

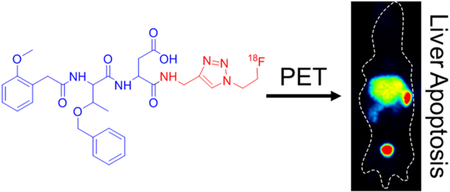
Abstract
Quantitative imaging of apoptosis in vivo could enable real-time monitoring of acute cell death pathologies such as traumatic brain injury, as well as the efficacy and safety of cancer therapy. Here, we describe the development and validation of F-18-labeled caspase-3 substrates for PET/CT imaging of apoptosis. Preliminary studies identified the O-benzylthreonine-containing substrate 2MP-TbD-AFC as a highly caspase 3-selective and cell-permeable fluorescent reporter. This lead compound was converted into the radiotracer [18F]-TBD, which was obtained at 10% decay-corrected yields with molar activities up to 149 GBq/μmol on an automated radiosynthesis platform. [18F]-TBD accumulated in ovarian cancer cells in a caspase- and cisplatin-dependent fashion. PET imaging of a Jo2-induced hepatotoxicity model showed a significant increase in [18F]-TBD signal in the livers of Jo2-treated mice compared to controls, driven through a reduction in hepatobiliary clearance. A chemical control tracer that could not be cleaved by caspase 3 showed no change in liver accumulation after induction of hepatocyte apoptosis. Our data demonstrate that [18F]-TBD provides an immediate pharmacodynamic readout of liver apoptosis in mice by dynamic PET/CT and suggest that [18F]-TBD could be used to interrogate apoptosis in other disease states.
Graphical Abstract
INTRODUCTION
Control of programmed cell death is essential for the development and maintenance of multicellular organisms with apoptosis playing an important role in organ development, immune system function,and tissue homeostasis.1 Aberrant apoptosis is associated with cardiovascular, neurological, and autoimmune diseases along with many cancers.2 Apoptosis is triggered by external receptor-mediated signaling events (extrinsic) or DNA damage (intrinsic) and is characterized by nuclear fragmentation, chromosomal fragmentation, and membrane blebbing.3 Both pathways activate regulatory caspases, which in turn converge to activate the executioner caspases, caspase-3, −6, and −7, which then process hundreds of intracellular substrates and commit the cell to apoptotic death.4,5 Caspases recognize a four amino acid sequence, denoted P4–P3–P2–1 with cleavage taking place after the C-terminal residue (P1), which is almost invariably aspartic acid.6 For caspase-3, the preferred epitope is Asp-Glu-Val-Asp (DEVD). While this tetrapeptide shows excellent caspase-3 activity in vitro, it has poor cell permeability due to high molecular weight and negative charge.7 Despite the challenges of developing cell permeable, potent caspase-3-selective compounds, the central role of caspase-3 in apoptosis makes it a natural target for noninvasive molecular imaging of cell death.
Molecular imaging of apoptosis can provide an immediate pharmacodynamic readout of therapeutic efficacy in cancer and a diagnostic tool to identify aberrant apoptotic cell death in acute pathologies such as traumatic brain injury.8 Previous efforts to image caspase-3 activity have struggled with low sensitivity, owing primarily to the intrinsic limitation to imaging signal resulting from stoichiometric (1-to-1) inhibition of caspase enzymes.9 In contrast to radiolabeled caspase inhibitors, e.g.,the isatin sulfonamides,10 the accumulation of a substrate-based radiotracer is not intrinsically limited by the number of active caspase-3 molecules in the cell. Therefore, this offers the potential for higher signal accumulation and greater sensitivity, even in the presence of low levels of active caspase-3. This is particularly important for visualizing apoptosis in tumors where only a fraction of the cells may be undergoing apoptosis at any given time.11 Another well-studied tracer, radiolabeled Annexin-V, binds to phosphatidylserine on the surface of apoptotic cells and has also been shown to undergo selective liver uptake in the Jo2 hepatotoxicity model.12–14 However, phosphatidylserine is also accessible in permeabilized necrotic cells making it difficult to distinguish apoptosis from necrosis with this tracer. This lack of selectivity prevents differentiation of dying tissue and dead tissue with Annexin-based tracers, and limits the ability of the imaging procedure to identify “sterile” apoptotic cell death from immunogenic necrotic cell death.
We hypothesized that a minimized substrate-based probe based on existing caspase-3 inhibitors would provide a starting point for the design of a cell-permeable radiotracer. Here we describe the design and validation of low-molecular-weight caspase-3 substrates as PET imaging probes for apoptosis. Structure optimization based on the M808 preclinical caspase- 3 inhibitor15 and informed by combinatorial studies of caspase-3 inhibitors or substrates16,17 led us to synthesize two lead compounds with either valine (V) or threonine O-benzyl ester (Tb) at the P2 position and capped at the N-terminus with a 2-methoxy phenyl group (2MP-VD and 2MP-TbD). The fluorescent probe 2MP-TbD-AFC showed improved caspase-3 specificity and similar cellular uptake relative to the canonical caspase-3 substrate, Ac-DEVD-AFC. Structural analysis of the caspase-3 binding site led to the synthesis of a series of fluorinated derivatives and the identification of the optimal linkage between the minimized dipeptide and the pendant fluorine. Using a newly described radiochemical methodology based on the [18F]-fluoroethylazide synthon, we generated the radiotracer, [18F]-16 ([18F]-TBd), at high yield, purity, and molar activity on an automated synthesis platform. Dynamic PET/CT showed that [18F]-TBD was preferentially accumulated and retained in the liver of a Jo2-induced mouse model of hepatic apoptosis. We conclude that [18F]-TBD is a promising substrate-based radiotracer for PET imaging of apoptosis in vivo.
RESULTS
Design and Characterization of Minimized Caspase-3 Fluorescent Substrates.
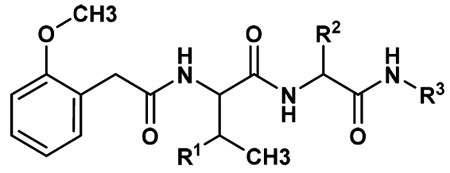
We synthesized two fluorescent substrates based on the irreversible caspase-3 inhibitor M808 (Figure S1).15 M808 is a valine-aspartic acid (VD) dipeptide capped at the N-terminal amine with an iodinated 2-methoxyphenyl (2MP) group and modified with a fluoromethylketone (FMK) at the C-terminus. Binding of M808 to the caspase-3 active site results in the formation of a covalent bond between the active site sulfhydryl and the FMK. To generate a caspase-3 substrate the FMK was replaced with 7-amino-4-(trifluoromethyl)coumarin (AFC). While AFC is essentially nonfluorescent when conjugated through its aromatic amine, proteolytic cleavage C-terminal to the P1 aspartic acid (red arrows, Figure 1a) liberates fluorescent AFC allowing correlation of fluorescent signal with caspase-3 activity. Initial lead compounds, 2MP-VD-AFC (1) and 2MP-TbD-AFC (2),were readily synthesized from t-butyl-protected dipeptide precursors and purified by RP-HPLC (Figure 1a, Scheme S1). Although the VD dipeptide was employed in the M808 inhibitor, previous combinatorial studies16 suggested that a valine to Tb substitution would improve substrate activity. Both probes were tested for caspase 3 activity, selectivity, and activity against apoptotic cells in culture.
Figure 1.

Selectivity and intracellular activity of fluorescent caspase-3 substrates 1 and 2. (a) Structures of 1 and 2. The red arrows indicate the peptide bond cleaved by caspase-3. (b) Caspase specificity of 1, 2, and Ac-DEVD-AFC. Substrate concentrations equal to 3 × Km were cleaved in vitro by 1 U each of a panel of caspase enzymes for 2 h. Data for each probe was normalized to the caspase-3 activity for that probe. Bars represent mean ± standard deviation, n = 3. Substrate 2 showed the highest selectivity for caspase-3. (c) OVCAR-5 (left panel) and OVCAR-8 ovarian cancer cells (right panel) were pretreated for 48 h with 20 μM cisplatin, 20 μM cisplatin and 20 μM Z-VAD-FMK, or PBS. Then, cells were treated with 1 μ/mL Annexin-V-AF568 (magenta) and 100 μM of Ac-DEVD-AFC, 1, or 2 (green) for 2 h at 37 °C followed by confocal microscopy. In contrast to 1, uptake and cleavage of compound 2 was sharply inhibited following caspase-3 inactivation by Z-VAD-FMK. Scale bar 50 μm.
Substrates 1, 2, and the canonical caspase-3/7 substrate Ac-DEVD-AFC were incubated with recombinant activated caspase-3 and fluorescence was monitored at λEx/λEm 380/500 nm. In this assay, substrate 2 showed a 4-fold higher cleavage by caspase-3 than substrate 1, suggesting that the Tb group positively influenced substrate activity. Kinetic analysis revealed that this enhancement was due almost entirely to an increase in kcat with little change in Km. As expected, both substrates showed significantly reduced activity relative to Ac-DEVD-AFC, due to a combination of a 100-fold lower Km and 10-fold higher kcat for the full-length peptide substrate (Table S1).
We next sought to determine the caspase specificity of 1 and 2 relative to Ac-DEVD-AFC. All substrates were incubated with recombinant caspases at 3 × Km to directly compare specificity at a constant point along the substrate vs velocity curve. Compound 2 showed excellent caspase-3 selectivity with minimal off-target activity observed for caspases 1 and 8. Compound 1 showed more modest caspase-3 selectivity with significant off-target caspase-8 activity. Interestingly, Ac-DEVD-AFC showed high substrate activity with caspases 3 and 7 along with off-target activity with caspases 8 and 10 (Figure 1b). These results suggested that the higher caspase-3 activity of 2 relative to 1 is accompanied by increased caspase-3 selectivity.
To demonstrate the cell permeability and caspase-3 substrate activity in living cells, we tested all three probes in a cell culture model of cisplatin-mediated apoptosis. We chose OVCAR-5 and OVCAR-8, two ovarian cancer cell lines with different sensitivities to cisplatin. Following treatment with cisplatin for 48 h, both lines show robust activation of caspase-3 and apoptotic phenotype in culture (Figure S2). Confocal microscopy after 120 min of treatment with 1, 2, or Ac-DEVD-AFC showed accumulation of fluorescent signal in cisplatin-treated OVCAR-5 and OVCAR-8 cells (Figure 1c). As AFC is nonfluorescent in the intact probe, the appearance of fluorescent signal within the cell is consistent with substrate cleavage and subsequent trapping of the free AFC fluorophore. Co-staining with Annexin-V confirmed extracellular exposure of phosphatidylserine, consistent with early apoptosis.18 Addition of the pan-caspase inhibitor Z-VAD-FMK reduced fluorescence of Ac-DEVD-AFC and 2 to background levels indicating that fluorescence accumulation is dependent on caspase activity. In contrast, Z-VAD-FMK treatment had less effect on the fluorescence of 1. This suggests that the combination of low substrate activity and poor target specificity of 1 may result in non-caspase-dependent cleavage and increased nonspecific background signal. Since cisplatin treatment in the presence of caspase inhibitors can trigger cellular necrosis, it is possible that 1 is cleaved by necrosis-related enzymes.19 We observed comparable or higher intracellular fluorescence with compound 2 compared to Ac-DEVD-AFC despite a >1000-fold reduction in catalytic efficiency. We suspect that this is due to improved cell permeability resulting from significant reduction in total probe mass, increased hydrophobicity from the 2MP and O-benzylthreonine groups, and the removal of two formal negative charges.20
Evaluation of Substrate-Based Radiotracer Candidates.
Given the encouraging performance of fluorescent substrates 1 and 2, we undertook synthesis of a series of fluorinated analogues. The goal of these studies was to evaluate the substrate activity of each analogue and to select the most potent candidates for fluorine-18 labeling and PET/CT. Analogues were prepared by conjugation of fluoroethylazide to a series of alkyne-modified 2MP-TbD and 2MP-VD peptides through copper-catalyzed azide–alkyne Husigen cycloaddition, known as “click chemistry”.21 This strategy was chosen to facilitate rapid translation of the most promising fluorinated variants to F-18-labeled PET tracers using well-established and automated [18F]-fluoroethylazide radiochemical protocols.22,23 Candidate fluorinated substrates were synthesized as described in Scheme S2 to produce compounds 3–21. A comparison of the physiochemical properties of all substrates is shown in Table S2. Substrates were assayed in vitro for caspase-3-mediated hydrolysis using analytical HPLC and the results reported as means ± standard deviation (n = 3, Table 1).
Table 1.
