

Abstract
Background
Cystic fibrosis (CF) is congenital multisystem disorder, that leads to gradual deterioration of pulmonary function. Advancements in therapy of CF-related lung disease have delayed its progression. However, lung transplantation remains the only therapeutic option for majority of such patients. Aim of the study was to assess qualification process and outcome of lung transplantation as a treatment of CF patients qualified in a single center between 2011 and 2018.
Material/Methods
This retrospective study assessed 41 patients who were qualified to be treated by means of lung transplantation due to CF in Lung Transplant Program of Silesian Center for Heart Diseases between 2011 and 2018. Analysis of patients during qualification process and after lung transplantation was performed. Lung recipients were observed during 1-year follow-up by means of pulmonary function tests.
Results
1-year survival was noted among 80% of the patients; 3-year survival and 5-year survival were noted among 70% of the recipients. Mean forced expiratory volume in 1 second (FEV1) increased after lung transplantation: 21.19% at qualification; and 76.67% at 12 months after lung transplantation. Mean forced vital capacity (FVC) results also improved: 34.18% at qualification and 78.34% at 12 months after lung transplantation. The 6-minute walk test (6MWT) before and after treatment noted an increase of 175.55 m.
Conclusions
Lung transplantation improves respiratory capacity of CF patients and prolongs their life.
MeSH Keywords: Cystic Fibrosis, Kaplan-Meier Estimate, Lung Transplantation, Respiratory Function Tests, Waiting Lists
Background
Cystic fibrosis (CF) is a congenital multisystem disorder caused by genetic mutation located on chromosome 7. The affected gene, called cystic fibrosis transmembrane conductance regulator (CFTR), is responsible for complex chloride channel and regulatory protein present in all exocrine tissues [1–3]. Deranged transport of CFTR-affected ions, such as chloride, sodium, and bicarbonate, can lead to excreting thick, viscous secretions in many organs [2,4,5]. Alteration in the mucus rheology is particularly problematic in respiratory system, due to the fact that viscid secretions obstruct small airways. Their chronic obstruction in this mechanism is soon followed by colonization with bacteria such as Hemophilus influenzae, Staphylococcus aureus, Burkholderia cepacia complex species, and Pseudomonas aeruginosa. It is estimated that 98% of the patients show either culture or serologic evidence of P. aeruginosa [6]. Frequent infections can cause damage to the bronchial walls, eventually leading to bronchiectasis. CF also includes acute exacerbations with typical pulmonary presentation of following symptoms: cough, tachypnea, dyspnea, increased sputum production, malaise, and weights loss. These conditions can cause a gradual decline in pulmonary function. Treatment consists of bronchodilators, antibiotics, agents to promote airway secretion clearance, vaccinations, and chest physiotherapy [7–14]. Supplemental oxygen and noninvasive positive pressure ventilation might be needed, as progression of CF is routinely accompanied by worsening hypoxemia [15,16]. Recently, a new class of drugs have emerged called CFTR modulators that act by improving production, processing, and function of CFTR protein. Pharmaceuticals, such as ivacaftor, tezacaftor, and lumacaftor has been approved in the United States of America [17]. Those drugs could improve pulmonary function and CF patient quality of life [18]. However, their application in treatment may be limited by adverse effects and economic barriers. Advancements in therapy of CF-related lung disease can delay disease progression, but unfortunately not stop it. Premature death due to respiratory failure occurs in at least 80% of CF patients [6]. As in other end-stage lung diseases, lung transplantation is a viable method of treatment. Double lung transplantation is required, as leaving a native lung would threaten the transplanted lung as a source of infection. Single lung transplantations among CF patients are a rarity and usually involve people who underwent pneumectomy a long time prior to new procedures. A thorough qualification process is a key to every successful treatment. In addition to general contraindications for any kind of transplantation, potential graft recipients due to CF undergo microbiological assessment, as it has been shown that patients with Burkholderia cenocepacia, Burkholderia gladioli and/or Mycobacterium abscessus have worse prognosis following lung transplantation [19–21]. The International Society for Heart and Lung Transplantation (ISHLT) has published a consensus statement, which recommends the following factors be used when considering the timing for the referral of a CF patient to a lung transplant facility: forced expiratory volume in 1 second (FEV1) below 30% of predicted value, rapid falling of this parameter despite optimal therapy among CF patients with advanced lung diseases, 6-minute walk distance of less than 400 m, development of pulmonary hypertension and clinical decline characterized by increasing frequency of exacerbations [22]. The literature has reported on lung transplantation as a treatment of CF and have shown significantly better outcome in comparison to lung recipients due to other causes [23,24]. The aim of this study was to assess the lung transplantation qualification process and the outcome of lung transplantation as a treatment of CF patients who qualified for a lung transplantation in a single center between 2011 and 2018.
