

Watch a video presentation of this article
Abbreviations
- AIH
autoimmune hepatitis
- AIP
autoimmune pancreatitis
- CBD
common bile duct
- CT
computed tomography
- ERCP
endoscopic retrograde cholangiopancreatography
- FNA
fine needle aspiration
- HPF
high‐power field
- HISORt
histology, imaging, serology, other organ involvement, and response to therapy
- IBD
inflammatory bowel disease
- IgG4‐RD
IgG4‐related disease
- IgG4‐SC
IgG4‐related sclerosing cholangitis
- LPCI
lymphoplasmacytic infiltrate
- MRCP
magnetic resonance cholangiopancreatography
- OOI
other organ involvement
- OP
obliterative phlebitis
- pANCA
perinuclear anti‐neutrophil cytoplasmic antibodies
- PSC
primary sclerosing cholangitis
- qPCR
quantitative polymerase chain reaction
- SF
storiform fibrosis
- sIgG4
serum IgG4
- SSC
secondary sclerosing cholangitis
BACKGROUND
IgG4‐related sclerosing cholangitis (IgG4‐SC) is the biliary manifestation of IgG4‐related disease (IgG4‐RD), a systemic fibroinflammatory condition that is characterized by mass lesions and/or strictures with classical histopathological findings in involved organs.1 IgG4‐SC can mimic primary sclerosing cholangitis (PSC), cholangiocarcinoma (CCA), and pancreatic adenocarcinoma. It is steroid‐responsive in its inflammatory phase; however, delays in treatment can lead to fibrosis, organ dysfunction and failure, and mortality.
Demographics and Epidemiology
IgG4‐SC has a male preponderance, usually presenting in the fifth and sixth decades of life.2 It is the most common extrapancreatic manifestation in patients with autoimmune pancreatitis (AIP type 1). Isolated IgG4‐SC accounts for only 8% of cases in Western cohorts.3 Epidemiological studies of IgG4‐SC are scarce. A nationwide survey of AIP in Japan estimated the annual incidence as 1.4/100,000 and prevalence as 4.6/100,000 of the population in 2011. Of the 918 patients with AIP, 95 (10.3%) had IgG4‐SC at the porta hepatis and 216 (23.5%) had intrahepatic disease.4
Risk Factors and Associations
IgG4‐SC has been linked to a history of chronic occupational exposure, especially “blue‐collar work,” in 60% to 88% of patients in United Kingdom and Dutch cohorts.5 Clinical history of allergy and/or atopy has been described in 40% to 60% of patients, in association with peripheral eosinophilia and elevated IgE levels.6 A coexistent history of other autoimmune diseases is found in up to 10% of patients.3
Diagnosis
Diagnosis relies on careful interpretation of histopathological appearances, radiological features, and serological abnormalities in an appropriate clinical scenario. Diagnostic criteria developed for IgG4‐SC are listed in Table 1.2
Table 1.
Diagnostic Criteria for IgG4‐Related Sclerosing Cholangitis
| HISORt Diagnostic Criteria for IgG4‐SCa | |
| Histology |
(i) Lymphoplasmacytic infiltrate with >10 IgG4+ cells per high‐power field within and around bile ducts (iii) Obliterative phlebitis (iv) Storiform fibrosis Bile duct biopsy specimens often do not provide sufficient tissue for a definitive diagnosis. IgG4 immunostaining of involved organs shows >10 IgG4+ cells per high‐power field. |
| Imaging |
Strictures of the biliary tree including: (i) intrahepatic ducts (ii) proximal extrahepatic ducts (iii) intrapancreatic ducts Fleeting and migrating biliary strictures |
| Serology | Serum IgG4 levels above the upper limit of normal |
| Organ involvement |
Including (but not restricted to): (i) Pancreas (focal pancreatic mass/enlargement without pancreatic duct dilatation, multiple pancreatic masses, focal pancreatic duct stricture without upstream dilatation, pancreatic atrophy) (ii) Retroperitoneal fibrosis (iii) Kidney (single or multiple parenchymal low‐attenuation lesions: round, wedge‐shaped, or diffuse patchy) (iv) Salivary or lacrimal gland |
| Response to steroid treatment |
Defined as: (i) normalization of liver enzymes (ii) stricture resolution Complete resolution of stricture may not be seen in all patients, especially those early in the course of treatment or with predominantly fibrotic strictures. |
