

Abstract
Most serum proteins are N-linked glycosylated, and therefore the glycoproteomic profiling of serum is essential for characterization of serum proteins. In this study, we profiled serum N-glycoproteome by our recently developed N-glycoproteomic method using solid-phase extraction of N-linked glycans and glycosite-containing peptides (NGAG) coupled with LC-MS/MS and site-specific glycosylation analysis using GPQuest software. Our data indicated that half of identified N-glycosites were modified by at least two glycans, with a majority of them being sialylated. Specifically, 3/4 of glycosites were modified by biantennary N-glycans and 1/3 of glycosites were modified by triantennary sialylated N-glycans. In addition, two novel atypical glycosites (with N–X–V motif) were identified and validated from albumin and α−1B-glycoprotein. The widespread presence of these two glycosites among individuals was further confirmed by individual serum analyses.
Graphical Abstract
Serum is one of the most valuable clinical sample sources for disease diagnostics and therapeutic monitoring.1 It contains not only the major plasma protein constituents, such as albumin, immunoglobulins, transferrin, haptoglobin, and lipoproteins, but also many other proteins that are synthesized and secreted, shed, or lost from various cells and tissues.2 The alteration of serum protein concentrations and post-translational modifications (PTMs) could potentially reflect the physiological and pathological status of individuals. Therefore, characterization of serum proteins as well as their post-translational modifications (PTMs) is an essential prerequisite for their clinical utilities.
A large amount of serum proteins are N-linked glycoproteins.3 Glycoproteins play important roles in various biological processes, including cell-to-cell recognition, growth, differentiation, and programmed cell death.4–7 Aberrant glycosylation is usually associated with the pathological progression of many diseases.8 Many serum glycoproteins have been used as biomarkers for diagnosis and monitoring of disease progression, such as PSA for prostate cancer, AFP for liver cancer, and CA-125 for ovarian cancer, as well as CA-199 and CEA for colon cancer.9 However, most of clinical tests measure serum glycoproteins at their expression level, while detailed glycosylation information could provide additional information for the studies of disease mechanisms and disease detection.10 It has been reported in previous studies that some distinctive glycoforms on specific glycoproteins could enhance the specificity and/or sensitivity of cancer diagnosis. For example, fucosylated AFP (AFP-L3), which is able to distinguish hepatocellular carcinoma (HCC) from benign liver diseases, has been used as a HCC biomarker.10 The fucosylation of glycans on GP73, kallikrein, and haptoglobin also showed great potentials for early detection of HCC as well as disease progression monitoring.11 Therefore, comprehensive glycoproteome profiling of serum is very important for its clinical studies.
During the past decade, thousands of N-linked glycoproteins have been identified from human serum by mass spectrometry based approaches.2,3,12–21 However, these studies focused on deglycosylated peptides (or released glycans) but lacked the site-specific glycosylation information. In this study, we used solid-phase extraction of N-linked glycans and glycosite-containing peptides method (NGAG),22 tandem mass spectrometry, and GPQuest software23 to profile serum N-glycoproteome at site-specific glycosylation level in addition to identification of glycans and glycosites. Especially, two atypical glycosylation sites from albumin (ALB) and α−1B-glycoprotein (A1BG) were identified from serum and verified from individual blood serum, revealing their widespread existence.
EXPERIMENTAL SECTION
Human Sera.
Human sera (pooled from >100 individuals) used for comprehensive glycosylation profiling were purchased from Sigma (S7023). Individual sera from 16 donors were obtained from The Johns Hopkins Hospital according to the Institutional Review Board strictures of the Johns Hopkins University. All sera were stored at −80 °C until use.
Protein Digestion.
Serum proteins (~75 mg/mL) were denatured in 8 M urea with 1 M NH4HCO3 buffer, reduced by 10 mM TCEP at 37 °C for 1 h, and alkylated by 15 mM iodoacetamide at room temperature in the dark for 30 min. The solutions were diluted 5-fold with deionized water. Sequencing grade trypsin (Promega, Madison, WI; protein, enzyme, 40:1, w/w) was then added to the samples and incubated at 37 °C overnight with shaking. Samples were centrifuged at 12,000g for 10 min to remove any particulate matter and then purified by a C18 column (Waters, Milford, MA, USA). Peptides were eluted from the C18 column in 60% ACN with 0.1% TFA, and the peptide concentrations were measured by BCA reagent (Thermo Fisher Scientific, Fair Lawn, NJ, USA).
Extraction of N-Linked Glycans and Glycosite-Containing Peptides Using the NGAG Method.
