

Abstract
Promiscuous inhibitors of tyrosine protein kinases, proteases and phosphatases are useful reagents for probing regulatory pathways and stabilizing lysates as well as starting points for the design of more targeted agents. Ubiquitination regulates many critical cellular processes, and promiscuous inhibitors of deubiquitinases (DUBs) would be similarly valuable. The currently available promiscuous DUB inhibitors are highly reactive electrophilic compounds that can crosslink proteins. Herein we introduce diarylcarbonate esters as a novel class of promiscuous DUB inhibitors that do not have the liabilities associated with the previously reported compounds. Diarylcarbonates stabilize the high molecular weight ubiquitin pools in cells and lysates. They also elicit cellular phenotypes associated with DUB inhibition, demonstrating their utility in ubiquitin discovery. Diarylcarbonates may also be a useful scaffold for the development of specific DUB inhibitors.
Keywords: DUB inhibitors, USP7, USP9, activity profiling, Bcr Abl, Mdm2, p53
Ubiquitination is involved in many essential cellular processes, including protein degradation, localization, autophagy, transcriptional regulation and DNA repair.1, 2 Ubiquitination is a dynamic process regulated by the opposing actions of the ubiquitin ligases and deubiquitinating enzymes (DUBs). Ubiquitin ligases attach ubiquitin to target proteins via isopeptide bonds between the C-terminus of ubiquitin and Lys residues of the target protein. Ligases can attach additional ubiquitins to form polyubiquitin chains at any of the seven ubiquitin Lys residues or the N-terminus. The most common polyubiquitin chains are linked at K48 and K63. K48 chains direct the target protein to the 26S proteasome for degradation, whereas K63 chains are involved in autophagy and a diverse array of other signaling functions. DUBs remove ubiquitin from target proteins and disassemble polyubiquitin chains.3-5 Approximately 115 DUBs exist in humans, most of which are cysteine proteases related to papain. The two largest classes are ubiquitin C-terminal hydrolases (UCHLs) and ubiquitin specific proteases (USPs). In general, UCHLs hydrolyze C-terminally modified ubiquitin, whereas USPs typically cleave ubiquitin chains, although individual DUBs within both classes have varying substrate repertoires and chain specificities.6, 7
The ubiquitin-proteasome system controls the levels of many critical regulatory proteins.8 For example, p53 is a substrate for the ubiquitin ligase Mdm2.9 Mdm2 levels are amplified in many cancers, causing a corresponding decrease in p53, ultimately promoting survival and proliferation. Mdm2 is itself degraded in a ubiquitin-dependent process. USP7 removes ubiquitin from Mdm2, protecting it from degradation. Inhibition of USP7 decreases Mdm2 levels resulting in a corresponding increase in p53, inducing apoptosis.10 Another example is Bcr-Abl kinase, the pathogenic fusion protein created by the chromosome translocation that causes chronic myelogenous leukemia. Bcr-Abl is degraded by ubiquitin-dependent autophagy.11 Bcr-Abl is protected from degradation by USP9x and the inhibition of USP9x results in the degradation of Bcr-Abl.12, 13 Thus USP7 and USP9x are promising targets for cancer-specific chemotherapy.14 DUBs have also emerged as potential targets for the treatment of neurodegeneration, immunosuppression and infection.15-17 DUBs, like other cysteine proteases, are challenging targets for drug discovery. As yet only a handful of selective DUB inhibitors have been reported, mainly for USP7.18-22
