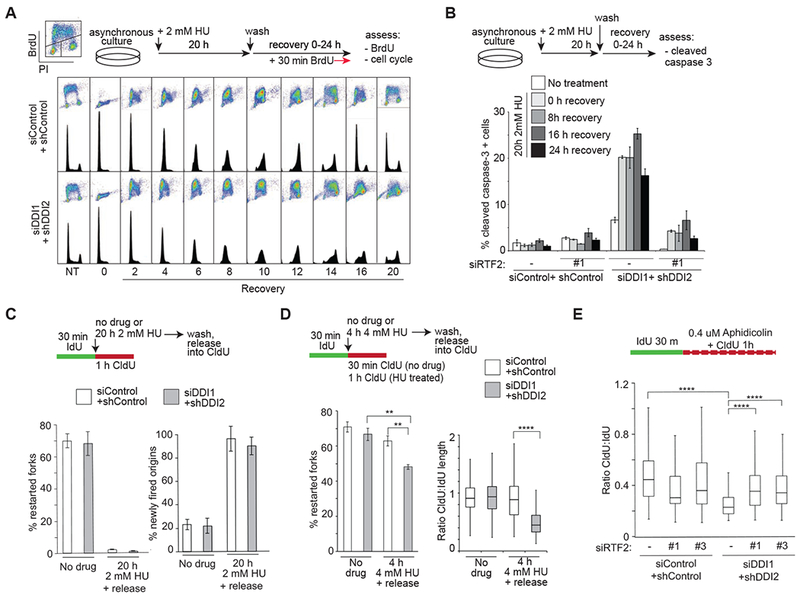
(A) Analysis of cell cycle progression in control and DDI1/2 depleted cells following synchronization by 20h 2mM HU treatment and release. 30 min prior to indicated timepoints, cells were labeled with BrdU. Images are representative of at least 3 independent experiments. (B) Analysis of the percentage of apoptotic cells as determined by cleaved caspase-3 staining measured by FACS. U2OS control and DDI1/2 depleted cells with or without knockdown of RTF2 were synchronization by 20 h 2 mM HU treatment and released for indicated times. Error bars represent SEM n=3. (C) Quantification of the percentage of restarted forks [restarted forks/(restarted forks plus non-restarted forks)] and newly-fired origins [newly fired origins/(continuing forks plus newly fired origins)] following 20 h of 2 mM HU in control and DDI1/2 knockdown cells. (D) Quantification of the percentage of restarted forks [restarted forks/(restarted forks plus non-restarted forks)] following 4h of 4 mM HU and of fork restart productivity defined by the ratio of CldU length to IdU length of restarted forks following 4h of 4 mM HU in control and DDI1/2 knockdown cells. (E) Analysis of fork processivity defined by the ratio of CldU to IdU label length of continuing forks treated with 0.4 uM aphidicolin during CldU labeling in control and DDI1/2 depleted cells with or without knockdown of RTF2. Each panel includes a schematic for experimental setup. Error bars represent SEM n=3. *p<0.05 **p<0.01 ***p<0.001 ****p<0.0001 by ANOVA. See also Figure S3
RTF2 removal is important for fork restart and progression
We next investigated the consequences of RTF2 persistence on replication at the level of individual replication forks using DNA fiber analysis, tracking the lengths and distributions of sequential IdU and CldU labels in the context of different drug treatments. To observe fork restart and origin firing, we incubated cells with CldU following treatment with HU. After 20 h replication stalling, both control and DDI1/2 depleted cells were unable to restart the replication forks that were active at the time of HU addition (Figure 3C). IdU incubation during the full 20h of HU treatment did not lead to a substantial increase in fork restart (Figure S3C), confirming that these forks are terminally stalled. IdU tract length before and after 20 h HU treatment was similar (Figure S3C), indicating that no significant net elongation occurred during prolonged replication stalling.
Consistent with the cell cycle analysis shown in Figure 3A, DDI1/2 depleted cells exhibited normal new origin firing after release from HU (Figure 3C). To assess fork behavior under conditions where control cells are competent for fork restart, we exposed cells to shorter (4 h) HU treatment. DDI1/2 knockdown cells exhibited impaired fork restart and shorter restarted tract length compared to controls (Figure 3D), consistent with DDI1/2 being necessary for fork function during acute stress. To assay fork progression proficiency during milder replication stress, we co-incubated the second CldU label with low-dose HU or aphidicolin. DDI1/2 depleted cells exhibited a significant decrease in the length of the CldU label compared to controls (Figure 3E and S3D). This phenotype was also rescued by depletion of RTF2 (Figure 3E, S3E), showing that excess RTF2 is detrimental to fork progression during replication stress.
Replication stress response deficiency secondary to RTF2 stabilization leads to chromosome damage
To determine if DNA replication stress ultimately results in genome instability if RTF2 is not degraded, we examined metaphase chromosomes following HU or aphidicolin treatment in DDI1/2 depleted cells. We were able to detect three types of metaphase: normal, damaged (>2 breaks), and highly damaged, in which chromosomes display uncountable breaks or gaps, which we could not score (Figure 4A and B). DDI1/2 depleted cells exhibited an increase in breaks in metaphases that we were able to score (Figure 4C and S4A) and an increase in levels of highly damaged metaphases compared to control cells after drug treatment (Figure 4B and S4A). Remarkably, the damage was completely ameliorated when RTF2 was also depleted (Figure 4D).
Figure 4: Replication stress response deficiency due to RTF2 stabilization leads to increased genome instability.