A 4 mg amount of peptides from pooled sera was (1 mL) used for extraction of N-linked glycans and glycosite-containing peptides using the NGAG method. The guanidination of lysine on tryptic peptides was performed by adding 1 mL of 2.85 M aqueous ammonia and 1 mL of 0.6 M o-methylisourea (Sigma-Aldrich, St. Louis, MO, USA) into 1 mL of peptide solution (final pH = 10.5) and incubating at 65 °C for 30 min.24 The reaction was stopped by adding 3 mL of 10% TFA (make sure the solution pH < 3). The sample was desalted by a C18 column and eluted in 1 mL of 60% ACN with 0.1% TFA.
The pH of the peptide solution was adjusted to 7.4 by adding 200 μL of 1 M phosphate buffer (PB buffer, pH 7.4). The peptides were incubated with 4 mL of equilibrated AminoLink resin (50% slurry, Pierce, Rockford, IL, USA) in the presence of 50 mM NaCNBH3 at room temperature for 2 h with shaking. The remaining aldehyde groups on the resin were blocked by 1 M Tris-HCl solution (pH 7.4) in the presence of 50 mM NaCNBH3 at room temperature for 30 min. The resin was washed 3 times with water. All carboxyl groups of the peptides, including the sialic acid of glycans, aspartic acid (D), and glutamic acid (E) residues and C-termini of peptides, were modified by aniline by incubating beads with 1 M aniline and 0.5 M carbodiimide EDC in 0.2 M MES buffer (~pH 5) at room temperature for 2 h or overnight. The EDC solution was added two more times (2–4 h intervals). The pH of the solution was checked frequently to ensure that it was 4–6.25,26
The resin was washed three times each with 50% ACN and 100 mM NH4HCO3 solution and resuspended in 400 μL of 100 mM NH4HCO3 solution. A 2 μg amount of Asp-N was added and incubated with resin overnight at 37 °C with shaking to remove all possibly unlabeled D/E containing peptides. The resin was washed three times each with 50% ACN, 1.5 M NaCl, H2O, and 0.1 M NH4HCO3 solution. The N-glycans were released from the resin by 4 μL of PNGase F (New England Biolab, Beverly, MA, USA) in 400 μL of 25 mM NH4HCO3 solution at 37 °C for 3 h or overnight with shaking. The resin was washed twice with 400 μL deionized water. The N-glycans were collected in the supernatant/wash solutions and desalted by Carbograph column (Grace, Deerfield, IL, USA) prior to being dried by vacuum.27
The resin was washed three times each with 50% ACN, 1.5 M NaCl, H2O, and 0.1 M NH4HCO3 solution. The former N-glycopeptides were released from the resin by 2 μg of Asp-N in 400 μL of 25 mM NH4HCO3 solution at 37 °C overnight with shaking. The resin was washed once each with 400 μL of D.I. water and 50% ACN, and the glycosite-containing peptides were collected in both supernatant and wash solutions. The glycosite-containing peptides were dried by vacuum and resuspended in 50 μL of 0.2% FA (make sure pH < 3) for HPLC fractionation.
Enrichment of Intact Glycopeptides Using Hydrophilic Interaction Column.
The intact glycopeptides were enriched from 4 mg of peptides from pooled sera or 0.2 mg of peptides from individual samples using a hydrophilic interaction column (HILIC) enrichment method.28 The tryptic peptides eluted from C18 column (in 60% ACN/0.1% TFA) were diluted by 95% ACN/1% TFA to a final solvent composition of 80% ACN with 1% TFA. The HILIC column (SeQuant, Southborough, MA, USA) was washed twice each by 1% TFA and 80% ACN with 1% TFA, followed by sample loading and three washes with 80% ACN with 1% TFA. The glycopeptides bound to the column were eluted in 0.8 mL of 1% TFA solution. The samples were dried via SpeedVac and resuspended in 50 μL of 0.2% FA for LC-MS/MS analysis.
Deglycosylation of Serum Peptides in Solution.
In order to estimate the glycosylation occupancy at the atypical glycosylation sites, we performed PNGase F treatment of pooled serum peptides in solution. The desalted peptides were dried and resuspended in 25 mM NH4HCO3 solution. The solution was then incubated with PNGase F at 37 °C overnight. The peptides were dried and resuspended in 0.2% FA for LC-MS/MS analysis.
Glycan Analysis Using MALDI-TOF-MS.
N-Glycans extracted from human serum using NGAG were analyzed by an Axima MALDI resonance mass spectrometer (Shimadzu). Dried N-glycans were resuspended in 20 μL of deionized water followed by spotting 1 μL of glycan sample with 1 μL DHB/ DMA matrix (100 mg/mL DHB, 2% DMA in 50% acetonitrile and 0.1 mM NaCl) onto a MALDI plate.27,29 The laser power was set to 100 for two shots each in 100 locations per spot. The average MS spectra (200 profiles) were used for glycan assignment by GlycoMod software (http://web.expasy.org/glycomod/) and comparison to human glycan database.27,30 The possible glycan structures based on identified glycan compositions and human glycan structure database were generated by GlycoWorkbench.31
Peptide Fractionation Using High-pH RPLC.
