

Abstract
Mutation at a single amino acid alters the isoprene donor specificity of prenyltransferases involved in the modification of ribosomally synthesized and post-translationally modified peptides (RiPPs). While most characterized RiPP prenyltransferases carry out the regiospecific transfer of C5 dimethylallyl donor to the side chain atoms on macrocyclic acceptor substrates, the elucidation of the cyanobactin natural product piricyclamide 70005E1 identifies an O-geranyl modification on Tyr, a reaction with little prior biochemical precedence. Reconstitution and kinetic studies of the presumptive geranyltransferase PirF shows that the enzyme utilizes a C10 donor, with no C5 transferase activity. The crystal structure of PirF reveals a single amino acid difference in the vicinity of the isoprene-binding pocket, relative to the C5 utilizing enzymes. Remarkably, only a single amino acid mutation is necessary to completely switch the donor specificity from a C5 to a C10 prenyltransferase, and vice versa. Lastly, we demonstrate that these enzymes may be used for the chemospecific attachment of C5 or C10 lipid groups on lanthipeptides, an unrelated class of RiPP natural products. These studies represent a rare example where prenyl donor specificity can be discretely altered, which expands the arsenal of synthetic biology tools for tuning biological activities of peptide natural products.
Graphical Abstract
One of the overarching goals of synthetic biology is to devise enzymatic platforms that may aid in the production of small molecules with pharmaceutical potential.1–4 Catalysts that affect such functionality on peptides are of particular interest, given the versatility with which large libraries of substrates with disparate chemically structures can be synthesized.5–6 Enzymes involved in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs) provide a rich source for such activities, particularly so given their inherent tolerance for a diverse range of substrates.7 For most RiPP enzymes, recognition of the peptide substrate occurs in a region (leader sequence) that is distinct from the site where chemistry occurs (core sequence). As the leader sequence is excised following the installation of the necessary modifications, the final product in RiPP pathways do not retain any necessary signature sequences. This demarcation of recognition sequences away from the site of modification results in the broad tolerance for substrates and protects against processing of non-cognate substrates. Some enzymes that catalyze tailoring modifications on RiPP substrates do not require the leader sequence.8–9
The F family of cyanobacterial prenyltransferases catalyzes the prenylation on Ser, Thr, or Trp residues situated in the cyanobactin class of RiPP substrates (Figure 1A).10 Unlike most small molecule prenyltransferases, the F enzymes catalyze electrophilic alkylation only on residues within linear or cyclic peptide substrates and not on free amino acids.11 We previously characterized PagF, a Tyr prenyltransferase from the prenylagaramide biosynthetic pathway of Oscillatoria agardhii.12 The PagF crystal structure revealed a truncated ABBA-fold that is only competent for catalysis upon binding of a suitably large substrate that can insulate the hydrophobic active site against solvent. Homologous genes from other cyanobactin pathways demonstrate diverse substrate scope, as LynF from the aestuaramide pathway11 (Lyngbya aestuarii) carries out reverse O-prenylation on Tyr, TruF1 from the trunkamide pathway10 (Procholoron sp.) catalyzes reverse O-prenylation on Thr/Ser, and KgpF from the kawaguchipeptin pathway13–14 (Microcystis aeruginosa NIES-88) catalyzes C-prenylation on Trp. These studies establish F enzymes as general, versatile tool for peptide lipidation.
Figure 1.

(A) Chemical structures of various prenylated cyanobactins, along with the corrsponding enzyme that installs the isoprene moiety. The chemical structures of DMAPP (C5 donor) and GPP (C10 donor) are also shown. (B, C) Kinetic curves showing the isoprene transferase activities of PagF and PirF using a synthetic tri-Tyr peptide as a substrate.
To date, all characterize homologs catalyze the transfer of 5-carbon dimethylallyl skeleton from DMAPP (dimethylallyl pyrophosphate), and cannot transfer large isoprenes such as (C10) geranyl or (C15) farnesyl units from GPP (geranyl pyrophosphate) or FPP (farnesyl pyrophosphate), respectively (Figure 1B, SI Table S2).11 Recent genome mining studies identified the piricyclamide cyclic peptides from Microcystis aeruginosa PCC7005.15 Heterologous expression of the cluster produced products (piricyclamides 7005E1 and E3) that were 136 Da larger than that of the expected cyclic product. Tandem mass-spectrometric analysis of fragment product ion spectra was consistent with either the loss of two dimethyallyl or one geranyl group from the parent compound. Subsequent structural characterization of piricyclamide 7005E1 demonstrated that the final product is a cyclic peptide (cyc-MSGVDYYNP) that is O-geranylated on Tyr, and in vitro reconstitution demonstrated that PirF is a geranyltransferase that can modify the hydroxyl oxygen of Tyr in peptide substrates.16
PirF showed activity with an artificial tri-Tyr substrate with only GPP (C10) as the alkyl donor, whereas DMAPP (C5) failed to modify the substrate (Figure 1C; Table 1). Kinetic characterization using this tri-Tyr substrate showed that the PirF geranyltransferase catalyzed C10 transfer from GPP with a (kcat/KM) value of 50.5 mM−1 min−1 but showed no activity using DMAPP (Table 1). Conversely, the PagF prenyltransferase catalyzed C5 transfer from DMAPP to the same substrate with a kcat/KM of 27.0 mM−1 min−1 but demonstrated no activity with GPP. These data indicate a strict specificity for the F enzymes for their respective donors, with no tolerance for alternative allylic substrates.