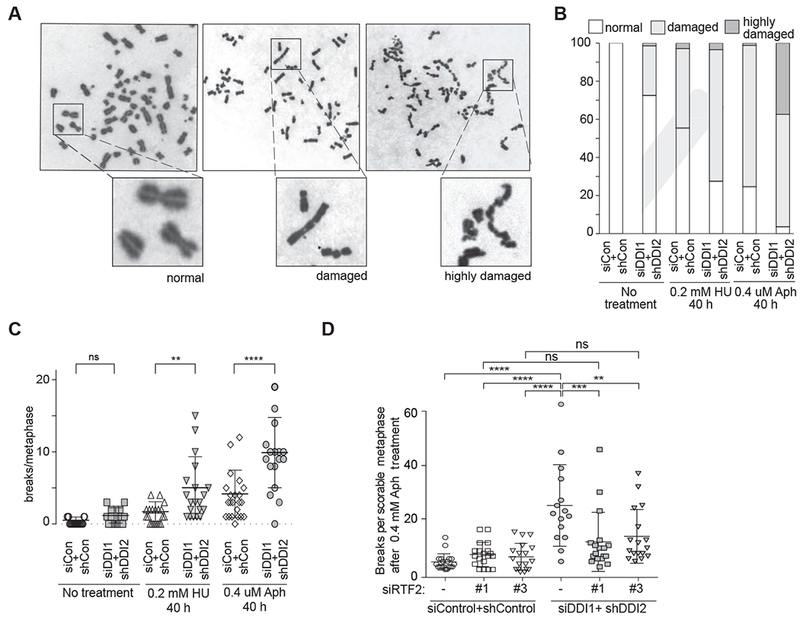
(A) Representative examples of chromosomes in the three classes of metaphases observed in U2OS cells after replication in the presence of low-dose HU or aphidicolin (Aph): normal, damaged, and highly damaged/uncountable. (B) Quantification of the proportion of each of these types of metaphase following 40 h of 0.2 mM HU or 0.4 μM Aph treatment in control and DDI1/2 depleted cells. (C) Quantification of the number of breaks per countable metaphase in U2OS control or DDI1/2 depleted cells. Cells were scored after no treatment, 40 h of 0.4 μM aphidicolin treatment or 40 h of 0.2 mM HU treatment. (D) Quantification of the number of breaks per countable metaphase in control or DDI1/2 depleted cells with or without knockdown of RTF2. Cells were scored after 40 h of 0.4 μM aphidicolin treatment. See also Figure S4
Removal of RTF2 limits accumulation of ssDNA during replication stress
To determine how DDI1/2–RTF2 interplay with DDR during replication stress, we investigated damage signaling in cells deficient for DDI1/2. The DDI1/2 depleted cells exhibited elevated and persistent levels of γH2AX following HU treatment by both western blot and immunofluorescence (IF) analysis, indicative of unrepaired damage (Figure S5A-C). DDI1/2 depleted cells exhibit normal-to-elevated levels of CHK1 phosphorylation during HU treatment, showing that ATR signaling remains intact (Figure S5A). These cells also showed elevated phospho-RPA and higher chromatin associated RPA staining (Figure 5A and B, S5D, S5H) suggestive of increased ssDNA. Assaying for the presence of ssDNA directly, by evaluating BrdU signal under non-denatured conditions, indeed revealed increased ssDNA in the DDI1/2 depleted cells (Figure S5E-F). Cell cycle progression in DDI1/2 knockdown cells following exposure to 0.5 mM HU was comparable to that seen in control cells, ruling out the possibility that differences are due to discrepancies in cell cycle (Figure S5G). In BJ fibroblasts, siRNA knockdown of DDI1 produced elevated levels of chromatin-bound RPA foci and siRNA knockdown of DDI2 had no significant effect, consistent with the sensitivity data (Figure S5H). Importantly, driving high exogenous overexpression of RTF2 is insufficient to increase phospho-RPA to the level seen in cells with DDI1/2 depletion, suggesting that levels of RTF2 at the replication fork in particular, are the key determinants of stress response (Figure S5I).
Figure 5: RTF2 promotes the formation of single stranded DNA.
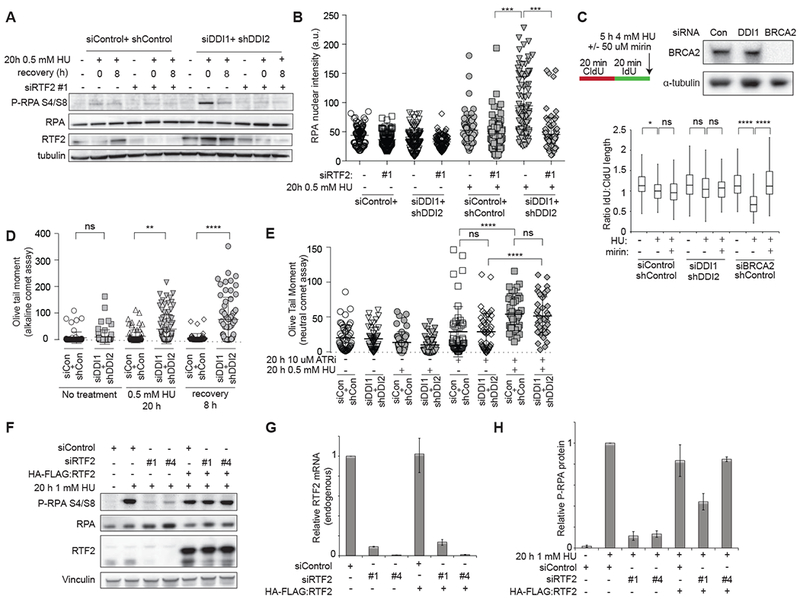
(A) Immunoblot analysis of whole cell extract to assess phosphorylated RPA32 levels. Control or DDI1/2 depleted cells with or without knockdown of RTF2 were treated with 0.5 mM HU for 20 h, released, and allowed to recover for 8 hours. (B) Quantification of mean nuclear intensity of chromatin-bound RPA staining in cells treated with 20h 0.5 mM HU and extracted with 0.25% Triton-X. (C) Analysis of nascent strand resection in control, DDI1/2-depleted, and BRCA2-depleted cells during 5 h of 4 mM HU treatment with or without the Mre11 inhibitor mirin. Cells were sequentially labeled for 20 minutes each with IdU and CldU. If no resection took place, the CldU/IdU ratio should be 1. Western blot shows levels of BRCA2 after siRNA-mediated depletion. (D) Analysis of DNA damage by alkaline comet assay. Control or DDI1/2 depleted cells were treated with 0.5 mM HU for 20 h, released, and allowed to recover for 8 hours. (E) Analysis of DNA damage by neutral comet assay. Control or DDI1/2 depleted cells were treated with 0.5 mM HU, 10 μM of the ATR inhibitor VE-821, or a combination of the two drugs for 20 h. (F) Western blot showing complementation by HA:RTF2 of the decrease in phospho-RPA signal following 20 h 1 mM HU in RTF2 depleted cells.(G) Graph showing relative endogenous RTF2 mRNA levels in cells treated with RTF2 siRNAs +/− complementation with HA-FLAG:RTF2. qPCR primer F recognizes the 5’ untranslated region of the endogenous transcript only. (H) Quantification of phosphorylated-RPA levels relative to total RPA. Error bars represent SEM n=3. *p<0.05 **p<0.01 ***p<0.001 ****p<0.0001 by ANOVA. See also Figure S5 and S6.
To determine if DDI1/2 depletion caused aberrant nucleolytic processing of newly replicated DNA, that would give rise to increased ssDNA, we labeled the nascent strand with consecutive pulses of CldU and IdU, treated with 4 mM HU for 5 hours, and assessed the relative lengths of the CldU and IdU tracts. In cells depleted of BRCA2, the HU treatment resulted in substantial shortening of signal from the second label compared to untreated cells. Addition of the MRE11 inhibitor mirin was sufficient to reverse the effect, as previously described (Schlacher et al., 2011; Schlacher et al., 2012) (Figure 5C). DDI1/2 knockdown cells showed no significant shortening of the nascent DNA strand after HU treatment compared to untreated or control cells (Figure 5C), suggesting that DDI1/2 do not affect MRE11-dependent nucleolytic degradation at stalled replication forks.