| Japanese Clinical Diagnostic Criteria of IgG4‐SC, 2012b | |
| 1. Hematological examination | Elevated serum IgG4 concentrations (≥135 mg/dL) |
| 2. Biliary tract Imaging | Diffuse or segmental narrowing of the intrahepatic and/or extrahepatic bile duct associated with thickening of the bile duct wall |
| 3. Coexistent organ involvement | Autoimmune pancreatitis, IgG4‐related dacryoadenitis/sialadenitis, or IgG4‐related retroperitoneal fibrosis |
| 4. Histopathological examination |
1. Marked lymphocytic and plasmacyte infiltration and fibrosis 2. Infiltration of IgG4+ plasma cells: >10 IgG4+ plasma cells per high‐power field 3. Storiform fibrosis Obliterative phlebitis |
| 5. Option: steroid therapy | Option to assess response to steroid therapy in a specialized facility, with access to endoscopic biliary biopsy and endoscopic ultrasound‐guided fine needle aspiration once pancreatic or biliary cancers has been ruled out |
| Exclusions | Malignant diseases such as pancreatic or biliary cancers, PSC, secondary sclerosing cholangitis caused by the diseases with obvious pathogenesis |
| Definite diagnosis |
(1) + (3) (1) + (2) + (4) a, b (4) a, b, c (4) a, b, d |
| Probable diagnosis | (1) + (2) + (5) option |
| Possible diagnosis | (1) + (2) |
The HISORt (histology, imaging, serology, other organ involvement, and response to therapy) criteria from the Mayo Clinic were originally developed for AIP. HISORt diagnostic criteria for IgG4‐SC were adapted from Ghazale et al.2
Japanese clinical diagnostic criteria for IgG4‐SC were adapted from Ohara et al. (2012). The Japanese Center for Disease Control for IgG4‐SC classifies the diagnosis as being definite, probable, or possible.
Both guidelines include typical imaging features of a thickened bile duct wall with segmental or diffuse biliary strictures, raised serum IgG4 levels, evidence of other organ involvement, and classical histological features. A radiological and biochemical response to corticosteroid therapy is supportive for diagnosis, with the caveat that steroids can improve the infiltrate around other malignant and inflammatory conditions.
Clinical Characteristics
Clinical presentation depends on disease activity and organ involvement. Patients usually present with obstructive jaundice (70% to 80%), weight loss, and abdominal pain.2, 3 No specific symptoms allow reliable differentiation from other causes of biliary obstruction. Those with concomitant AIP can have symptomatic pancreatic exocrine and endocrine insufficiency.3
Radiological Features
IgG4‐SC can involve any part of the biliary tree, and it is best characterized by cholangiography7 (Figure 1). Particular features include long (>1/3 length) and multifocal strictures, mild upstream dilatation, and proximal biliary disease with diffuse pancreatic swelling and a thin, narrowed pancreatic duct. However, endoscopic retrograde cholangiopancreatography (ERCP) alone is a poor discriminator (88% specificity, 45% sensitivity) to differentiate IgG4‐SC from PSC and CCA.8 Cross‐sectional imaging is a vital part of assessment and can show thickened bile duct walls, a liver/hilar mass, and evidence of other organ involvement.
Figure 1.
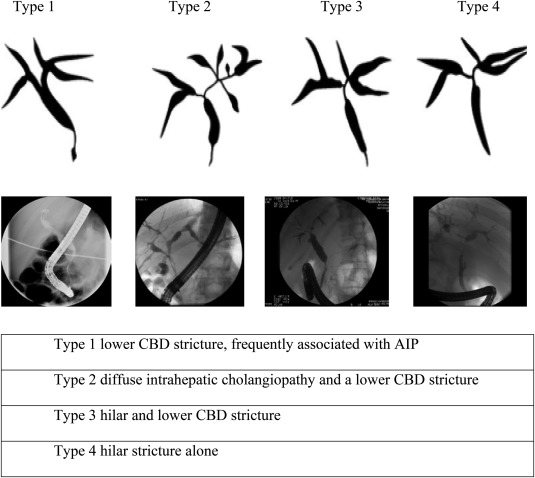
Classification of IgG4‐SC by cholangiographic features. Type 1 IgG4‐SC is defined by a low bile duct stricture, which is most frequently associated with AIP, and corresponds with compression of the intrapancreatic bile duct by inflammation/fibrosis within the head of pancreas. Type 2 IgG4‐SC is defined by both a diffuse intrahepatic cholangiopathy and a lower common bile duct (CBD) stricture. Type 3 IgG4‐SC includes a hilar and lower CBD stricture. Type 4 IgG4‐SC is defined by hilar stricturing alone. The type determines the differential diagnosis for IgG4‐SC: type 1, pancreatic cancer, distal CCA, chronic pancreatitis; type 2, PSC and those for type 1; type 3, hilar CCA and those for type 1; and type 4, hilar CCA, which is the most difficult to differentiate. Adapted with permission from Pancreas.7 Copyright 2006, Wolters Kluwer Health.