The isolated glycosite-containing peptides and enriched intact glycopeptides from pooled sera were injected via a 1220 Series HPLC (Agilent Technologies, Inc., CA) into a Zorbax Extend-C18 analytical column containing 1.8 μm particles at a flow rate of 0.2 mL/min. The mobile-phase A consisted of 10 mM ammonium formate (pH 10) and B consisted of 10 mM ammonium formate and 90% ACN (pH 10). Sample separation was accomplished using the following linear gradient: 0–2% B, 10 min; 2–8% B, 5 min; 8–35% B, 85 min; 35–95% B, 5 min; 95–95% B, 15 min. Peptides were detected at 215 nm, and 96 fractions were collected along with the LC separation in a time-based mode from 16 to 112 min. The 96 fractions of the glycosite-containing peptide and intact glycopeptide samples were concatenated into 12 fractions by combining fractions 1, 13, 25, 37, 49, 61, 73, 85; 2, 14, 26, 38, 50, 62, 74, 86, and so on. The samples were then dried in a Speed-Vacuum and stored at −80 °C until LC-MS/MS analysis.
LC-MS/MS Analysis.
All N-linked glycosite-containing peptide and intact glycopeptide samples underwent one LC-MS/MS run per sample (or fraction) on a Q-Exactive mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Peptides were separated on a Dionex Ultimate 3000 RSLC nano system with a 75 μm × 15 cm Acclaim PepMap100 separating column protected by a 2 cm guard column (Thermo Scientific). The mobile phase flow rate was 300 nL/min and consisted of 0.1% formic acid in water (A) and 0.1% formic acid/95% acetonitrile (B). The gradient profile was set as follows: 4–7% B for 5 min, 7–40% B for 90 min, 40–90% B for 5 min, 90% B for 10 min, and equilibrated in 4% B for 10 min. MS analysis was performed using a Q-Exactive mass spectrometer. The spray voltage was set at 2.2 kV. Orbitrap MS1 spectra (AGC 1 × 106) were collected from 400 to 1800 m/z at a resolution of 60K followed by data-dependent HCD MS/MS (resolution, 7500; collision energy, 45%; activation time, 0.1 ms) of the 20 most abundant ions using an isolation width of 2.0 Da. Charge state screening was enabled to reject unassigned and singly charged ions. A dynamic exclusion time of 25 s was used to discriminate against previously selected ions. For intact glycopeptide samples, 100 m/z was set as the fixed first mass in MS/MS fragmentation to include all oxonium ions of glycopeptides.
Database Search.
The LC-MS/MS data of glycosite-containing peptides from sera were searched against UniProt human protein databases (downloaded from http://www.uniprot.org, August 2016) by Sequest in Proteome Discoverer V1.4 (Thermo Fisher Scientific). The search parameters for glycosite-containing peptides isolated by the NGAG method were set as follows: up to two missed cleavages were allowed for semi C-term trypsin digestion (C-termini of peptides must be tryptic end); 10 ppm and 0.06 Da mass tolerance are set for precursor and MS.MS ions, respectively; carbamidomethylation (C) was set as a static modification, deamidation (N), oxidation (M), aniline (D, E, C-termini of peptides; +75.0473 Da), guanidination (K, +42.0218 Da), and aniline + guanidination (K at the C-termini of peptides, +117.0691 Da) were set as dynamic modifications. The search parameters for glycosite-containing peptides isolated by the SPEG method and in solution deglycosylated peptides were set as follows: up to two missed cleavages were allowed for full trypsin digestion; carbamidomethylation (C) was set as a static modification, and deamidation (N) and oxidation (M) were set as dynamic modifications. All results were filtered with a 1% false discovery rate (FDR).
Intact Glycopeptide Analysis.
The intact N-glycopeptide data were first converted to “mzML” format using Trans-Proteome Pipline (TPP)32 and searched by GPQuest.23 The search parameters were set as follows: at least two oxonium ions out of the top 10 fragment ions in the MS/MS spectra were used for extraction of intact glycopeptide MS/MS spectra. The identified N-linked glycans from human sera by the NGAG method were used for the N-glycan database, and all identified serum glycosite-containing peptides by all different methods (including the glycosite-containing peptides identified in this study by the NGAG method and all other serum glycosite-containing peptides reported previously2,3,12–21) were used for the serum-specific glycosite-containing peptide database. The mass tolerances of 10 and 20 ppm were allowed for precusors and fragmentation ions. The false discovery rate of identified intact glycopeptides was estimated by the decoy peptide method, and 1% FDR was used for identification cutoff.