To determine if the increase in ssDNA was secondary to the generation of DNA DSB, we next assayed for the presence of damage in HU-synchronized S-phase cells by performing comet assays (Collins, 2004). Using an alkaline comet assay, which detects both double- and single-stranded breaks as well as alkali-labile base-damaged sites, we observed increased DNA damage in DDI1/2 knockdown cells immediately following HU treatment and 8 hours after release (Figure 5D) consistent with the increased damage burden previously observed in these cells. Since alkaline comet may be indirectly detecting ssDNA, we used the neutral comet assay to assess the presence of DSB specifically. By neutral comet, no significant increase in damage was seen in either control or DDI1/2 knockdown cells following treatment with HU. However, if cells were co-incubated with HU and an ATR inhibitor, a combination previously shown to cause global fork collapse, we observed a significant increase in DSBs in both control and DDI1/2 knockdown cells (Figure 5E). By IF, both control and DDI1/2 knockdown cells treated with ATR inhibitor and HU exhibited the pannuclear gH2AX and RPA signal typical of cells undergoing “replication catastrophe” (Figure S6A-B) (Toledo et al., 2013). In contrast, DDI1/2 knockdown cells treated with HU alone did not exhibit pannuclear γH2AX/RPA (Figure S6B). Taken together, these data suggest that loss of DDI1/2 and persistence of RTF2 does not lead to canonical replication fork collapse, but rather to the accumulation of ssDNA at stalled forks in the context of functional ATR.
RTF2 promotes ssDNA accumulation and ATR activation during replication stress
During the rescue experiments, we saw that the only observable phenotype of RTF2 depletion in control cells was a modest decrease in phospho-RPA, consistent with decrease in ssDNA being produced when RTF2 was depleted from the replication fork. To extend this observation, we exposed U2OS cells treated with two independent siRNAs against RTF2 to HU and complemented these cells with siRNA-resistant HA-FLAG-RTF2. RTF2 depleted cells showed lower phospho-RPA signal when compared to control cells following HU exposure, and HA-FLAG:RTF2 expression rescued phospho-RPA levels (Figure 5F-H). Similar effects were seen in BJ cells upon RTF2 depletion (Figure S6C). UV treatment, which in unsynchronized cells leads to activation of ATR via NER pathway independent of RTF2 (Giannattasio et al., 2010; Marini et al., 2006) resulted in normal phospho-RPA signal in RTF2 depleted cells (Figure S6D-E). These data suggest that RTF2 may be necessary to promote ssDNA generation upon replication stress.
Discussion
Replication fork stalling is a major source of genomic instability. Investigation of events at the stressed replication fork is essential for understanding the complex mechanisms cells have evolved to deal with the challenging DNA landscape faced during replication. Such investigation informs our understanding of the etiology of diseases driven by changes to genomic structure, including cancer. Here, we introduce DDI1/2-dependent removal of human RTF2 from the replication fork as a critical process in protecting the replicating cell from genomic instability.
DDI-RTF2 axis of replication stress response
We have identified RTF2 as a key replisome component that needs to be removed during replication stress in a DDI1 and 2- dependent event. Without DDI1 and DDI2, RTF2 accumulates at the replisome resulting in an inappropriate increase of ssDNA at the replication fork (Figure 6). Under conditions of improper RTF2 enrichment, transiently stalled forks become progressively more difficult to restart, and forks that have become inactive from long-term stalling cannot be properly processed. The resulting unresolved replication intermediates trigger apoptosis during late S phase or lead to chromosome breakage that we observe in metaphases.
Figure 6: Model for DDI and RTF2 function at the replication fork.
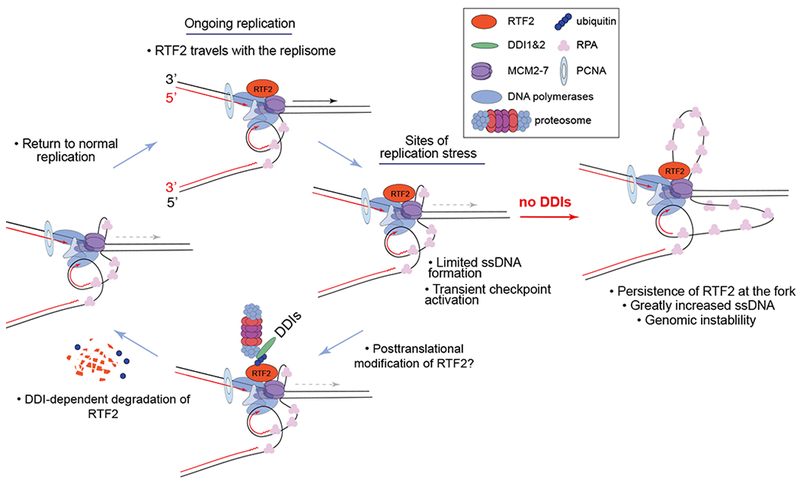
Human RTF2 travels with the replisome. We show that when RTF2 is depleted, there is less RPA phosphorylation under replication stress conditions, consistent with a decrease of uncoupling of the replicative helicase and polymerase. Under physiological conditions, RTF2’s function is limited by its DDI1/2-dependent turnover. Both mouse and human RTF2 have been shown to be ubiquitinated and SUMOylated in vivo in high throughput assays. RTF2 also has a putative SUMO interacting domain. It remains to be determined what posttranslational modifications of RTDC1 are necessary for its degradation. Eventually, the turnover signal is turned off and normal replication resumes. In the absence of functional DDI1 and DDI2, RTF2 is not removed from the stressed fork and excess ssDNA accumulates, leading to functional deactivation of that fork and subsequent genomic instability when the replication cannot be completed.
Accumulation of ssDNA during S-phase can be attributed to two major pathways: helicase uncoupling and fork reversal/resection (Byun et al., 2005; Zellweger et al., 2015). Because DDI1/2 depleted cells, which have an excess of RTF2 at the fork, exhibit no defects in nascent strand resection following HU, we favor the idea that RTF2 functions in a pathway that uncouples the replicative helicase from the replicative polymerases. We propose that in DDI1/2 depleted cells, high levels of RTF2 at the replication fork results in unrestrained uncoupling, leading to excessive ssDNA. This hypothesis remains to be tested experimentally.
Our data indicate that the physiological amounts of RTF2 traveling with the replication fork are desirable for robust ATR activation following replication fork stalling. Such activity might be necessary for transient response to endogenous stress, but would need to be curbed by a feedback mechanism, which we propose is mediated by DDI1/2. A plausible trigger for this feedback mechanism might involve reaching a specific ssDNA threshold, which in turn signals for RTF2 degradation. In our model, DDI1/2 are necessary to maintain a balance between generating enough ssDNA to allow checkpoint activation, but not so much as to hinder normal restart and repair of the stalled fork.