Material and Methods
This retrospective study assessed 41 patients (20 females and 21 males) with mean age at qualification of 25.21 years (range: 14–47 years), who were qualified to be treated by means of lung transplantation due to CF in the Lung Transplant Program of Silesian Center for Heart Diseases between 2011–2018. The number of transplanted patients was 32 (17 females and 15 males, with mean age at transplantation of 26 years). All patients except one were double lung transplantation (DLT) recipients. Single lung transplantation was performed on one female recipient, who underwent pneumonectomy 3 years prior to the procedure of transplantation, so DLT was not possible due to technical aspects. The qualification process of this facility required multiple assessments. During the qualification process, kidney function was evaluated by serum creatinine (mg/dL) and estimated glomerular filtration rate (mL/min/1.73 m2). Liver function was evaluated by serum bilirubin (mg/dL), aspartate transaminase (UI/L), alanine transaminase (UI/L), and N terminal pro B type natriuretic peptide (pg/mL). Pulmonary function tests included spirometry (forced expiratory volume in first second: FEV1%, actual FEV1 [l], and forced vital capacity: FVC%, and actual FVC), were performed as well as arterial blood gases assessment. Six-minute walk test (6MWT) results (distance [m], Borg’s scale, oxygen saturation (SpO2 [%]) were analyzed. Cardiac evaluation included echocardiography (left ventricle ejection fraction [%], right ventricular systolic pressure [mm Hg], tricuspid annular plane systolic excursion [mm] acceleration time [ms]). Survival on National Lung Transplantation Waitlist (NLTWL) was assessed by means of Kaplan Meier curve. After the lung transplantation, survival was estimated by Kaplan Meier curve. The 1-year follow-up assessments were performed, including spirometry and 6MWT results. Follow-up was assessed among DLT recipients, who has already reached at least 3-month survival. Qualified patients with unknown end point (other than one of following: lung transplantation or death without procedure as a qualified candidate) were excluded from the study. Additional exclusion criterion was intraoperative death during transplantation itself.
Statistical analysis
Data was expressed as mean ± standard deviation. The Mann-Whitney U test, and chi-squared test and Yates test were used, as appropriate. Kaplan-Maier curves were used for depiction of the survival time. Statistica 10.0 statistical software (StatSoft Inc., Tulsa, OK, USA) was used for statistical analysis. All findings with P<0.05 were considered statistically significant.
Results
Qualification process
Between 2011 and 2018, 41 patients with CF were referred to Silesian Center for Heart Diseases’ Lung Transplant Ward and were finally qualified to be treated by means of lung transplantation. At qualification, average actual FEV1 was 0.74±0.23l. Patients obtained 21.19±6.21% of predicted value. Percentage of predicted FVC was higher (35.3±13.3%). Mean value of this parameter at qualification was 1.48 ± 0.66l. The 6MWT revealed that patients were able to walk 343.28±144.17 m on average. Performing it was judged as mildly tiring (mean Borg scale 4.72±2.59). However, mean oxygen saturation drop was 12.125 percentage points with mean oxygen saturations before and after the test being respectively 88.91% and 76.79%. It is consistent with the fact that congenital lung diseases make patients more adaptable to insufficient oxygen supply. All of the patients had proper left ventricle ejection fraction (62.15±6.92%; min: 52% max: 74%). NT-proBNP was 90.39±55.51 pg/mL on average. Mean glycemia was almost at the upper limit (5.48±1.53 mmol/L).
Lung plethysmography demonstrated that even though patients usually had proper total lung capacity (TLC: mean result 106.91±22.80%; median: 112.15%), their residual volume was significantly increased (RV: mean 296.78±68.51%; min: 134%; max: 444%).