Estimation of the Glycosylation Occupancy.
The glycosylation occupancies at the atypical glycosites were estimated by using the following formula:
wherein Pglyco represents the N-linked glycosylation occupancy, Adeamidation_1 and Anonglyco_1 represent the peak areas of the same peptides with and without deamidation (N) in serum peptide samples, respectively. Adeamidation_2 and Anonglyco_2 represent the peak areas of the same peptides with and without deamidation (N) in PNGase F treated serum peptide samples, respectively. Although the method is based on the assumption of the same peptides with and without deamidation (N) have the same ionization efficiencies by mass spectrometry (which might not be the case), it could still facilitate the estimation of the glycosylation occupancy.
RESULTS AND DISCUSSION
Identification of N-Linked Glycans and Glycosite-Containing Peptides from Human Sera Using the NGAG Method.
In this study, we intend to characterize serum glycoproteins to identify their glycans, glycosites, and site-specific glycosylation. In order to achieve these goals, we applied our newly developed solid-phase extraction NGAG method22 into a human serum sample.
We first analyzed N-linked glycans and glycosite-containing peptides from human serum by using NGAG method22 (Figure 1A). Briefly, human serum proteins from a commercial source (>100 individual pool) were first digested into peptides using trypsin, and the ε-amino groups on the side chain of lysine residues were modified by guanidination.24 After being desalted, peptides were covalently conjugated to the AminoLink beads (aldehyde-functionalized) via their N-termini. After modification of carboxyl groups (including aspartic acid (D), glutamic acid (E), and peptide C-termini, as well as the sialic acid of the glycans) of the peptides using aniline, N-glycans were released from the beads via PNGase F digestion. The released N-glycans were then analyzed by MALDI-TOF-MS. From one MALDI-TOF-MS analysis, 65 N-linked glycans with different compositions were detected (Figure 1B and Supporting Information Figure S1 and Table S1). Among these, seven were oligo-mannose type, 35 were fucosylated glycans, and 30 were sialylated glycans (17 contained both fucose and sialic acid residues).
Figure 1.

Profiling of N-linked glycans, glycosites, and intact glycopeptides from human sera. (A) Workflow of the study. N-linked glycans and glycosite-containing peptides were first extracted from human sera (pooled from >100 individuals) by the NGAG method and identified using mass spectrometry. The enriched intact glycopeptides were then identified by searching GPQuest against identified glycans and glycosite-containing peptides from sera. (B) Glycan types identified from serum by the NGAG method. (C) Glycosites containing either N−X−S or N−X−T motif using the NGAG method.
We then released N-linked glycosite-containing peptides from the AminoLink Beads through Asp-N digestion. Asp-N specifically cleavages the peptide bonds at the N-termini of asparagine acid (D) residues of the peptides. In NGAG method, as the original aspartic acid residues of the peptides have been modified by aniline, only newly generated aspartic acid residues at the N-glycosites after PNGase F treatment can be cleaved by Asp-N at this step. The released glycosite-containing peptides were analyzed by Q-Exactive mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), which resulted in the identification of 560 glycosite-containing peptides from 331 N-glycoproteins (Figure 1C and Table S2). Among these, 240 glycosite-containing peptides (43%) contained the N−X−S motif, while the other 360 peptides (57%) contained the N−X−T motif.
Profiling of Site-Specific Glycosylation from Human Sera.
In order to obtain the site-specific glycosylation information, we identified intact glycopeptides from pooled sera. Briefly, the intact N-glycopeptides were enriched from pooled human sera using HILIC and were analyzed by 2D-LC-MS/MS. The mass spectrometry data were then searched against the serum intact glycopeptide candidate database using GPQuest software23 (precursor mass matching option). In the intact glycopeptide candidate database, we included not only all possible combinations of identified N-glycans and glycosite-containing peptides from human sera by NGAG method but also the combination of identified glycans by NGAG with all other identified human serum glycosite-containing peptides (all were converted to tryptic peptides using an in-house program) reported previously.2,3,12–21 Therefore, the intact glycopeptide candidate database comprises 65 N-glycans and 4028 unique glycosite-containing peptides representing 3792 glycosites from 2129 glycoproteins (Table S3).