Our work suggests that DDI-RTF2 axis is distinct from the BRCA2/RAD51-dependent block to nucleolytic degradation at stalled forks. No degradation of newly replicated DNA is detectable in the DDI1/2 depleted cells, although the MRE11 nuclease is enriched on the stressed fork when the DDIs are depleted. The function of the enriched MRE11, although difficult to study due to the pleiotropic effects of this nuclease, will have to be analyzed further. Because we observe no obvious nucleolytic processing of nascent DNA during replication stress, we hypothesize that the bulk of the ssDNA observed in the DDI1/2 knockdown model is a consequence of helicase unwinding ahead of the fork. However, it remains a possibility that the ssDNA is formed behind the replication fork when RTF2 is inappropriately stabilized. Further studies using electron microscopy to directly visualize the aberrant fork structures following replication fork stalling, will allow us to observe ssDNA localization in relation to the replication fork (Hashimoto et al., 2010; Neelsen et al., 2014).
Another set of enzymes that are important for proper response to replication stress are structure specific nucleases, including Mus81-Eme1, which process unreplicated DNA at common fragile sites during mitosis to elicit completion of outstanding replication and prevent chromosome missegregation (Minocherhomji et al., 2015). Depletion of DDI1/2 leads to genomic instability under conditions of mild replication stress akin to those which elicit common fragile site expression. Our data show that the majority of the DNA damage that occurs in S phase in the absence of DDI/2, does not involve DSBs. However, metaphases display high numbers of breaks raising the possibility that aberrant fork structures created in the absence of DDI1/2 transit into mitosis and are processed nucleolytically. Alternatively, structural aberrancies due to ssDNA accumulation could make these substrates inaccessible to nuclease activity. How the mitotic DSBs are formed in DDI-depleted cells remains to be determined.
There is a striking similarity between the cellular consequences of DDI1/2 depletion and the phenotypes caused by knockdown of the fork protection complex factors TIM and TIPIN. Both DDI1/2 and TIM-TIPIN depletion lead to fork restart defects, a dramatic accumulation of ssDNA which is converted to DSB by ATR inhibition, and chromosomal fragmentation following exposure to replication stress drugs (Errico et al., 2007; Leman et al., 2010; Smith et al., 2009). Biochemically, TIM-TIPIN complex has been shown to promote replication fork coupling by stimulating DNA polymerase activity and constraining the MCM helicase from its unwinding activity (Cho et al., 2013). It will be of interest to explore the relationship of RTF2 and TIM-TIPIN.
Physiological role of RTF2
Excess RTF2 at the replisome is detrimental to normal processing of the stalled fork, and we characterized it mainly as a negative factor in the maintenance of genomic stability. However, its identification as a component of the elongating replisome by us and in a previous study (Dungrawala et al., 2015) as well as the decreased signaling through the ATR pathway we observe upon depletion of RTF2, suggests that it will have a positive activity during DNA replication. It might function to facilitate fork pausing at replication fork barriers like the rDNA, or it may be more globally required to stimulate ATR signaling after the fork stalls or encounters a lesion. Moreover, we cannot exclude the possibility that RTF2 is necessary even for normal replication and future studies will be necessary to explore its function during unperturbed replication.
Implications of DDI-RTF2 axis identification
Our work identifies a pathway that may serve as a potential therapeutic target for cancer treatment. In adult humans and mice, DDI2 is expressed broadly in tissues, while DDI1 shows enriched expression in the testes (Consortium, 2013). However, DDI1 expression is increased in a variety of cancer cell lines (Barretina et al., 2012), suggesting that it may be required during the increased replication stress characteristic of oncogenic transformation. Our experiments show that increased RTF2 is particularly toxic when cells are subjected to increased replication stress. Inhibiting the putative ubiquitin ligase responsible for RTF2 modification, or directly inhibiting DDI1/2, should the protease activity of DDI1/2 prove important, could contribute to selective death of tumor cells by capitalizing on their endogenous replication stress while leaving even the actively dividing normal cells relatively unaffected.
Our work also proposes an approach for studying protein dynamics at the replication fork. It is difficult to detect a decrease of a protein at a site of interest especially for non-abundant proteins. While RTF2 was previously detected by iPOND (Dungrawala et al., 2015), its contribution to the cellular response to replication stress was only revealed when it was inappropriately stabilized by DDI1/2 depletion. This study highlights that the examination of the replication fork proteome upon
the inhibition of specific members of the UPS system may identify other proteins that must be removed during replication stress response. It will be of interest to survey the other protein turnover and proteasome shuttle proteins for their effects on the replisome dynamics under replication stress conditions.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Agata Smogorzewska (asmogorzewska@rockefeller.edu).
CELL LINES
U2OS cells (female), HeLa cells (female) and BJ cells (male) were used. BJ cells were immortalized in-house by transduction with E6/E7 and human telomerase reverse transcriptase. Cell lines were not authenticated by STR analysis.
VIRUSES
Retroviruses for mammalian infection of plasmids were packaged in 293T cells transfected with Gag/Pol and VSVG-expressing plasmids via TransIT 293T reagent (Mirus Bio). Lentiviruses for mammalian infection of plasmids were packaged in 293T cells transfected with Gag/Pol, VSVG, Rev, and Tat-expressing plasmids via TransIT 293T reagent (Mirus Bio).
METHOD DETAILS
Cell culture, transfection and viral transduction:
U2OS cells (ATCC) and HeLa 1.2.11 cells (de Lange lab) were maintained in Dulbecco Modified Eagle Medium supplemented with 15% (v/v) fetal bovine serum (Atlanta Biologicals), non-essential amino acids, glutamax and 100 units of penicillin/streptomycin per ml, and 0.1 mg streptomycin per ml (all from Invitrogen). BJ foreskin fibroblasts (ATCC) were transformed by HPV16 E6E7 expression and/or immortalized by expression of catalytic subunit of human telomerase (hTERT), and were maintained as above except with 15% (v/v) fetal bovine serum and nonessential amino acids (Invitrogen).
Transfection of fibroblasts with siRNAs was performed using Lipofectamine RNAiMax (Invitrogen) as according to manufacturer’s instruction with final siRNA concentration of 25nM. DDI1 siRNAs are Stealth HSS181016, HSS140552, and HSS140553 (Invitrogen); DDI2 siRNAs are Silencer Select s38861, s38862, and s38863 (Ambion); and RTF2 siRNAs are Stealth MSS227432, MSS227433, and MSS227434 and Silencer Select s226737 (all from Invitrogen). DDI2 shRNAs V3LHS_328065, V3LHS_32806, and V3LHS_328069 (Open Biosystems) were cloned into pMSCV-PM mir30. All sequences are in Supplemental Table 1. Viruses for transducing shRNA or expression constructs into fibroblasts were first packaged in HEK293T cells using TransIT reagent (MirusBio) as according to manufacturer’s protocol. Viral supernatants collected were used to infect fibroblasts in the presence of 4μg polybrene per ml of media. Cells were selected in the appropriate antibiotics for at least one week post viral infection.
cDNA and plasmids:
DDI2 cDNA was obtained from Origene (SC313304) and was confirmed to match the published reference sequence with the exception of one SNP (C1124T). DDI1 cDNA was obtained from Open Biosystems (MHS1010-7295269). RTF2 cDNA was obtained from TransOMIC (TCH1303 Clone BC003359). DDI1, DDI2, and RTF2 cDNAs were PCR amplified and cloned into pDONR223 using BP clonase (both from Invitrogen). pDONR223 derivatives encoding DDI2 were cloned into retroviral pEA59 N-GFP (gift from the Kapoor laboratory) for mass spectrometry; those encoding both DDI1 and DDI2 were cloned into pHAGE-N-eGFP for complementation. pDONR223-RTF2 was cloned into MSCV-HA:FLAG for complementation and pHAGE CMV-N-eGFP for overexpression.