There were 32 qualified patients who received lung transplantations. The remaining 9 patients died on the waiting list without the procedure. Statistical analysis showed no significant differences among those 2 group at qualification.
Survival on the NLTWL was estimated by Kaplan-Meier curve for all qualified patients. Figure 1 shows that a 50% chance of survival was noted at 17 months. As has been previously established, 50% survival on the NLTWL should be 2 years [22]. This suggests that patients might be sent to referral too late. However, due to the small group limitations, this problem requires additional research.
Figure 1.
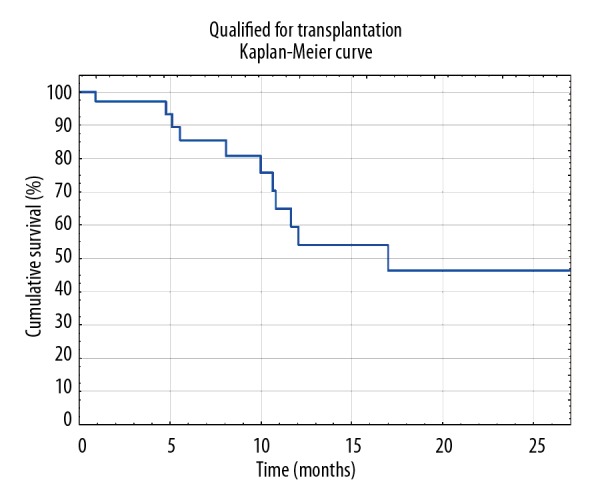
Survival on National Lung Transplantation Waitlist estimated by Kaplan-Meier curve.
Lung transplantation
Thirty-two patients underwent lung transplantation as a treatment, among which 7 patients are now deceased. During the 7-year study period, 78.05% of qualified patients became transplant recipients. Patients transplanted due to CF were 27.59% of all lung transplant recipients in our facility between 2011 and 2018. Statistical analysis revealed that the deceased recipients were on the NLTWL significantly longer than their living counterparts (522.25±260.77 days versus 253.320±255.4 days; P value=0.046292). However, mean time between transplantation and death was 180 days. Results from our center also showed that there was virtually no difference between mean time from qualification for lung transplantation to the endpoint regardless of its type (qualification to transplantation was 290.00 days versus qualification to death without procedure was 290.41 days). Survival after transplantation was estimated by Kaplan-Meier curve and presented in Figure 2. One-year survival was noted among 80% of the lung transplant recipients. Three-year survival and 5-year survival were noted among 70% of the lung transplant recipients.
Figure 2.
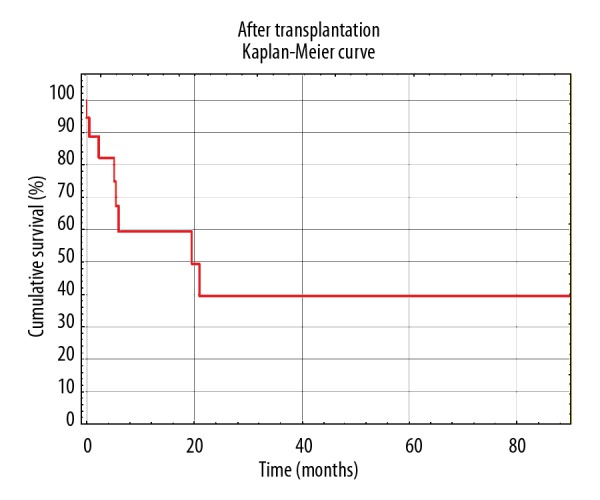
Survival after lung transplantation estimated by Kaplan-Meier curve.
During every check-up in Silesian Center for Heart Diseases, patients have their graft function evaluated by means of spirometry and 6MWT. Due to the fact that one of the patients underwent single lung transplantation, she was only included in the Kaplan-Meier survival curves, but was excluded from post-transplant analysis. Only the results of double lung recipients with follow-up of at least 3 months were assessed. Improvement of their functional status is presented by means of FEV1% increase during 1 year of follow-up. Detailed depictions are shown in Figure 3. Patients improved their results of approximately 55 percentage points at 12 months after lung transplantation in comparison to qualification score. Increase of FVC% was also noted. Even though it was slightly smaller (43 percentage points more), progress was visible. Detailed results are shown in Figure 4. Mean 6MWT distance results are shown in Figure 5. Approximately 175.55 m on average as an improvement may not seem like a lot. However, CF as a congenital disease with early onset of symptoms will make patients more adapted, so he/she could be walking longer distance with significant desaturation during 6MWT. After receiving the graft, patients were able to maintain proper oxygen saturation with slight, insignificant drops during 6MWT. Oxygen saturation levels during this examination before and after double lung transplantation are presented in Figure 6.
