

Abstract
Headaches are highly disabling and are among the most common neurological disorders worldwide. Despite the high prevalence of headache, therapeutic options are limited. We recently identified the delta opioid receptor (DOR) as an emerging therapeutic target for migraine. In this study, we examined the effectiveness of a hallmark DOR agonist, SNC80, in disease models reflecting diverse headache disorders including: chronic migraine, post-traumatic headache (PTH), medication overuse headache by triptans (MOH), and opioid-induced hyperalgesia (OIH). To model chronic migraine C57BL/6 mice received chronic intermittent treatment with the known human migraine trigger, nitroglycerin. PTH was modeled by combining the closed head weight drop model with the nitroglycerin model of chronic migraine. For MOH and OIH, mice were chronically treated with sumatriptan or morphine, respectively. The development of periorbital and peripheral allodynia was observed in all four models; and SNC80 significantly inhibited allodynia in all cases. In addition, we also determined if chronic daily treatment with SNC80 would induce MOH/OIH, and we observed limited hyperalgesia relative to sumatriptan or morphine. Together, our results indicate that DOR agonists could be effective in multiple headache disorders, despite their distinct etiology, thus presenting a novel therapeutic target for headache.
Keywords: hypersensitivity, mTBI, hyperalgesia, pain
1. Introduction
Headache disorders are ranked as the third highest worldwide for years lost to disability (2015; 2017). Primary headaches are due to the headache condition itself, and include migraine. Although episodic migraine is more common, chronic migraine is more debilitating, and these patients experience at least 15 or more headache days/month (2013). While a number of preventives are available, they are not highly effective and have low tolerability (Blumenfeld et al., 2013). Secondary headaches are defined as headaches that are due to another medical condition (2013), and common causes include traumatic brain injury and medication overuse. Post-traumatic headache (PTH) is highly prevalent, and more than 50% of mild traumatic brain injury (mTBI) patients go on to develop PTH, which can last for up to 5 years post-injury (Stacey et al., 2017). Medication overuse headache (MOH) is observed following chronic use of medications prescribed for headache which paradoxically exacerbate and increase the frequency of headache (2013). For example, MOH has been reported for triptan overuse, a commonly prescribed class of acute migraine medications (Katsarava et al., 2004; Limmroth et al., 2002). Similarly, chronic use of opioids results in opioid induced hyperalgesia (OIH), a phenomenon where pain severity increases beyond the original pain, and expands in area (Hayhurst and Durieux, 2016). Opioids are commonly prescribed for migraine, and can produce headache that is more frequent and severe, and refractory to other treatment (Bigal and Lipton, 2009; Buse et al., 2012; Thorlund et al., 2016). Currently, the first line of treatment for MOH and OIH is withdrawal of the overused drug (Diener et al., 2016), but this has low patient compliance, and adjunct therapies that are mechanistically distinct from the MOH-causing drug would be helpful.
Despite the high prevalence of headache disorders, patients have limited therapeutic options. Our group recently identified the delta opioid receptor (DOR) as a promising target for migraine (Charles and Pradhan, 2016; Pradhan et al., 2014b). We found that in a nitroglycerin (NTG) preclinical model of migraine, DOR agonists significantly inhibited migraine-associated allodynia and conditioned place aversion, a correlate of migraine-associated negative affect (Pradhan et al., 2014b). In addition, we also observed that DOR activation decreased the number of cortical spreading depression events in a model of migraine aura (Pradhan et al., 2014b). Our group recently developed a model of post-traumatic migraine (Moye et al., 2018), the most severe form of PTH (Theeler et al., 2013); and again DOR activation effectively prevented the development of chronic migraine induced by mTBI.
Clinically, headache disorder patients present with already established headache. To date, we have shown that DOR agonists can alleviate the development of migraine or PTH, but it is unknown if DOR activation can effectively block already established pain associated with primary and secondary headaches. The aim of this study was to determine if the DOR agonist, SNC80, could alleviate established cephalic and peripheral allodynia in models of chronic migraine, PTH, MOH associated with triptans, and OIH. Further, we also determined if chronic DOR activation could itself produce a MOH/OIH state.
2. Methods
2.1. Animals
Experiments were performed on male and female C57BL6/J mice (Jackson Laboratories, Bar Harbor, ME. USA), weighing 20-30g, and no sex differences were observed. Mice were group housed in a 12h-12h light-dark cycle, where the lights were turned on at 07:00 and turned off at 19:00. Food and water were available ad libitum. All responses were conducted in a blinded fashion by 1-3 experimenters. Weight was recorded on each test day for all experiments. Neither treatments nor drugs significantly affected weight gain or mortality. All experimental procedures were approved by the University of Illinois at Chicago Office of Animal Care and Institutional Biosafety Committee, in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) guidelines and the Animal Care Policies of the University of Illinois at Chicago. All results are reported according to Animal Research: reporting of In Vivo Experiments (ARRIVE) guidelines.
2.2. Sensory sensitivity testing
Separate groups of animals were used for hind paw and cephalic experiments. For all behavioral experiments, mice were counterbalanced into groups following the first basal test for mechanical sensitivity. The experimenter was blinded to the drug condition being tested. No adverse effects were observed in any of the experiments. All mice were tested in a separate behavior room with low-light (~35-50 lux) and low-noise conditions, between 09:00 and 16:00. For all behavioral tests, mice were habituated to the testing rack for 2 days prior to the first test day, and on each test day for 20 minutes prior to the first measurement. For peripheral measurements, the plantar surface of the mouse hind paw was tested. For cephalic testing, mice were tested in 4 oz paper cups, to which they had been previously habituated for 1 hour over 2 days. The periorbital region caudal to the eyes and near the midline was tested. To assess mechanical sensitivity, the threshold for responses to punctate mechanical stimuli (mechanical allodynia) was tested according to the up-and-down method (Chaplan et al., 1994). The region of interest was stimulated with a series of eight von Frey hair filaments (bending force ranging from 0.00g to 2g). A response was defined as a lifting, shaking, or licking of the hind paw or head, depending on the region tested. The first filament tested was 0.4g. In the absence of a response, a heavier filament (up) was tried, and in the presence of a response, a lighter filament (down) was tested. This pattern was followed for a maximum of four filaments following the first response.