Cell cycle, breakage and cell survival analyses:
Cell cycle analyses were performed per instructions from BD Biosciences (FITC-anti-BrdU BD 559619). Cells were analyzed on the Accuri cytometer (BD). For cell cycle/apoptosis analysis, cells were labeled with EdU and processed with the Click-iT® EdU AlexaFluor® 488 Flow Cytometry Assay Kit (Invitrogen) per manufacturer’s instructions, hybridized with cleaved caspase 3-Alexa Fluor® 647 Conjugate (Cell Signaling), and stained with DAPI (Sigma Aldrich). Cells were analyzed on the LSR-II cytometer (BD). Further data analysis was done using FlowJo software. Analyses of chromosomal breakage following treatment with DNA damaging agents were performed as previously described (Garner et al., 2013). Cells treated with indicated doses of HU or aphidicolin were arrested with 0.1 ug colcemid per ml of media for 90 min, trypsinized, incubated at 37 C for 10 min in 0.075 M KCl, and fixed in 3:1 methanol:acetic acid. Metaphase spreads were prepared by dropping the cell suspension onto slides pre-wetted with ddH20. Slides were dried at 42°C overnight before staining with KaryoMAX Giemsa (Invitrogen) in Gurr Buffer for 3 min. After rinsing with fresh Gurr Buffer followed by distilled water, the slides were fully dried then scanned unmounted using the Metafer (Metasystems). Metaphases were scored for presence of abnormalities by a person blinded to the experimental sample . For cell survival assays, cells were transfected with siRNAs once 72 hours prior to drug treatment, and seeded overnight before treatment with drugs at indicated concentrations. Cells were allowed to grow to near confluency and passaged once at appropriate ratios to prevent overgrowth. Once cells reached near confluency after passaging, cell number was determined by using a Z2 Coulter counter (Beckman Coulter).
Antibodies:
The following antibodies were used at the indicated dilutions: PCNA PC-10 (Santa Cruz, s-56, 1:500), MCM6 C-20 (Santa Cruz, sc-9843, 1:500), RPA (Bethyl A300-244A, 1:2000, and Calbiochem NA18, 1:500), GFP (in house and Roche 11814460001, 1:1000), RPN2, RPN10, RPN12, and RPN7 (sampler pack, Enzo Life Sciences BML-PW8965, 1:1000), alpha-tubulin (Sigma Aldrich #T9026, 1:3000), vinculin (Sigma Aldrich V9131, 1:2500), FITC-anti-BrdU (BD 559619, 1:50), cleaved caspase-3 (Asp175) (Cell Signaling, #9661, 1:500), Cleaved Caspase-3 (Asp175) (D3E9)-Alexa Fluor® 647 Conjugate (Cell Signaling #9602, 1:50), BrdU B44 (BD 347580, 1:50), BrdU BU1/75 (ABD Serotec/BioRad OBT0030CX, 1:200), BRCA2 Ab-1 (Millipore OP95, 1:2000), phospho-RPA S4/S8 (Bethyl A300-245A, 1:1000), phospho-Chk1 S345 (Cell Signaling 2341, 1:500), γH2AX S136 JB301 (Millipore 05-636, 1:2000), H2AX (Bethyl A300-082A, 1:1000), CHK1 (Cell Signaling 2360S, 1:1000), Mre11 and Nbs1 (kind gifts from John Petrini, 1:3000 and 1:5000 respectively), RTF2 (LS Bio 340588, 1:1000), H2B (abcam ab52599, 1:1000), DDB2 (abcam ab51017, 1:250), TONSL (kind gift from Daniel Durocher, 1:1000). For generation of anti-DDI, antibody was raised in rabbit against a GST-tagged C-terminal fragment of DDI2 (a.a. 201-399) by Covance, and serum was purified against a His-tagged C-terminal fragment of DDI2 (1:500).
Immunoprecipitation experiments:
For GFP:DDI2 mass spectrometry, pellets from GFP:DDI2 or GFP expressing cells was resuspended in IP buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 2 mM MgCl2, 0.1% Tween-20) supplemented with protease inhibitors (Roche) and 2 mM DTSSP (Thermo), sonicated for 3×20 s at setting 15 (Misonix), and microcentrifugated at maximum speed. Crosslinker was quenched with addition of 20 uM Tris. Lysate was incubated with m270-Epoxy resin (Invitrogen) coupled to rabbit anti-GFP (in house) overnight at 4C. Resin was washed 6X with IP buffer, eluted in 0.5N NH4OH + 0.5mM EDTA, and eluate was dried down O/N in a vacufuge. Protein was resuspended in 1X Laemmli buffer, boiled for 30 min to reverse crosslinks, and run on a 4-12% Bis-Tris gel (Invitrogen) for in-gel digestion. For IP-western blot, cell pellets were resuspended in modified MCLB buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors and +/− 2mM DTSSP where indicated, and IPed as above. Resin was boiled for 30 min in 1X Laemmli buffer and run on a 4-12% Bis-Tris gel (Invitrogen) for immunoblotting.
Immunoblotting and immunofluorescence:
Whole cell extracts were prepared by directly lysing cell pellets in modified RIPA buffer (25 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% Triton-X) supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Calbiochem). Samples were sonicated 15s, Laemmli buffer was added to 1X concentration, and samples were boiled at 95°C for 10 min before proteins were separated on 4-12% gradient Bis-Tris gels (Invitrogen) by SDS-PAGE, and transferred to PVDF membrane by electrophoresis. Immunoblotting analyses were performed using antibodies listed above. For immunofluorescence, cells seeded on coverslips were washed once with PBS and fixed in 3.7% (w/v) formaldehyde in PBS for 10 min at room temperature before permeablized with 0.5% (v/v) NP-40 in PBS for 20 min at room temperature. For pre-extraction, cells were treated with 0.25% (v/v) Triton-X in PBS for 5 min at RT prior to fixation. Coverslips were then blocked 1 h with 5% (v/v) fetal bovine serum in PBS at room temperature. Primary antibody incubation was performed for 2 hours at room temperature before coverslips were washed 3 times in PBS + 0.05% (v/v) Tween. Coverslips were then stained with secondary antibodies conjugated with Alexa Fluor® 488 or 594 (Invitrogen) for 2 h at room temperature and washed 3 times with PBS + 0.05% Tween, 1× with PBS, and 1× with ddH2O before being mounted in Vectashield (Vector Laboratories) containing DAPI or DAPI-Fluoromount (Southern Biotech). Image analyses of the stained slides were performed using the Axio Observer A1 fluorescence microscope (Carl Zeiss), equipped with a Plan- Apochromat 63× NA-1.4 oil objective, the AxioCam CCD camera, and the AxioVision Rel Version 4.7 software. For fluorescence intensity analysis, images were analyzed by Image J.