Figure 3.
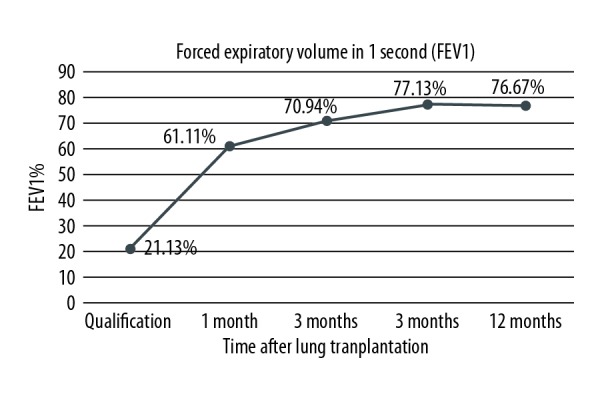
Improvement of forced expiratory volume (FEV) in 1 second expressed as percentage of predicted value (FEV1%) after lung transplantation of double lung transplant recipients.
Figure 4.
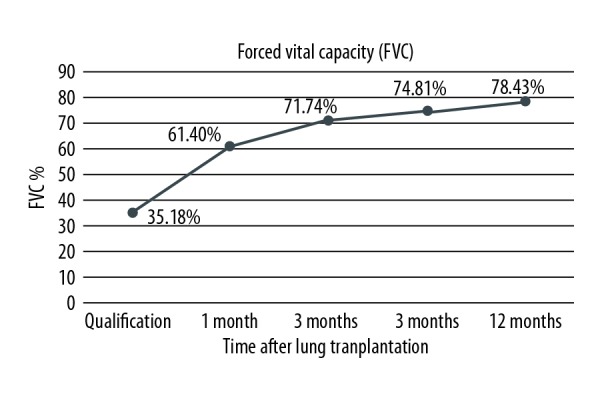
Improvement of forced vital capacity (FVC) expressed as percentage of predicted value (FVC%) after lung transplantation of double lung transplant recipients.
Figure 5.
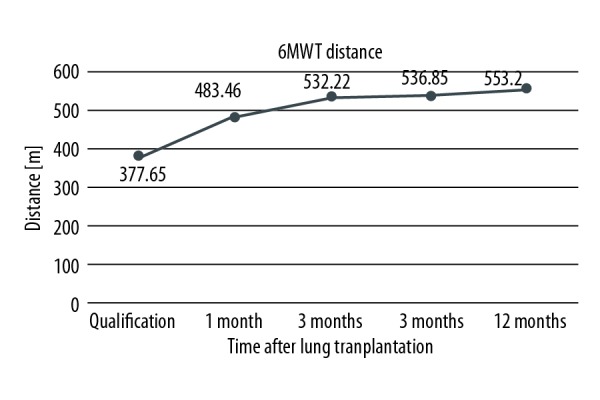
Improvement of distance obtained during 6-minute walk test (6MWT) after lung transplantation of double lung transplant recipients.
Figure 6.
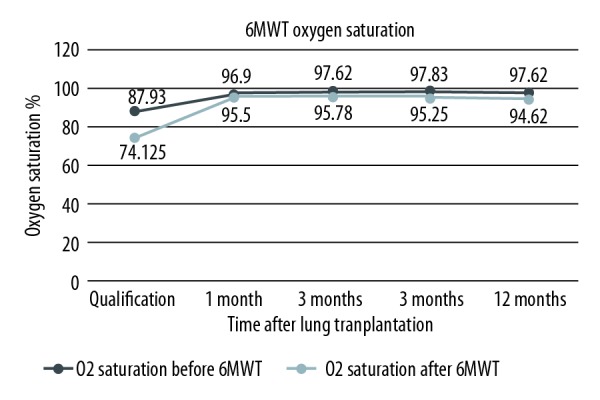
Improvement of oxygen saturation during 6-minute walk test (6MWT) after lung transplantation of double lung transplant recipients.