Promiscuous inhibitors can also be useful tools as well valuable starting points for selective inhibitor design, as illustrated with staurosporine in protein kinases.23 Staurosporine is a promiscuous cell permeable protein kinase inhibitor that is used to block protein phosphorylation. A response to staurosporine can be diagnostic for the presence of a protein tyrosine kinase in a regulatory pathway. Staurosporine also inspired the development of midostaurin, a multi-target protein kinase inhibitor recently approved for the treatment of acute myeloid leukemia.24 as well as selective protein kinase inhibitors such as the drug imatinib (Gleevec). Similarly, promiscuous protease inhibitors such as pepstatin are routinely used to stabilize cell lysates, while the statin pharmacophore is utilized in AIDS drugs targeting HIV protease.25 Promiscuous DUB inhibitors would be similarly useful for stabilizing ubiquitination both in vitro and in vivo, as well as for the development of more selective inhibitors with therapeutic potential. Further, many DUBs have semi-redundant functions, which suggests that multi-target DUB inhibitors may be required for therapeutic efficacy.26, 27 Indeed, the cellular effects of the natural products phenethylisothiocyanate (PEITC), Δ−12-prostaglandin J2 (15dPGJ2) and curcumin can be attributed, at least in part, to multi-target DUB inhibition.13, 28-33 Several promiscuous DUB inhibitors are known (Figure 1A).5 Some of these compounds were initially reported as selective inhibitors, but are now known to inhibit several DUBs and some even have other enzyme targets.7, 33-37 The diaryl dienone found in G5 isopeptidase inhibitor 1 (G5) and curcumin is a particularly successful DUB pharmacophore. Derivatives AC17, b-AP15 and VLX1570 display in vivo anticancer activity, and VLX1570 advanced to clinical trials (now discontinued).27, 30, 38, 39 However, these compounds, together with PR-619 and WP1130, contain multiple electrophillic “warheads” that can react with other cellular targets and even result in protein crosslinking (Figure 1A).34, 37, 40 Indeed, insoluble protein aggregates have been observed in cells treated with WP1130, presumably the result of such cross-linking.41 Moreover, the enone reaction is effectively irreversible, amplifying off-target effects.40 New promiscuous DUB inhibitors are desired that do not have these liabilities.
Figure 1. DUB inhibitors.

A. Promiscuous DUB inhibitors. B. Proposed mechanism of diaryl carbonate inhibition. C. General scheme for synthesis of diarylcarbonates.
Carbonate esters inhibit chymotrypsin by forming a stable carbonylated enzyme that mimics the acylenzyme intermediate formed during the catalytic cycle.42 The carbonylated enzyme eventually hydrolyzes, but this process requires hours.42 These compounds do not inhibit the prototypical cysteine protease papain42 and therefore carbonate esters have not been considered a useful warhead for cysteine protease inhibition. Recognizing the success of the diaryl dienone pharmacophore, we surmised that diarylcarbonate esters might inhibit DUBs via an analogous reaction to form a kinetically stable thiocarbonate (Figure 1B). We screened a small set of diphenyl carbonate esters for the ability to stabilize high molecular weight ubiquitinated proteins (HMW-Ub) in lysates (Table 1).32, 43 Lysates were prepared from HEK 293T cells expressing N-terminally HA-tagged ubiquitin (HA-Ub) to facilitate the observation of ubiquitinated proteins. HMW-Ub proteins were readily observable, but decomposed with a half-life of 30 min (Figure 2A,B). Note that this decomposition would be expected to impact global surveys of ubiquitination (one potential application of a promiscuous DUB inhibitor). The proteasome inhibitor bortezomib failed to stabilize the HMW-Ub proteins, indicating that decomposition was the result of DUB activity (Figure 2C).44, 45 In contrast, the promiscuous DUB inhibitor G5 stabilized the HMW-Ub pool (Figure 2A and C).34 Similar stabilization of HMW-Ub was observed with two other promiscuous DUB inhibitors, NSC-632839 and ubiquitin-aldehyde (Figure 2C).34 In contrast, the USP14-specific inhibitor IU1 had little effect on decomposition of HMW-Ub pools.46 These observations demonstrate that the stabilization of HMW-Ub can be used to screen for DUB inhibition, as well as the value of promiscuous DUB inhibitors for stabilizing ubiquitinated proteins.
Table 1.
Structure-activity relationship of DUB inhibition.