RNA preparation, reverse transcription, and real time PCR:
Total messenger RNA was extracted by using RNeasy plus kit (Qiagen) and reverse transcribed to synthesize cDNA by using SuperScript III Reverse Transcriptase (Invitrogen). The relative transcript levels of genes of interest were determined by quantitative real-time PCR using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) and normalized against actin. The primers for specific gene of interest are listed in Table S1. RNAi and primer sequences
DNA fiber analysis:
Exponentially growing cells were labelled with 50-100 μM IdU and 50-100 μM CldU, for time periods indicated in each individual experiment. Labelled cells were trypsinized, washed once in ice-cold PBS, and resuspended in ice-cold PBS at 1 χ 106cells/mL. Two microliters of this suspension were spotted onto a Silane-Prep slide (Sigma Aldrich) and lysed with 10 μl of spreading buffer* (0.5% SDS in 200 mM Tris-HCl (pH 7.4) and 50 mM EDTA). After 6 min, the slides were tilted at 15° to horizontal, allowing the DNA to spread. Slides were air-dried and fixed in methanol and acetic acid (3:1) for 2 min. DNA was denatured with 2.5 M HCl in PBS+0.1% Triton X-100 for 30 min at room temperature. Slides were rinsed three times in PBS and blocked in PBS + 0.1% Triton X-100 (PBS-T) + 10% goat serum for 1 h at room temperature. Rat anti-BrdU (BU1/75, Novus or Serotec, 1:200) and mouse anti-BrdU (Becton Dickinson 347580, 1:50) were then applied to detect CldU and IdU, respectively. After a 1-h incubation, slides were washed three times in PBS + 0.05% Tween, stained with Alexa Fluor 488-labelled goat anti-mouse IgG1 antibody and Alexa Fluor 555-labelled goat anti-rat antibody (Invitrogen, 1:350 each) for 1 h at room temperature, and washed 2× with PBS + 0.05% Tween, 1× with PBS, 1× with ddH2O, and allowed to air dry. Slides were mounted in Vectashield (Vector Labs) and stored at 4 C. Replication tracks were imaged as immunofluorescence experiments (above) and measured using Image J software.
DNA comet assays:
Cells were transfected once with siRNAs 72 hours prior to collection. Comet assays were performed using the CometAssay kit (Trevigen) per manufacturer’s instructions. Briefly, cells were washed once in ice-cold PBS, resuspended to 1×105 cells/mL in PBS, mixed 1:10 (v/v) with molten low-melt agarose, spread onto Comet slides, allowed to set at 4C for 10 min, and lysed in Lysis Solution for 30 min-1 h. For alkaline comet assay, slides were transferred to Alkaline Unwinding Solution for 20 min and subjected to electrophoresis in Alkaline Electrophoresis Solution at 21V for 30 min at 4C. Slides were washed 2× 5 min in ddH2O, and immersed 1× 5 min in 70% ethanol then dried at 37 for 15 min. For neutral comet assay, slides were immersed in Neutral Electrophoresis Buffer for 30 min, subjected to electrophoresis in the same buffer at 21 V for 45 min at 4C, and immersed in DNA Precipitation Solution for 30 min at RT. Slides were immersed for 30 min in 70% ethanol then dried at 37 for 15 min. Dried slides were stained with a 1:30000 dilution of SYBR-GOLD and visualized and analyzed by Metasystems CometScan software.
SILAC labeling:
Cells were grown in DMEM F12 medium for SILAC (Pierce) supplemented with 10% (v/v) dialyzed FBS (Pierce), glutamax and 100 units of penicillin/streptomycin per ml (Invitrogen). Cells designated as “heavy” were given media supplemented with 13C6 L-Lysine and 3C615N4 L-Arginine while cells designated as “light” were given media supplemented with unlabelled L-Lysine and L-Arginine (all from Pierce). Cells were passaged using enzyme-free Cell Dissociation Buffer (Thermo) for at least 7 population doublings. Cells were transfected twice with siRNAs 72 and 48 hours prior to collection.
iPOND:
Cells were treated with 0.4 uM aphidicolin for 3 hours, and EdU was added at 10 uM for 50 min (DDI-depleted) or 40 min (control) to yield a labeled length similar to a 10 min pulse in untreated cells. For chase, cells were treated with 10 uM EdU for 10 min, washed 3× in PBS, and treated with 20 uM thymidine for 50 min. Cells were washed 1× in PBS, crosslinked 20 min with 1% formaldehyde in PBS at RT, quenched for 20 min with 0.125 M glycine in PBS, washed 3× in cold PBS, and collected by scraping. For SILAC, heavy and light cells were pooled 1:1 at this stage (quantified by unfixed count plate). Pellets were resuspended in 5 mL permeabilization buffer (20 mM Hepes pH 7.5, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5 % NP40 (IGEPAL)), incubated on ice for 15 min, spun at 2500 g for 10 min. These pellets were washed 1× in PBS, resuspended in Click Reaction Buffer (8.8 mL PBS, 5 uL biotin azide, 1 mL 100 mM ascorbate, 200 ul 100 mM CuSO4), incubated for 1 h at 4C, and washed in PBS. Pellets were resuspended in 500 ul Nuclear Lysis buffer (25 mM NaCl, 2 mM EDTA, 50 mM TrisHCl pH 8.0, 1% NP40), incubated on ice for 15 min, sonicated for a total of 120 seconds at setting 13, and spun in a microcentrifuge at maximum speed for 10 min. Lysate was diluted 1:1 with Wash Buffer (150 mM NaCl, 2 mM EDTA, 50 mM TrisHCl pH 8.0, 0.5% NP40) and subjected to pulldown with 50 ul streptavidin beads (Agilent) O/N at 4C. Beads were washed 4× with Wash Buffer, eluted for 30 min at 95C in 2X Laemmli buffer, and eluate was run ~1 cm into a 4-12% Bis-Tris gel (Invitrogen) for mass spec analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
All ANOVA and t-test analysis was done using Graphpad Prism software. Error bars represent SEM n ≤ 3 where n represents biological replicates except for those on survival curves which represent SEM n = 3 where n represents technical replicates. For fiber analysis, at least 150 fibers were measured per condition and statistical significance was derived by ANOVA analysis of one biological replicate. For nuclear intensity and comet assay, at least 50 cells were measured per condition and statistical significance was derived by ANOVA analysis of one biological replicate. The graphs for fiber analysis, nuclear intensity, and comet assay are representative of at least 3 independent experiments.