Discussion
Cystic fibrosis is a viable treatment for patients with respiratory failure. According to ISHLT registry, 50% survival after lung transplantation was assessed to be approximately 9.5 years. These results were even better after assessment of survival conditional to 3 months survival, as it reached 10.9 years [25]. Mearchery et al. (British researchers) also assessed that approximately 51% of patients live approximately 10 years [26]. Snell et al. claimed that 45% of CF patients after lung transplantation reached 10-year survival [27]. However, a Swedish team published excellent results which stated that in their lung transplant program 62,4% of CF patients reached 10-year survival [28]. In our center, the first patient transplanted due to CF still lives and has reached 7-year follow-up. Kaplan-Meier survival estimation based on our results have not reached 50% survival yet, but it only makes us hopeful for the future. Graft recipients due to this disease also have the best survival outcome among all lung transplant recipients from the Lung Transplant Program of Silesian Center for Heart Diseases. This finding is consistent with ISHLT reports as well [25]. One-year survival was reached by 80% of patients from our center. The aforementioned British researchers also assessed these parameters and it was revealed that their 1-year survival was 82% [26]. The Swedish study again reported the best 1-year survival at 86.4% [28]. Patients with CF have a good chance of receiving lung transplant in our facility with 78,05% of them eventually becoming a recipient. Such result is slightly better than Italian study by Borchi et al. who assessed that 76.08% of CF patient were able to be transplanted among all patients with known endpoints from this study [29]. Kourliouros et al. reported that according to the UK Transplant Registry, only 61% of CF patients will become graft recipients 3 years after registration in the United Kingdom’s Waitlist for Lung Transplantation [30]. In our facility, 21.95% of patients with CF died on the NLTWL with 50% of patients being on it for almost 1.5 years. The Italian study reported its mortality ratio to be 19.3%. However, if counted in the same way as our study, it would be 23.92% of their CF candidates who died. CF was also the most common indication for transplantation in their program [29]. Double lung recipients at qualification were able to obtain 21% of their predicted FEV1; at 12 months after lung transplantation, such percentage was approximately 77%. Those results were similar to those reported by Mearchery et al., whose patients presented the same FEV1% at qualification and reached 78% of predicted FEV1 at 12 months after lung transplantation [26]. ISHLT Registries and Lung Transplant Programs all over the world report better and better outcomes of CF patients, who undergo lung transplantation. Hayes et al. reported that outcomes of CF lung transplant recipients do not depend on overall volume of the facility, but on CF-specific volume [31]. This means that even smaller Lung Transplant Programs can obtained great outcomes, provided they will transplant CF patients frequently. However, it is important to remember that CF is a disease with many comorbidities. It also affects the liver and pancreas. Jardel et al. published a study assessing extrapulmonary conditions like diabetes, renal insufficiency, metabolic bone disease, hypertension, and liver disease as they considered these co-morbidities to be important [32]. It is worth remembering that immunosuppressive treatment after lung transplantation is burdened by adverse effects. The liver and pancreas of CF patients can also undergo irreversible damage that could lead to the need for transplantation of those solid organs. Novel approaches for liver-lung transplantation with good outcomes have been published recently by Salman et al. [33]. Also, pancreatic islet-lung transplantation has also been performed [34]. New possibilities await CF patients and transplantology specialists as the first combined pancreatic islet-lung-liver transplantation in a 14-year-old adolescent was also reported this year with positive outcomes [35].
Conclusions
Lung transplantation is a viable treatment for CF patients with respiratory failure. It not only has the ability to prolong life of these young patients, but it also improves their functional status measures by means of spirometry and 6MWT. It seems that patients should be referred to lung transplant centers sooner.
Footnotes
Source of support: Departmental sources
Conflict of interest
None.