2.3. NTG model of chronic migraine
NTG was purchased at a concentration of 5.0 mg/mL, in 30% alcohol, 30% propylene glycol and water (American Reagent, NY, USA). NTG was freshly diluted on each test day in 0.9% saline to a concentration of 1mg/mL for a dose of 10 mg/kg. The vehicle (VEH) used in these experiments was 0.9% saline. We previously found that there was no significant difference in mechanical thresholds between 0.9% saline, and the solution in which NTG was dissolved in (6% propylene glycol, 6% alcohol, 0.9% saline) (Pradhan et al., 2014a). Mice were treated every second day for 9 days with vehicle or NTG (10 mg/kg, ip). For hind paw experiments, basal thresholds were assessed on days 1, 3, 5, 7, and 9. For cephalic experiments, basal thresholds were assessed on days 1, 5, and 9. On test days, mechanical thresholds were measured prior to vehicle/NTG injection.
2.4. Model of post-traumatic headache
Mild traumatic brain injury (mTBI) was induced by using the closed head weight-drop method, as described previously (Moye et al., 2018; Zohar et al., 2003). Briefly, mice were mildly anesthetized with 2.5% isoflurane with an oxygen flow rate of 0.6-0.8 liters per minute. Mice were placed chest down on a foam sponge (dimensions: 7-1/2 in. × 5-1/2 in. × 1-7/8 in) to support the head and body, which allowed for anterior-posterior motion without any rotational movement at the moment of impact. The mouse and sponge were placed directly underneath the weight-drop device which consisted of a hollow cylindrical tube (inner diameter 2.54 cm, 80 cm height) placed approximately 1cm vertically over the mouse’s head, in between the ear and eye. To induce mTBI, a 30g weight (13 mm diameter, 34 mm height) was dropped through the tube, striking the mouse and causing a closed head injury. Immediately after mTBI, mice were returned to their home cages for recovery for 2 weeks. Sham animals were anesthetized but not subjected to the weight-drop. To model post-traumatic headache, a low dose of NTG (0.1 mg/kg IP) was used relative to the chronic model describe above. We have previously shown that this dose does not cause basal hypersensitivity in intact or sham mice, but does in mTBI animals (Moye et al., 2018; Pradhan et al., 2014a). NTG was freshly diluted on each test day in 0.9% saline to a concentration of 0.01 mg/mL for a dose of 0.1 mg/kg. Two weeks following mTBI or a sham procedure, mice were treated every other day for 9 days with vehicle or low dose NTG (0.1 mg/kg, ip), and tested in the same time frames as described above.
2.5. Model of medication overuse headache (MOH)
Sumatriptan (SUMA; 0.6 mg/kg, ip; Sandoz, NC, USA) was purchased at a concentration of 12 mg/mL and diluted to 0.06 mg/mL in 0.9% saline. Mice were treated once daily with vehicle or SUMA (0.6 mg/kg, ip) over 11 days, and tested on days 1, 3, 5, 7, 9, and 11 for hind paw testing, and on days 1, 5, and 9 for cephalic testing. On test days, mechanical thresholds were measured 30 minutes after injection.
2.6. Model of opioid induced hyperalgesia (OIH)
A stock concentration of 10 mg//ml morphine (MORPH) was diluted fresh daily with saline. Mice were treated twice daily with vehicle or MORPH over 4 days (20 mg/kg SC days 1-3, 40 mg/kg s.c. day 4), and tested on days 1-4 for hind paw testing, and on days 1 and 3 for cephalic testing. Basal mechanical thresholds were measured daily prior to MORPH injections.
2.7. Acute treatment with DOR agonist
Eighteen to twenty-four hours after the last drug administration day we determined the effect of SNC80. On this challenge test day, basal hind paw and cephalic mechanical thresholds were determined, after which mice received either vehicle (VEH) or SNC80 (10 mg/kg, ip; Tocris Bioscience, Bristol, UK). SNC80 was diluted to 1 mg/mL in 0.33% 1N HCl/0.9% saline. Post-SNC80 thresholds were assessed 2 hours after basal testing, and 45 minutes after SNC80 injection.
2.8. Chronic treatment with DOR agonist
To determine whether chronic DOR activation caused hypersensitivity similar to MOH, Mice were treated once daily with vehicle, SUMA (0.6 mg/kg, ip), or SNC80 (10 mg/kg, ip) over 11 days. For hind paw experiments, mice were tested on days 1, 3, 5, 7, 9, and 11, and another cohort of mice were tested on days 1 and 11. For cephalic experiments, mice were tested on days 1 and 11, and days 1, 5 and 11 depending on the experiment.
2.9. Statistical Analysis
Data are expressed as mean ± s.e.m. All mice tested were included in the analysis. All statistical analyses were performed by SigmaStat software, and graphs were generated using GraphPad Prism. For all behavioral experiments, a two-way repeated-measures analysis of variance (ANOVA) was performed, with treatment (vehicle/SUMA/MORPH/NTG/SNC80) and time (days) as factors. When a significant interaction occurred, subsequent Holm-Sidak post-hoc analysis was performed. A significance level of p<0.05 was used.
3. Results
3.1. DOR activation inhibits established chronic migraine-associated pain
To determine the effect of DOR activation in a model of chronic migraine, we tested whether an acute dose of SNC80 could reverse established mechanical allodynia to chronic intermittent administration of the human migraine trigger NTG (Moye and Pradhan, 2017b; Pradhan et al., 2014a). To model chronic migraine, NTG (10 mg/kg, ip) or a vehicle was given every other day for 9 days (5 test days total). Hind paw thresholds were taken prior to NTG administration on days 1, 3, 5, 7, and 9, and in a separate group of mice cephalic thresholds were taken on days 1, 5, and 9 (Figure 1A). Mice chronically treated with NTG developed a basal peripheral and cephalic hypersensitivity, an effect not seen in the VEH treated groups (Figure 1B and D). Twenty-four hours after the final NTG/VEH administration (day 10), basal responses were measured and NTG-treated mice continued to show peripheral and cephalic allodynia (Figure 1C and E: basal). Mice were treated acutely with vehicle or SNC80 (10 mg/kg IP), and post-treatment thresholds were measured 45 minutes later. SNC80 had no effect on mechanical responses in animals chronically treated with vehicle (Figure 1C,E: post-drug). However, SNC80 significantly attenuated peripheral and cephalic allodynia induced by chronic NTG. These data demonstrate that DOR activation can block established chronic migraine-associated pain.