Caspase-3 Activity of Fluorinated Substrates Based on Compound 1
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Hydrolyzed Product(ng) |
| 3 | CH3 | Asp | 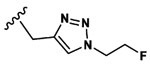 |
32.6±1.0 |
| 4 | CH3 | Asp | 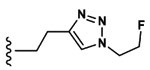 |
7.0±1.1 |
| 5 | CH3 | Asp | 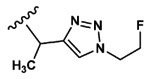 |
3.3±1.0 |
| 6 | CH3 | Asp | 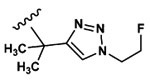 |
8.3±5.0 |
| 7 | CH3 | Asp | 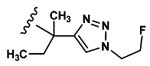 |
0 |
| 8 | CH3 | Asp | 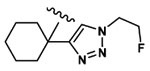 |
0 |
| 9 | CH3 | Asp | 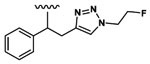 |
4.2±0.7 |
| 10 | CH3 | Asp | 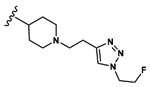 |
0 |
| 11 | CH3 | Asp | 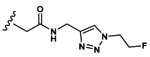 |
27.3±0.6 |
| 12 | CH3 | Asp | 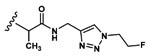 |
11.0±1.2 |
| 13 | CH3 | Asp | 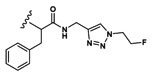 |
51.7±3.8 |
| 14 | CH3 | Asp | 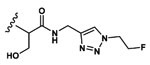 |
22.7±7.8 |
| 15 |  |
Asp | 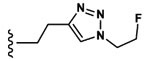 |
450±9.3 |
| 16 | 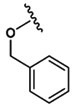 |
Asp | 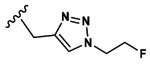 |
471±4.1 |
| 17 | 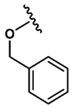 |
Asp(OMe) | 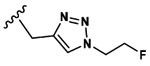 |
0.7±0.7 |
| 18 | 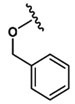 |
Ala | 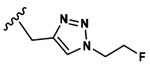 |
0 |
| 19 | 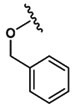 |
Asp | 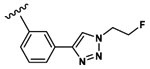 |
66.4±3.9 |
| 20 |  |
Asp |  |
404±10.8 |
| 21 |  |
Asp |  |
321±2.1 |
Several structure—activity relationships emerged from these studies. First, variation of the linker (R3) in the series of compounds based on 1 (3–14) shows that incorporation of phenylalanine between the scissile amide bond and the triazole resulted in the highest substrate activity (13). However, this large linker could be effectively replaced with a single methylene group leading to only a 40% reduction in activity (3). Interestingly, adding additional methylene groups (4) or substituting the single methylene with methyl (5), dimethyl (6), cyclohexyl (8), or phenyl (9) groups dramatically reduced substrate activity, in some cases below the assay’s limit of detection. Substitution of Tb for V elicited a 14-fold increase in caspase-3 activity in 16 relative to 3, a greater improvement than the 4-fold enhancement observed between 1 and 2. As was the case with the fluorescent substrates, this increase was largely driven through kcat rather than Km (Table S3). Based on the activity of the AFC-conjugated fluorescent substrates, we replaced the minimal aliphatic linker in 16 with two aniline moieties. Interestingly, the para-substituted aniline substrate 20 showed comparable activity to 16 while the activity of the meta-substituted analog 19 was approximately 6-fold lower. Replacement of the aspartic acid residue with alanine produced the uncleavable probe, 18, which was used as a chemical control in the PET/CT experiments. It has been shown that 1,2,3 triazoles can serve as rigid substitutes for amide bonds and have been used extensively in peptidomimetic design.24 Indeed, the linker in 16 may function as a glycine isostere which is supported by the nearly identical caspase-3 substrate activities of compounds 3 and 11 and the 30% reduction in substrate activity when the triazole is eliminated in 21.
The most promising fluorinated candidates were incubated with a panel of caspases to determine caspase specificity (Figure 2). Given a constant triazole linker, Tb in the P2 position (16) substantially improves caspase-3 specificity relative to valine (3). Addition of a second methylene group in the linker region (15) increases off-target caspase-8 activity by a factor of 4. When valine is present in the P2 position, specificity is reduced by replacement of the single methylene in the linker with glycine or phenylalanine (11 and 13). Substitution of the para-aniline in the linker (20) increased caspase-7 hydrolysis by a factor of 2. These data confirm the high selectivity of 16 for caspase-3 despite significant overlap of substrate sequence preference within the caspase family. This may also enhance probe selectivity for apoptotic versus necrotic cell death as cathepsin and calpain proteases, which play a key role in programmed necrosis, have distinct substrate profiles and do not cleave the amide bond c-terminal to aspartic acid.25,26 The replacement of valine at the P2 position with the bulky, unnatural amino acid O-benzylthreonine may also enhance apoptotic selectivity by precluding off-target cleavage at this site (see also Figure 1).
Figure 2.

Caspase-3 specificity of radiotracer candidates. 1 mM of 3 (a), 11 (b), 13 (c), 15 (d), 16 (e), or 20 (f) was incubated with a panel of caspase enzymes at 37 °C for 2 h. Cleavage products were measured by HPLC and peak areas were compared with a standard curve of 2MP-VD-OH or 2MP-TbD-OH to calculate the yield of hydrolyzed product in ng. Bars represent mean ± standard deviation, n = 3.
Structural Basis for the Caspase-3 Substrate Activity and Selectivity of Compound 16.
To gain structural insights into the activity and selectivity of 16, we docked selected caspase-3 substrates into the binding pocket of a human caspase-3 crystal structure17 (PDB ID: 1RHQ). Of particular interest were the interactions between the P2 position of the substrate and the S2 pocket of the enzyme. In caspase-3, the S2 pocket comprises 3 aromatic ring-containing residues (Y204, W206, and F256). As expected, docking results showed that the valine side chain of compounds 1 and 3–14 showed similar interactions with the S2 pocket as published crystal structures. In compounds 2 and 15–20, which contained the Tb group at P2, docked poses revealed a bent conformation and the presence of π–π interactions between the O-benzyl ring and the phenolic F256 side chain (Figure 3a).
Figure 3.

Active site modeling of caspase-3 with substrates. (a) Active site of caspase-3 with pharmacophore docked pose of 16 in the closed configuration. Green represents hydrophobic and magenta hydrophilic surfaces. (b) Flexibility of the caspase-3 S2 pocket observed in crystal structures compared to the modeled closed conformation (green residues). (c) Binding of compound 16 in a flexible model of the caspase-3 active site. (d) Sequence alignment of the residues that define the S2 pocket of human caspases.
Comparison of caspase-3 crystal structures from the Protein Data Bank shows that the Y204 side chain can exist in two conformations (Figure 3b), suggesting that the S2 subsite may be conformationally flexible. In the apo structures, the Y204 side chain occludes part of the S2 pocket (closed conformation). In ligand bound conformations, Y204 rotates to accommodate larger hydrophobic groups (open conformation). The side chain of F256 was also found to be similarly flexible. Using this information, we carried out additional computational binding studies using a flexible receptor docking protocol. The results showed that the O-benzyl group of 16 was readily accommodated in the open conformation of the S2 pocket and was stabilized by extensive π–π interactions with the aromatic side chains of Y204 and F256 (Figure 3c). Empirical scoring of the closed configuration shows little difference in binding enthalpy between 3 and 16 (−9.7 and −9.45 kcal/mol, respectively). In the flexible model, compound 3 affinity is unchanged whereas 16 improves to − 11.29 kcal/mol. This model suggests that the increased binding energy of 16 could account for its significantly higher substrate activity relative to 3. This flexible S2 site model also provides a foundation for further compound optimization to take advantage of the remaining space within the pocket.
The flexibility of the caspase-3 S2 site may also explain the increased selectivity of O-benzylthreonine-containing substrates. Alignment of human caspase sequences shows that only caspase-3 and caspase-7 have tyrosine, phenylalanine, and tryptophan in the S2 pocket (Figure 3d). From this we would predict that 16 would show “off-target” substrate activity with caspase-7, which was found to be the case (see Figure 2e). In contrast, caspases 1, 2, 9, and 10 have valine or alanine at position 204 and significant residue variability at positions 206 and 256 which may account for the almost complete absence of activity of 16 for these enzymes.
The linker region in the P1′ position was also found to modulate activity and selectivity. Compounds with more than one methylene group, alanine, or glycine (as in 4, 11, and 12) at this position generated docking poses with a wide range of root-mean-square deviations. Compounds 5–10 that had rigid or bulky groups at this position showed stearic clashes that affected positioning of carbonyl oxygen at the oxyanion hole. A single methylene group at this position in compound 16 may allow sufficient substrate flexibility for carbonyl oxygen positioning and catalytic turnover while avoiding excessive flexibility and reduced specificity.
Radiosynthesis of Fluorine-18-Labeled Substrates and Cell-Based Uptake Studies.
Since substrate 16 showed the highest caspase-3 activity and specificity, it was taken forward for radiotracer development. Compounds were radiolabeled with aqueous 18-fluorine using an automated protocol on a GE Tracerlab as previously described.23 Briefly, Kryptofix 222 was used to catalyze tosyl displacement of 2-azidoethyltosylate with aqueous fluorine-18 to produce 2-[18F]-fluoroethylazide. This was distilled into the click reaction vial containing click solution (CuSO4, sodium ascorbate, TBTA, piperidine) and alkyne precursor. After 20 min, the reaction was transferred to a scavenging azide-functionalized resin to remove unreacted alkyne precursor. The labeled product was then purified with a C18 cartridge and eluted with 100% ethanol through a 0.2 μm filter (Scheme S3). All products were >99% radiochemical purity as monitored by radio-HPLC. Product identity was confirmed by comparison of retention time to that of the “cold” standard. Decay corrected yields and molar activities were [18F]-16 (hereafter [18F]-TBD) 9.84 ± 4.21%, 17.7–149.5 GBq/μmol (n = 12); [18F]-17 10.51 ± 4.46%, 224.4–512.0 GBq/μmol (n = 2); and [18F]-18 (hereafter [18F]-TBA) 12.6%, 35 GBq/μmol (n = 1) (Figures S3-S5). This high molar activity can be attributed to removal of ≫99% of the unreacted precursor through our previously described azide stripping protocol. [18F]-TBD was hydrolyzed by caspase-3 in vitro (Figure S6) confirming retention of substrate activity. Incubation of the product in ethanol at room temperature for 4 h showed no change in radiochemical purity (Figure S7).
To test the accumulation of [18F]-TBD in apoptotic cells, we pretreated OVCAR-5 cells with 8 μM cisplatin alone, cisplatin with 20 μM Z-VAD-FMK, or PBS for 48 h followed by incubation with 0.4, 2.5, or 25 μCi of [18F]-TBD for 30 min at 37 °C. After washing at 37 °C and cell lysis, lysate activity was normalized to the total sample protein concentration (Figure 4a). Z-VAD-FMK and PBS treated cells retained significantly less activity than cells treated with cisplatin alone, indicating that retention of the radiotracer was dependent on caspase activity.
Figure 4.

Accumulation of [18F]-TBD in cisplatin-treated OVCAR-5 cells. (a) OVCAR-5 cells were pretreated for 48 h with 8 μM cisplatin, 8 μM cisplatin and 20 μM Z-VAD-FMK, or PBS. Then, cells were treated with 0.44, 2.5, or 25 μCi of [18F]-TBD for 30 min. Normalized activity (counts per minute, CPM) was graphed (mean ± standard deviation) against the input activity and fit to a linear regression for each cell treatment. ANCOVA analysis of the slopes shows that probe uptake in cells treated with cisplatin is significantly different (p = 0.022, n = 3 for each condition) than uptake in untreated cells or cisplatin-treated cells where apoptosis is pharmacologically inhibited. (b) Retention of [18F]-TBD in OVCAR-5 cells treated with cisplatin for 48. The reported cisplatin IC50 value is indicated by a dotted line.27 Normalized counts were graphed (mean ± standard deviation) against cisplatin concentration and fit to a nonlinear regression. A statistical F-test finds that the slope is nonzero with high certainty (p < 0.001, n = 3 for each condition).
In order to measure the sensitivity of our probe to changes in cisplatin concentration, OVCAR-5 cells were pretreated with 0–50 μM cisplatin for 48 h followed by incubation with 5 μCi of [18F]-TBD for 1 h and washing at 37 °C. As expected, we observed a statistically significant increase in probe uptake at cisplatin concentrations above the reported IC50 for OVCAR-5 (3.6 μM, Figure 4b). Analogous results were also obtained with the less sensitive cell line, OVCAR-8 (11.1 μM, Figure S8).27 Washing apoptotic OVCAR-5 cells at 4 °C resulted in increased retention of activity but a similar cisplatin dose–response curve (Figure S9). A methyl ester prodrug variant ([18F]-17) with a formal charge of zero showed approximately 2-fold higher tracer uptake and a similar cisplatin dose–response curve (Figure S10). Taken together, these data indicate that radiotracer uptake and retention is proportional to apoptotic activity.
PET/CT with [18F]-TBD.
Having established that [18F]-TBD accumulates in cancer cells in an apoptosis- and caspase-dependent manner, we next tested the extent of radiotracer uptake and retention in apoptotic tissues in vivo using a Jo2-induced hepatic cytotoxicity model. Jo2 is a monoclonal antibody that acts as a Fas receptor (CD95) agonist, triggering apoptosis through the extrinsic pathway, followed by secondary necrosis.28,29 After intravenous injection, Jo2 accumulates in the liver and induces widespread and near-uniform hepatocyte apoptosis within 2 h (Figure S11).29,30 Female athymic nude mice were injected with 10 μg of Jo2 i.v. (tail vein). After approximately 2 h, 85–123 μCi of [18F]-TBD was injected via tail vein followed by a 30 min dynamic PET and 5 min CT. After 7.5 min, we observed increased accumulation in the livers of Jo2-treated mice compared to control (Figure 5a). To control for changes in vascular perfusion and liver metabolism from Jo2 treatment, we administered the caspase-3-insensitive probe [18F]-TBA. No increase in the liver uptake of [18F]-TBA was observed following Jo2 treatment (Figure 5b). Probe clearance was primarily renal with ~30% ID/cc accumulation in the bladder at 30 min post-injection, along with significant hepatobiliary excretion.