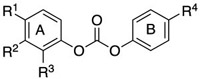 |
|||||
|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | R4 | EC50 (μM) |
| C1 | H | H | H | H | >150 a |
| C2 | H | OMe | H | OMe | >150 a |
| C3 | H | H | H | CH2NHBoc | >150 a |
| C4 | H | H | H | CH2NH2 | 210 ± 40 [250 ± 70] |
| C5 | H | H | H | CH2NH-4-ClBn | 300 ± 100 |
| C31 | H | Me | H | Me | >150 a |
| C11 | F | H | H | CH2NH2 | 300 ± 100 |
| C12 | CH3 | H | H | CH2NH2 | 250 ± 20 |
| C13 | CH3O | H | H | CH2NH2 | 260 ± 40 |
| C14 | Cl | H | H | CH2NH2 | 30 ± 10 [50 ± 10] |
| C15 | Br | H | H | CH2NH2 | 30 ± 10 [40 ± 10] |
| C16 | H | H | Cl | CH2NH2 | 60 ± 20 |
| C17 | 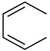 |
H | CH2NH2 | 20 ± 10 | |
| C18 | H | 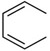 |
CH2NH2 | 40 ± 20 | |
| C23 | Cl | H | H | CH2NHBoc | >150 a |
| C19 | 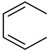 |
H | CH2NHBoc | >150 a | |
| C20 | 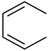 |
H | CH2NH-neo-Pent | 30 ± 10 | |
| C21 | 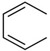 |
H | CH2NH-i-Bu | 70 ± 10 | |
| C22 | 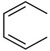 |
H | CH2-NH-(C=NH)NH2 | 50 ± 20 | |
| C6 | 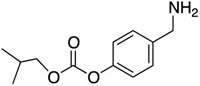 |
>500 | |||
 |
|||||
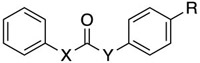
| Cmpd | X | Y | R | EC50 (μM) | |
| C8 | O | NH | CH2NH2 | >150 a | |
| C7 | NH | O | CH2NH2 | >150 a | |
| C10 | O | CH2 | CH2NH2 | >150 a | |
| C9 | CH2 | O | CH2NH2 | >150 a | |
The values of EC50 for the inhibition of the decomposition of high molecular weight ubiquitinated proteins (HMW-Ub) in lysates prepared from HEK 293T cells expressing HA-Ub. The values of EC50 are the mean ± s.e.m. of at least 3 independent experiments (see Figure 2 for representative experiment). Brackets denote the values of EC50 for the inhibition of the decomposition of HMW-Ub in lysates prepared from Cos-1 cells expressing HA-Ub.
No effect was observed at 150 μM, the highest concentration tested (the solubility limit).
Figure 2. Diarylcarbonates inhibit the decomposition of high molecular weight ubiquitinated proteins (HMW-Ub).

Lysates were prepared from HEK 293T cells expressing HA-ubiquitin. Samples were incubated at 37 °C and reactions were quenched by the addition of reducing Laemmli buffer. HMW-Ub was assessed by SDS-PAGE and immunoblotting with anti-HA antibody. A. Representative immunoblots measuring the decomposition of HMW-Ub in lysates treated with either the DMSO vehicle, G5 (10 μM) or C4 (500 μM). B. Plot of the decomposition of HMW-Ub in lysates treated with DMSO and C4 (500 μM) (N = 2; error bars denote range). C. The effect of proteasome and DUB inhibitors on the decomposition of HMW-Ub (N=2, average and range are shown). D = DMSO, B = bortezomib (a proteasome inhibitor); N = NSC 632839 (a broad spectrum DUB inhibitor); G5 = G5 isopeptidase inhibitor 1 (a broad spectrum DUB inhibitor); relative to the control (DMSO alone at time = 0). D. Representative decomposition of HMW-Ub after 2 h incubation in the presence of varying concentrations of C4. E. Quantitation of D.