By using 1% FDR as the identification cutoff, we assigned 7748 oxonium ion-containing MS/MS spectra to 1359 unique intact glycopeptides, which consist of 63 N-glycan compositions and 370 unique glycosites from 248 glycoproteins (Figure 2a and Table S4). The majority of glycosites were occupied by complex glycans (88% of glycosites, Figure 2A) and/or modified by at least one sialylated glycan (80% of glycosites and 86.9% of intact glycopeptide PSMs, Figure 2B), while only 24 glycosites from 22 glycoproteins were modified by oligo-mannose glycans. In addition, 3/4 of glycosites were modified by at least one of the glycans that contain biantennary glycans (N4H5, N4H5F1, N4H5S1, N4H5S2, N4H5F1S1, and N4H5F1S2), and 1/3 of glycosites were modified by triantennary glycans that contain N5H6 with 1–3 sialic acids (N5H6S1, N5H6S2, N5H6S3, N5H6F1S1, N5H6F1S2, and N5H6F1S3) (Figure 2C).
Figure 2.

Site-specific glycosylation profiling of human serum glycoproteins. (A) Heat map of identified intact glycopeptides from serum. The peptide-spectrum matches (PSMs) of the intact glycopeptides, comprising different glycans (upper) and glycosites (left), were exhibited in the heat map. The numbers of glycosites modified by each glycan and of glycans at each glycosite were summarized at the bottom and right parts of the figure, respectively. N, HexNAc; H, Hex; F, fucose; S, sialic acid. (B) PSMs of different glycans with 0–4 sialic acids. (C) Top ten glycans detected on serum glycoproteins.
When classifying the intact glycopeptides based on their peptide backbone, we found that the majority of the glycosites (80%) were solely occupied by complex glycans, and six glycosites were modified by all three different types of glycans (Figure 2A). Half of glycosites were modified by at least two glycans, and 17 glycosites were even occupied by more than 10 glycans, among which glycosite Asn-98 in apolipoprotein D (APOD) was modified by 24 different glycans, glycosite Asn-241 in haptoglobin (HP) was modified by 23 glycans, and glycosite Asn-340 in Ig α−1 chain C region was modified by 21 different glycans (Figure 2A). It can be expected that multiple glycan attachment should be the case for most of the N-linked glycosite when deeper intact glycopeptide analysis was performed. As expected, the diversity of the glycan compositions at different glycosites of the same glycoproteins was also observed. For example, among three N-glycosites identified from Complement C3, oligo-mannose (N2H4–N2H7) and hybrid (N3H3–N3H5) glycan types were detected at the glycosite Asn-85, while only N4H5S2 glycan was detected at the glycosite Asn-939 and N6H7F1S4 glycan at glycosite Asn-1617 (Table S4).
Identification of Atypical N-Glycosites on Albumin and α−1B-Glycoprotein.
It has been reported that N-linked glycosylation could occur at the atypical glycosylation sites other than N−X−S/T sequon.33–35 In this study, we also identified some peptides with deamidation (N) sites at asparagine but a lack of typical N−X−S/T sequon during the glycosite-containing peptide analysis. As the identification of deamidation at asparagines could happen due to the chemical deamidation if the glycosites were identified based on their deglycosylated peptide only,36,37 the direct identification of intact glycopeptide that forms at the atypical glycosites was needed for their verification.
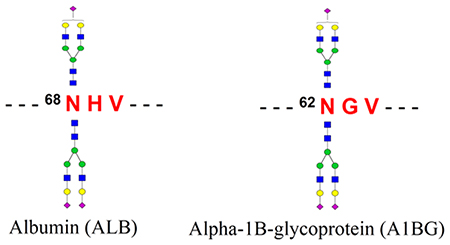
To identify the intact glycopeptide forms of the atypical glycosite-containing peptides, the mass spectrometry data of HILIC enriched samples were searched against the intact glycopeptide candidate database comprising identified glycans and the peptides with deamidation (N) but lacking a typical N−X−S/T sequon. By using 1% FDR as a cutoff, we identified four new intact glycopeptides using our GPQuest software.23 Among these intact glycopeptides, both peptides LV 6 8 N#EVTEFAK from albumin (ALB) and 62N#GVAQEPVHLDSPAIK from α−1B-glycoprotein (A1BG) were modified by complex glycans N4H5S1 and N4H5S2 (Figure 3 and Figure S2)
Figure 3.

Identification and validation of an atypical N-glycosite on human serum albumin using site-specific glycosylation analysis. (A) Spectrum of the intact glycopeptide LV68N#EVTEFAK + HexNAc4Hex5Ac1 from albumin. (B) A spectrum of the intact glycopeptide LV68N#EVTEFAK + HexNAc4Hex5Ac2 from albumin. The oxonium ions (green) were used to extract the intact glycopeptide spectrum, the masses of the precursor and peptide/(peptide + glycan fragment) ions (orange) as well as the b-/y-ions of the peptide portion (b-ions, blue; y-ions, red) were used for intact glycopeptide identification. (C) Spectrum of the deglycosylated form of the peptide LV68N#EVTEFAK. 68Asn on the peptide was identified as the glycosylation site based on the deamidation.