Figure 1:
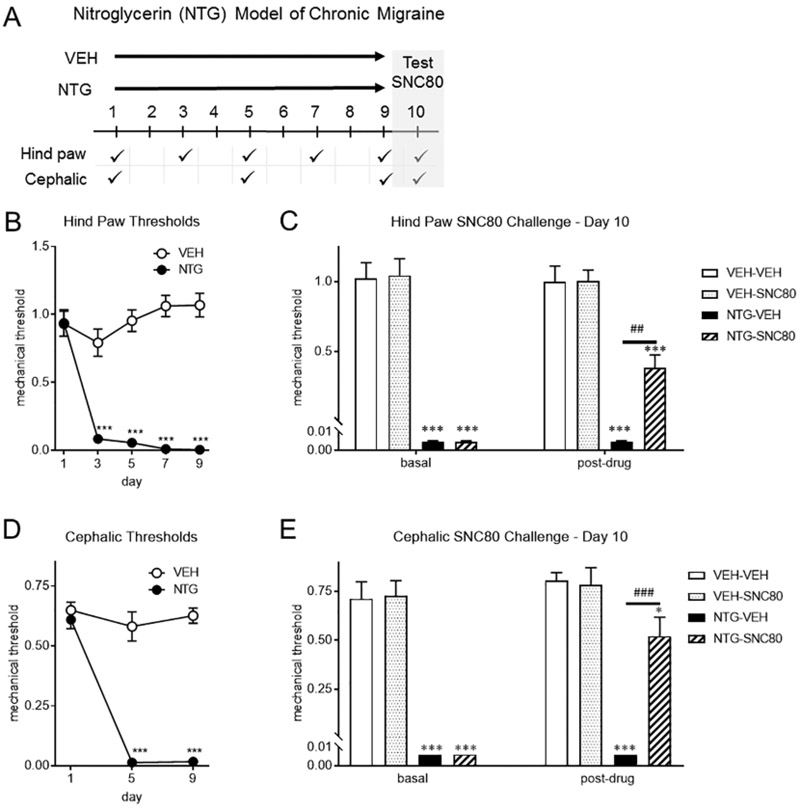
SNC80 treatment attenuates chronic NTG-induced allodynia. (a) Experimental outline. Separate groups of mice were tested for hind paw or cephalic allodynia. C57BL6/J mice were treated with vehicle (0.9% NaCl, VEH) or NTG (10 mg/kg, IP) every second day for 9 days. Baselines were measured prior to VEH/NTG administration. NTG produced a basal hypersensitivity in hind paw (b) and cephalic (d) regions, an effect not observed in vehicle (VEH) treated mice. p<0.001 effect of drug, time, and interaction, two-way RM ANOVA and Holm-Sidak post hoc analysis. ***p<0.001 relative to vehicle on day 1. On day 10, hind paw (c) or cephalic (e) basal responses were measured and NTG-treated mice had significantly lower thresholds compared to VEH (basal). SNC80 (10 mg/kg IP, post-drug) was administered and animals were tested 45 min later. SNC80 significantly inhibited allodynia in both regions. p<0.05 effect of treatment, drug, and interaction, two-way ANOVA and Holm-Sidak post hoc analysis. ***p<0.001 relative to veh-veh, ## p<0.01, ###p<0.001 relative to NTG-VEH. n=6/group. DOR activation blocks chronic migraine-associated pain.
3.2. DOR activation inhibits post-traumatic headache
We have previously developed a model of PTH by combining the closed head weight drop method and the NTG model of chronic migraine (Moye et al., 2018). Mice underwent a closed head injury or sham procedure followed by a 2 week recovery period. At 2 weeks post-injury, low dose NTG (0.1 mg/kg, ip) or a vehicle was administered every other day over 9 days (5 test days total). Similar to the chronic migraine model, hind paw thresholds were taken prior to NTG administration every other day, and in a separate group of mice cephalic thresholds were taken on days 1, 5, and 9 (Figure 2A). mTBI mice developed a basal peripheral and cephalic hypersensitivity following chronic intermittent treatment with low dose NTG, an effect not observed in the sham controls (Figure 2B and D). On day 10, 24h after the final NTG/VEH administration, mTBI-NTG-treated mice continued to show peripheral and cephalic allodynia (Figure 2C and E: basal). Animals were treated with either vehicle or SNC80 (10 mg/kg IP), and post-treatment thresholds were measured 45 minutes later. Acute treatment with SNC80 effectively inhibited hind paw and cephalic allodynia (Figure 2C and E: post-drug); and did not affect general nociception in the mice chronically treated with vehicle. These data demonstrate that DOR activation can alleviate established PTH-associated pain.
Figure 2:

SNC80 treatment acutely reverses allodynia in a model of PTH. (a) Experimental outline. Separate groups of mice were tested for hind paw or cephalic allodynia. C57BL6/J mice either underwent a closed head weight drop (mTBI) or sham procedure, and 2 weeks later were treated with vehicle (0.9% NaCl, VEH) or low dose NTG (0.1 mg/kg, IP) every second day for 9 days. Baselines were measured prior to VEH/NTG administration. NTG produced a basal hypersensitivity in hind paw (b) and cephalic (d) regions, an effect not observed in vehicle (VEH) treated mice. p<0.001 effect of drug, time, and interaction, two-way RM ANOVA and Holm-Sidak post hoc analysis. ***p<0.001 relative to vehicle on day 1. On day 10, hind paw (c) or cephalic (e) basal responses were measured and NTG-treated mice had significantly lower thresholds compared to VEH (basal). SNC80 (10 mg/kg IP, post-drug) was administered and animals were tested 45 min later. SNC80 significantly inhibited allodynia in both regions. p<0.05 effect of treatment, drug, and interaction, two-way ANOVA and Holm-Sidak post hoc analysis. **p<0.01, ***p<0.001 relative to VEH-VEH, ###p<0.001 relative to NTG-VEH. n=6/group. DOR activation blocks pain associated with post-traumatic headache.
3.3. DOR activation inhibits medication overuse headache to sumatriptan
Overuse of sumatriptan (SUMA), an acute migraine medication, can lead to MOH. We tested whether an acute dose of SNC80 would inhibit chronic SUMA-induced allodynia. To model MOH, SUMA or a vehicle was given once daily for 11 days (Tipton et al., 2015). Hind paw thresholds were tested 30 minutes before VEH or SUMA injection every other day, and cephalic thresholds on days 1, 5, and 9 (Figure 3A). Mice chronically treated specifically with SUMA developed basal hind paw and cephalic hypersensitivity (Figure 3B and D). Twenty-four hours after the last SUMA/VEH injection (day 12), basal hind paw and cephalic thresholds continued to be low in the SUMA treated groups (Figure 2C and E; basal). Mice were treated with SNC80 (10 mg/kg IP) or vehicle, and SNC80 significantly attenuated this allodynia (Figure 3C and E; postdrug). To determine the duration of the anti-allodynic effect of SNC80 we tested mice 24h following injection of vehicle or SNC80, and the mechanical thresholds for both groups were comparably low (SUMA-VEH vs. SUMA-SNC80; 0.11 ± 0.07 vs. 0.13 ± 0.04, mean ± SEM). These data suggest that DOR activation can acutely inhibit MOH caused by overuse of sumatriptan.