Diphenylcarbonates C1-C3, C6 and C31 failed to substantially stabilize the HMW-Ub pool (Table 1). Intriguingly, the methylaminodiphenylcarbonate C4 (500 μM) prevented decomposition of the HMW-Ub pool, increasing half-life to ≥150 min (Figure 2A,B,D,E). This effect was dose-dependent, with an EC50 value of 210 μM. The analogous carbamates, C7 and C8, did not stabilize the HMW-Ub pool indicating that the carbonate is required for DUB inhibition (Table 1). The substitution of p-F (C11), p-Me (C12) and p-MeO (C13) on the A ring had no effect on inhibitory activity (Table 1). In contrast, the p-Cl (C14) and p-Br (C15) increased inhibitory potency by a factor of approximately 10 (Table 1). The half-life of HMW-Ub pool in HEK 293T cell lysates treated with C14 (250 μM) was >6 h. C14 and C15 were similarly more effective than C4 in Cos-1 lysates (Table 1). C14, C15 and the naphthyl compounds C17 and C18 all had values of EC50 less than 50 μM for stabilization of the endogenous K48-linked HMW ubiquitin pool in untransfected HEK 293T lysates. The superiority of p-Cl over the isosteric p-Me substitution suggested that electronic properties, rather than steric interactions, account for the improved activity of C14 and C15.
The screening results suggested that the amine group on ring B is required for activity (Table 1). Further exploration of the SAR of the B ring confirmed this finding. Modification of the amino group with a benzyl (C5) retained DUB inhibitory activity, while activity was lost with Boc modification (C3). Inhibitory activity was not recovered when ring A contained p-Cl (C23) or was replaced with naphthyl (C19). Inhibitory activity was retained with neopentyl substitution (compare C20 to C17), but isobutyl substitution was somewhat deleterious (C21). Lastly, replacement of the amine with a guanidinium group was also efficacious (C22). These observations suggest that the positive charge is required for effective DUB inhibition.
To assess the selectivity of DUB inhibition, lysates from HEK 293T cells treated with diarylcarbonates were analyzed by HA-Ub-VS activity profiling.38, 40, 47, 48 Treatment of HEK 293T cell lysates with HA-Ub-VS produced the characteristic pattern of protein bands at 300, 160-100, 45, 38 and 36 kDa, generally ascribed to USP9x (292 kDa), USP19 (146 kDa), USP7/8 (128 and 127 kDa, respectively), USP28/15 (122 and 112 kDa, respectively), UCHL5 (38 kDa), UCHL3 (26 kDa) and UCHL1 (25 kDa) (Figure 3A).48 C17 was a promiscuous DUB inhibitor, decreasing the labeling of all the bands. The bands attributed to USP9x, USP19, USP7/8 and USP28/17 appeared to be more sensitive to C17 than UCHL1 and UCHL3. The inhibition of USP7 was confirmed by immunoblotting (Figure 3B).
Figure 3. Characterization of DUB inhibition by C17.

A. A lysate of HEK 293T cells (1.5 mg/ml) was treated with C17 (25 μM) or DMSO followed by HA-Ub-VS (1.5 μM). Aliquots were removed and analyzed for HA. B. A HEK 293T cell lysate was incubated with C17, treated with HA-UbVS, analyze by SDS-PAGE and immunoblotted with anti-USP7. USP7* denotes USP7 modified by HA-Ub-VS. An intervening lane was removed for clarity. C. A lysate of HEK 293T cells (1.5 mg/ml) was treated with C17 (25 μM) or DMSO simultaneously with HA-Ub-VS (1.5 μM). Aliquots were removed and analyzed for HA. Intervening lane removed for clarity. D. A lysate of HEK 293T cells (15 mg/ml) was treated with either C17 (250 μM) or DMSO for 30 min. After this time, lysate was diluted tenfold and HA-Ub-VS (1.5 μM) was added. Aliquots were removed and analyzed by HA blot. E. Representative progress curve. Purified recombinant His6USP9x (0.7 nM) was mixed with Ub-AMC (300 nM) and C17 in 1% DMSO and reaction was monitored by measuring AMC release on a 96 well plate reader. Lines denote fits to a single step inactivation mechanism as described in Material and Methods.