Figure 5.

[18F]-TBD accumulates and is retained in apoptotic livers of Jo2-treated mice. (a) Representative PET 7.5 min post i.v. injection of female athymic nude mice with [18F]-TbD in Jo2 treated (left) versus untreated (right) mice. Open arrow indicates liver, closed arrow GI. (b) Representative PET 7.5 min post i.v. injection of of female athymic nude mice with [18F]-TBA in Jo2 treated (left) versus untreated (right) mice. Open arrow indicates liver, closed arrow GI. (c) Three-tissue compartment model used for dynamic PET analysis. (d) Kinetic parameter k3 [18F]-TBD (left) or [18F]-TBA (right) treatment. Student’s two-tailed t test p-value 0.029 for Jo2 (n = 7) vs control (n = 4), ns = not significant. (e) ROC analysis of the [18F]-TBD (left) or [18F]-TBA (right) probes.
Pharmacokinetic Modeling of PET Data.
Hepatobiliary excretion can complicate the interpretation of PET/CT imaging. This is of particular concern in the hepatotoxicity model used here. Standard multicompartment analysis tools provided in commercial packages (such as PMOD) are designed for tumor and brain analysis with transport only via the blood pool. These analyses are therefore not ideal for modeling coupled transport between compartments independent of the blood pool, i.e., hepatobiliary excretion to the gastrointestinal tract (GI). To better understand our imaging results, we modeled the pharmacokinetics of [18F]-TBD and [18F]-TBA using a modified 3-compartment model (Figure 5c). This model describes the fraction flux of radioactivity per second between volumes of interest encompassing the heart (blood pool), the liver, and the GI. In the case of [18F]-TBD, we found that k3 (the rate of probe washout from the liver to the GI) was 40% lower following Jo2 treatment showing decreased tracer washout in apoptotic livers. No difference was observed for [18F]-TBA (Figure 5d). These results are well-supported by ex vivo biodistribution studies (Figure S12). ROC analysis shows that probe [18F]-TBD has a strong predictive value for detecting liver cell death whereas probe [18F]-TBA does not (Figure 5e). A summary of all kinetic constants can be found in Table S4. Comparison of PET data from [18F]-TBD and [18F]-TBA demonstrates that caspase activity is required for probe retention and that enhanced liver uptake in Jo2 treated mice is not simply a function of collapsed vasculature or nonspecific uptake in apoptotic or necrotic hepatocytes. Given the dramatic physiological changes that occur within apoptotic tissue, the inclusion of [18F]-TBA as a “chemical control” allows us to conclusively link decreased washout rate with caspase-3 activity rather than cellular necrosis. These imaging experiments and the corresponding pharmacokinetic modeling confirm our in vitro observations that tracer retention is driven through caspase-3 activity and that [18F]-TBD possesses sufficient cell permeability and caspase-3 activity to report on liver cell death in vivo.
CONCLUSION
We have developed a minimized dipeptide caspase-3 substrate suitable for imaging apoptosis in vivo. We have demonstrated that by leveraging substrate turnover, acquisition of dynamic PET, and careful pharmacokinetic modeling, it is possible to detect and quantitate a robust PET signal even in the context of unfavorable biodistribution, i.e., high tracer flux through the liver. Our data demonstrates that the use of radiolabeled substrates for molecular imaging of caspase-3 activation in conjunction with dynamic PET can provide immediate pharmacodynamic readout of apoptotic cell death in vivo.
EXPERIMENTAL PROCEDURES
Cell Culture.
OVCAR-5 (from the Developmental Therapeutics Program at National Cancer Institute) and OVCAR-8 (from ATCC) cells were maintained in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (Sigma), 1% (v/v) penicillin-streptomycin (Corning, Inc., Corning, NY, USA), and 1 mM sodium pyruvate (Corning). Cells were maintained at 37 °C in a humidified incubator supplemented with 5% (v/v) CO2. Cells were tested for mycoplasma contamination at least every 6 months.
Animals.
Experiments with athymic nu/nu-Foxn1 mice (Envigo) were reviewed and approved by the Institutional Animal Care and Use Committee of M. D. Anderson Cancer Center (IACUC ID: 00000825).
Solid Phase Peptide Synthesis.
All peptides were synthesized manually using 5 g solid sample cartridges and a vacuum manifold. Amino acids were purchased from Advanced ChemTech (Louisville, KY, USA). 2-Chlorotrityl resin (600 mg, 0.84 mmol binding capacity, AnaSpec, Inc., Fremont, CA, USA) was swelled in DMF for 60 min rotating and then washed twice with 6 mL DMF. Initial C-terminal amino acid coupling was performed with 0.8 equiv (0.627 mmol) of fluorenylmethyloxycarbonyl chloride (Fmoc)-protected amino acid (199.8 mg Fmoc-Gly-OH, 224 mg Fmoc-Ser(tBu)-OH, 221.3 mg Fmoc-Ala-OH, 257.7 mg Fmoc-Phe-OH, 292.5 mg Fmoc-Asp(tBu)-OH, or 262.6 mg Fmoc-Asp(OMe)-OH) in 6 mL DMF. To this was added 2 equiv of N,N-diisopropylethylamine (DIEA, 1.68 mmol, 295 μL). After 5 min, 3 equiv of DIEA (2.52 mmol, 442 μL) was added and incubated rotating for 60 min. Free chloride was capped by addition of 1 mL methanol for 15 min. Resin was then washed 5× with 6 mL DMF. Fmoc deprotection was performed with 20% (v/v) piperidine in DMF for 6 s, 10 s, and 10 min followed by 5× wash with DMF. Additional amino acid couplings were performed with 3 equiv (2.52 mmol) of Fmoc-protected amino acid (855 mg Fmoc-Val-OH, 1035 mg Fmoc-Asp(tBu)-OH, or 1086 mg Fmoc-Thr(Bzl)-OH), 3 equiv of N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 2.52 mmol, 892 mg) and 3 equiv of DIEA in 8 mL DMF. After 1 h, resin was washed and Fmoc deprotection performed as described above. N-Terminal 2-methoxyphenyl capping was performed by addition of 3 equiv of 2-methoxyphenyl acetic acid (2.52 mmol, 418.7 mg), with 3 equiv of HBTU and DIEA in 8 mL DMF. When appropriate, N-terminal acylation was performed with the addition of 200 μL of acetic anhydride in 4 mL DMF for 30 min with the addition of 3 equiv of DIEA after the first 5 min. Resin was then washed 5× with DMF and 5× with DCM and dried under vacuum for 30 min. Peptides were cleaved from the resin by addition of 8 mL of 1% trifluoroacetic acid (TFA, v/v) in DCM for 90 s and gravity filtered into a flask on ice. Cleavage repeated four times. Filtrate was concentrated by rotary evaporator. TFA was coevaporated with 2 × 25 mL DCM. Crude product was purified by reverse phase HPLC (Luna 5 μm C18(2), LC Column 250 × 21.2 mm; Phenomenex, Torrance, CA) using gradient elution (20–55% Buffer B over 10 min, 55–65% Buffer B over 15 min; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). After lyophilization, peptides were obtained as a white solid. Analysis by ESI+: 2MP-ValAsp(tBu)Ala-OH expected [M + Na]+ = 530.24, observed [M + Na]+ = 530.39); 2MP-ValAsp(tBu)Gly-OH expected [M + Na]+ = 516.23, observed [M + Na]+ = 516.2); 2MP-ValAsp(tBu)Phe-OH expected [M + H]+ = 584.29, observed [M + H]+ = 584.45); 2MP-ValAsp(tBu)Ser(tBu)-OH expected [M + Na]+ = 602.31, observed [M + Na]+ = 601.87); 2MP-Thr(Bzl)Asp(tBu)-OH expected [M + Na]+ = 551.24, observed [M + Na]+ = 551.18); Ac-Thr(Bzl)Asp(tBu)-OH expected [M + Na]+ = 445.2, observed [M + Na]+ = 445.1); 2MP-Thr(Bzl)Ala-OH expected [M + Na]+ = 451.17, observed [M + Na]+ = 452.82); 2MP-Thr(Bzl)Asp(OMe)-OH expected [M + Na]+ = 487.2, observed [M + Na]+ = 488.91).
Synthesis of 3-(2-(2-(2-Methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxo-4-((2-oxo-4-(trifluoromethyl)-2H-chromen-7-yl)amino)butanoic Acid (1).
Commercially synthesized (GenScript, Piscataway, NJ) 4-(tert-butoxy)-2-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic acid (2MP-ValAsp(tBu)-OH, 30 mg, 69 μmol) was dissolved in 200 μL of DMF. To this was added 2 equiv of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]-pyridinium 3-oxid hexafluorophosphate (HATU, 50 mg, 128 μmol) and DIEA (24 μL). After 5 min, 1.5 equiv of 7-amino-4-(trifluoromethyl)coumarin (AFC, 24 mg, 103.5 μmol) was added, and it was incubated overnight rotating at RT. The crude product was purified by reverse phase HPLC using gradient elution (20–55% Buffer B over 10 min, 55–65% Buffer B over 15 min; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). Collected fractions were lyophilized and mass spec was used to confirm intermediate. The tert-butyl protecting group was removed by treatment with of a 95:5 mixture of TFA:H2O (367.5 μL). After 1 h, the reaction was neutralized with 10 N NaOH (400 μL). Precipitate was dissolved in a 1:1 mixture of CH3CN:H2O (400 μL) and purified by reverse phase HPLC using gradient elution (20–55% Buffer B over 10 min, 55–65% Buffer B over 15 min; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). After lyophilization, the title compound was obtained as a white solid (11.7 mg, 28.9%). Analysis by ESI+ (Expected [M + H]+ = 592.18 Observed [M + H]+ = 592.24. 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.44 (s, 1H), 10.07 (d, J = 155 Hz, 1H), 8.46 (dd, J = 165.9 Hz, 1H), 8.26 (d, J = 6.3 Hz, 1H), 7.95 (s, 1H), 7.56 (dd, J = Hz, 1H), 7.47 (d, J = 9.5 Hz, 1H), 7.40 (d, J = 8.9Hz, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.13 (t, J = 7.7, 8.0 Hz, 1H), 6.84 (m, 2H), 4.81 (m, J = 73.2, 2H), 3.68 (s, 3H), 3.52 (m, 2H), 2.84 (m, 1H), 2.61 (m, 2H), 0.94 (d, J = 6.6, 3H), 0.86 (d, J = 6.7, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.76, 171.64, 171.51, 171.18, 168.02, 158.55, 158.38, 157.96, 157.19, 154.42, 150.81, 144.36, 143.21, 142.66, 141.96, 140.66, 134.57, 133.22, 132.62, 130.86, 127.76, 124.38, 119.93, 110.44, 108.34, 106.45, 59.81, 55.17, 50.20, 36.41, 35.96, 29.41, 18.98, 18.00.
Synthesis of 3-(3-(Benzyloxy)-2-(2-(2-methoxyphenyl)-acetamido)butanamido)-4-oxo-4-((2-oxo-4-(trifluoromethyl)-2H-chromen-7-yl)amino)butanoic acid (2).
2MP-Thr(Bzl)Asp(tBu)-OH (34 mg, 69 μmol) was dissolved in 200 μL of DMF. To this was added 2 equiv of HATU (50 mg, 128 μmol) and DIEA (24 μL). After 5 min, 1.5 equiv of AFC (24 mg, 103.5 μmol) was added. Incubated overnight rotating at RT. The crude product was purified by reverse phase HPLC using gradient elution (20–55% Buffer B over 10 min, 55–65% Buffer B over 15 min; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). Collected fractions were lyophilized and mass spec was used to confirm intermediate. The tert-butyl protecting group was removed by treatment with of a 95:5 mixture of TFA:H2O (367.5 μL). After 1 h, the reaction was neutralized with 10 N NaOH (400 μL). Precipitate was dissolved in a 1:1 mixture of CH3CN:H2O (400 μL) and purified by reverse phase HPLC using gradient elution (20–55% Buffer B over 10 min, 55–65% Buffer B over 15 min; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). After lyophilization, the title compound was obtained as a white solid (7.1 mg, 15%). Analysis by ESI+ (Expected [M + H]+ = 684.21 Observed [M + H]+ = 684.44). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.48 (s, 1H), 10.26 (d, J = 98.3 Hz, 1H), 8.46 (dd, J = 162.9 Hz, 1H), 7.95 (dd, J = 67.74 Hz, 1H), 7.84 (d, J = 29.8 Hz, 1H), 7.58 (q, J = 27, 8.2, 1.7 Hz, 1H), 7.49 (m, 1H), 7.31 (d, J = 4.6 Hz, 2H), 7.19 (m, 5H), 6.91 (m, 2H), 6.83 (m, 1H), 4.78 (m, 1H), 4.44 (m, 2H), 3.86 (m, 1H), 3.68 (d, J = 10.9 Hz, 3H), 3.53 (dt, J = 40.1, 2H), 2.83 (td, J = 16.8, 15.6 Hz, 1H), 1.09 (dd, J = 6.2 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.08, 171.88, 170.90, 170.76, 170.48, 159.09, 157.66, 139.10, 131.40, 131.20, 128.54, 128.47, 128.44, 128.42, 128.08, 127.89, 127.77, 127.64, 124.29, 120.66, 120.58, 116.76, 116.74, 111.09, 108.89, 106.97, 74.89, 70.93, 70.57, 58.73, 55.76, 37.39, 37.15, 16.61.