Table 1.
Kinetic parameters for F enzyme-catalyzed prenylation using a tri-Tyr substrate.
| Enzyme | Donor | kcat (min−1) | KM (mM) |
kcat/KM min−1 mM−1 |
|---|---|---|---|---|
| PagF wt | DMAPP GPP |
7.30 ± 0.14 — a |
0.27 ± 0.03 — |
27.0 — |
| PirF wt | DMAPP GPP |
— 23.2 ± 1.1 |
— 0.46 ± 0.08 |
— 50.5 |
| F222A PagF |
DMAPP GPP |
2.54 ± 0.18 7.31 ± 0.26 |
1.10 ± 0.24 0.19 ± 0.03 |
2.31 38.5 |
| F222G PagF |
DMAPP GPP |
1.15 ± 0.09 27.5 ± 1.1 |
1.25 ± 0.30 0.37 ± 0.06 |
0.92 74.3 |
—, No reaction.
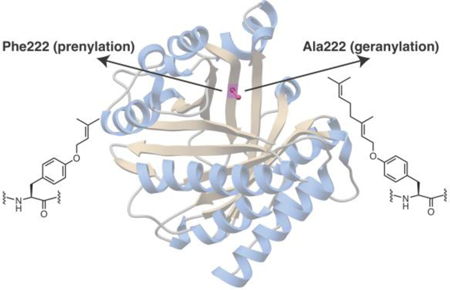
The preference of PirF for GPP as the alkyl donor is intriguing given that the enzyme shows greater than 70% identity in primary sequence to the well characterized F enzymes like PagF, LynF, AcyF, and others, all of which use DMAPP as the allylic donor, and show no activity using GPP. Moreover, a sequence alignment shows that all of the residues in PagF that are implicated in catalysis are conserved in PirF (SI Figure S1). In order to elucidate the molecular rationale for this altered donor preference, we determined the 2.3 Å resolution crystal structure of PirF that was expressed and purified from an Escherichia coli heterologous system. The structure recapitulates the truncated ABBA-like prenyltransferase fold, consisting of 10 antiparallel β-strands surrounded by 10 α-helices, previously observed in the PagF structures.12 A comparison of the PirF structure with that of PagF shows that nearly all residues in the active site are highly conserved with one notable exception. In the PagF-DMSPP co-crystal structures, Phe222 is located at the apex of the active site cavity, where it forms part of the hydrophobic tunnel necessary to stabilize the allylic carbocation against solvent quenching. In PirF, the equivalent residue is Gly221, and this substitution results in the enlargement of the binding site (Figure 2A, 2B, SI Figure S2).
Figure 2.

A view along the hydrophobic active site tunnel of the (A) DMAPP-dependent PagF, as compared that of the (B) GPP-dependent PirF. Note that the most significant difference between the two enzymes is that F222 in the PagF active site is altered to a G221 in PirF. Active site residues identified by prior mutational analysis are shown as sticks.
We next sought to test if indeed this single residue could serve as the selectivity switch for F enzymes in selecting for a C5 vs. C10 donor. To that end, we generated the Phe222→Ala and Gly variants of PagF, and tested each of these for prenylation on the tri-Tyr substrate using either DMAPP or GPP as the isoprene donor. Surprisingly, while wild-type PagF showed no activity using GPP, both the Phe222→Ala and Phe222→Gly variants showed robust geranyltransferase activity (Figure 3A; Table 1).
Figure 3.

(A) Extracted ion chromatograms from LC/MS analysis of F enzyme assays using a tri-Tyr substrate. The top trace is for a control tri-Tyr substrate. The data show the isoprene donor preference of PagF for DMAPP, PirF for GPP, and Phe222→Ala PagF for GPP. Green trace shows single and double prenylation (1.5:2), orange and purple traces show 100% double geranylation. (B) Kinetic curves showing the activities of Phe222→Gly PagF and Phe222→Ala PagF using either DMAPP or GPP as the isoprene donor. Note that this single residue change resulted in a variant in which the donor specificity is switched completely. (C) Structure of Phe222→Ala PagF superimposed on a difference Fourier electron density map, calculated with coefficient |Fobs – Fcalc| using phases from the final model with the coordinates of GPP removed prior to a round of simulated annealing refinement.