ADDITIONAL RESOURCES
Table S1. RNAi and primer sequences
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse alpha-tubulin | Sigma Aldritch | Cat# T9026 |
| Rabbit BRCA2 Ap1 | Millipore | Cat# OP95 |
| Mouse BrdU Clone B44 FITC conjugate | BD | Cat# 559619 |
| Mouse BrdU Clone B44 | BD | Cat# 347580 |
| Rat BrdU Clone BU1/75 | ABD Serotec/BioRad | Cat# OBT0030CX |
| Rabbit CHK1 | Cell Signaling | Cat# 2360 |
| Mouse phospho-Chk1 S345 | Cell Signaling | Cat# 2341 |
| Rabbit cleaved caspase-3 (Asp175) | Cell Signaling | Cat# 9661 |
| Rabbit cleaved caspase-3 (Asp175) (D3E9)-Alexa Fluor® 647 Conjugate | Cell Signaling | Cat# 9602 |
| Mouse DDB2 [2246C4a] | AbCam | Cat# ab51017 |
| Rabbit DDI1/2 polyclonal | This paper | n/a |
| Mouse GFP | Roche | Cat# 11814460001 |
| Rabbit GFP polyclonal | This paper | n/a |
| H2AX | Bethyl | Cat# A300-082A |
| γH2AX S136 JB301 | Millipore | Cat# 05-636 |
| Rabbit H2B [EP957Y] | AbCam | Cat# ab52599 |
| MCM6 C-20 | Santa Cruz | Cat# sc-9843 |
| Rabbit Mre11 | Petrini Lab | n/a |
| Rabbit Nbs1 | Petrini Lab | n/a |
| Mouse PCNA PC-10 | Santa Cruz | Cat# sc-56 |
| Rabbit RPA | Bethyl | Cat# A300-244A |
| Mouse RPA | Calbiochem | Cat# NA18 |
| Mouse RPN2/S1 (112-1) | Enzo Life Sciences | Cat# BML-PW9270 |
| Rabbit RPN7/S10 | Enzo Life Sciences | Cat# BML-PW8225 |
| Mouse RPN10/S5A (S5a-18) | Enzo Life Sciences | Cat# BML-PW9250 |
| Rabbit RPN12/S18 | Enzo Life Sciences | Cat# BML-PW8815 |
| Mouse RTF2 | LS Bio | Cat# 340588 |
| Rabbit RTF2 | Proteintech | Cat# 16633-1-AP |
| Bacterial and Virus Strains | ||
| n/a | ||
| Biological Samples | ||
| n/a | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Biotin Azide (PEG4 carboxamide-6-Azidohexanyl Biotin) | Thermo Fisher | Cat# B10184 |
| DTSSP (3,3′-dithiobis(sulfosuccinimidyl propionate) | Thermo Fisher | Cat# 21578 |
| L-Arginine-HCl 13C6 15N4 for SILAC | Thermo Fisher | Cat# 88434 |
| Mirin | Sigma Aldrich | Cat# M9948 |
| VE-821 | Sigma Aldrich | Cat# SML1415 |
| Critical Commercial Assays | ||
| Click-It EdU Flow Assay Kit | Thermo Fisher | Cat# C10632 |
| Comet Assay Kit | Trevigen | Cat# 4250-050-K |
| SILAC Protein Quantitation Kit DMEM | Thermo Fisher | Cat# A33969 |
| Deposited Data | ||
| n/a | ||
| Experimental Models: Cell Lines | ||
| BJ | ATCC | CRL-2522 |
| HeLa 1.2.11 | de Lange lab | n/a |
| U2OS | ATCC | HTB-96 |
| Experimental Models: Organisms/Strains | ||
| n/a | ||
| Oligonucleotides | ||
| RNAi sequences in Supplementary Table 1 | ||
| Beta-Actin qPCR F gctacgagctgcctgacg | This paper | n/a |
| Beta-Actin qPCR R ggctggaagagtgcctca | This paper | n/a |
| DDI1 qPCR F tggaacacaacgtgctacct | This paper | n/a |
| DDI1 qPCR R atctgtctgggggctgtct | This paper | n/a |
| DDI2 qPCR F tcgatgtagtgtgtgtgtactgc | This paper | n/a |
| DDI2 qPCR R ccagtgaggtagattctttaccactt | This paper | n/a |
| RTF2 qPCR F tgctgaagacaaggatggag | This paper | n/a |
| RTF2 qPCR R tgaaacagactctgctgcct | This paper | n/a |
| RTF2 endo qPCR F agtgacagctttgggggttt | This paper | n/a |
| RTF2 endo qPCR R ttccattgggccactaattc | This paper | n/a |
| Recombinant DNA | ||
| DDI2 cDNA | Origene | SC313304 |
| DDI1 cDNA | Open Biosystems | MHS1010-7295269 |
| RTF2 cDNA | TransOMIC | TCH1303 |
| Software and Algorithms | ||
| CometScan | MetaSystems | N/A |
| FlowJo | FlowJo LLC | Version 9 |
| Graphpad | Prism | Version 6 |
| Other | ||
Highlights.