General Synthesis of Cold Radiotracer Substrates (Compounds 3–20).
69 μmol of precursor was dissolved in 700 μL of DMF with HBTU (52 mg, 138 μmol). DIEA (24 μL, 138 μmol) was added and allowed to react at 25 °C for 5 min. Next, peptide coupling was performed with addition of 1.5 equiv (103.5 μmol) of the amine-alkyne, incubated at 25 °C with end-over-end rotation. After 6 h, the reaction was mixed with 10 mL ethyl acetate and the organic phase washed twice with 1 N HCl, twice with saturated NaHCO3, and twice with brine. The organic phase was dried over MgSO4. After filtration of the salts and evaporation under vacuum, the crude product was dissolved in 200 μL DMF. To this was added 500 μL of click solution (16 mg/mL CuSO4·2H2O and 30 mg/mL l-ascorbic acid), 2 μL of tris(benzyltriazolylmethyl)amine (TBTA, 100 mg/mL), and 500 μL of 2-fluoroethylazide (0.2 M in DMF). The reaction proceeded overnight at 25 °C with end-over-end rotation. The crude product was purified by reverse phase HPLC using gradient elution (20–55% Buffer B over 10 min, 55–65% Buffer B over 15 min; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). Collected fractions were lyophilized and mass spec was used to confirm intermediate. The tert-butyl protecting group was removed by treatment with of a 95:5 mixture of TFA:H2O (367.5 μL). After 1 h, the reaction was neutralized with 10 N NaOH (400 μL). Precipitate, if present, was dissolved in a 1:1 mixture of CH3CN:H2O (400 μL) and purified by reverse phase HPLC using gradient elution (20–55% Buffer B over 10 min, 55–65% Buffer B over 15 min; Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN + 0.1% (v/v) TFA). Collected fractions were lyophilized and ESI+ mass spectrometry used to confirm purified product.
Synthesis of 4-(((1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)-methyl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (3).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and propargylamine (103.5 μmol, 6.8 μL). After lyophilization, the title compound was obtained as a white solid (7.6 mg, 16.8%). Analysis by ESI+ (Expected [M + H]+ = 507.23. Observed [M + H]+ = 507.21). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.33 (s, 1H), 8.44 (d, J = 8.2 Hz, 1H), 8.33 (t, J = 5.7, 5.8 Hz, 1H), 7.20 (t, J = 7.2, 8.4 Hz, 1H), 7.13 (d, J = 7.1 Hz, 1H), 6.93 (d, J = 5.8, 8.2 1H), 6.85 (t, J = 7.2, 7.2, 1H), 4.71 (dt, J = 4.7, 4.9 Hz, 2H), 4.62 (t, J = 11.6, 10.3 Hz, 2H), 4.57 (m, 1H), 4.11 (dd, J = 5.6, 5.7 Hz, 1H), 4.00 (t, J = 7.3, 7.4 Hz, 1H), 3.72 (s, 3H), 3.39 (dd, J = 69.3 Hz, 2H), 2.71 (dd, J = 4.6, 5.1 Hz, 1H), 2.54 (m, 2H), 0.84 (d, J = 6.4 Hz, 3h), 0.81 (d, J = 7.2 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.18, 171.73, 171.32, 157.58, 145.49, 130.99, 128.37, 125.05, 123.55,120.57, 111.01, 83.02, 81.68, 59.19, 55.73, 50.41, 49.95, 37.06, 35.00, 30.47, 19.47, 18.93,18.29.
Synthesis of 4-((2-(1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)ethyl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (4).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and 1-amino-3-butyne (103.5 μmol, 9 μL). After lyophilization, the title compound was obtained as a white solid (2.9 mg, 7.3%). Analysis by ESI+ (Expected [M + H]+ = 521.24. Observed [M + H]+ = 521.19). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 1H NMR (500 MHz, DMSO) δ 12.37 (s, 1H), 8.53 (d, J = 8.3 Hz, 1H), 8.14 (d, J = 7.0 Hz, 1H), 8.01 (t, J = 5.6, 5.6 Hz, 1H), 7.88 (s, 1H), 7.25 (m, 2H), 7.01 (d, J = 8.2 Hz, 1H), 6.92 (t, J = 7.4, 7.4 Hz, 1H), 4.93 (t, J = 4.7, 4.7 Hz, 1H), 4.83 (t, J = 4.7, 4.7 Hz, 1H), 4.76 (q, J = 4.6, 4.3, 4.3 Hz, 1H), 4.70 (q, J = 4.6, 4.2, 4.2 Hz, 1H), 4.02 (t, J = 7.4, 7.4 Hz, 1H), 3.81 (s, 3H), 3.61 (d, J = 15.2 Hz, 2H), 3.29 (dp, J = 19.4, 6.2, 6.2, 6.2, 6.2 Hz, 3H), 2.83 (d, J = 4.5 Hz, 0H), 2.79 (t, J = 5.5, 5.5 Hz, 1H), 2.73 (m, 2H), 2.52 (dd, J = 16.4, 9.4 Hz, 1H), 0.98 (d, J = 6.7 Hz, 3H), 0.92 (d, J = 6.6 Hz, 4H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.29, 171.86, 171.56, 170.76, 157.54, 130.99, 128.34, 127.07, 124.89, 123.10, 120.53, 111.00, 81.71, 59.78, 55.73, 50.38, 50.22, 39.45, 39.03, 36.58, 29.47, 19.42, 19.21.
Synthesis of 4-((1-(1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)ethyl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (5).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and 1-methyl-prop-2-ynylamine (103.5 μmol, 7.2 mg). After lyophilization, the title compound was obtained as a white solid (12.5 mg, 31.5%). Analysis by ESI+ (Expected [M + H]+ = 521.24. Observed [M + H]+ = 521.15). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.33 (s, 1H), 8.44 (d, J = 8.1 Hz, 1H), 8.16 (d, J = 7.6 Hz, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.76 (s, 1H), 7.18 (t, J = 8.5, 6.5 Hz, 1H), 7.14 (d, J = 7.9 Hz, 1H), 6.91 (d, J = 7.9 Hz, 1H), 6.80 (t, J = 7.6, 7.3 Hz, 1H), 4.98 (m, 2H), 4.83 (m, 2H), 4.59 (m, 2H), 4.11 (dd, J = 5.6, 5.7 Hz, 1H), 4.10 (q, J = 7.9, 7.5, 6.3, 1H), 4.00 (t, J = 7.4, 7.5 Hz, 1H), 4.02 (dd, J = 4.5, 4.7 Hz, 1H), 3.71 (s, 3H), 3.40 (q, J = 5.5, 5.6, 9.3 Hz, 2H), 2.72 (m, 1H), 1.35 (m, 3H), 0.86 (d, J = 10.3 Hz, 3H), 0.80 (d, J = 10.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6-(CD3)2SO) δ 172.18, 171.73, 171.32, 170.88, 157.58, 145.48, 130.99, 128.37, 125.01, 123.55, 120.57, 111.01, 83.02, 81.68, 59.19, 55.73, 50.41, 49.95, 37.06, 36.62, 35.00, 30.47, 19.47, 18.93.
Synthesis of 4-((2-(1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)propan-2-yl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (6).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and 2-methyl-3-butyn-2-amine (103.5 μmol, 6.8 μL). After lyophilization, the title compound was obtained as a white solid (5.5 mg, 13.5%). Analysis by ESI+ (Expected [M + H]+ = 535.26. Observed [M + H]+ = 535.23). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.27 (s, 1H), 8.39 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.76 (s, 2H), 7.19 (t, J = 7.9 Hz, 1H), 7.13 (d, J = 7.4 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.83 (t, J = 7.4 Hz, 1H), 4.77 (dt, J = 47.2, 4.8 Hz, 2H), 4.61 (t, J = 4.7 Hz, 1H), 4.57 (p, J = 6.1 Hz, 2H), 4.00 (t, J = 7.5 Hz, 1H), 3.72 (s, 3H), 3.46 (m, 2H), 2.67 (dd, J = 16.4, 4.9 Hz, 1H), 2.43 (dd, J = 16.6, 9.0 Hz, 2H), 1.50 (d, J = 9.3 Hz, 6H), 0.86 (d, J = 6.8 Hz, 3H), 0.82 (d, J = Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.27, 171.82, 171.05, 170.15, 157.57, 153.51, 130.95, 128.28, 125.01, 121.81, 120.53, 110.99, 81.62, 59.21, 55.73, 51.23, 50.30, 50.20, 36.96, 36.57, 30.50, 28.80, 28.37, 19.45, 19.02.
Synthesis of 4-((2-(1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)butan-2-yl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (7).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and 3-methyl-1-pentyn-3-amine hydrochloride (103.5 μmol, 14.3 mg). After lyophilization, the title compound was obtained as a white solid (14.5 mg, 34.7%). Analysis by ESI+ (Expected [M + H]+ = 549.29. Observed [M + H]+ = 549.2). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.31 (s, 1H), 8.43 (t, J = 8.4, 8.4 Hz, 1H), 8.30 (d, J = 7.5 Hz, 1H), 7.91 (dd, J = 13.2, 7.9 Hz, 1H), 7.61 (m, 1H), 7.20 (dd, J = 9.7, 7.4 Hz, 1H), 7.14 (q, J = 8.3, 8.3, 8.2 Hz, 1H), 6.95 (dd, J = 13.2, 8.2 Hz, 1H), 6.86 (m, 1H), 4.83 (m, 1H), 4.75 (m, 1H), 4.63 (m, 2H), 4.20 (m, 1H), 4.05 (dt, J = 8.4, 6.1, 6.1 Hz, 1H), 3.74 (d, J = 11.3 Hz, 3H), 3.47 (m, 2H), 2.69 (m, 1H), 2.46 (m, 1H), 2.29 (t, J = 7.4, 7.4 Hz, 1H), 1.92 (ddt, J = 39.4, 25.4, 6.4, 6.4 Hz, 2H), 1.50 (m, 3H), 0.84 (m, 6H), 0.69 (m, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.27, 171.90, 170.90, 170.12, 157.58, 130.97, 128.28, 125.00, 122.28, 122.19, 120.57, 111.04, 81.61, 58.98, 57.85, 55.74, 54.59, 50.30, 36.98, 36.40, 32.53, 31.25, 30.66, 19.48, 18.95, 8.29.
Synthesis of 4-((1-(1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)cyclohexyl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic acid (8).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and 1-ethynylcyclohexylamine (103.5 μmol, 14.3 μL). After lyophilization, the title compound was obtained as a white solid (5.6 mg, 12.9%). Analysis by ESI+ (Expected [M + H]+ = 575.29. Observed [M + H]+ = 575.21). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.32 (s, 1H), 9.08 (t, J = 1.3, 1.3 Hz, 1H), 8.44 (d, J = 7.9 Hz, 1H), 8.27 (m, 1H), 7.53 (s, 1H), 7.26 (m, 1H), 7.11 (dd, J = 7.5, 1.8 Hz, 1H), 6.92 (dd, J = 8.3, 1.1 Hz, 1H), 6.83 (dd, J = 7.4, 1.1 Hz, 1H), 4.81 (t, J = 4.9, 4.9 Hz, 1H), 4.71 (t, J = 4.9, 4.9 Hz, 1H), 4.58 (m, 2H), 4.06 (t, J = 7.7, 7.7 Hz, 2H), 3.71 (s, 3H), 3.54 (dd, J = 15.1, 10.9 Hz, 2H), 2.99 (t, J = 7.3, 7.3 Hz, 1H), 2.71 (d, J = 5.4 Hz, 1H), 2.67 (d, J = 5.3 Hz, 1H), 1.93 (m, 2H), 1.76 (dd, J = 16.9, 7.9 Hz, 2H), 1.46 (dddd, J = 33.1, 17.1, 7.8, 3.7 Hz, 6H), 0.86 (d, J = 6.7 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H). 13C NMR (151 MHz, DMSO, d6-(CD3)2SO): δ 171.75, 171.46, 170.31, 169.73, 157.05, 130.58, 130.44, 127.79, 124.47, 121.66, 120.04, 119.30, 110.53, 81.21, 58.39, 57.44, 56.69, 53.53, 36.48, 35.83, 34.94, 30.24, 28.98, 24.99, 21.11, 18.98, 18.43.