Kinetic characterization of both variants showed that the enzymes catalyzed geranyl transfer with (kcat/KM) value of 38.5 mM−1 min−1 and 74.3 mM−1 min−1, for the Ala222 and Gly222 PagF variants (Figure 3B; Table 1). Of note, these were not simple gain of function variants, as both Ala222 and Gly222 PagF showed a 10 to 20-fold decrease in C5 isoprene transferase activity, relative to the wild-type (kcat/KM values of 2.31 mM−1 min−1 and 0.92 mM−1 min−1, respectively) (Table 1). Hence, the Phe222→Ala/Gly mutations in PagF were sufficient to prompt a complete switch in isoprene donor specificity.
To understand the basis for the donor specificity switch induced by this single amino acid mutation at Phe222, we focused structural biological efforts on the two PagF variants at this position. While the Phe222→Gly variant failed to crystallize, we obtained crystals of Phe222→Ala PagF in complex with GPP and MgCl2 that diffracted to 1.85 Å resolution. Clear and continuous electron density corresponding to the isoprene can be visualized in the active site at the isoprene donor-binding site (Figure 3C). The C10 moiety of GPP extends out and occupies the volume that is created by the Phe222→Ala substitution (SI Figure S4). A superposition of the structure of this variant with that of wild-type PagF bound to DMSPP and the cyclic[INPYLYP] peptide shows that the additional 5-carbon skeleton can be accommodated in the variant without significantly affecting substrate binding (SI Figure S2, S4). Presumably, the Gly222 variant would provide even lesser steric hindrance, explaining the greater catalytic efficiency of this variant using GPP as the allylic donor.
We, and others, have shown that the F enzymes are broadly tolerant of acceptor substrates, provided that the size of the substrate is sufficient to encapsulate the isoprene donor-binding site and provide a solvent-excluded hydrophobic environment for effective prenyl transfer.11–12, 14 Studies of PagF showed that an N-terminal Tyr-aliphatic dipeptide motif was sufficient for substrate recognition, and two physiologically relevant non-cognate peptides bearing this sequence were both substrates for the enzyme. Given the preference of these enzymes for cyclic peptide substrates, it seemed plausible that they may be utilized to attach lipid groups to an unrelated class of RiPP natural products, namely lanthipeptides.17
The class II lanthipeptide synthase ProcM is a catalytically promiscuous catalyst that functions on over 30 different substrates, and one of which (prochlorosin 1.1; Proc1.1) is processed to produce a bicyclic peptide with a Phe-Phe motif at the amino terminus.18–19 The first Phe in the Proc1.1 precursor was changed to a Tyr, and incubation of this variant precursor with ProcM resulted in a species containing both thioether rings (SI Figure S5). Proteolytic cleavage of the leader sequence yielded a lanthipeptide product (YF-Proc1.1) that contains an N-terminal Tyr-Phe motif. We then tested this lanthipeptide as a substrate for both wild-type and Phe222→Ala PagF. Notably, incubation of YF-Proc1.1 with wild-type PagF, GPP and MgCl2 produced a isoprenylated lanthipeptide, and incubation with either PirF or Phe222→Ala PagF, GPP, and MgCl2 yielded a geranylated lanthipeptide (Figure 4, SI Figure S6). These data show that F family enzymes can function on products from other RiPP pathways and sets the stage for future efforts aimed at RiPP diversification.
Figure 4.

(A) Chemical structure of the YF-Proc1.1 variant, and YF-Proc1.1 that is prenylated using either wild-type or Phe222→Ala variant PagF or PirF as the catalyst. (B) Extracted ion chromatograms from LC/MS analysis of F enzyme assays using the lanthipeptide variants YF-Proc1.1 as a substrate.
Prior studies on small molecule secondary metabolite prenyltransferases, as well as the cyanobacterial F family peptide prenyltransferases demonstrate that these enzymes can function on a diverse range of acceptor substrates, but show strict specificity for the prenyl donor.20–24 The A. terreus aromatic prenyltransferase is tolerant for a range of donors ranging from C5 to C20 but may be too permissive for biotechnology usage.25 The F enzymes present examples of biocatalysts that are strictly tolerant for the allylic donor but, in which donor specificity may be altered entirely. Our determination of the molecular rationale for the C10 transferase activity of PirF, and subsequent switch in the specificity of PagF from a selective DMAPP utilizing C5 transferase to a selective GPP-dependent C10 transferase presents a rare example of structure-guided donor selectivity reversal. These studies add to the growing toolkit of enzymatic catalysts that can be used for the production of peptide-based small molecule libraries with more drug-like properties.