References
- 1.Ren CL, Fink AK, Petren K, et al. Outcomes of infants with indeterminate diagnosis detected by cystic fibrosis newborn screening. Pediatrics. 2015;135:e1386–92. doi: 10.1542/peds.2014-3698. [DOI] [PubMed] [Google Scholar]
- 2.Levy H, Nugent M, Schneck K, et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin Genet. 2016;89:539–49. doi: 10.1111/cge.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ooi CY, Castellani C, Keenan K, et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics. 2015;135:e1377–85. doi: 10.1542/peds.2014-2081. [DOI] [PubMed] [Google Scholar]
- 4.Kharrazi M, Yang J, Bishop T, et al. Newborn screening for cystic fibrosis in California. Pediatrics. 2015;136:1062–72. doi: 10.1542/peds.2015-0811. [DOI] [PubMed] [Google Scholar]
- 5.Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–89. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- 6.Vigneswaran WT, Garrity Er, Jr, Odell JA. Chapter: Lung transplantation for cystic fibrosis and bronchiectasis. CRC Press Taylor and Francis Group; Boca Raton, FL, USA: 2016. Lung transplantation. Principles and practice; pp. 207–8. [Google Scholar]
- 7.Konstan MW, Wagener JS, Pasta DJ, et al. Clinical use of dornase alpha is associated with a slower rate of FEV1 decline in cystic fibrosis. Pediatr Pulmonol. 2011;46:545–53. doi: 10.1002/ppul.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain M, Thomson AH. Palivizumab, pneumococcal and influenza vaccination in cystic fibrosis. J R Soc Med. 2009;102(Suppl 1):23. doi: 10.1258/jrsm.2009.s19006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malfroot A, Adam G, Ciofu O, et al. Immunisation in the current management of cystic fibrosis patients. J Cyst Fibros. 2005;4:77–87. doi: 10.1016/j.jcf.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Groves HE, Jenkins L, Macfarlane M, et al. Efficacy and long-term outcomes of palivizumab prophylaxis to prevent respiratory syncytial virus infection in infants with cystic fibrosis in Northern Ireland. Pediatr Pulmonol. 2016;51:379–85. doi: 10.1002/ppul.23376. [DOI] [PubMed] [Google Scholar]
- 11.Moss RB. Allergic bronchopulmonary aspergillosis and Aspergillus infection in cystic fibrosis. Curr Opin Pulm Med. 2010;16:598–603. doi: 10.1097/MCP.0b013e32833e24a6. [DOI] [PubMed] [Google Scholar]
- 12.Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: A randomized controlled trial. JAMA. 2003;290:1749–56. doi: 10.1001/jama.290.13.1749. [DOI] [PubMed] [Google Scholar]
- 13.Nikolaizik WH, Schöni MH. Pilot study to assess the effect of inhaled corticosteroids on lung function in patients with cystic fibrosis. J Pediatr. 1996;128:271–74. doi: 10.1016/s0022-3476(96)70407-9. [DOI] [PubMed] [Google Scholar]
- 14.van Haren EH, Lammers JW, Festen J, et al. The effects of the inhaled corticosteroid budesonide on lung function and bronchial hyperresponsiveness in adult patients with cystic fibrosis. Respir Med. 1995;89:209–14. doi: 10.1016/0954-6111(95)90249-x. [DOI] [PubMed] [Google Scholar]
- 15.Elphick HE, Mallory G. Oxygen therapy for cystic fibrosis. Cochrane Database Syst Rev. 2013;(7):CD003884. doi: 10.1002/14651858.CD003884.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young AC, Wilson JW, Kotsimbos TC, Naughton MT. Randomised placebo controlled trial of non-invasive ventilation for hypercapnia in cystic fibrosis. Thorax. 2008;63(1):72–77. doi: 10.1136/thx.2007.082602. [DOI] [PubMed] [Google Scholar]
- 17.Durmowicz AG, Lim R, Rogers H, et al. The U.S. Food and Drug Administration’s experience with ivacaftor in cystic fibrosis. Establishing efficacy using in vitro data in lieu of a clinical trial. Ann Am Thorac Soc. 2018;15(1):1–2. doi: 10.1513/AnnalsATS.201708-668PS. [DOI] [PubMed] [Google Scholar]
- 18.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray S, Charbeneau J, Marshall BC, LiPuma JJ. Impact of Burkholderia infection on lung transplantation in cystic fibrosis. Am J Respir Crit Care Med. 2008;178(4):363–71. doi: 10.1164/rccm.200712-1834OC. [DOI] [PubMed] [Google Scholar]
- 20.Boussaud V, Guillemain R, Grenet D, et al. Clinical outcome following lung transplantation in patients with cystic fibrosis colonised with Burkholderia cepacia complex: results from two French centres. Thorax. 2008;63(8):732–37. doi: 10.1136/thx.2007.089458. [DOI] [PubMed] [Google Scholar]
- 21.Lobo LJ, Chang LC, Esther CR, Jr, et al. Lung transplant outcomes in cystic fibrosis patients with pre-operative Mycobacterium abscessus respiratory infections. Clin Transplant. 2013;27(4):523–29. doi: 10.1111/ctr.12140. [DOI] [PubMed] [Google Scholar]
- 22.Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014 – an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2015;34(1):1–15. doi: 10.1016/j.healun.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 23.The International Society for Heart and Lung Transplantation, 2017 slides. Adult lung transplantation statistics. Available at: https://ishltregistries.org/registries/slides.asp.