Synthesis of 4-((2-(1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)-1-phenylethyl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (9).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and 1-phenylbut-3-yn-1-amine hydrochloride (103.5 μmol, 18.9 mg). After lyophilization, the title compound was obtained as a white solid (24.4 mg, 31.9%). Analysis by ESI+ (Expected [M + H]+ = 597.28. Observed [M + H]+ = 597.24). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.17 (s, 1H), 8.51 (d, J = 8.5 Hz, 0H), 8.33 (d, J = 8.5 Hz, 1H), 8.22 (m, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.75 (s, 1H), 7.48 (d, J = 7.6 Hz, 4H), 7.26 (d, J = 2.8 Hz, 2H), 7.14 (d, J = 1.8 Hz, 1H), 6.94 (dd, J = 8.2, 1.1 Hz, 1H), 6.85 (dd, J = 7.3, 1.2 Hz, 1H), 6.75 (td, J = 7.4, 7.3, 1.1 Hz, 1H), 5.01 (q, J = 8.0, 7.8, 7.8 Hz, 1H), 4.81 (m, 1H), 4.59 (t, J = 4.6, 4.6 Hz, 2H), 4.50 (m, 1H), 4.08 (dd, J = 7.6, 6.1 Hz, 1H), 3.81 (dd, J = 8.3, 5.9 Hz, 1H), 3.72 (s, 3H), 3.42 (dd, J = 15.1, 3.6 Hz, 1H), 2.99 (t, J = 6.9, 6.9 Hz, 2H), 2.78 (d, J = 7.5 Hz, 1H), 2.54 (m, 2H), 0.84 (m, 6H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.21, 171.55, 171.45, 170.08, 157.60, 142.94, 138.09, 131.12, 128.66, 128.40, 127.33, 127.22, 126.92, 125.97, 124.92, 120.60, 111.09, 83.05, 58.82, 55.76, 55.62, 53.14, 53.01, 37.28, 34.86, 32.82, 30.75, 19.51, 18.37.
Synthesis of 4-((1-(2-(1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)ethyl)piperidin-4-yl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (10).
Peptide coupling performed with 2MP-ValAsp(tBu)-OH, (30 mg, 69 μmol) and 1-(but-3-yn-1-yl)piperidin-4-amine (103.5 μmol, 15.8 mg). After lyophilization, the title compound was obtained as a white solid (6.4 mg, 14%). Analysis by ESI+ (Expected [M + H]+ = 604.32. Observed [M + H]+ = 604.65). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.41 (s, 1H), 8.17 (dd, J = 120 Hz, 1H), 8.03 (s, 1H), 7.89 (dd, J = 1H), 7.78 (dd, J = 32.9, 1H), 7.21 (t, J = 7.6, 7.9, 1H), 7.15 (d, J = 6.6 Hz, 1H), 6.94 (d, J = 7.9 Hz, 1H), 6.87 (t, J = 7.0, 7.3 Hz, 1H), 4.76 (dt, J = 47.5 Hz, 2H), 4.67 (dt, J = 28.0 Hz, 2H), 4.50 (m, 1H), 3.95 (dt, J = 76.3 Hz, 1H), 3.68 (m, 1H), 3.50 (t, J = 14.9, 16.6 Hz, 2H), 3.45 (d, J = 25.4 Hz, 2H), 3.06 (m, 2H), 2.68 (m, 1H), 1.91 (m, 2H), 1.61 (m, 2H), 0.85 (d, J = 10.10 Hz, 3H), 0.81 (d, J = 6.82 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.68, 171.19, 157.94, 157.14, 142.39, 130.54, 127.94, 124.53, 123.22, 120.19, 110.66, 82.63, 81.29, 55.34, 54.59, 50.85, 49.94, 47.43, 47.28, 36.87, 36.41, 29.86, 29.02, 28.70, 25.73, 20.17, 18.95, 18.62, 17.87.
Synthesis of 4-((2-(((1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-2-oxoethyl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (11).
Peptide coupling performed with 2MP-ValAsp(tBu)Gly-OH, (30 mg, 60.8 μmol) and propargylamine (91.2 μmol, 5.8 μL). After lyophilization, the title compound was obtained as a white solid (7.8 mg, 20.2%). Analysis by ESI+ (Expected [M + H]+ = 564.25. Observed [M + H]+ = 563.86). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.38 (s, 1H), 8.36 (dd, J = 56.9 Hz, 1H), 8.20 (t, J = 5.8, 5.6 Hz, 1H), 8.06 (dt, J = 61.4 Hz, 1H), 7.91 (s, 1H), 7.78 (dd, J = 61.1 Hz, 1H), 7.21 (t, J = 7.0, 8.4 Hz, 1H), 7.12 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.86 (t, J = 7.3, 7.5 Hz, 1H), 4.75 (dt, J = 47.5 Hz, 2H), 4.64 (dt, J = 27.2 Hz, 2H), 4.51 (q, J = 5.7, 7.5, 7.6 Hz, 1H), 4.31 (d, J = 5.7 Hz, 2H), 4.12 (t, J = 7.1, 7.4 Hz, 1H), 3.74 (s, 3H), 3.64 (d, J = 5.9 Hz, 2H), 2.72 (m, 1H), 2.54 (m, 2H), 3.41 (d, J = 15.1 Hz, 2H), 0.81 (d, J = 7.3 Hz, 3H), 0.78 (d, J = 6.7 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.85, 171.37, 170.71, 170.52, 168.53, 157.09, 145.00, 130.58, 127.89, 124.49, 123.23, 120.13, 110.58, 82.45, 81.34, 57.68, 55.27, 49.98, 42.28, 36.79, 35.72, 34.20, 30.54, 29.00, 19.06, 17.85.
Synthesis of 4-((1-(((1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-1-oxopropan-2-yl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (12).
Peptide coupling performed with 2MP-ValAsp(tBu)Ala-OH, (30 mg, 59 μmol) and propargylamine (88.5 μmol, 5.7 μL). After lyophilization, the title compound was obtained as a white solid (8.9 mg, 22.3%). Analysis by ESI+ (Expected [M + H]+ = 578.27. Observed [M + H]+ = 578.84). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.36 (s, 1H), 8.41 (m, 1H), 8.19 (dd, J = 72.14 Hz, 1H), 7.89 (s, 1H), 7.85 (t, J = 9.5, 5.5 Hz, 1H), 7.76 (dd, J = 7.07 Hz, 1H), 7.20 (m, 1H), 7.08 (m, 1H), 6.94 (m, 1H), 6.84 (m, 1H), 4.74 (dq, J = 47.30 Hz, 2H), 4.65 (dt, J = 27.89 Hz, 2H), 4.48 (dq, J = 30.3 Hz, 1H), 4.30 (m, 2H), 4.07 (m, 2H), 3.74 (s, 3H), 3.52 (m, 1H), 3.38 (t, J = 16.8, 15.0 Hz, 1H), 2.73 (m, 1H), 2.54 (m, 2H), 1.14 (d, J = 4.70 Hz, 3H), 0.83 (d, J = 5.3 Hz 3H), 0.81 (d, J = 7.2 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 173.62, 172.13, 171.74, 170.81, 170.11, 130.60, 127.89, 124.51, 123.05, 110.56, 99.55, 90.99, 82.46, 81.35, 55.28, 49.28, 40.41, 34.36, 34.30, 30.72, 25.91, 19.05, 19.00, 17.96.
Synthesis of 4-((1-(((1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (13).
Peptide coupling performed with 2MP-ValAsp(tBu)Phe-OH, (30 mg, 51 μmol) and propargylamine (76.5 μmol, 4.88 μL). After lyophilization, the title compound was obtained as a white solid (3.7 mg, 8.2%). Analysis by ESI+ (Expected [M + H]+ = 654.3. Observed [M + H]+ = 654.81). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.37 (s, 1H), 8.39 (dd, J = 46.6 Hz, 1H), 8.02 (s, 1H), 7.93 (dd, J = 27.5 Hz, 1H), 7.80 (dd, J = 51.1 Hz, 1H), 7.73 (t, J = 7.8, 9.5 Hz, 1H), 7.16 (m, 5H), 7.12 (d, J = 8.7 Hz, 1H), 7.09 (d, J = 7.1 Hz, 1H), 6.93 (t, J = 6.6, 8.4 Hz, 1H), 6.84 (t, J = 7.2, 8.4 Hz, 1H), 4.74 (dt, J = 47.8 Hz, 2H), 4.64 (dt, J = 27.8 Hz, 2H), 4.50 (q, J = 6.8, 7.2, 7.6 Hz, 1H), 4.35 (m, 2H), 4.29 (m, 2H), 4.05 (dt, J = 43.9 Hz, 1H), 3.72 (s, 3H), 3.57 (d, J = 15.4 Hz, 2H), 2.98 (m, 1H), (m, 1H), 2.54 (m, 2H), 0.81 (dd, J = 6.4 Hz, 3H), 0.76 (dd, J = 9.1 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.47, 171.31, 171.03, 170.80, 170.46, 168.55, 158.61, 158.40, 158.20, 157.99, 157.16, 130.70, 129.12, 128.08, 120.27, 118.29, 116.30, 114.30, 110.62, 82.50, 81.38, 55.32, 50.04, 49.92, 40.41, 36.94, 36.72, 30.69, 30.32, 19.07, 17.97, 17.75.
Synthesis of 4-((1-(((1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-3-hydroxy-1-oxopropan-2-yl)amino)-3-(2-(2-(2-methoxyphenyl)acetamido)-3-methylbutanamido)-4-oxobutanoic Acid (14).
Peptide coupling performed with 2MP-ValAsp(tBu)Ser(tBu)-OH, (30 mg, 52 μmol) and propargylamine (78 μmol, 5 μL). After lyophilization, the title compound was obtained as a white solid (5.6 mg, 13.8%). Analysis by ESI+ (Expected [M + H]+ = 594.26. Observed [M + H]+ = 594.88). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.32 (s, 1H), 8.39 (dd, J = 25.7 Hz, 1H), 8.28 (m, 1H), 8.17 (dt, J = 19.3 Hz, 1H), 7.82 (s, 1H), 7.76 (m, 1H), 7.16 (t, J = 7.0, 7.0 Hz, 1H), 7.07 (dd, J = 26.5 Hz, 1H), 6.90 (t, J = 6.0, 8.2 Hz, 1H), 6.81 (dd, J = 17.9 Hz, 1H), 4.78 (dt, J = 22.1 Hz, 2H), 4.70 (t, J = 4.9, 4.5 Hz, 1H), 4.60 (dt, J = 27.9 Hz, 2H), 4.28 (t, J = 5.1, 7.9 Hz, 2H), 4.16 (t, J = 6.2, 5.8 Hz, 1H), 4.08 (m, 1H), 3.70 (s, 3H), 3.51 (m, 2H), 3.32 (m, 2H), 2.69 (m, 1H), 0.78 (d, J = 7.5 Hz, 6H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.08, 171.78, 171.54, 170.66, 169.66, 157.08, 145.19, 130.58, 127.88, 120.12, 123.04, 110.56, 82.81, 81.32, 69.26, 61.36, 58.89, 55.26, 49.89, 36.77, 34.33, 28.99, 28.67, 19.03, 17.85.
Synthesis of 3-(3-(Benzyloxy)-2-(2-(2-methoxyphenyl)-acetamido)butanamido)-4-((2-(1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)ethyl)amino)-4-oxobutanoic Acid (15).
Peptide coupling performed with 2MP-Thr(Bzl)Asp(tBu)-OH, (36 mg, 69 μmol) and 1-amino-3-butyne (103.5 μmol, 9 μL). After lyophilization, the title compound was obtained as a white solid (10.3 mg, 22.2%). Analysis by ESI+ (Expected [M + H]+ = 613.27. Observed [M + H]+ = 613.29). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.43 (s, 1H), 8.26 (dd, J = 89.4 Hz, 1H), 7.90 (m, 1H), 7.83 (s, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.30 (m, 5H), 7.21 (t, J = 8.4, 8.2 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 6.93 (d, J = 8.0 Hz, 1H), 6.86 (t, J = 7.6, 7.4 Hz, 1H), 4.74 (dt, J = 47.3 Hz, 2H), 4.60 (dt, J = 27.7 Hz, 2H), 4.56 (m, 1H), 4.47 (t, J = 10.8, 12.2 Hz, 2H), 4.40 (q, J = 4.3, 3.9, 3.9 Hz, 1H), 3.83 (dt, J = 41.9 Hz, 2H), 3.71 (s, 3H), 3.48, (m, 2H), 3.22 (q, J = 6.4, 7.5, 8.1 Hz, 2H), 2.66 (m, 2H0 1.06 (d, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.84, 170.88, 170.15, 169.63, 158.03, 157.11, 144.32, 138.52, 130.72, 128.11, 127.56, 124.31, 122.68, 120.24, 110.70, 82.61, 81.27, 74.84, 74.53, 70.47, 70.10, 57.82, 57.70, 55.32, 49.79, 49.66, 36.96, 36.07, 25.17, 16.22.
Synthesis of 3-(3-(Benzyloxy)-2-(2-(2-methoxyphenyl)-acetamido)butanamido)-4-(((1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-4-oxobutanoic Acid (16).