The spectra of these glycopeptides were manually confirmed using the following procedure. (1) All these spectra contain at least seven oxonium ions out of the top ten fragment ions, which confirmed that these spectra were generated from intact glycopeptides. (2) In these MS/MS spectra, we detected two peaks with 203 Da increment each after the peak at 1149.6 Da (for ALB glycopeptides, Figure 3) or after the peak at 1878 Da (for A1BG glycopeptides, Figure S2), followed by three peaks with 162 Da increment per peak. These features confirmed that the last five peaks were the peptide + HexNAc to peptide + HexNAc2Hex3 (N2H3) fragment ions (y-ions), and therefore these peaks were the intact peptide ions from ALB and A1BG glycopeptides, respectively. (3) Glycan composition of each glycopeptide was determined based on mass difference between the precursor and the glycosite-containing peptide, while the glycosylation site was identified with high confidence by the deamidated asparagine site in the glycosite-containing peptide. (4) Other fragment ions (excluding oxonium ions and y-ions) in the spectra were matched to the peptide portion of the glycopeptides. In the spectra in both panels A and B of Figure 3, 11 b/y fragment ions were matched to the peptide LV68N#EVTEFAK from ALB, which covered 80% amino acids in the peptide. In the spectra in Figure S2, up to 26 b/ y fragment ions were matched to the peptide 62N#GVAQEPVHLDSPAIK from A1BG. This information indicated the high confidence of these intact glycopeptide identifications. (5) As had been reported previously,38 different glycopeptides that contain the same peptide backbone (attached different glycans) shared similar fragmentation patterns. Similar y-ion fragment profiles of the peptide portion (prior to the glycosite) were also observed between glycosylated and deglycosylated forms of the same glycosite-containing peptides. These features enhanced the reliability of the identification of these atypical glycosite-containing glycopeptides.
Glycosylation Stoichiometries at the Atypical N-Glycosites.
In order to confirm whether the glycosylation at these two atypical glycosites were partially or completely glycosylated, we performed the global proteomic analysis of tryptic peptides from a pooled sera sample. The non-glycosylated forms of both peptides were identified, indicating their partially glycosylated status in human sera. To estimate their glycosylation stoichiometries, an aliquot of tryptic peptide sample was further treated by PNGase F prior to LC-MS/MS analysis. The glycosylation stoichiometries were then estimated by comparing the intensities between deglycosylated and nonglycosylated forms of the same peptides in the sera sample. The tryptic peptide sample (no PNGase F treatment) was used as a control to deduct the affects from possible natural or chemical deamidation reaction. Using this method, the glycosylation stoichiometries at atypical N-glycosites LV68N#EVTEFAK from ALB and 62N#GVAQEPVHLDSPAIK from A1BG were calculated to be 1.8% and 16.7%, respectively (Figure 4A,B). Low abundance of glycosylated forms of LV68N#EVTEFAK from ALB might be one of the reasons for their missing identification in previous studies.
Figure 4.

Distribution of Albumin Atypical Glycosylation Sites among Population.
Albumin is the most abundant protein in sera, and it does not contain any N−X−S/T motif for typically N-linked glycosylation; therefore it has been considered as not N-linked glycosylated. In order to confirm whether N-linked glycosylation at the atypical glycosite Asn-68 commonly existed among people, we analyzed the intact glycopeptides at the atypical glycosites Asn-68 of ALB from 16 individual serum samples. The intact glycopeptides were enriched from tryptic peptides of these individual serum samples using HILIC column and were analyzed by LC-MS/MS. By using GPQuest software and the same parameter as set above, we quantitatively detected both intact glycopeptides at the atypical glycosite (Figure 5A) from all 16 samples: the intensity of glycan N4H5S2 was usually more than 2-fold of the intensity of glycan N4H5S1 at the glycosite.
Figure 5.

Widespread distribution of N-glycosylation at the atypical glycosites Asn-68 of albumin and Asn-62 of A1BG among the population. The distribution of the intact glycopeptides from albumin and from A1BG among 16 individual serum samples are shown in panels A and B, respectively. The total intensities of each sample were used for normalization by SEIVE (Thermo Fisher), and the intensities of the intact glycopeptides were used to exhibit their relative abundances among samples.