Figure 3:

SNC80 treatment attenuates hind paw and cephalic allodynia induced by chronic sumatriptan. (a) Experimental outline. Separate groups of mice were tested for hind paw or cephalic allodynia. C57BL6/J mice were treated with vehicle (0.9% NaCl, VEH) or sumatriptan (SUMA, 0.6 mg/kg, IP) every day for 11 days. Baselines were measured prior to VEH/SUMA administration. SUMA produced a basal hypersensitivity in hind paw (b) and cephalic (d) regions, an effect not observed in vehicle (VEH) treated mice. p<0.001 effect of drug, time, and interaction, two-way RM ANOVA and Holm-Sidak post hoc analysis. ***p<0.001 relative to vehicle on day 1. On day 10, hind paw (c) or cephalic (e) basal responses were measured and SUMA-treated mice had significantly lower thresholds compared to VEH (basal). SNC80 (10 mg/kg IP, postdrug) was administered and animals were tested 45 min later. SNC80 significantly inhibited allodynia in both regions. p<0.05 effect of treatment, drug, and interaction, twoway ANOVA and Holm-Sidak post hoc analysis. ***p<0.001 relative to VEH-VEH, ###p<0.001 relative to SUMA-VEH. n=8-9/group (hind paw), n=11/group (cephalic). DOR activation blocks pain associated with medication overuse headache.
3.4. DOR activation inhibits opioid-induced hyperalgesia to morphine
To determine the effect of DOR activation on OIH, we tested whether an acute dose of SNC80 would reverse established peripheral and cephalic allodynia induced by chronic morphine (MORPH). To model OIH, MORPH (20 mg/kg, SC days 1-3; 40 mg/kg, SC day 4) or vehicle was given twice a day for 4 days. All basal response were determined in the AM before the morning injection which occurred 2 hours after testing (Figure 4A). Only mice chronically treated with morphine developed a basal hind paw or cephalic hypersensitivity (Figure 4B and D), an effect that was still observed 18-24h after the final VEH/MORPH injection (Figure 4C and E; basal). Animals were treated with either vehicle or SNC80 (10 mg/kg IP), and SNC80 significantly attenuated hind paw and cephalic allodynia in chronic morphine treated animals (Figure 4C and E: post-drug). These data demonstrate that DOR activation can inhibit OIH-associated pain, and suggests that DOR and MOR regulate pain through different mechanisms.
Figure 4:

SNC80 treatment attenuates allodynia induced by chronic morphine. (a) Experimental outline. Separate groups of mice were tested for hind paw or cephalic allodynia. C57BL6/J mice were treated with vehicle (0.9% NaCl, VEH) or morphine (MORPH, 20 mg/kg SC days 1-3; 40 mg/kg SC day 4) twice a day for 4 days. Injections occurred in the morning and late afternoon. Baselines were measured prior to the VEH/MORPH administration in the morning. MORPH produced a basal hypersensitivity in hind paw (b) and cephalic (d) regions, an effect not observed in vehicle (VEH) treated mice. p<0.001 effect of drug, time, and interaction, two-way RM ANOVA and Holm-Sidak post hoc analysis. ***p<0.001 relative to vehicle on day 1. On day 10, hind paw (c) or cephalic (e) basal responses were measured and MORPH-treated mice had significantly lower thresholds compared to VEH (basal). SNC80 (10 mg/kg IP, postdrug) was administered and animals were tested 45 min later. SNC80 significantly inhibited allodynia in both regions. p<0.001 effect of treatment, drug, and interaction, two-way ANOVA and Holm-Sidak post hoc analysis. *p<0.05, ***p<0.001 relative to VEH-VEH, ###p<0.001 relative to MORPH-VEH. n=8-9/group (hindpaw), n=6/group (cephalic). DOR activation blocks pain associated with opioid induced hyperalgesia.
3.5. Chronic DOR activation produces limited OIH
We determined if chronic daily administration of SNC80 would cause OIH-associated allodynia. As a positive control, we concurrently tested a group of mice with sumatriptan (Figure 5A). Mice were given vehicle, SUMA or SNC80 once a day for 11 days, and initially tested on days 1, 3, 5, 7, 9, and 11 for the development of peripheral allodynia. Mice chronically treated with SUMA or SNC80 developed a basal hind paw hypersensitivity, an effect not seen in the vehicle control group (Figure 5B). Along with the pharmacological effects, repeated testing can produce associative learning, which can be a major component of drug tolerance and hyperalgesia. To determine whether the basal hypersensitivity in SUMA- and SNC80-treated mice was pharmacologically induced or learned, mice were given vehicle, SUMA, or SNC80 once a day for 11 days, but only tested on days 1 and 11. Mice chronically treated with SUMA developed a basal peripheral allodynia, an effect not seen in vehicle or SNC80-treated mice (Figure 5C). To determine if chronic DOR activation would cause cephalic allodynia, a separate group of mice were similarly treated daily with SNC80 for 11 days and cephalic thresholds were measured on days 1 and 11. There was no significant difference between VEH- and SNC80-treated mice (Figure 5D). To further characterize when mice would develop MOH to SNC80, we injected mice with vehicle or SNC80 every day for 11 days, and tested on days 1, 5 and 11. On day 5, there was no significant difference between VEH- and SNC80-treated mice, in contrast to VEH- and SUMA-treated mice. However, by the third test day (day 11) SNC80 and SUMA-treated animals both showed periorbital allodynia relative to controls (Figure 5E). These data indicate that repeated use of a DOR agonist produces a limited form of MOH that is less severe than sumatriptan.
Figure 5:

Chronic SNC80 administration induces limited OIH/MOH. (a) Experimental outline. Separate groups of mice were tested for hind paw or cephalic allodynia. C57BL6/J mice were treated with vehicle (0.9% NaCl, VEH), SNC80 (10 mg/kg IP), or SUMA (0.6 mg/kg IP) daily for 11 days. Baselines were measured prior to the VEH/SNC80/SUMA administration. (b) SUMA- and SNC80-treated animals had significantly lower hind paw mechanical responses relative to VEH controls. p<0.001 effect of drug, time, and interaction, two-way RM ANOVA and Holm-Sidak post-hoc analysis. n=8-12/group. (c) Male C57Bl/6J mice were treated with VEH, SUMA, or SNC80 every day for 11 days, but only tested on days 1 and 11. SUMA-treated animals had significantly lower hind paw mechanical responses on day 11, an effect not observed in VEH- or SNC80-treated mice. p<0.001 effect of drug, time, and interaction, two-way RM ANOVA and Holm-Sidak post-hoc analysis. n=8-10/group. (d) Mice were treated with VEH or SNC80 every day for 11 days, and tested on days 1 and 11 for cephalic responses. SNC80 did not induce cephalic hypersensitivity, p=0.359 effect of drug and time, two-way RM ANOVA, n=8/group. (e) Mice were treated with VEH, SUMA, or SNC80 every day for 11 days, and tested on days 1, 5 and 11 for cephalic responses. SNC80 induced cephalic hypersensitivity, but at a slower rate to sumatriptan. p<0.001 effect of drug, time, and interaction, two-way RM ANOVA and Holm-Sidak post-hoc analysis. n=8-12/group. Chronic DOR activation does not induce OIH/MOH as rapidly as sumatriptan, and this effect may be through increased associative learning.