Peptide coupling performed with 2MP-Thr(Bzl)Asp(tBu)-OH, (36 mg, 69 μmol) and propargylamine (103.5 μmol, 6.8 μL). After lyophilization, the title compound was obtained as a white solid (7.4 mg, 21.3%). Analysis by ESI+ (Expected [M + H]+ = 599.26. Observed [M + H]+ = 599.3). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 12.39 (s, 1H), 8.40 (t, J = 6.9, 8.3 Hz, 1H), 8.27 (m, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.78 (s, 1H), 7.30 (m, 5H), 7.22 (t, J = 7.0, 8.2 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.85 (t, J = 8.4, 8.5 Hz, 1H), 4.71 (dt, 47.2 Hz, 2H), 4.46 (m, 2H), 4.29 (m, 2H), 4.19 (dd, J = 15.6 Hz, 1H), 3.83 (dt, J = 37.4 Hz, 1H), 3.70 (s, 3H), 3.46 (dd, J = 68.5 Hz, 2H), 2.70 (m, 1H), 2.54 (m, 1H), 1.05 (d, J = 6.3 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.76, 170.71, 170.31, 169.80, 157.73, 157.08, 145.05, 138.62, 130.61, 128.08, 127.53, 124.31, 123.06, 120.19, 116.32, 110.64, 82.56, 81.22, 80.62, 74.64, 70.36, 70.13, 57.32, 55.27, 49.95, 49.56, 49.51, 40.43, 36.80, 36.08, 34.57, 34.48, 16.11.
Synthesis of Methyl 3-(3-(benzyloxy)-2-(2-(2-methoxyphenyl)acetamido)butanamido)-4-(((1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-4-oxobutanoate (17).
Peptide coupling performed with 2MP-Thr(Bzl)Asp(OMe)-OH, (33 mg, 69 μmol) and propargylamine (103.5 μmol, 6.8 μL). Post-click TFA deprotection was omitted. After lyophilization, the title compound was obtained as a white solid (22.3 mg, 50.7%). Analysis by ESI+ (Expected [M + H]+ = 635.25. Observed [M + H]+ = 635.72). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 8.42 (m, 1H), 8.27 (m, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.80 (dd, J = 47.9, 8.1 Hz, 1H), 7.30 (m, 5H), 7.23 (t, J = 7.9 Hz, 1H), 7.17 (m, 1H), 6.96 (d, J = 8.2 Hz, 1H), 6.87 (t, J = 7.4 Hz, 1H), 4.81 (m, 1H), 4.73 (q, J = 4.3, 3.7 Hz, 1H), 4.66 (dq, J = 9.5, 5.5, 4.4 Hz, 2H), 4.60 (q, J = 4.6 Hz, 1H), 4.47 (m, 2H), 4.36 (m, 2H), 4.20 (ddd, J = 40.0, 15.3, 5.7 Hz, 1H), 3.88 (dt, J = 42.1, 5.7 Hz, 1H), 3.71 (d, J = 2.0 Hz, 3H), 3.54 (d, J = 9.9 Hz, 3H), 3.50 (m, 2H), 2.69 (m, 2H), 1.05 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, DMSO-d6-(CD3)2SO) δ 172.22, 171.18, 170.78, 170.26, 157.54, 145.52, 145.43, 139.09, 131.15, 131.08, 128.54, 128.49, 127.99, 127.77, 124.77, 123.53, 120.65, 111.12, 83.02, 81.68, 75.11, 70.60, 57.79, 55.74, 50.26, 49.98, 37.27, 36.55, 35.04, 16.58.
Synthesis of 3-(Benzyloxy)-N-(1-(((1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-1-oxopropan-2-yl)-2-(2-(2-methoxyphenyl)acetamido)butanamide (18).
Peptide coupling performed with 2MP-Thr(Bzl)Ala-OH, (29 mg, 69 μmol) and propargylamine (103.5 μmol, 6.8 μL). Post-click TFA deprotection was omitted. After lyophilization, the title compound was obtained as a white solid (19 mg, 53.7%). Analysis by ESI+ (Expected [M + H]+ = 555.27. Observed [M + H]+ = 555.70). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 8.37 (dt, J = 39.2 Hz, 1H), 8.00 (dd, J = 63.3 Hz, 1H), 7.87 (s, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.28 (m, 5H), 7.22 (t, J = 8.1, 7.3 Hz, 1H), 7.16 (d, J = 7.8 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.86 (t, J = 7.3, 7.5 Hz, 1H), 4.73 (dt, J = 47.5 Hz, 2H), 4.62 (dt, J = 28.1 Hz, 2H), 4.49 (t, J = 12.0, 8.9 Hz, 1H), 4.42 (m, 2H), 4.31 (m, 2H), 3.71 (s, 3H), 3.48 (m, 2H), 3.88 (dt, J = 32.6 Hz, 1H), 1.17 (dd, J = 31.7 Hz, 3H), 1.05 (t, J = 3.2, 5.4 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.91, 170.52, 169.33, 157.06, 138.57, 130.66, 128.04, 127.40, 127.37,127.24, 124.31, 120.14, 110.59, 82.43, 81.32, 74.90, 74.60, 70.34, 69.99, 56.68, 55.23, 49.93, 48.15, 39.92, 39.50, 36.90, 34.24, 18.26, 16.07.
Synthesis of 3-(3-(Benzyloxy)-2-(2-(2-methoxyphenyl)-acetamido)butanamido)-4-((3-(1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)phenyl)amino)-4-oxobutanoic Acid (19).
Peptide coupling performed with 2MP-Thr(Bzl)Asp(tBu)-OH, (36 mg, 69 μmol) and 3-ethynylaniline (103.5 μmol, 12.1 mg). After lyophilization, the title compound was obtained as a white solid (2.6 mg, 8.3%). Analysis by ESI+ (Expected [M + H]+ = 661.27. Observed [M + H]+ = 662.37). 1H NMR (500 MHz, DMSO) δ 12.44 (s, 1H), 9.94 (d, J = 18.5 Hz, 1H), 8.59 (d, J = 8.0 Hz, 1H), 8.38 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 7.3 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.50 (q, J = 9.9, 9.2, 9.2 Hz, 2H), 7.25 (m, 8H), 6.93 (dd, J = 8.1, 2.9 Hz, 1H), 6.84 (m, 1H), 4.92 (t, J = 4.7, 4.7 Hz, 1H), 4.83 (t, J = 4.7, 4.7 Hz, 1H), 4.78 (m, 2H), 4.73 (t, J = 4.7, 4.7 Hz, 1H), 4.48 (m, 2H), 4.34 (m, 1H), 3.90 (m, 1H), 3.69 (d, J = 5.0 Hz, 3H), 3.60 (d, J = 15.3 Hz, 1H), 3.49 (dd, J = 15.1, 6.1 Hz, 1H), 2.82 (m, 1H), 2.62 (ddd, J = 32.1, 16.6, 7.8 Hz, 1H), 1.09 (dd, J = 6.3, 2.9 Hz, 3H). 13C NMR (125 MHz, DMSO) δ 172.11, 171.34, 169.77, 168.59, 157.56, 146.88, 139.07, 138.98, 131.45, 131.16, 129.67, 128.53, 128.48, 128.46, 128.42, 128.04, 127.98, 127.74, 127.68, 122.24, 120.63, 119.48, 116.58, 111.12, 81.65, 75.44, 70.64, 58.13, 55.74, 50.73, 50.57, 39.49, 37.43, 37.28, 16.65.
Synthesis of 3-(3-(Benzyloxy)-2-(2-(2-methoxyphenyl)-acetamido)butanamido)-4-((4-(1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)phenyl)amino)-4-oxobutanoic Acid (20).
Peptide coupling performed with 2MP-Thr(Bzl)Asp(tBu)-OH, (36 mg, 69 μmol) and 4-ethynylaniline (103.5 μmol, 12.1 mg). After lyophilization, the title compound was obtained as a white solid (2.9 mg, 9.3%). Analysis by ESI+ (Expected [M + H]+ = 661.27. Observed [M + H]+ = 662.37). 1H NMR (500 MHz, DMSO) δ 12.46 (d, J = 18.5 Hz, 1H), 9.88 (d, J = 30.3 Hz, 1H), 8.64 (d, J = 8.1 Hz, 1H), 8.38 (d, J = 7.6 Hz, 1H), 7.98 (d, J = 6.9 Hz, 1H), 7.80 (d, J = 8.2 Hz, 1H), 7.74 (d, J = 8.6 Hz, 1H), 7.70 (d, J = 4.2 Hz, 2H), 7.65 (d, J = 8.5 Hz, 1H), 7.32 (d, J = 4.3 Hz, 3H), 7.27 (d, J = 4.4 Hz, 3H), 7.18 (m, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.87 (dt, J = 14.8, 7.4, 7.4 Hz, 1H), 4.91 (m, 1H), 4.82 (dt, J = 6.5, 3.8, 3.8 Hz, 1H), 4.77 (m, 2H), 4.72 (m, 1H), 4.49 (m, 2H), 3.90 (m, 1H), 3.70 (d, J = 4.2 Hz, 3H), 3.50 (dd, J = 15.1, 8.4 Hz, 2H), 2.82 (td, J = 16.1, 16.1, 5.7 Hz, 1H), 2.62 (m, 1H), 1.09 (d, J = 6.3 Hz, 3H). 13C NMR (125 MHz, DMSO) δ 172.13, 172.08, 171.31, 170.51, 157.56, 146.77, 139.10, 138.82, 131.29, 131.18 128.54, 128.51, 128.49, 128.46, 128.08, 128.01, 127.77, 127.73, 125.94, 125.91, 121.71, 120.14, 120.07, 111.15, 83.03, 78.68, 70.94, 58.49, 55.76, 55.73, 50.99, 36.56, 36.50, 16.62.
Synthesis of 3-(3-(Benzyloxy)-2-(2-(2-methoxyphenyl)-acetamido)butanamido)-4-oxo-4-(prop-2-yn-1-ylamino)-butanoic Acid (21, Alkyne Precursor to [18F]-16).
Peptide coupling performed with 2MP-Thr(Bzl)Asp(tBu)-OH, (36 mg, 69 μmol) and propargylamine (103.5 μmol, 6.8 μL). Click reaction was omitted, tert-butyl deprotection and purification performed as described above. After lyophilization, the title compound was obtained as a white solid (17.1 mg, 37.8%). Analysis by ESI+ (Expected [M + Na]+ = 532.21. Observed [M + Na]+ = 531.98). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 10.02 (s, 1H), 8.31 (dt, J = 41.8 Hz, 1H), 7.76 (q, J = 3.6, 4.9, 7.5 Hz, 1H), 7.29 (m, 5H), 7.22 (t, J = 8.0, 7.8 Hz, 1H), 7.17 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 8.9 Hz, 1H), 6.87 (t, J = 7.3, 7.3 Hz, 1H), 4.43 (t, J = 7.8, 5.3 Hz, 2H), 4.50 (d, J = 11.8 Hz, 2H), 4.30 (m, 1H), 3.88 (dd, J = 35.4 Hz, 2H), 3.82 (m, 1H), 3.78 (dt, J = 15.11 Hz, 1H), 3.72 (s, 3H), 3.49 (m, 2H), 1.15 (dd, J = 30.3 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 171.72, 170.48, 169.31, 157.08, 138.60, 130.68, 128.07, 128.04, 127.44, 127.39, 127.27, 124.35, 120.18, 110.62, 80.90, 74.99, 74.67, 73.07, 70.40, 70.05, 56.46, 55.27, 48.02, 36.94, 27.93, 18.24, 16.10.
Synthesis of Methyl 3-(3-(Benzyloxy)-2-(2-(2-methoxyphenyl)acetamido)butanamido)-4-oxo-4-(prop-2-yn-1-ylamino)butanoate (Alkyne PrecuRsor to [18F]-17).
Peptide coupling performed with 2MP-Thr(Bzl)Asp(OMe)-OH, (33 mg, 69 μmol) and propargylamine (103.5 μmol, μL). Click reaction was omitted, tert-butyl deprotection and purification performed as described above. After lyophilization, the title compound was obtained as a white solid (18.6 mg, 52.5%). Analysis by ESI+ (Expected [M + Na]+ = 546.22. Observed [M + Na]+ = 547.18). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 8.36 (t, J = 7.6, 9.4 Hz, 1H), 8.20 (m, 1H), 7.76 (dd, J = 32.2 Hz, 1H), 7.30 (m, 5H), 7.22 (t, J = 7.6, 7.6 Hz, 1H), 7.17 (d, J = 3.8 Hz, 1H), 6.94 (d, J = 8.3, 1H), 6.87 (t, J = 7.6, 7.0 Hz, 1H), 4.65 (m, 1H), 4.47 (dd, J = 20.0 Hz, 2H), 4.37 (dq, J = 27.8 Hz, 1H), 3.90 (m, 2H), 3.71 (s, 3H), 3.53 (s, 3H), 3.06 (t, J = 2.5, 2.6 Hz, 1H), 2.73 (dd, J = 16.6 Hz, 1H), 2.54 (m, 2H), 1.03 (dd, J = 15.5 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 170.58, 170.41, 169.71, 169.62, 157.05, 138.51, 130.61, 128.07, 127.45, 127.27, 124.26, 120.18, 110.63, 80.72, 80.60, 74.76, 73.02, 70.37, 70.13, 57.07, 56.48, 55.27, 51.50, 49.27, 36.84, 35.88, 28.08, 15.97.