Supplementary Material
Acknowledgements
We thank Prof. Wilfred van der Donk and members of the van der Donk laboratory with assistance in production of the procholorosin lanthipeptide.
Funding Sources
Supported by grants from NIH (GM122521 and GM102602 to E.W.S.) and (GM079038 to S.K.N.). P.E. is supported in part by the Carter Fellowship from the Department of Biochemistry, UIUC. M.M. is supported by JSPS Overseas Research Fellowships (Japan), and Uehara Memorial Foundation.
Footnotes
ASSOCIATED CONTENT
Supporting Information. A PDF file of experimental methods and additional figures. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Erb TJ; Jones PR; Bar-Even A, Current opinion in chemical biology 2017, 37, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).O’Connor SE, Annu Rev Genet 2015, 49, 71–94. [DOI] [PubMed] [Google Scholar]
- (3).Tan GY; Deng Z; Liu T, F1000Res 2015, 4. [DOI] [PMC free article] [PubMed]
- (4).Walker MC; Chang MC, Chem Soc Rev 2014, 43 (18), 6527–36. [DOI] [PubMed] [Google Scholar]
- (5).Ruffner DE; Schmidt EW; Heemstra JR, ACS synthetic biology 2015, 4 (4), 482–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang L; Bulaj G, Current medicinal chemistry 2012, 19 (11), 1602–18. [DOI] [PubMed] [Google Scholar]
- (7).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian KD; Fischbach MA; Garavelli JS; Goransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Muller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJ; Rebuffat S; Ross RP; Sahl HG; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Sussmuth RD; Tagg JR; Tang GL; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA, Natural product reports 2013, 30 (1), 108–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Funk MA; van der Donk WA, Acc Chem Res 2017, 50 (7), 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhu S; Hegemann JD; Fage CD; Zimmermann M; Xie X; Linne U; Marahiel MA, J Biol Chem 2016, 291 (26), 13662–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Donia MS; Ravel J; Schmidt EW, Nat Chem Biol 2008, 4 (6), 341–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McIntosh JA; Donia MS; Nair SK; Schmidt EW, J Am Chem Soc 2011, 133 (34), 13698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hao Y; Pierce E; Roe D; Morita M; McIntosh JA; Agarwal V; Cheatham TE 3rd; Schmidt EW; Nair SK, Proceedings of the National Academy of Sciences of the United States of America 2016, 113 (49), 14037–14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Okada M; Sugita T; Akita K; Nakashima Y; Tian T; Li C; Mori T; Abe I, Org Biomol Chem 2016, 14 (40), 9639–9644. [DOI] [PubMed] [Google Scholar]
- (14).Parajuli A; Kwak DH; Dalponte L; Leikoski N; Galica T; Umeobika U; Trembleau L; Bent A; Sivonen K; Wahlsten M; Wang H; Rizzi E; De Bellis G; Naismith J; Jaspars M; Liu X; Houssen W; Fewer DP, Angew Chem Int Ed Engl 2016, 55 (11), 3596–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Leikoski N; Fewer DP; Jokela J; Alakoski P; Wahlsten M; Sivonen K, PLoS One 2012, 7 (8), e43002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Morita M; Hao Y; Jokela JK; Sardar D; Lin Z; Sivonen K; Nair SK; Schmidt EW, J Am Chem Soc 2018, 140 (19), 6044–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Repka LM; Chekan JR; Nair SK; van der Donk WA, Chem Rev 2017, 117 (8), 5457–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li B; Sher D; Kelly L; Shi Y; Huang K; Knerr PJ; Joewono I; Rusch D; Chisholm SW; van der Donk WA, Proceedings of the National Academy of Sciences of the United States of America 2010, 107 (23), 10430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mukherjee S; van der Donk WA, J Am Chem Soc 2014, 136 (29), 10450–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chooi YH; Wang P; Fang J; Li Y; Wu K; Wang P; Tang Y, J Am Chem Soc 2012, 134 (22), 9428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Heide L, Current opinion in chemical biology 2009, 13 (2), 171–9. [DOI] [PubMed] [Google Scholar]
- (22).Tanner ME, Natural product reports 2015, 32 (1), 88–101. [DOI] [PubMed] [Google Scholar]
- (23).Tello M; Kuzuyama T; Heide L; Noel JP; Richard SB, Cell Mol Life Sci 2008, 65 (10), 1459–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Winkelblech J; Fan A; Li SM, Applied microbiology and biotechnology 2015, 99 (18), 7379–97. [DOI] [PubMed] [Google Scholar]
- (25).Chen R; Gao B; Liu X; Ruan F; Zhang Y; Lou J; Feng K; Wunsch C; Li SM; Dai J; Sun F, Nat Chem Biol 2017, 13 (2), 226–234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.