4. Discussion
Despite the extraordinary disability caused by headache disorders, these patients have remarkably few effective treatment options. Chronic migraine poses a significant clinical burden, and is experienced by 1-2% of the population (May and Schulte, 2016), with approximately 3% of episodic migraine patients converting to chronic migraine per year (Scher et al., 2003). Chronic migraine is treated with preventives from varying drug classes, including tricyclic antidepressants, anti-convulsants and beta blockers (Charles, 2017). However, these treatments only work for a subset of patients and are associated with a number of adverse effects resulting in less than 50% of patients being satisfied with their treatment (Bigal et al., 2008). Patients suffering from PTH face similar limitations. There are no specific pharmacological treatments for PTH, and there has never been a large scale clinical trial to test PTH-specific therapies (Monteith and Borsook, 2014; Moye and Pradhan, 2017a). PTH with a migraine-like phenotype is usually treated with the same category of drugs as migraine without injury, with comparably poor success rates (Moye and Pradhan, 2017a). A further complication associated with the treatment of headache is the phenomenon of MOH. Overuse of medications prescribed to treat migraine, such as triptans and opioids, have been associated with MOH, and an estimated 15% of migraine patients go on to develop this disorder (Schwedt et al., 2018). Furthermore, MOH results in central sensitization that produces increased pain responsivity in both cephalic and peripheral regions (Munksgaard et al., 2013). The primary treatment for MOH is withdrawal from the overused medication, however, clinical studies have reported a 20-50% relapse rate within the first year of withdrawal (Katsarava et al., 2003; Zidverc-Trajkovic et al., 2018), with most patients relapsing within the first 6 months of withdrawal (Katsarava et al., 2003). There is clearly a need to diversify the tool box of pharmacotherapies available for the treatment of primary and secondary disorders. The results from our study indicate that DOR could be a promising addition to this tool box and could be effective for multiple types of headache disorders.
We have previously identified DOR as a novel therapeutic target for migraine (Charles and Pradhan, 2016); and shown that DOR agonists, when administered shortly after NTG administration, block acute NTG-evoked peripheral allodynia (Pradhan et al., 2014b). We have also previously demonstrated that when SNC80 is administered with low-dose NTG in a model of post-traumatic migraine, DOR activation can prevent the development to chronic PTH (Moye et al., 2018). Patients usually present with well-established headache, and the goal of this study was to determine the utility of DOR agonists in models that reflect this clinically significant state. We tested the DOR agonist, SNC80, in several models of headache. SNC80 is highly selective for DOR (Vicente-Sanchez et al., 2016), and we previously demonstrated that its effects in the NTG model were blocked by the DOR antagonist, naltrindole (Pradhan et al., 2014b). We found that the DOR agonist, SNC80, blocked established pain associated with chronic migraine, PTH, and MOH to chronic sumatriptan or morphine. In all models, we measured mechanical allodynia in both peripheral and cephalic regions, and DOR activation was anti-allodynic regardless of area tested. In addition, we observed that unlike sumatriptan or morphine, chronic DOR stimulation did not pharmacologically result in MOH/OIH, thus strengthening the case for DOR as a promising target for drug development.
In the United States, MOR-based therapies such as morphine, hydrocodone, and oxycodone, are still regularly prescribed for headache (Bigal and Lipton, 2009; Buse et al., 2012; Thorlund et al., 2016). Paradoxically, while opioids provide acute relief, chronic use results in refractory headache and contributes to the progression of migraine from an episodic to a chronic state (Bigal and Lipton, 2009; Buse et al., 2012; Thorlund et al., 2016), a condition associated with OIH (Chu et al., 2008; Roeckel et al., 2016). Additionally, MOR agonists are highly addictive, and over-prescription has led to a devastating public health crisis. In order to circumvent this cycle of sensitization and drug abuse, pharmacotherapies that are mechanistically different from MOR are required. Although DOR is a member of the opioid receptor family and is antihyperalgesic in preclinical models, it is physically and functionally a distinct protein. MOR and DOR are expressed in different cellular and anatomical brain regions. For example, MOR is highly expressed in pain processing regions such as the thalamus and periaqueductal grey, while DOR expression is higher in the striatum and cortical regions (Le Merrer et al., 2009; Mansour et al., 1995). Even in areas that express both receptors, MOR and DOR are often expressed on different cell types. Only a small percentage of cells in pain processing regions such as the dorsal root ganglia, spinal cord, and lateral parabrachial nucleus showed co-expression of MOR and DOR (Bardoni et al., 2014; Erbs et al., 2015; Scherrer et al., 2009; Wang et al., 2018); and MOR and DOR were differentially expressed on dural projections from the trigeminal ganglia (Rice et al., 2017). Our finding that SNC80 can inhibit OIH-associated pain supports the idea that MOR and DOR distinctly regulate pain processing. OIH is caused by maladaptations in pain circuits induced by chronic morphine treatment. Our studies would suggest that despite this chronic exposure to morphine, DOR functionality remains intact and could be an effective treatment for OIH, acting as an adjunct therapy during opioid withdrawal/weaning.
DOR agonists are not effective in most preclinical models of acute pain, but rather gain functionality in chronic conditions. Multiple publications show that DOR is dynamic, and increased functionality is observed following chronic stimuli such as pain (Cahill et al., 2003; Huang et al., 2015; Kabli and Cahill, 2007; Pradhan et al., 2013), intestinal inflammation (DiCello et al., 2018), morphine administration (Cahill et al., 2001), and ethanol exposure (van Rijn et al., 2012). In keeping with these findings, our results also show that DOR agonists are effective in multiple models of headache-associated pain, in which each model requires long term exposure to NTG, morphine, or sumatriptan. How DOR functionality increases in chronic pain states is a topic of intense study (Vicente-Sanchez et al., 2016). DOR mRNA levels have been found to be upregulated in animal models of peripheral inflammatory pain (Cahill et al., 2003) and acute pulpitis (Huang et al., 2015), indicating that pain can cause increased DOR expression. In addition, microscopic studies indicate that DORs are predominantly located on intracellular compartments, and that tissue injury redistributes these receptors to the cell membrane (Bao et al., 2003; Cahill et al., 2003; Gendron et al., 2006; Guan et al., 2005; Patwardhan et al., 2005; Zhang et al., 1998). However, there is some debate regarding the specificity of the DOR antibodies used in these studies (Bardoni et al., 2014; Scherrer et al., 2009; Wang et al., 2010). Electrophysiological studies have also revealed that chronic peripheral pain can also increase DOR coupling to Ca2+ channels in dorsal root ganglia (Pradhan et al., 2013). There are a number of lines of evidence to indicate that DOR functionality is upregulated following chronic pain; and future studies will focus on where and how this increase occurs.