A1BG is expressed at high levels in the adult and fetal liver; it is also present in normal adult serum or plasma.39 Elevated levels of A1BG had been reported in cancers.39 Three N−X−S/ T motifs containing glycosites and one N−X−V motif containing glycosite from A1BG were identified in this study. To confirm whether the atypical glycosite Asn-62 from A1BG was glycosylated in 16 individual serum samples, we also quantified their intact glycopeptides from these samples. Our results showed that both glycans were detected in all samples again (Figure 5B). Although the variations of these glycopeptide intensities among people could be up to 5-fold, the intensity of glycan N4H5S2 was about 4-fold of the glycan N4H5S1. These results confirmed the widespread existence of N-glycosylation at the glycosite Asn-68 of ALB and the glycosite Asn-62 of A1BG among the population.
CONCLUSIONS
Due to the significant clinical utilities, serum glycomic and glycoproteomic have been reported in several previous studies.3 However, the analyses of intact glycopeptides with site-specific glycan information allow one to obtain a comprehensive glycosylation information on serum glycoproteome. In this study, we performed the comprehensive glycoproteome analysis of pooled sera using our NGAG method, LC-MS/ MS, and GPQuest software. Using the method, the N-linked glycan and glycosite-containing peptide were first isolated and identified by mass spectrometry; the intact glycopeptides were then enriched and analyzed for their site-specific glycan determination. Importantly, our approaches identified two atypical N-linked glycosylation sites from albumin and α−1B-glycoprotein. Both atypical glycosylation sites were partially glycosylated and contained the N−X−V sequon. Further evidence showed that these two atypical glycosites commonly existed among individuals. The site-specific glycosylation map of serum glycoproteins and two atypical glycosites newly identified in this study would be a valuable resource for the studies of blood compositions and functions as well as for biomarker discovery research.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health, National Cancer Institute, the Early Detection Research Network (EDRN; Grant U01CA152813), the Clinical Proteomic Tumor Analysis Consortium (CPTAC; Grant U24CA210985), National Heart Lung and Blood Institute, Programs of Excellence in Glycosciences (PEG; Grant P01HL107153), and the National Institute of Allergy and Infectious Diseases (Grant R21AI122382). The work was also supported by National Natural Science Foundation of China (Grant Nos. 81773180 and 21705127).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b01051.
Identified glycans coupled with MALDI-TOF-MS analysis and identification and validation of atypical Nglycosite, (PDF)
Table S1 listing N-linked glycans indentified from human serum using NGAG methode (XLSX)
Table S2 listing glycosite-containing peptides identified from human sera by NGAG method (XLSX)
Table S3 listing previously reported glycosite-containing peptides identified from human sera (XLSX)
Table S4 listing intact glycopeptides identified from human sera (XLSX)
REFERENCES
- (1).Anderson NL; Anderson NG Mol. Cell. Proteomics 2002, 1, 845–867. [DOI] [PubMed] [Google Scholar]
- (2).Farrah T; Deutsch EW; Omenn GS; Campbell DS; Sun Z; Bletz JA; Mallick P; Katz JE; Malmström J; Ossola R; et al. Mol. Cell. Proteomics 2011, 10 (9), [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu T; Qian W-J; Gritsenko MA; Camp DG; Monroe ME; Moore RJ; Smith RD J. Proteome Res 2005, 4, 2070–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rudd PM; Elliott T; Cresswell P; Wilson IA; Dwek RA Science 2001, 291, 2370–2376. [DOI] [PubMed] [Google Scholar]
- (5).Lis H; Sharon N Eur. J. Biochem 1993, 218, 1–27. [DOI] [PubMed] [Google Scholar]
- (6).Tian Y; Zhang H Proteomics: Clin. Appl 2010, 4, 124–132. [DOI] [PubMed] [Google Scholar]
- (7).Haltiwanger RS; Lowe JB Annu. Rev. Biochem 2004, 73, 491–537. [DOI] [PubMed] [Google Scholar]
- (8).Drake PM; Cho W; Li B; Prakobphol A; Johansen E; Anderson NL; Regnier FE; Gibson BW; Fisher SJ Clin. Chem 2010, 56, 223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Crutchfield CA; Thomas SN; Sokoll LJ; Chan DW Clin. Proteomics 2016, 13, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li D; Mallory T; Satomura S Clin. Chim. Acta 2001, 313, 15–19. [DOI] [PubMed] [Google Scholar]