Synthesis of 3-(Benzyloxy)-2-(2-(2-methoxyphenyl)-acetamido)-N-(1-oxo-1-(prop-2MP-yn-1-ylamino)propan-2-yl)-butanamide (Alkyne Precursor to [18F]-18).
Peptide coupling performed with 2MP-Thr(Bzl)Ala-OH, (29 mg, 69 μmol) and propargylamine (103.5 μmol, 6.8 μL). Click reaction was omitted, tert-butyl deprotection and purification performed as described above. After lyophilization, the title compound was obtained as a white solid (24.8 mg, 78.7%). Analysis by ESI+ (Expected [M + Na]+ = 488.22. Observed [M + Na]+ = 488.61). 1H NMR (600 MHz, DMSO, d6-(CD3)2SO): δ 8.37 (t, J = 5.6, 5.6 Hz, 1H), 8.30 (t, J = 5.5, 5.5 Hz, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.30 (m, 5H), 7.22 (m, 1H), 6.96 (d, J = 1.2 Hz, 1H), 6.94 (d, J = 1.2 Hz, 1H), 6.86 (d, J = 1.1 Hz, 1H), 4.52 (s, 1H), 4.49 (s, 1H), 4.44 (d, J = 11.8 Hz, 2H), 4.30 (m, 1H), 3.94 (dt, J = 6.3, 3.2, 3.2 Hz, 1H), 3.88 (m, 1H), 3.72 (d, J = 2.2 Hz, 3H), 3.48 (dd, J = 15.0, 4.8 Hz, 2H), 3.32 (s, 1H), 1.18 (dd, J = 25.1, 7.0 Hz, 3H), 1.06 (dd, J = 8.5, 6.2 Hz, 3H). 13C NMR (125 MHz, DMSO, d6-(CD3)2SO): δ 172.15, 170.92, 169.78, 157.55, 139.08, 131.15, 128.55, 128.52, 128.46, 127.92, 127.87, 127.74, 124.83, 120.66, 111.11, 81.32, 75.31, 73.59, 70.70, 57.01, 55.76, 48.49, 37.43, 28.38, 18.73, 16.64.
HPLC in Vitro Caspase Cleavage Assay.
All caspases were purchased from BioVision, Inc. (Milpitas, CA, USA). Triplicate 1 mM solutions of each substrate were made in 5 μL total volume of DMSO in 1.5 mL microcentrifuge tubes. To this was added 45 μL of hydrolysis buffer (50 mM 4-[2-hydroxyethyl]piperazine-1-ethanesulfonic acid [HEPES], 50 mM NaCl, 10 mM ethylenedinitrilotetraacetic acid [EDTA], 10 mM DL-dithiothreitol [DTT], 0.1% 3-[[3-cholamidopropyl] dimethylammonio]-1-propanesulfonate [CHAPS], and 5% glycerol, pH 7.2) with or without 1 U of caspase enzyme per reaction. Reactions proceeded at 37 °C for 2 h. Caspases then were inactivated by 5 min incubation at 95 °C. Hydrolyzed substrate was detected using an Agilent 1100 Series HPLC equipped with a Phenomenex Gemini 5u C18 110A column. Substrates analyzed by gradient elution: ValAsp-containing substrates with 20–23% B From 0 to 27 min, 23–30%B from 27 to 30 min; Thr(Bzl)Asp-containing substrates with 30–40% B from 0 to 30 min (Buffer A: dH2O with 0.1% (v/v) TFA, Buffer B: CH3CN). Hydrolyzed 2MP-VD-OH and 2MP-TbD-OH had retention times of approximately 18.2 and 16.8 min, respectively. Total product was calculated by comparing to standard curves of 2MP-VD-OH or 2MP-TbD-OH as applicable. For calculating the Km of substrates, 0.125 to 3 mM of substrate was incubated with 300 nM caspase-3 enzyme. At time 0 and 30 min, the reaction volume was inactivated and hydrolysis measured as described above. Total hydroxide produced per min from 0 to 30 min was calculated and Km, Vmax, and kcat were determined by nonlinear regression (Michaelis–Menten) in GraphPad Prism 6.
Fluorescent in Vitro Caspase Cleavage Assay.
1 mM solutions of each substrate in 10 μL total volume of DMSO were transferred to triplicate wells of a Costar clear bottom black side 96 well plate. To each well was added 90 μL of hydrolysis buffer (50 mM HEPES, 50 mM NaCl, 10 mM EDTA, 10 mM DTT, 0.1% CHAPS, and 5% glycerol, pH 7.2) with or without 1 U of caspase enzyme per reaction. Fluorescence (380 ex, 500 em) was measured using a BioTek Synergy H4 plate reader at 37 °C for 2 h with 5 min reads. The fluorescence of the no caspase control was subtracted from all samples. Total free AFC was calculated by generating a standard curve from 1 × 10−3 to 1 × 10−7 M AFC. For calculating the Km of substrates, 0.125 to 2 mM of substrate was incubated with 525.5 nM caspase-3 enzyme. Total AFC produced per min from 0 to 30 min was calculated and Km, Vmax and kcat were determined by nonlinear regression (Michaelis–Menten) in GraphPad Prism 6.
Cell-Based Apoptosis Assay.
OVCAR-5 and OVCAR-8 cells were seeded at 50,000 cells per well into 8 well Nunc Lab-Tek II Chamber Slides (Thermo Fisher Scientific, Waltham, MA, USA). After 24 h, cells were treated with 8 μM (OVCAR-5) or 20 μM (OVCAR-8) cisplatin in the presence or absence of 20 μM pan-caspase inhibitor Z-VAD-FMK (Enzo Life Sciences, Inc., Exeter, UK). Negative control wells were treated with PBS. After 2 days, cells were treated with 50 μM of 1, 2, or Ac-DEVD-AFC and 8 μM Annexin-V-CF568 (Biotium, Fremont, CA, USA). After 2 h, cells were imaged in a humidified 37 °C chamber on an Olympus FV1000 with a 20× long working distance objective. AFC spectral capture range 478–529 nm, excitation 405 nm laser HV = 673, offset 7%. Annexin-V excitation 568 nm laser HV 429, offset 6%.
Immunoblot.
OVCAR-5 and OVCAR-8 cells were treated with various concentrations of cisplatin in cell growth media for different intervals. Cells were washed with PBS and incubated for 1 h in lysis buffer (50 mM HEPES, pH 7.0; 150 mM NaCl; 1.5 mM MgCl2; 1 mM EGTA; 10 mM NaF; 10 mM sodium pyrophosphate; 10% (v/v) glycerol; 1% (v/v) Triton X-100) plus protease and phosphatase inhibitors (1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 1 mM Na3VO4) on ice, and then centrifuged at 17,000g for 30 min at 4 °C. The protein concentration was assessed using a bicinchoninic acid (BCA) protein assay (Thermo Scientific, Waltham, MA). Total cell lysates were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were incubated with specific antibodies against caspase-3 and cleaved caspase-3 and actin as a loading control (Cell Signaling, Danvers, MA). Immunoblot analysis was performed with the indicated antibodies and visualized with an ECL enhanced chemiluminescence detection kit (GE Healthcare, Pittsburgh, PA).
Immunohistochemistry.
Mouse tissue slides were subjected to immunohistochemical staining according to the manufacturer’s protocol (Biocare Medical, Concord, CA). Sections 6 μm thick were cut from each mouse liver tissue block. Incubation at 60 °C for 20 min was used to restore antigenic reactivity followed by two 20 min incubations in xylene. After slides were rehydrated, antigen retrieval was performed in rodent de-cloaking for 10 min at 100 °C. Sections were incubated in rodent blocking for 60 min and then incubated with anticleaved caspase-3 (1:100, Cell Signaling, Danvers, MA) at 4 °C overnight. Antirabbit immunoglobulin secondary antibodies were then applied for 1 h at room temperature followed by washing 3 times in PBS for 10 min. DAB chromagen was added for 1 min per slide followed by 3 additional washes in PBS for 10 min and then hematoxylin staining was performed for 1 min per slide followed by 3 additional washes in PBS for 10 min. Finally, images were taken with an Olympus microscope.
Molecular Modeling.
Caspase-3 structure (PDB ID 1RHQ) was imported and chains A and B were prepared for docking studies using Protein Preparation Wizard of Molecular Operating Environment (MOE) software (version 2016.08). Ligand 2D structures were generated in ChemDraw Professional 16.0 and prepared for docking studies using MOE. A pharmacophore model based on crystallographic ligand was generated to assist docking of the ligands. Pharmacophore guided docking experiment was carried out where binding site was defined using crystallographic ligand position, placement settings were changed to pharmacophore with timeout set to 10,000 and number of return poses were set to 10,000. For flexible docking, refinement settings were changed to Induced Fit method with cutoff of 5 Å and side chains were set to tethered setting with weight of 1. For termination criterion, gradient was set to 0.1 and iterations were set to 1000. Force constant of 100 and radius offset of 0.5 were used as pharmacophore restraints. Out of the 250 final poses, 25 poses were reported for the analysis.
Radiochemistry.
Radiosyntheses were performed with a TracerLab FX (General Electric Healthcare, Münster, Germany) automatic module as previously described.23 Briefly, aqueous [18F]-fluoride was acquired from the MD Anderson Cyclotron Radiochemistry Facility and adsorbed onto a preconditioned Sep-PAK Light QMA Cartridge (ABX GmbH, Radeberg, Germany). [18F]-fluoride was eluted into the reaction vial with 700 μL of a potassium carbonate Kryptofix 2.2.2. solution (52.8 mg of K2CO3, 240.1 mg of Kryptofix 2.2.2. in 4 mL of water, 16 mL of CH3CN). The solution was dried under vacuum and nitrogen flow at 60 °C for 2 min. 500 μL of dry CH3CN was added and then the mixture was azeotropically dried at 120 °C for 3 min. Addition of 2-azidoethyltosylate precursor (5 mg) in 500 μL CH3CN to the dried [18F]-fluoride and stirring at 80 °C for 15 min produced [18F]2-fluoroethylazide. The volatile [18F]2-fluoroethylazide was distilled under N2 flow for 5–10 min at 60 °C into a receiving vial containing click solution (CuSO4 [50 μL; 35 mg/mL in water], sodium ascorbate [50 μL; 174 mg/mL in PBS], TBTA [13 μL; 100 mg/mL in DMF], piperidine [13 μL; 20% in DMF]) and the alkyne precursor (830 nmol in 25 μL of DMF). Click reaction proceeded at room temperature for 20 min. The mixture was transferred into a plastic solid phase scavenging reactor containing an azide scavenging resin (64 mg) with the click solution described above. The slurry was agitated for 20 min at room temperature after which the filtrate was collected and resin washed with 700 μL DMF. The combined filtrate was transferred into a quenching vial containing 0.085% (v/v) H3PO4 in water (15 mL) before being loaded onto a light C18 cartridge (Sep-PAK Light, Waters, Milford, USA) prewashed with EtOH (3 mL) and water (6 mL). The cartridge was washed with 6 mL of water, dried under nitrogen, and eluted with 1 mL of ethanol through a 0.2 μm syringe filter. Activity was determined by dose calibrator and quality control performed by analytical radio-HPLC on a C18 column (Econosil C18, 10 μm, 250 mm, 4.6 mm), with a water (0.1% (v/v) TFA) and CH3CN (0.1% (v/v) TFA) gradient (5% B → 60% B in 30 min) and a flow rate of 1 mL/min. The identity of the radiolabeled compound was confirmed by coelution of the cold standard (synthesis described above). A standard curve was constructed on the same column using a series of starting material concentrations to determine the relationship between peak area (UV detection) and precursor concentration. This standard curve was then used to determine the specific activity of the final formulated product.
Synthesis of 2-Fluoroethylazide.
2-Fluoroethylazide was synthesized according to the protocol described by Glaser et al.22 2-Fluoroethyltosylate (128 mg, 586 μmol) was dissolved in anhydrous DMF (10 mL) and reacted with NaN3 (113 mg, 1.79 mmol) for 48 h under nitrogen. The residual solid was filtered off and 2-fluoroethylazide stored under nitrogen and used directly with no additional purification. Analysis by ESI+ (Expected [M + H]+ = 90.0. Observed [M + H]+ = 90.7). Warning - this compound is potentially explosive and should not be concentrated to dryness.
Synthesis of 2-Azidoethyl Tosylate.
2-Azidoethyl tosylate was synthesized according to the protocol described by Demko and Sharpless with minor modifications.21 2-Bromoethanol (2.5 g, 20 mmol) was refluxed with NaN3 (1.56 g, 24 mmol) in H2O (6 mL) overnight. The reaction was cooled to room temperature, dried over MgSO4, and extracted twice with DCM. The organic layer was dried over MgSO4 and filtered to give crude 2-azidoethanol in DCM. To this, p-toluenesulfonyl chloride (3.9 g, 20 mmol) and triethylamine (4 mL, 28 mmol) were added. The reaction was stirred for 90 min under nitrogen at room temperature and then quenched with glycine (300 mg) for 2 h at room temperature. The organic layer was washed twice with 1 M NaOH, dried over MgSO4, and concentrated by rotary evaporation. The crude product was purified by flash chromatography on silica gel (EtOAc:Hexanes, 3:7) to afford the title compound as a pale yellow oil (892 mg, 19% yield). Analysis by ESI+ (Expected [M + H]+ = 242.05. Observed [M + H]+ = 241.85).