One of our aims was to determine if chronic treatment with a DOR agonist would result in OIH/MOH. Interestingly, we found that daily treatment and testing with SNC80 resulted in subsequent hyperalgesia, but that daily treatment alone did not result in increased pain sensitivity, unlike sumatriptan. These results suggest that pharmacological activation of DOR does not produce OIH/MOH, however, if chronic SNC80 is paired with repeated testing then DOR activation might facilitate associative learning resulting in behavioral sensitization. The DOR is expressed in a number of brain regions that can regulate different kinds of learning; including the hippocampus, amygdala, and striatum (Le Merrer et al., 2009; Pellissier et al., 2016; Pradhan et al., 2011). Knockout of DOR results in impairment in object recognition tasks (Le Merrer et al., 2013), as well as deficiencies in place conditioning tasks (Le Merrer et al., 2011). In addition, DORs in the nucleus accumbens shell have been shown to modulate predictive learning (Bertran-Gonzalez et al., 2013; Laurent et al., 2014; Laurent et al., 2015a; Laurent et al., 2015b). We have also previously demonstrated that tolerance to SNC80 is significantly dependent on associative learning (Pradhan et al., 2010; Vicente-Sanchez et al., 2018), and environmental cues related to memory and learning can modulate behavioral outcomes to repeated exposure to opioids (Gamble et al., 1989; Mitchell et al., 2000). Our results should be considered during the development of DOR agonists for headache, as chronic DOR activation could facilitate associated learned behavior in migraineurs.
5. Conclusion
We demonstrate that DOR is a promising therapeutic for several established headache disorders, including MOH, OIH and PTH. Unlike the mu opioid receptor, DOR agonists have low abuse liability as they are not readily self-administered in animal models and do not cause physical dependence (Brandt et al., 2001; Negus et al., 1998; Stevenson et al., 2005). DOR agonists also do not produce significant adverse effects such as respiratory depression or constipation (Codd et al., 2009; Gallantine and Meert, 2005). An additional benefit of DOR-based therapeutics is that activation of DOR can positively regulate emotional tone (Lutz and Kieffer, 2013), and have been advanced to clinical trials for the treatment of anxiety and depression (2012; Richards et al., 2016). This affect may be particularly important considering the high co-morbidity between headache disorders and psychiatric conditions (Minen et al., 2016), and the negative emotional state induced by withdrawal from opioids (Koob and Volkow, 2016). Headaches disorders are incredibly disabling, and DOR is a novel mechanisticallydistinct option that could expand the treatment portfolio.
Highlights.
Delta opioid receptor activation inhibits headache-associated pain in preclinical models of chronic migraine, post-traumatic headache, and medication overuse headache.
Mu opioid receptor agonists are commonly prescribed for headache and result in opioid inducted hyperalgesia, which is inhibited by a delta opioid receptor agonist.
Chronic treatment with a delta opioid receptor agonist produces limited medication overuse headache relative to sumatriptan.
ACKNOWLEDGEMENTS
LM is a part of the Graduate Program in Neuroscience at UIC.
FUNDING
Research was supported by DA040688 (AP) and Diversity Supplement for LM DA040688-02S1.
Footnotes
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. J.Violin and A. Crombie are current or former employees of Trevena Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 2012. Astra-Zeneca-Pharmaceuticals: A phase IIa, multicenter, randomized, double-blind, double-dummy, active and placebo controlled, parallel group study to assess the efficacy and safety of azd7268 in patients with major depressive disorder, clinical study report synopsis. https://clinicaltrials.gov/ct2/show/NCT01020799?term=AZD7268&rank=3.
- 2013. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 33, 629–808. [DOI] [PubMed] [Google Scholar]
- 2015. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386, 743–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2017. Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol 16, 877–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Jin SX, Zhang C, Wang LH, Xu ZZ, Zhang FX, Wang LC, Ning FS, Cai HJ, Guan JS, Xiao HS, Xu ZQ, He C, Hokfelt T, Zhou Z, Zhang X, 2003. Activation of delta opioid receptors induces receptor insertion and neuropeptide secretion. Neuron 37, 121–133. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Tawfik VL, Wang D, Francois A, Solorzano C, Shuster SA, Choudhury P, Betelli C, Cassidy C, Smith K, de Nooij JC, Mennicken F, O'Donnell D, Kieffer BL, Woodbury CJ, Basbaum AI, MacDermott AB, Scherrer G, 2014. Delta opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron 81, 1312–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Laurent V, Chieng BC, Christie MJ, Balleine BW, 2013. Learning-related translocation of delta-opioid receptors on ventral striatal cholinergic interneurons mediates choice between goal-directed actions. J Neurosci 33, 16060–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal ME, Lipton RB, 2009. Excessive opioid use and the development of chronic migraine. Pain 142, 179–182. [DOI] [PubMed] [Google Scholar]
- Bigal ME, Serrano D, Reed M, Lipton RB, 2008. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology 71,559–566. [DOI] [PubMed] [Google Scholar]
- Blumenfeld AM, Bloudek LM, Becker WJ, Buse DC, Varon SF, Maglinte GA, Wilcox TK, Kawata AK, Lipton RB, 2013. Patterns of use and reasons for discontinuation of prophylactic medications for episodic migraine and chronic migraine: results from the second international burden of migraine study (IBMS-II). Headache 53, 644–655. [DOI] [PubMed] [Google Scholar]
- Brandt MR, Furness MS, Rice KC, Fischer BD, Negus SS, 2001. Studies of tolerance and dependence with the delta-opioid agonist SNC80 in rhesus monkeys responding under a schedule of food presentation. J.Pharmacol.Exp.Ther 299, 629–637. [PubMed] [Google Scholar]