We examined the kinetics of HA-Ub-VS labeling in order to determine if C17 forms kinetically stable DUB complexes as proposed in Figure 1. In the absence of C17, at least eight bands were observed when HEK 293T cell lysates were treated with HA-Ub-VS (Figure 3C). Labeling was largely complete within 5 min. When C17 (25 μM) and HA-Ub-VS were added simultaneously to the lysate, no inhibition of DUB labeling was observed (Figure 3C), indicating that HA-Ub-VS (1.5 μM) out-competed C17 (25 μM) under these conditions. However, labeling was reduced when lysate was pre-incubated with C17 (250 μM), then diluted 10-fold prior to HA-Ub-VS treatment (Figure 3D). Thus the DUB•C17 complexes were stable to dilution. HA-Ub-VS labeling recovered with longer incubation times (Figure 3D). These observations are consistent with the hypothesis that inhibition involves the formation and subsequent decomposition of thiocarbonylated enzymes. The labeling of UCHL5, UCHL3 and UCHL1 recovered within 15 min. Labeling of USP9x, USP19 and USP7/8 did not recover in 2 h (Figure 3D). These observations suggest that the selective inhibition of USPs over UCHLs may be due to different stabilities of the thiocarbonylated enzymes.
Inhibition was confirmed with USP7 catalytic domain (USP7CD) and USP9x recombinant enzymes. The diarylcarbonates were time dependent inhibitors of both DUBs (representative progress curve shown in Figure 3E). The progress curves were well described by a single step inactivation mechanism. In most cases, the values of kon for USP7CD were lower than those for USP9x (Table 2). Comparable values of kon were observed for commercially available DUB inhibitors, WP1130, NSC-632839, b-AP15 and PR619 (range 49-140 M−1s−1). None of the compounds were effective inhibitors of the prototypical cysteine protease papain or the related protease ficin (Figure 4A,B). C14, C15, and C22 also failed to inhibit SENP6-catalyzed deSUMOylation of RanGap (Figure 4C). The diaryl carbonates did not inhibit porcine pancreatic elastase but were substrates of the prototypical serine proteases trypsin and chymotrypsin (Figure 4D-F). C17 and C22 were equivalent substrates for chymotrypsin (Km = ~ 1 μM, kcat = 0.03 s−1). C17 was also a good substrate for trypsin (Km = 1 μM, kcat = 0.02 s−1). Surprisingly, C22, which contained a guanidinium moiety associated with trypsin substrates, displayed relatively poor turnover (Km = 0.2 μM, kcat = 0.001 s−1).
Table 2.
Kinetic parameters for the inhibition of recombinant USP7 catalytic domain (USP7CD) and USP9x.
| Cmpd | USP7CD | USP9x |
|---|---|---|
|
kon
(M−1 s−1) |
kon (M−1 s−1) |
|
| C14 | 13 ± 4 | 18 ± 2 |
| C15 | 8 ± 3 | 6 ± 1 |
| C17 | 9 ± 4 | 49 ± 3 |
| C18 | NI | 17 ± 1 |
| C20 | 21 ± 2 | NI |
| C22 | NI | 58 ± 3 |
| WP1130 | 66 ± 4 | 50 ± 30 |
| b-AP15 | n.d. | 140 ± 10 |
| PR619 | n.d. | 90 ± 20 |
| D12 prost | n.d. | NI |
| BITC a | n.d. | 170 ± 40 |
| PEITC a | n.d. | 150 ± 20 |
Data were fit to a single step inactivation mechanism as described in Material and Methods. NI, no inhibition; n.d., not determined. The values of kon for benzylisothiocyanate (BEITC) and phenethylisothiocyante (PEITC) are included for comparison.
Values from 13.
Figure 4. Specificity of diarylcarbonates.

A. Papain was preincubated with inhibitor (100 μM) for 30 min prior to addition of Z-Arg-AMC (300 μM). B. Ficin was preincubated with inhibitor (100 μM) for 30 min prior to addition of Z-Arg-AMC (300 μM). C. His6-tagged catalytic domain of SENP6 (aa 628-1112) (200 nM) was treated with inhibitor for 30 min at room temperature followed by addition of SUMOylated His6-tagged catalytic domain of RanGap1 containing the SUMO modification site (aa 418-588) (3 μM) for 10 min at 37 °C after which time lysates were quenched with Laemeli buffer and resolved by SDS-PAGE then blotted and probed with anti-His6 antibodies. D, E. Serine proteases catalyze the hydrolysis of C17 (squares) and C22 (circles). D. Diarylcarbonates were incubated in the presence and absence of chymotrypsin (80 nM). E. Velocity versus substrate concentration plot for trypsin catalyzed hydrolysis. F. Velocity versus substrate concentration plot for chymotrypsin catalyzed hydrolysis. The fits to the Michaelis-Menten equation are shown.