In Vitro Hydrolysis of [18F]-16.
After radiosynthesis, 50 μCi (25 μL) of [18F]-16 was added to 75 μL of hydrolysis buffer containing 2 μg/mL caspase-3 enzyme in triplicate. This mixture was incubated at 37 °C for 1 h and enzyme inactivated by raising the temperature to 95 °C for 5 min. Then, 20 μL of each reaction was analyzed by radio-HPLC on a C18 column (Econosil C18, 10 μm, 250 mm, 4.6 mm), a water (0.1% (v/v) TFA) and CH3CN (0.1% (v/v) TFA) gradient (5% B → 60% B in 15 min) with a flow of 1 mL/min.
Detection of Apoptosis in Cisplatin-Treated Cells with [18F]-16 or [18F]-17.
OVCAR-5 and OVCAR-8 cells were seeded at 100,000 cells per well into 12 well plates. After 24 h, cells were treated with 0–50 μM cisplatin in the presence or absence of 20 μM pan-caspase inhibitor Z-VAD-FMK. Negative control wells were treated with PBS. After 2 days of treatment, 5 μCi of [18F]-16 or [18F]-17 in 1 mL serum free media was added per well, followed by 1 h incubation at 37 °C. Cells then were washed 3 × 5 min with 1 mL warmed (37 °C) or cold (4 °C) serum free media. Cells then were lysed by addition of 1 mL of 1N NaOH with 0.5% (w/v) SDS and orbital shaking at 220 rpm for 5 min. Then, 800 μL of lysate was transferred to scintillation vials and decay events measured using a Packard Cobra Quantum Gamma Counter. After decay of the radioisotope, lysate A280 protein concentration was measured using a NanoDrop1000. All CPM data was normalized to the sample protein concentration.
PET/CT of Jo2-Treated Mice.
Female athymic nude mice were anesthetized and then injected intravenously with 10 μg of Jo2 antibody to trigger liver apoptosis.30 After 2 h, mice were anesthetized and intravenously injected with ~100 μCi of [18F]-16 or [18F]-18. Dynamic PET images were acquired for 30 min followed by a 5 min CT scan on a Bruker Albira PET/CT/SPECT Preclinical Imaging System and reconstructed using an iterative MLEM algorithm. Volumes of interest (VOI), representative images, and time-activity curves were generated using PMOD v 3.610 software. VOI were drawn manually to encompass the heart, liver, kidneys, gastrointestinal tract, and bladder. These VOIs were used to calculate the %ID/g for each organ during the course of the dynamic PET acquisition.
Mathematical Modeling of Dynamic PET/CT Data.
A coupled multicompartment model of transfer of radiotracer from the blood pool to the liver and from the liver to the large intestine was built. A nonlinear ODE solver with boundary conditions that all rate constants must be >0 was applied (Mathematica) to extract the influx and efflux rates from the liver and large intestine. Biological parameters supplied to the model included VOI volume, average tracer concentration, animal weight. Adjusted R2 were typically >0.9. The model was trained and validated on one mouse that was injected with 98 μCi of [18F]-16 and treated with vehicle and one mouse that was injected with 95 μCi of [18F]-16 and treated with Jo2. The initial conditions and modeling for each individual animal was then done blinded to the tracer injected and the treatment. Rate constants were compared using a priori selected test for difference between liver efflux rates in Jo2 treated vs untreated animals.
Code Availability.
Mathematica notebooks are supplied in the Supporting Information.
Statistical Analysis.
Statistical analysis was performed using Graphpad Prism 6. All bar graphs represent means ± standard deviation. Slope comparisons between linear regressions performed with ANCOVA tests. Comparison of slopes of nonlinear regressions to zero performed with an F test. Student’s two-tailed t test used for comparison of kinetic constants.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by UTMDACC startup funds (SWM), a UTMDACC Moonshot Knowledge Gap Pilot Project (SWM), 1R21CA181994-01 (SWM, ZL), 2R44CA206771 (SWM), and a G.E. In-kind Multi-investigator Imaging (MI2) Research Award (SWM). AO was supported by a cancer prevention educational award (R25T CA057730, Shine Chang). ZL, HY, and RB were supported by the MD Anderson SPORE in Ovarian Cancer NCI P50 CA 83639, the National Foundation for Cancer Research, the philanthropic support from generous donations from Stuart and Gaye-Lynn Zarrow, the Mossy foundation and the Roberson endowment. MD Anderson’s Nuclear Magnetic Resonance Facility and Small Animal Imaging Facility (SAIF) are supported by the MD Anderson Cancer Center Support Grant CA016672 (Pisters).
ABBREVIATIONS USED
- Tb
threonine O-benzyl ester
- FMK
fluoromethylketone
- AFC
7-amino-4-(trifluoromethyl)coumarin
- GI
gastrointestinal tract
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.8b00514.
Structure and synthesis of compoounds, kinetic parameters, physicochemical properties, RadioHPLC analysis, bodistribution of F18-labeled probes (PDF) Mathematica notebooks (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Fuchs Y, and Steller H (2011) Programmed Cell Death in Animal Development and Disease. Cell 147 (4), 742–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Favaloro B, Allocati N, Graziano V, Di Ilio C, and De Laurenzi V (2012) Role of Apoptosis in Disease. Aging 4 (5), 330–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).He B, Lu N, and Zhou Z (2009) Cellular and Nuclear Degradation during Apoptosis. Curr. Opin. Cell Biol 21 (6), 900–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).de Almagro MC, and Vucic D (2012) The Inhibitor of Apoptosis (IAP) Proteins Are Critical Regulators of Signaling Pathways and Targets for Anti-Cancer Therapy. Exp. Oncol 34 (3), 200–211. [PubMed] [Google Scholar]
- (5).Walsh JG, Cullen SP, Sheridan C, Lüthi AU, Gerner C, and Martin SJ (2008) Executioner Caspase-3 and Caspase-7 Are Functionally Distinct Proteases. Proc. Natl. Acad. Sci. U. S. A 105 (35), 12815–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, et al. (1997) A Combinatorial Approach Defines Specificities of Members of the Caspase Family and Granzyme B. Functional Relationships Established for Key Mediators of Apoptosis. J. Biol. Chem 272 (29), 17907–17911. [DOI] [PubMed] [Google Scholar]
- (7).Micale N, Vairagoundar R, Yakovlev AG, and Kozikowski AP (2004) Design and Synthesis of a Potent and Selective Peptidomimetic Inhibitor of Caspase-3. J. Med. Chem. 47 (26), 6455–6458. [DOI] [PubMed] [Google Scholar]
- (8).Bramlett HM, and Dietrich WD (2015) Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 32 (23), 1834–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Neves AA, and Brindle KM (2014) Imaging Cell Death. J. Nucl. Med 55 (1), 1–4. [DOI] [PubMed] [Google Scholar]
- (10).Zhou D, Chu W, Rothfuss J, Zeng C, Xu J, Jones L, Welch MJ, and Mach RH (2006) Synthesis, Radiolabeling, and in Vivo Evaluation of an 18F-Labeled Isatin Analog for Imaging Caspase-3 Activation in Apoptosis. Bioorg. Med. Chem. Lett 16 (19), 5041–5046. [DOI] [PubMed] [Google Scholar]
- (11).Liu S, Edgerton SM, Moore DH, and Thor AD (2001) Measures of Cell Turnover (Proliferation and Apoptosis) and Their Association with Survival in Breast Cancer. Clin. Cancer Res 7 (6), 1716–1723. [PubMed] [Google Scholar]
- (12).Bauwens M, De Saint-Hubert M, Devos E, Deckers N, Reutelingsperger C, Mortelmans L, Himmelreich U, Mottaghy FM, and Verbruggen A (2011) Site-Specific 68Ga-Labeled Annexin A5 as a PET Imaging Agent for Apoptosis. Nucl. Med. Biol 38 (3), 381–392. [DOI] [PubMed] [Google Scholar]
- (13).Reshef A, Shirvan A, Akselrod-Ballin A, Wall A, and Ziv I (2010) Small-Molecule Biomarkers for Clinical PET Imaging of Apoptosis. J. Nucl. Med 51 (6), 837–840. [DOI] [PubMed] [Google Scholar]
- (14).Bullok KE, Gammon ST, Violini S, Prantner AM, Villalobos VM, Sharma V, and Piwnica-Worms D (2006) Permeation Peptide Conjugates for in Vivo Molecular Imaging Applications. Mol. Imaging 5 (1), 1–15. [PubMed] [Google Scholar]
- (15).Méthot N, Vaillancourt JP, Huang J, Colucci J, Han Y, Ménard S, Zamboni R, Toulmond S, Nicholson DW, and Roy S (2004) A Caspase Active Site Probe Reveals High Fractional Inhibition Needed to Block DNA Fragmentation. J. Biol. Chem 279 (27), 27905–27914. [DOI] [PubMed] [Google Scholar]
- (16).Poreba M, Kasperkiewicz P, Snipas SJ, Fasci D, Salvesen GS, and Drag M (2014) Unnatural Amino Acids Increase Sensitivity and Provide for the Design of Highly Selective Caspase Substrates. Cell Death Differ. 21 (9), 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Becker JW, Rotonda J, Soisson SM, Aspiotis R, Bayly C, Francoeur S, Gallant M, Garcia-Calvo M, Giroux A, Grimm E, et al. (2004) Reducing the Peptidyl Features of Caspase-3 Inhibitors: A Structural Analysis. J. Med. Chem 47 (10), 2466–2474. [DOI] [PubMed] [Google Scholar]
- (18).Elmore S (2007) Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol 35 (4), 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang LJ, Hao YZ, Hu CS, Ye Y, Xie QP, Thorne RF, Hersey P, and Zhang XD (2008) Inhibition of Apoptosis Facilitates Necrosis Induced by Cisplatin in Gastric Cancer Cells. Anti-Cancer Drugs 19 (2), 159–166. [DOI] [PubMed] [Google Scholar]
- (20).Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ (2001) Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev 46 (1–3), 3–26. [DOI] [PubMed] [Google Scholar]
- (21).Demko ZP, and Sharpless KB (2001) An Intramolecular [2 + 3] Cycloaddition Route to Fused 5-Heterosubstituted Tetrazoles. Org. Lett 3 (25), 4091–4094. [DOI] [PubMed] [Google Scholar]
- (22).Glaser M, and Arstad E (2007) Click Labeling” with 2-[18F]Fluoroethylazide for Positron Emission Tomography. Bioconjugate Chem. 18 (3), 989–993. [DOI] [PubMed] [Google Scholar]
- (23).Pisaneschi F, Kelderhouse LE, Hardy A, Engel BJ, Mukhopadhyay U, Gonzalez-Lepera C, Gray JP, Ornelas A, Takahashi TT, Roberts RW, et al. (2017) Automated, Resin-Based Method to Enhance the Specific Activity of Fluorine-18 Clicked PET Radiotracers. Bioconjugate Chem. 28 (2), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pedersen DS, and Abell A (2011) 1,2,3-Triazoles in Peptidomimetic Chemistry. Eur. J. Org. Chem 2011 (13), 2399–2411. [Google Scholar]
- (25).Biniossek ML, Nägler DK, Becker-Pauly C, and Schilling O (2011) Proteomic Identification of Protease Cleavage Sites Characterizes Prime and Non-Prime Specificity of Cysteine Cathepsins B, L, and S. J. Proteome Res 10 (12), 5363–5373. [DOI] [PubMed] [Google Scholar]
- (26).Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Brömme D, Ellman JA, and Craik CS (2006) Substrate Profiling of Cysteine Proteases Using a Combinatorial Peptide Library Identifies Functionally Unique Specificities. J. Biol. Chem 281 (18), 12824–12832. [DOI] [PubMed] [Google Scholar]
- (27).Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al. (2012) Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 41, D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, and Nagata S (1993) Lethal Effect of the Anti-Fas Antibody in Mice. Nature 364 (6440), 806–809. [DOI] [PubMed] [Google Scholar]
- (29).Goetz M, Ansems JV, Galle PR, Schuchmann M, and Kiesslich R (2011) In Vivo Real-Time Imaging of the Liver with Confocal Endomicroscopy Permits Visualization of the Temporospatial Patterns of Hepatocyte Apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol 301 (5), G764–72. [DOI] [PubMed] [Google Scholar]
- (30).Nishimura Y, Hirabayashi Y, Matsuzaki Y, Musette P, Ishii A, Nakauchi H, Inoue T, and Yonehara S (1997) In Vivo Analysis of Fas Antigen-Mediated Apoptosis: Effects of Agonistic Anti-Mouse Fas MAb on Thymus, Spleen and Liver. Int. Immunol 9 (2), 307–316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.