- Buse DC, Pearlman SH, Reed ML, Serrano D, Ng-Mak DS, Lipton RB, 2012. Opioid use and dependence among persons with migraine: results of the AMPP study. Headache 52, 18–36. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A, 2003. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain 101, 199–208. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A, 2001. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J.Neurosci 21, 7598–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL, 1994. Quantitative assessment of tactile allodynia in the rat paw. J.Neurosci.Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Charles A, 2017. Migraine. N Engl J Med 377, 553–561. [DOI] [PubMed] [Google Scholar]
- Charles A, Pradhan AA, 2016. Delta-opioid receptors as targets for migraine therapy. Curr Opin Neurol 29, 314–319. [DOI] [PubMed] [Google Scholar]
- Chu LF, Angst MS, Clark D, 2008. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain 24, 479–496. [DOI] [PubMed] [Google Scholar]
- Codd EE, Carson JR, Colburn RW, Stone DJ, Van Besien CR, Zhang SP, Wade PR, Gallantine EL, Meert TF, Molino L, Pullan S, Razler CM, Dax SL, Flores CM, 2009. JNJ-20788560 [9-(8-azabicyclo[3.2.1]oct-3-ylidene)-9Hxanthene-3-carboxylic acid diethylamide], a selective delta opioid receptor agonist, is a potent and efficacious antihyperalgesic agent that does not produce respiratory depression, pharmacologic tolerance, or physical dependence. J.Pharmacol.Exp.Ther 329, 241–251. [DOI] [PubMed] [Google Scholar]
- DiCello JJ, Saito A, Rajasekhar P, Eriksson EM, McQuade RM, Nowell CJ, Sebastian BW, Fichna J, Veldhuis NA, Canals M, Bunnett NW, Carbone SE, Poole DP, 2018. Inflammation-associated changes in DOR expression and function in the mouse colon. Am J Physiol Gastrointest Liver Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener HC, Holle D, Solbach K, Gaul C, 2016. Medication-overuse headache: risk factors, pathophysiology and management. Nat Rev Neurol 12, 575–583. [DOI] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, Koch M, Kessler P, Hentsch D, Birling MC, Koutsourakis M, Vasseur L, Veinante P, Kieffer BL, Massotte D, 2015. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct 220, 677–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallantine EL, Meert TF, 2005. A comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Basic Clin.Pharmacol.Toxicol 97, 39–51. [DOI] [PubMed] [Google Scholar]
- Gendron L, Lucido AL, Mennicken F, O'Donnell D, Vincent JP, Stroh T, Beaudet A, 2006. Morphine and pain-related stimuli enhance cell surface availability of somatic delta-opioid receptors in rat dorsal root ganglia. J.Neurosci 26, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Xu ZZ, Gao H, He SQ, Ma GQ, Sun T, Wang LH, Zhang ZN, Lena I, Kitchen I, Elde R, Zimmer A, He C, Pei G, Bao L, Zhang X, 2005. Interaction with vesicle luminal protachykinin regulates surface expression of delta-opioid receptors and opioid analgesia. Cell 122, 619–631. [DOI] [PubMed] [Google Scholar]
- Hayhurst CJ, Durieux ME, 2016. Differential Opioid Tolerance and Opioid-induced Hyperalgesia: A Clinical Reality. Anesthesiology 124, 483–488. [DOI] [PubMed] [Google Scholar]
- Huang J, Lv Y, Fu Y, Ren L, Wang P, Liu B, Huang K, Bi J, 2015. Dynamic Regulation of Delta-Opioid Receptor in Rat Trigeminal Ganglion Neurons by Lipopolysaccharide-induced Acute Pulpitis. J Endod. [DOI] [PubMed] [Google Scholar]
- Kabli N, Cahill CM, 2007. Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 127, 84–93. [DOI] [PubMed] [Google Scholar]
- Katsarava Z, Limmroth V, Finke M, Diener HC, Fritsche G, 2003. Rates and predictors for relapse in medication overuse headache: a 1-year prospective study. Neurology 60, 1682–1683. [DOI] [PubMed] [Google Scholar]
- Katsarava Z, Schneeweiss S, Kurth T, Kroener U, Fritsche G, Eikermann A, Diener HC, Limmroth V, 2004. Incidence and predictors for chronicity of headache in patients with episodic migraine. Neurology 62, 788–790. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND, 2016. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent V, Bertran-Gonzalez J, Chieng BC, Balleine BW, 2014. delta-opioid and dopaminergic processes in accumbens shell modulate the cholinergic control of predictive learning and choice. J Neurosci 34, 1358–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent V, Morse AK, Balleine BW, 2015a. The role of opioid processes in reward and decision-making. Br J Pharmacol 172, 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent V, Wong FL, Balleine BW, 2015b. delta-Opioid receptors in the accumbens shell mediate the influence of both excitatory and inhibitory predictions on choice. Br J Pharmacol 172, 562–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Merrer J, Becker JA, Befort K, Kieffer BL, 2009. Reward processing by the opioid system in the brain. Physiol Rev. 89, 1379–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Merrer J, Plaza-Zabala A, Boca CD, Matifas A, Maldonado R, Kieffer BL, 2011. Deletion of the delta Opioid Receptor Gene Impairs Place Conditioning But Preserves Morphine Reinforcement. Biol.Psychiatry. [DOI] [PubMed] [Google Scholar]
- Le Merrer J, Rezai X, Scherrer G, Becker JA, Kieffer BL, 2013. Impaired hippocampus-dependent and facilitated striatum-dependent behaviors in mice lacking the delta opioid receptor. Neuropsychopharmacology 38, 1050–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limmroth V, Katsarava Z, Fritsche G, Przywara S, Diener HC, 2002. Features of medication overuse headache following overuse of different acute headache drugs. Neurology 59, 1011–1014. [DOI] [PubMed] [Google Scholar]
- Lutz PE, Kieffer BL, 2013. Opioid receptors: distinct roles in mood disorders. Trends Neurosci 36, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ, 1995. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 18, 22–29. [DOI] [PubMed] [Google Scholar]
- May A, Schulte LH, 2016. Chronic migraine: risk factors, mechanisms and treatment. Nat Rev Neurol 12, 455–464. [DOI] [PubMed] [Google Scholar]
- Minen MT, Begasse De Dhaem O, Kroon Van Diest A, Powers S, Schwedt TJ, Lipton R, Silbersweig D, 2016. Migraine and its psychiatric comorbidities. J Neurol Neurosurg Psychiatry 87, 741–749. [DOI] [PubMed] [Google Scholar]
- Monteith TS, Borsook D, 2014. Insights and advances in post-traumatic headache: research considerations. Curr Neurol Neurosci Rep 14, 428. [DOI] [PubMed] [Google Scholar]