DDIs are required for cellular survival following replication stress
DDIs remove the replisome component C20orf43/RTF2 from stalled forks
RTF2 removal from stalled forks is required for genome stability
Acknowledgments
We thank the members of the Smogorzewska lab for their comments and suggestions during the manuscript preparation. A.S. is grateful to Titia de Lange for frequent discussion of this project. We are grateful to Henrik Molina and the staff of the proteomics resource center for the advice on the SILAC experiments and the mass spectrometry analysis presented in the paper. We also acknowledge the genomics and flow resource centers at the Rockefeller University. We thank Titia de Lange, John Petrini, Tarun Kapoor, Daniel Durocher for reagents and Armin Gamper and Christopher Bakkinist for the advice on the iPOND protocol. MCK was supported by the Women and Science Fellowship from the Rockefeller University and by the American Cancer Society-J.T.Tai Postdoctoral Fellowship. This work was supported by the Pershing Square Sohn Prize for Young Investigators in Cancer Research and The Gabrielle H. Reem and Herbert J. Kayden Early-Career Innovation Award. AS is an HHMI Faculty Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, and Dantuma NP (2011). The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol 18, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, and Cortez D (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, and Foiani M (2009). The checkpoint response to replication stress. DNA Repair (Amst) 8, 1038–1046. [DOI] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, and Cimprich KA (2005). Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19, 1040–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WH, Kang YH, An YY, Tappin I, Hurwitz J, and Lee JK (2013). Human Tim-Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. Proc Natl Acad Sci U S A 110, 2523–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, and Elledge SJ (2006). Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc Natl Acad Sci U S A 103, 18143–18147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AR (2004). The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 26, 249–261. [DOI] [PubMed] [Google Scholar]
- Consortium GT (2013). The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, and Elledge SJ (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, and Gautier J (2003). An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11, 203–213. [DOI] [PubMed] [Google Scholar]
- Dalgaard JZ GE, MacFarlane RJ (2011). Eukaryotic Replication Barriers: How, Why and Where Forks Stall. DNA Replication-Current Advances, 269–304. [Google Scholar]
- Dantuma NP, Heinen C, and Hoogstraten D (2009). The ubiquitin receptor Rad23: at the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair (Amst) 8, 449–460. [DOI] [PubMed] [Google Scholar]
- De Piccoli G, Katou Y, Itoh T, Nakato R, Shirahige K, and Labib K (2012). Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell 45, 696–704. [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser S, Gali RR, Schwickart M, Larsen CN, Leggett DS, Muller B, Feng MT, Tubing F, Dittmar GA, and Finley D (2002). Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat Cell Biol 4, 725–730. [DOI] [PubMed] [Google Scholar]
- Errico A, Costanzo V, and Hunt T (2007). Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc Natl Acad Sci U S A 104, 14929–14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz A, Ackermann L, and Hoppe T (2016). Ring of Change: CDC48/p97 Drives Protein Dynamics at Chromatin. Front Genet 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel AM, Pike BL, and Gasser SM (2009). ATR/Mec1: coordinating fork stability and repair. Curr Opin Cell Biol 21, 237–244. [DOI] [PubMed] [Google Scholar]
- Giannattasio M, Follonier C, Tourriere H, Puddu F, Lazzaro F, Pasero P, Lopes M, Plevani P, and Muzi-Falconi M (2010). Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell 40, 50–62. [DOI] [PubMed] [Google Scholar]
- Gomez TA, Kolawa N, Gee M, Sweredoski MJ, and Deshaies RJ (2011). Identification of a functional docking site in the Rpn1 LRR domain for the UBA-UBL domain protein Ddi1. BMC Biol 9, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Suppa C, and Emanuel BS (2007). Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J Mol Biol 366, 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Raschle M, and Mailand N (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Hamperl S, and Cimprich KA (2016). Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 167, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Zhu Q, Wani G, Sharma N, Han C, Qian J, Pentz K, Wang QE, and Wani AA (2014). Ubiquitin-specific protease 7 regulates nucleotide excision repair through deubiquitinating XPC protein and preventing XPC protein from undergoing ultraviolet light-induced and VCP/p97 protein-regulated proteolysis. J Biol Chem 289, 27278–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills SA, and Diffley JF (2014). DNA replication and oncogene-induced replicative stress. Curr Biol 24, R435–444. [DOI] [PubMed] [Google Scholar]
- Huang P, Chubb S, Hertel LW, Grindey GB, and Plunkett W (1991). Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res 51, 6110–6117. [PubMed] [Google Scholar]
- Inagawa T, Yamada-Inagawa T, Eydmann T, Mian IS, Wang TS, and Dalgaard JZ (2009). Schizosaccharomyces pombe Rtf2 mediates site-specific replication termination by inhibiting replication restart. Proc Natl Acad Sci U S A 106, 7927–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Vossler RA, Diaz-Martinez LA, Winter NS, Clarke DJ, and Walters KJ (2006). UBL/UBA ubiquitin receptor proteins bind a common tetraubiquitin chain. J Mol Biol 356, 1027–1035. [DOI] [PubMed] [Google Scholar]
- Kim JC, Harris ST, Dinter T, Shah KA, and Mirkin SM (2016). The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, and Carr AM (2013). Replication stress and genome rearrangements: lessons from yeast models. Curr Opin Genet Dev 23, 132–139. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, and Lupski JR (2007). A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247. [DOI] [PubMed] [Google Scholar]
- Leman AR, Noguchi C, Lee CY, and Noguchi E (2010). Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J Cell Sci 123, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- Marini F, Nardo T, Giannattasio M, Minuzzo M, Stefanini M, Plevani P, and Muzi Falconi M (2006). DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc Natl Acad Sci U S A 103, 17325–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, and Hickson ID (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. [DOI] [PubMed] [Google Scholar]
- Morawe T, Honemann-Capito M, von Stein W, and Wodarz A (2011). Loss of the extraproteasomal ubiquitin receptor Rings lost impairs ring canal growth in Drosophila oogenesis. J Cell Biol 193, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, and Lopes M (2014). Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol 1094, 177–208. [DOI] [PubMed] [Google Scholar]
- Nowicka U, Zhang D, Walker O, Krutauz D, Castaneda CA, Chaturvedi A, Chen TY, Reis N, Glickman MH, and Fushman D (2015). DNA-damage-inducible 1 protein (Ddi1) contains an uncharacteristic ubiquitin-like domain that binds ubiquitin. Structure 23, 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi M, Taguchi T, and Ikegami S (1978). Aphidicolin: a specific inhibitor of DNA polymerases in the cytosol of rat liver. Biochem Biophys Res Commun 82, 1084–1090. [DOI] [PubMed] [Google Scholar]
- Ruthemann P, Balbo Pogliano C, and Naegeli H (2016). Global-genome Nucleotide Excision Repair Controlled by Ubiquitin/Sumo Modifiers. Front Genet 7, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, and Cortez D (2013). Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288, 31458–31467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirkis R, Gerst JE, and Fass D (2006). Ddi1, a eukaryotic protein with the retroviral protease fold. J Mol Biol 364, 376–387. [DOI] [PubMed] [Google Scholar]
- Siva M, Svoboda M, Veverka V, Trempe JF, Hofmann K, Kozisek M, Hexnerova R, Sedlak F, Belza J, Brynda J, et al. (2016). Human DNA-Damage-Inducible 2 Protein Is Structurally and Functionally Distinct from Its Yeast Ortholog. Sci Rep 6, 30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Fu MA, and Brown EJ (2009). Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. J Cell Biol 187, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JY, Hwang BJ, Ford JM, Hanawalt PC, and Chu G (2000). Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol Cell 5, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Trempe JF, Saskova KG, Siva M, Ratcliffe CD, Veverka V, Hoegl A, Menade M, Feng X, Shenker S, Svoboda M, et al. (2016). Structural studies of the yeast DNA damage-inducible protein Ddi1 reveal domain architecture of this eukaryotic protein family. Sci Rep 6, 33671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal-Kacmaz K, Mullen TE, Kaufmann WK, and Sancar A (2005). Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol 25, 3109–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, Wolf M, Weimann L, Schulze N, Li F, Kaschani F, Riemer A, Zierhut C, Kaiser M, Iliakis G, et al. (2016). VCP/p97 Extracts Sterically Trapped Ku70/80 Rings from DNA in Double-Strand Break Repair. Mol Cell 64, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell 67, 882–890 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa-Sugata N, and Masai H (2007). Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem 282, 2729–2740. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, and Lupski JR (2009). The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 41, 849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.