Laboratory Evaluation
Patients with IgG4‐SC may be diagnosed during investigation of abnormal liver function tests, usually in the context of cholestasis.2 Elevated inflammatory markers are nonspecific, and serum protein electrophoresis reveals a polyclonal hypergammaglobulinemia. Antinuclear antibody titers can be positive (almost 50%), although no specific autoantibody has been identified. Total serum IgG concentrations are increased (>16 g/L) in more than 50%, yet can be normal despite an elevated IgG4 subclass.9 Serum IgE levels are raised (>125 kU/L) in 35% to 60%, and peripheral eosinophilia is present in up to one third of these.6
Serum IgG4 concentrations are raised (>1.4 g/L) in 65% to 80% at diagnosis.2, 3 However, elevated IgG4 is seen in 5% to 25% of inflammatory, autoimmune, and malignant conditions and 5% of healthy individuals.9 Serum IgG4 levels greater than 5.6 g/L increase the specificity and positive predictive value for differentiating IgG4‐SC from PSC and CCA to 100%.9, 10 Patients with a normal serum IgG4 have a distinct clinical phenotype, with a reduced risk for relapse and fewer organs involved.9
Tissue Sampling
Cytological samples, from brush cytology of biliary strictures or fine needle aspiration guided by intraductal or endoscopic ultrasound, are useful to exclude malignancy (sensitivity 20% to 50%), but are nondiagnostic of IgG4‐SC. Intrabiliary biopsies, fluoroscopically guided or visually directed at cholangioscopy, yield small samples but can show characteristic histological features.11, 12 Ampullary biopsies from the major papilla may demonstrate an IgG4+ infiltrate (50% to 80% of AIP/IgG4‐SC type I). Liver biopsy can confirm involvement of small intrahepatic bile ducts (26% of cases). Analysis of biliary fluid may show elevated IgG4 levels compared with other biliary disorders but is nonspecific.
Histology
Similar histopathological findings (lymphoplasmacytic infiltrate, obliterative phlebitis, and storiform fibrosis) are evident in all organs affected by IgG4‐RD.11 In IgG4‐SC, more than 10 IgG4+ cells/HPF in a biopsy or more than 50 IgG4+ cells/HPF in a resection specimen, and an IgG4/IgG ratio greater than 40%, in the context of two of the earlier features, are considered diagnostic11 (Figure 2). Limitations to interpretation include patchy distribution of disease, insufficient tissue sampling, and fibrotic phenotype.12 An abundance of IgG4+ cells alone is not sufficient for diagnosis and can also be seen in PSC and CCA.12
Figure 2.
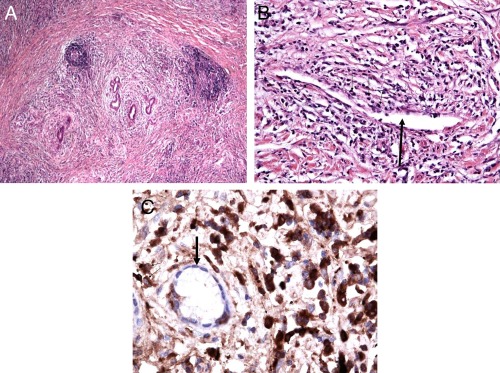
Histological features in IgG4‐SC. Bile duct resection in IgG4‐SC. (A) Lymphoplasmacytic infiltrate with periductal distribution and a storiform pattern of fibrosis (original magnification ×10). (B) Obliterative phlebitis: a thin‐walled vessel (thick black arrow) surrounded by numerous plasma cells and lymphocytes (thin black arrows) that appear to partially compress the luminal diameter (original magnification ×20). (C) IgG4+ plasma cell infiltration: immunohistochemical staining of the bile duct (thick black arrow) surrounded by abundant IgG4+ plasma cells (thin black arrows; dark brown cells), greater than 50 per high‐power field (original magnification ×40).
Differential Diagnosis
Differentiating IgG4‐SC from other benign and malignant causes is challenging.1 Diagnostic clues are provided in Table 2.
Table 2.