- Moye LS, Novack ML, Tipton AF, Krishnan H, Pandey SC, Pradhan AA, 2018. The development of a mouse model of mTBI-induced post-traumatic migraine, and identification of the delta opioid receptor as a novel therapeutic target. Cephalalgia, 333102418777507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moye LS, Pradhan AA, 2017a. From blast to bench: A translational mini-review of posttraumatic headache. J Neurosci Res 95, 1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moye LS, Pradhan AAA, 2017b. Animal Model of Chronic Migraine-Associated Pain. Curr Protoc Neurosci 80, 9 60 61–69 60 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munksgaard SB, Bendtsen L, Jensen RH, 2013. Modulation of central sensitisation by detoxification in MOH: results of a 12-month detoxification study. Cephalalgia 33, 444–453. [DOI] [PubMed] [Google Scholar]
- Negus SS, Gatch MB, Mello NK, Zhang X, Rice K, 1998. Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J.Pharmacol.Exp.Ther 286, 362–375. [PubMed] [Google Scholar]
- Patwardhan AM, Berg KA, Akopain AN, Jeske NA, Gamper N, Clarke WP, Hargreaves KM, 2005. Bradykinin-induced functional competence and trafficking of the delta-opioid receptor in trigeminal nociceptors. J Neurosci 25, 8825–8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellissier LP, Pujol CN, Becker JA, Le Merrer J, 2016. Delta Opioid Receptors: Learning and Motivation. Handb Exp Pharmacol. [DOI] [PubMed] [Google Scholar]
- Pradhan A, Smith M, McGuire B, Evans C, Walwyn W, 2013. Chronic inflammatory injury results in increased coupling of delta opioid receptors to voltagegated Ca2+ channels. Mol Pain 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Befort K, Nozaki C, Gaveriaux-Ruff C, Kieffer BL, 2011. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci 32, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Smith ML, McGuire B, Tarash I, Evans CJ, Charles A, 2014a. Characterization of a novel model of chronic migraine. Pain 155, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Smith ML, Zyuzin J, Charles A, 2014b. delta-Opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. Br J Pharmacol 171,2375–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A, Evans C, Kieffer BL, 2010. Ligand-directed trafficking of the delta-opioid receptor in vivo: two paths toward analgesic tolerance. J.Neurosci. 30, 16459–16468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice FL, Xie JY, Albrecht PJ, Acker E, Bourgeois J, Navratilova E, Dodick DW, Porreca F, 2017. Anatomy and immunochemical characterization of the nonarterial peptidergic diffuse dural innervation of the rat and Rhesus monkey: Implications for functional regulation and treatment in migraine. Cephalalgia 37, 1350–1372. [DOI] [PubMed] [Google Scholar]
- Richards EM, Mathews DC, Luckenbaugh DA, Ionescu DF, Machado-Vieira R, Niciu MJ, Duncan WC, Nolan NM, Franco-Chaves JA, Hudzik T, Maciag C, Li S, Cross A, Smith MA, Zarate CA Jr., 2016. A randomized, placebocontrolled pilot trial of the delta opioid receptor agonist AZD2327 in anxious depression. Psychopharmacology (Berl) 233, 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeckel LA, Le Coz GM, Gaveriaux-Ruff C, Simonin F, 2016. Opioid-induced hyperalgesia: Cellular and molecular mechanisms. Neuroscience 338, 160–182. [DOI] [PubMed] [Google Scholar]
- Scher AI, Stewart WF, Ricci JA, Lipton RB, 2003. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 106, 81–89. [DOI] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, Kieffer BL, Basbaum AI, 2009. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137, 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwedt TJ, Alam A, Reed ML, Fanning KM, Munjal S, Buse DC, Dodick DW, Lipton RB, 2018. Factors associated with acute medication overuse in people with migraine: results from the 2017 migraine in America symptoms and treatment (MAST) study. J Headache Pain 19, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey A, Lucas S, Dikmen S, Temkin N, Bell KR, Brown A, Brunner R, Diaz-Arrastia R, Watanabe TK, Weintraub A, Hoffman JM, 2017. Natural History of Headache Five Years after Traumatic Brain Injury. J Neurotrauma 34, 1558–1564. [DOI] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Rice KC, Negus SS, 2005. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenz yl]-N,N-diethylbenzamide] and heroin. J.Pharmacol.Exp.Ther 314, 221–231. [DOI] [PubMed] [Google Scholar]
- Theeler B, Lucas S, Riechers RG, 2nd, Ruff RL, 2013. Post-traumatic headaches in civilians and military personnel: a comparative, clinical review. Headache 53, 881–900. [DOI] [PubMed] [Google Scholar]
- Thorlund K, Sun-Edelstein C, Druyts E, Kanters S, Ebrahim S, Bhambri R, Ramos E, Mills EJ, Lanteri-Minet M, Tepper S, 2016. Risk of medication overuse headache across classes of treatments for acute migraine. J Headache Pain 17, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton AF, Tarash I, McGuire B, Charles A, Pradhan AA, 2015. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL, 2012. Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol Psychiatry 71, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Sanchez A, Dripps IJ, Tipton AF, Akbari H, Akbari A, Jutkiewicz EM, Pradhan AA, 2018. Tolerance to high-internalizing delta opioid receptor agonist is critically mediated by arrestin 2. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Sanchez A, Segura L, Pradhan AA, 2016. The delta opioid receptor tool box. Neuroscience 338, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Tawfik VL, Corder G, Low SA, Francois A, Basbaum AI, Scherrer G, 2018. Functional Divergence of Delta and Mu Opioid Receptor Organization in CNS Pain Circuits. Neuron 98, 90–108.e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q, Lu YJ, Zhang ZN, He SQ, Zheng HC, Wu SX, Hokfelt TG, Bao L, Zhang X, 2010. Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. Proc.Natl.Acad.Sci.U.S.A 107, 13117–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bao L, Arvidsson U, Elde R, Hokfelt T, 1998. Localization and regulation of the delta-opioid receptor in dorsal root ganglia and spinal cord of the rat and monkey: evidence for association with the membrane of large dense-core vesicles. Neuroscience 82, 1225–1242. [DOI] [PubMed] [Google Scholar]
- Zidverc-Trajkovic JJ, Pekmezovic T, Jovanovic Z, Pavlovic A, Mijajlovic M, Radojicic A, Sternic N, 2018. Long-term predictors of remission in patients treated for medication-overuse headache at a specialized headache center: A prospective cohort study. Cephalalgia 38, 265–273. [DOI] [PubMed] [Google Scholar]
- Zohar O, Schreiber S, Getslev V, Schwartz JP, Mullins PG, Pick CG, 2003. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience 118, 949–955. [DOI] [PubMed] [Google Scholar]