- (11).Zhou J; Yang W; Hu Y; Hoti N; Liu Y; Shah P; Sun S; Clark D; Thomas S; Zhang H Anal. Chem 2017, 89, 7623–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang H; Chan DW Cancer Epidemiol., Biomarkers Prev 2007, 16, 1915–1917. [DOI] [PubMed] [Google Scholar]
- (13).Yang W; Laeyendecker O; Wendel SK; Zhang B; Sun S; Zhou J-Y; Ao M; Moore RD; Jackson JB; Zhang H Theranostics 2014, 4, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu Y; Huettenhain R; Surinova S; Gillet LCJ; Mouritsen J; Brunner R; Navarro P; Aebersold R Proteomics 2013, 13, 1247–1256. [DOI] [PubMed] [Google Scholar]
- (15).Pan C; Zhou Y; Dator R; Ginghina C; Zhao Y; Movius J; Peskind E; Zabetian CP; Quinn J; Galasko D; Stewart T; Shi M; Zhang JJ Proteome Res. 2014, 13, 4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Whitmore TE; Peterson A; Holzman T; Eastham A; Amon L; McIntosh M; Ozinsky A; Nelson PS; Martin DB J. Proteome Res 2012, 11, 2653–2665. [DOI] [PubMed] [Google Scholar]
- (17).Tan ZJ; Yin HD; Nie S; Lin ZX; Zhu JH; Ruffin MT; Anderson MA; Simeone DM; Lubman DM J. Proteome Res 2015, 14, 1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Song E; Zhu R; Hammoud ZT; Mechref YJ Proteome Res. 2014, 13, 4808–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zeng X; Hood BL; Sun M; Conrads TP; Day RS; Weissfeld JL; Siegfried JM; Bigbee WL J. Proteome Res 2010, 9, 6440–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Calvano CD; Zambonin CG; Jensen ON J. Proteomics 2008, 71, 304–317. [DOI] [PubMed] [Google Scholar]
- (21).Qiu RQ; Regnier FE Anal. Chem 2005, 77, 7225–7231. [DOI] [PubMed] [Google Scholar]
- (22).Sun S; Shah P; Eshghi ST; Yang W; Trikannad N; Yang S; Chen L; Aiyetan P; Hoti N; Zhang Z; Chan DW; Zhang H Nat. Biotechnol 2016, 34, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Toghi Eshghi, S.; Shah P; Yang W; Li X; Zhang H Anal. Chem 2015, 87, 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brancia FL; Oliver SG; Gaskell SJ Rapid Commun. Mass Spectrom 2000, 14, 2070–2073. [DOI] [PubMed] [Google Scholar]
- (25).Panchaud A; Hansson J; Affolter M; Bel Rhlid R; Piu S; Moreillon P; Kussmann M Mol. Cell. Proteomics 2008, 7, 800–812. [DOI] [PubMed] [Google Scholar]
- (26).Schilling O; Barré O; Huesgen PF; Overall CM Nat. Methods 2010, 7, 508–511. [DOI] [PubMed] [Google Scholar]
- (27).Yang SJ; Li Y; Shah PK; Zhang H Anal. Chem 2013, 85, 5555–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hägglund P; Bunkenborg J; Elortza F; Jensen ON; Roepstorff P J. Proteome Res 2004, 3, 556–566. [DOI] [PubMed] [Google Scholar]
- (29).Shah P; Yang S; Sun S; Aiyetan P; Yarema KJ; Zhang H Anal. Chem 2013, 85, 3606–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yang S; Zhang H Anal. Chem 2012, 84, 2232–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ceroni A; Maass K; Geyer H; Geyer R; Dell A; Haslam SM J. Proteome Res 2008, 7, 1650–1659. [DOI] [PubMed] [Google Scholar]
- (32).Keller A; Eng J; Zhang N; Li X. j.; Aebersold R Mol. Syst. Biol 2005, 1 (1), [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Baycin-Hizal D; Tian Y; Akan I; Jacobson E; Clark D; Wu A; Jampol R; Palter K; Betenbaugh M; Zhang H Anal. Chem 2011, 83, 5296–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zielinska DF; Gnad F; Wisniewski JR; Mann M Cell 2010, 141, 897–907. [DOI] [PubMed] [Google Scholar]
- (35).Sun S; Zhang H Anal. Chem 2015, 87, 11948–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Palmisano G; Melo-Braga MN; Engholm-Keller K; Parker BL; Larsen MR J. Proteome Res 2012, 11, 1949–1957. [DOI] [PubMed] [Google Scholar]
- (37).Hao P; Ren Y; Alpert AJ; Sze SK Mol. Cell. Proteomics 2011, 10, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yang W; Shah P; Toghi Eshghi S; Yang S; Sun S; Ao M; Rubin A; Jackson JB; Zhang H Anal. Chem 2014, 86, 6959–6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Piyaphanee N; Ma Q; Kremen O; Czech K; Greis K; Mitsnefes M; Devarajan P; Bennett MR Proteomics: Clin. Appl 2011, 5, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.