Differential Diagnosis of IgG4‐Related Sclerosing Cholangitis Types 1 to 4
| Category | Feature | Details |
|---|---|---|
| PSC | ||
| Clinical | Age of onset (y) |
PSC: <40 IgG4‐SC: 50 to 60 |
| Gender |
Males predominate PSC: ratio 1.5:1 IgG4‐SC: ratio 7:1 |
|
| IBD |
PSC: 75% IgG4‐SC/AIP: 5% |
|
| Autoimmune disease |
PSC: 70% IgG4‐SC: 10% |
|
| Allergy |
PSC: <40% IgG4‐SC: 60% |
|
| Laboratory values | pANCA |
PSC: 40% IgG4‐SC: <10% |
| sIgG4 > 1.4 g/L |
PSC: 9% to 18% IgG4‐SC: 65% to 80% |
|
| sIgG4 > 5.6 g/L | 100% specificity for IgG4‐SC versus PSC | |
| Eosinophilia (>5%) |
PSC: 20% IgG4‐SC: 40% |
|
| sIgG1/IgG4 ratio (sIgG4 1.4‐2.8 g/L) | 95% specificity of ratio >0.24 for IgG4‐SC versus PSC‐high IgG4 | |
| IgG4/IgG RNA ratio | 94.0% sensitivity and 98.7% specificity for IgG4‐SC versus PSC using qPCR cutoff >5% IgG4/IgG RNA ratio | |
| Imaging | Cholangiogram (MRCP and ERCP) |
PSC: beaded or pruned‐tree, short bandlike strictures IgG4‐SC: long, continuous strictures with prestenotic dilatation, pancreatic mass, and irregular pancreatic duct |
| CT scan C/A/P |
PSC: <5% pancreatitis IgG4‐SC: >90% AIP and OOI |
|
| Histology | Morphology |
PSC: onion‐skin fibrosis and periportal sclerosis IgG4‐SC: LPCI, SF, OP, and presence of eosinophils |
| IgG4+ plasma count in liver biopsy |
PSC: <10/HPF (but >10/HPF can be seen in PSC/AIH overlap, in PSC high sIgG4) IgG4‐SC: >10/HPF (>50/HPF in resection) |
|
| IgG4/IgG ratio |
PSC: <40% IgG4‐SC: >40% |
|
| Treatment | Steroids |
PSC: no response; variable response in PSC/AIH overlap and PSC high sIgG4 IgG4‐SC: response in two‐thirds of strictures |
| SSC | ||
| Clinical | Clinical history guides investigation |
Infection, e.g., AIDS cholangiopathy Vascular, e.g., hepatic artery thrombosis Toxic, e.g., postchemotherapy Congenital, e.g., Caroli disease Infiltrative, e.g., histiocytosis X Trauma, e.g., postbiliary trauma Immunological, e.g., eosinophilic cholangitis |
| Cholangiocarcinoma | ||
| Laboratory values | Ca19‐9 levels (>37 kU/L) |
CCA: 77% elevated IgG4‐SC: 63% elevated |
| sIgG4 > 5.6 g/L | 100% specificity for IgG4‐SC versus CCA | |
| IgG4/IgG RNA ratio | 94.0% sensitivity and 98.7% specificity for IgG4‐SC versus CCA using qPCR cutoff >5% IgG4/IgG RNA ratio | |
| Imaging | Cholangiogram |
CCA: distal, mid, or hilar short and tight bile duct stricture, ductal or liver mass IgG4‐SC: long, continuous strictures with prestenotic dilatation, often in association with a pancreatic mass |
| CT scan C/A/P |
CCA: metastatic disease IgG4‐SC: OOI |
|
| Cytology | Bile duct brushings or FNA |
CCA: dysplastic/malignant cells in CCA (20%‐50%) IgG4‐SC: lymphocytes and reactive cells |
| Histology | Morphology |
CCA: adenocarcinoma IgG4‐SC: LPCI, SF, OP, and presence of eosinophils |
| IgG4+ plasma count in biliary/liver biopsy |
CCA: <10/HPF (>10/HPF surrounding some lesions) IgG4‐SC: >10/HPF (>50/HPF in resection specimens) |
|
| IgG4/IgG ratio |
CCA: ratio <40% IgG4‐SC: ratio >40% |
|
| Treatment | Steroids |
CCA: inflammatory area around malignant strictures can improve but not resolve IgG4‐SC: response in two‐thirds of strictures |
Differential diagnosis of IgG4‐SC types 1 to 4 (adapted from Culver and Chapman1). Diagnostic clues to differentiate IgG4‐SC from PSC and secondary (SSC) sclerosing cholangitis, and CCA.