We performed cysteine reactivity profiling of HeLa lysates to further evaluate the promiscuity of diarylcarbonate C22.49 Cysteine reactivity profiling can assess reactivity changes to over 700 cysteines in the presence of the inhibitor, although few DUBs are detected because they react poorly with the cysteine-reactive iodoacetamide-alkyne (IAA) probe used for these studies. In this method, a lysate is treated with either DMSO (control) or inhibitor, followed by the IAA probe and incorporation of isotopic labels (DMSO light: inhibitor heavy). Samples are mixed, processed, and analyzed by mass spectrometry to monitor changes in cysteine reactivity between DMSO and inhibitor-treated samples. Light:heavy ratios (R) are obtained for each cysteine-containing peptide that is identified and R values ≥3 indicate a cysteine that is a significant target of the inhibitor. Eight hundred and eighty cysteines were detected in these experiments (Supporting Table 1), including thirty-three active site cysteines (e.g., OTUB1, tRNA cytosine methyl transferase, IMPDH2, CASP7, thioredoxin, GAPDH, peroxiredoxin, an E2 ligase and GST-omega), as well as twenty-eight metal binding sites and nitrosation sites. Only three cysteine residues were identified as targets in at least one of two independent experiments: C369 in D-3-phosphoglycerate dehydrogenase, C101 in DNA replication licensing factor MCM6 (observed in only one experiment), and C46 in proteasome subunit beta type-2 (observed in only one experiment). Modification of the 20S proteasome is unlikely to affect function/association with the 19S subunit as we found that the compounds had no effect on proteasome activity (see below). Thus these global cysteine reactivity profiling data show that C22, and presumably other diarylcarbonates, are not promiscuous electrophillic agents.
We also investigated the activity of the diarylcarbonates in the G76VUb-GFP assay, which monitors flux through the ubiquitin-proteasome system.50 The G76V mutation prevents DUB cleavage, such that GFP remains fused to Ub. As reported by others.50, 51 GFP fluorescence significantly increased when HEK 293T cells expressing G76VUb-GFP were treated with the proteasome inhibitor bortezomib. The magnitude of this increase is similar to that previously reported.51 No significant increase in GFP fluorescence was observed in HEK 293T cells treated with G5 isopeptidase inhibitor 1. C14, C15, C17 and C18 also failed to increase GFP fluorescence (Figure 5F). Thus these compounds do not inhibit the 26S proteasome.
Figure 5. C17 causes the accumulation of HMW-Ub and reduce the levels of Bcr-Abl in K562 cells.

A. The accumulation of K48-linked Ub in K562 leukemia cells treated with diphenyl carbonates (50 μM) for 2 h. B. As in A, but Bcr-Abl was measured by immunoblotting with anti-Abl antibodies. C. Dose dependence of the C17-induced depletion of Bcr-Abl. Intervening lane removed for clarity. D. Quantification of blots as in C. E. K562 cells were treated with C17 (50 μM) in the presence and absence of bortezomib (6 μM) for 4 h and Bcr-Abl was measured by immunoblotting with anti-Abl antibodies. F. Quantitation of blot in E. Significance: DMSO relative to C17 and bortezomib p=0.002; DMSO relative to C17, p = 0.03. G. Effect of C17 on the cell cycle as determined by FACS. Black bars represent apoptotic cells (sub G1) and grey bars show cells in G1.