- 24.Yusen RD, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-first adult lung and heart-lung transplant report – 2014; focus theme: retransplantation. J Heart Lung Transplant. 2014;33:1009–24. doi: 10.1016/j.healun.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Khush KK, Cherikh WS, Chambers DC, et al. International Society for Heart and Lung Transplantation. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-fifth adult heart transplantation report-2018; Focus theme: Multiorgan transplantation. J Heart Lung Transplant. 2018;37(10):1155–68. doi: 10.1016/j.healun.2018.07.022. [DOI] [PubMed] [Google Scholar]
- 26.Meachery G, De Soyza A, Nicholson A. Outcomes of lung transplantation for cystic fibrosis in a large UK cohort. Thorax. 2008;63:725–31. doi: 10.1136/thx.2007.092056. [DOI] [PubMed] [Google Scholar]
- 27.Snell G, Reed A, Stern M, Hadjiliadis D. The evolution of lung transplantation for cystic fibrosis: A 2017 update. J Cyst Fibros. 2017;16(5):553–64. doi: 10.1016/j.jcf.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Gilljam M, Nyström U, Dellgren G, et al. Survival after lung transplantation for cystic fibrosis in Sweden. L Eur Cardiothorac Surg. 2017;51(3):571–76. doi: 10.1093/ejcts/ezw328. [DOI] [PubMed] [Google Scholar]
- 29.Borchi B, Barao Ocampo M, Cimino G, et al. Italian Cystic Fibrosis Lung Transplantation Group. Mortality rate of patients with cystic fibrosis on the waiting list and within one year after lung transplantation: a survey of Italian CF centers. Ital J Pediatr. 2018;44(1):72. doi: 10.1186/s13052-018-0512-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kourliouros A, Hogg R, Mehew J, et al. Patient outcomes from time of listing for lung transplantation in the UK: Are there disease-specific differences? Thorax. 2019;74(1):60–68. doi: 10.1136/thoraxjnl-2018-211731. [DOI] [PubMed] [Google Scholar]
- 31.Hayes D, Jr, Sweet SC, Benden C, et al. Transplant center volume and outcomes in lung transplantation for cystic fibrosis. Transpl Int. 2017;30(4):371–77. doi: 10.1111/tri.12911. [DOI] [PubMed] [Google Scholar]
- 32.Jardel S, Reynaud Q, Durieu I. Long-term extrapulmonary comorbidities after lung transplantation in cystic fibrosis: Update of specificities. Clin Transplant. 2018;32(6):e13269. doi: 10.1111/ctr.13269. [DOI] [PubMed] [Google Scholar]
- 33.Salman J, Grannas G, Ius F, et al. The liver-first approach for combined lung and liver transplantation. Eur J Cardiothorac Surg. 2018;54(6):1122–27. doi: 10.1093/ejcts/ezy217. [DOI] [PubMed] [Google Scholar]
- 34.Kessler L, Bakopoulou S, Kessler R, et al. Combined pancreatic islet-lung transplantation: A novel approach to the treatment of end-stage cystic fibrosis. Am J Transplant. 2010;10(7):1707–12. doi: 10.1111/j.1600-6143.2010.03143.x. [DOI] [PubMed] [Google Scholar]
- 35.Klee P, Dirlewanger M, Lavallard V, et al. Combined pancreatic islet-lung-liver transplantation in a pediatric patient with cystic fibrosis-related diabetes. Horm Res Paediatr. 2018;90(4):270–74. doi: 10.1159/000488107. [DOI] [PubMed] [Google Scholar]