Abbreviations: AIH, autoimmune hepatitis; CT, computed tomography; FNA, fine needle aspiration; HPF, high‐power field; IBD, inflammatory bowel disease; LPCI, lymphoplasmacytic infiltrate; MRCP, magnetic resonance cholangiopancreatography; OOI, other organ involvement; OP, obliterative phlebitis; pANCA, perinuclear anti‐neutrophil cytoplasmic antibodies; qPCR, quantitative polymerase chain reaction; SF, storiform fibrosis; sIgG4, serum IgG4; SSC, secondary sclerosing cholangitis.
Treatment
Urgent treatment is appropriate in IgG4‐SC, even when asymptomatic, to prevent infectious cholangitis and permanent fibrosis. Spontaneous improvement of IgG4‐SC type 1 may be seen, often related to reduced pancreatic inflammation, but is less likely in types 2 to 4.13 The mainstay of treatment is systemic corticosteroids, despite an absence of randomized placebo‐controlled trials. Steroid use has been shown to induce remission quicker, more consistently, and with a lower relapse rate than a conservative approach.13 International consensus suggests initiation with prednisolone 30 to 40 mg daily for 4 weeks, reducing by 5 mg every 2 weeks, depending on response.14 The concept of a “steroid trial to confirm diagnosis” in those with high suspicion of IgG4‐SC should only be performed under close observation in experienced centers, after thorough exclusion of malignancy. Response should be seen within 4 to 6 weeks and confirmed by repeat biochemistry and imaging. Serum IgG4 levels decrease, but this is not disease‐specific.9 Remission with steroids, defined as complete resolution of strictures and/or normalization of liver biochemistry, was reported in two‐thirds of patients with IgG4‐SC.2 Nonresponse may represent burnt‐out disease, fibrotic phenotype, or an alternative diagnosis.
Biliary stenting is useful to decompress the biliary tree in those with symptomatic obstructed biliary strictures.2 Stents can be removed and/or fall out spontaneously once steroid therapy is effective.
Relapse
Patients with IgG4‐SC are at high risk for relapse (50% to 60%), most within 6 months of discontinuing or tapering steroid treatment.3 Factors predictive of a relapse include proximal strictures and high serum IgG4 (>2.8 g/L) and IgE (>380 kIU/L) levels at diagnosis.3, 6, 9 Treatment is with a second course of corticosteroids, with or without the initiation of immunosuppressive therapy, such as azathioprine (Table 3). However, the benefits of immunosuppressive agents over low‐dose steroids alone in reducing time to further relapses are uncertain.15 Predictors of resistance to immunosuppressive therapy include other organ involvement and retroperitoneal fibrosis.15 Rituximab, a CD20‐depletion agent, can be used in IgG4‐SC with incomplete remission, steroid dependency, or intolerance.15
Table 3.
Immunosuppressive Therapy in IgG4‐Related Sclerosing Cholangitis
| Agent | Regimen |
|---|---|
| Prednisolonea |
30 to 40 mg for 2 to 4 weeks Taper by 10 mg every 2 weeks until 20 mg, then 5 mg every 2 weeks |
| Azathioprineb | 2 mg/kg per day in a single dose |
| Mycophenolate mofetilc | 750 to 1000 mg twice per day |
| Mercaptopurinec | 2.5 mg/kg per day in two divided doses |
| Methotrexated | 10 to 25 mg/week plus folic acid |
| Tacrolimusd | Adjusted to a target blood level range of 4 to 11 ng/mL |
| Cyclophosphamidee | 15 mg/kg every 2 weeks for three doses and every 3 weeks thereafter by intravenous infusions |
| Rituximab | 1,000 mg at week 0 and week 2 by intravenous infusions; further infusions at 3 to 6 months guided by response and relapse |
Second‐line immunosuppressive agents used in those with intolerance, adverse effects, or relapse despite corticosteroids.
Prednisolone dose can be adjusted to body weight and disease aggressiveness.
Azathioprine is the most widely used second‐line immunosuppressive agent in IgG4‐SC.
Mycophenolate and mercaptopurine are recommended in treatment of extrapancreatic manifestations and in those intolerant of azathioprine.
Methotrexate and tacrolimus used in case report/series only in IgG4‐SC.
Cyclophosphamide has been used in multisystem IgG4‐RD, including IgG4‐SC.
Outcome
If diagnosed and treated early, IgG4‐SC has a favorable prognosis. Delayed therapy and progressive disease can lead to complications including venous thrombosis, portal hypertension, liver cirrhosis, and mortality.3 An all‐cause increased risk for malignancy (>2‐fold) has been shown.3 Adverse effects of immunosuppressive treatment, including diabetes, osteoporosis, and opportunistic infections, can occur and patients require careful monitoring.
Conclusions
IgG4‐SC remains a diagnostic challenge; the key issue being differentiation from other forms of sclerosing cholangitis and pancreatobiliary malignancy. Current therapy is guided by international expert consensus but is not supported by randomized controlled trials. Rituximab provides an option for those experiencing adverse effects and/or refractory to conventional therapy, and it gives clues to disease pathogenesis. The longer‐term consequences of irreversible fibrosis, cirrhosis, and risk for malignancy are now becoming apparent. Further work to establish risk factors and determinants of fibrotic disease and the mechanisms underlying this are essential.
Potential conflict of interest: Nothing to report.
REFERENCES
- 1. Culver EL, Chapman RW. IgG4‐related hepatobiliary disease: an overview. Nat Rev Gastroenterol Hepatol 2016;13:601–612. [DOI] [PubMed] [Google Scholar]
- 2. Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, et al. Immunoglobulin G4‐associated cholangitis: clinical profile and response to therapy. Gastroenterology 2008;134:706–715. [DOI] [PubMed] [Google Scholar]
- 3. Huggett MT, Culver EL, Kumar M, Hurst JM, Rodriguez‐Justo M, Chapman MH, et al. Type 1 autoimmune pancreatitis and IgG4‐related sclerosing cholangitis is associated with extrapancreatic organ failure, malignancy, and mortality in a prospective UK cohort. Am J Gastroenterol 2014;109:1675–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanno A, Masamune A, Okazaki K, Kamisawa T, Kawa S, Nishimori I, et al. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2011. Pancreas 2015;44:535–539. [DOI] [PubMed] [Google Scholar]
- 5. de Buy Wenniger LJM, Culver EL, Beuers U. Exposure to occupational antigens might predispose to IgG4‐related disease. Hepatology 2014;60:1453–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Culver EL, Sadler R, Bateman AC, Makuch M, Ferry B, Aalberse R, et al. Immunoglobulin E, eosinophils and mast cells in atopic individuals provide novel insights in IgG4‐related disease. J Hepatol 2016;64:S646. [Google Scholar]
- 7. Nakazawa T, Ohara H, Sano H, Ando T, Joh T. Schematic classification of sclerosing cholangitis with autoimmune pancreatitis by cholangiography. Pancreas 2006;32:229. [DOI] [PubMed] [Google Scholar]
- 8. Kalaitzakis E, Levy M, Kamisawa T, Johnson GJ, Baron TH, Topazian MD, et al. Endoscopic retrograde cholangiography does not reliably distinguish IgG4‐associated cholangitis from primary sclerosing cholangitis or cholangiocarcinoma. Clin Gastroenterol Hepatol 2011;9:800–803.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Culver EL, Sadler R, Simpson D, Cargill T, Makuch M, Bateman AC, et al. Elevated serum IgG4 levels in diagnosis, treatment response, organ involvement, and relapse in a prospective IgG4‐related disease UK cohort. Am J Gastroenterol 2016;111:733–743. [DOI] [PubMed] [Google Scholar]
- 10. Boonstra K, Culver EL, de Buy Wenniger LM, van Heerde MJ, van Erpecum KJ, Poen AC, et al. Serum immunoglobulin G4 and immunoglobulin G1 for distinguishing immunoglobulin G4‐associated cholangitis from primary sclerosing cholangitis. Hepatology 2014;59:1954–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4‐related disease. Mod Pathol 2012;25:1181–1192. [DOI] [PubMed] [Google Scholar]
- 12. Bateman AC, Culver EL. IgG4‐related disease—experience of 100 consecutive cases from a specialist centre. Histopathology 2017;70:798–813. [DOI] [PubMed] [Google Scholar]
- 13. Hart PA, Kamisawa T, Brugge WR, Chung JB, Culver EL, Czaká L, et al. Long‐term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut 2013;62:1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of IgG4‐related disease. Arthritis Rheumatol 2015;67:1688–1699. [DOI] [PubMed] [Google Scholar]
- 15. Hart PA, Topazian MD, Witzig TE, Clain JE, Gleeson FC, Klebig RR, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut 2013;62:1607–1615. [DOI] [PubMed] [Google Scholar]