We tested the effects of diarylcarbonates on K562 leukemia cells, MCF7 breast cancer cells and B16/F10 melanoma cells to demonstrate the utility of promiscuous DUB inhibitors in uncovering ubiquitin–mediated regulation. K562 cells depend on the oncogenic fusion protein Bcr-Abl kinase for survival.52 Four compounds, C14, C15, C17 and C18, caused the accumulation of K48-linked HMW-Ub in K562 cells (Figure 5A) as well as a decrease in Bcr-Abl (Figure 5B-D; C22 was not effective in HEK 293T cells, presumably because it was not permeable). The presence of bortezomib did not prevent Bcr-Abl degradation induced by C17, as expected for an autophagy-mediated process (Figure 5E,F). C17 also caused a dose-dependent increase in G1 and apoptotic (sub G1) cells after a 24 h incubation (Figure 6G). Importantly, the lysates used in our experiments were prepared with sonication in SDS to solubilize protein aggregates prior to PAGE analysis. Therefore the decrease in Bcr-Abl levels cannot be attributed to sequestration into insoluble aggregates as has been observed with WP1130.41
Figure 6. C17 reduces the levels of Mdm2 and cause the accumulation of P53 and P21 in MCF7 cells.

Intervening lanes were removed for clarity. A. MCF7 cells were treated with C17 (50 μM) for 4 h and K48 ubiquitin chains were measured by immunoblotting. B-F. MCF7 cells were treated with C17 for 4 h and the levels of Mdm2 (B), p53 (C, D and E) and p21 (F) were measured by immunoblotting. Note: error bars in E are too small to be clearly observable. G. MCF7 and B16/F10 cells were treated with C17 every 24 h. After 72 h, viable cells were measured by Alamar Blue®. H. The effects of C17 on the cell cycle as measured by FACS. I. The viability of MCF7 cells treated with products of C17 hydrolysis.
Both MCF7 and B16/F10 cells depend on Mdm2 for survival. As expected, C17 caused the accumulation of soluble HMW-Ub in MCF7 cells (Figure 6A). Mdm2 levels decreased with a concomitant increase in p53, as expected when USP7 is inhibited (Figure 6B-E). A robust increase in p21/WAF1 levels was also observed (Figure 6F), and cell viability decreased (Figure 6G).53 Similar results were obtained when B16/F10 cells were treated with C17 (Figure 6G,H). The hydrolysis products of C17, 2-naphthol and 4-aminomethylphenol were not cytotoxic (the remaining hydrolysis product is carbonic acid/CO2, which is already present in culture medium) (Figure 6I). Together, these experiments demonstrate that the diarylcarbonate inhibitors recapitulate known DUB inhibitor phenotypes, and will therefore be a useful tool for the discovery of new ubiquitin regulation.
The experiments described above demonstrate that diarylcarbonates inhibit DUBs and recapitulate cellular phenotypes reported for other DUB inhibitors. Previously reported promiscuous DUB inhibitors are reactive Michael acceptors, and many modify other cellular proteins. These off-target interactions are exacerbated by the irreversible nature of Michael adducts. In contrast, diarylcarbonates inhibit DUBs transiently. Although they can react with serine proteases (above and 54), diarylcarbonates have a relatively small off-target spectrum as observed in cysteine activity profiling. Importantly diarylcarbonates do not induce the accumulation of insoluble ubiquitin aggregates even at high concentrations. The diaryl carbonate motif is a malleable framework for inhibitor design, so further development may be able to focus the spectrum of inhibition and perhaps also reveal inhibitors of other cysteine proteases. Such specific inhibitors would also be valuable as research tools and potential drug candidates.
Supplementary Material
ACKNOWLEDGEMENTS
MJCL acknowledges a Howard Hughes Medical Institute International Student Fellowship and a Brandeis Sprout grant. This work was supported by the National Institutes of Health R01 GM100921 to LH. FEO is a co-founder and shareholder of UbiQ Bio BV.
Footnotes
A. Supplementary data
Supplementary data associated with this article can be found, in the online version.
Current address: Quality Assurance Technical Services, Bioverativ, a Sanofi Company, 225 Second Avenue, Waltham, MA 02451
REFERENCES AND NOTES
- 1.Ciechanover A Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2012;1824(1): 3–13. [DOI] [PubMed] [Google Scholar]
- 2.Komander D, Rape M. The Ubiquitin Code. Annual Review of Biochemistry. 2012;81(1): 203–229. [DOI] [PubMed] [Google Scholar]
- 3.Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17(1): 57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ndubaku C, Tsui V. Inhibiting the deubiquitinating enzymes (DUBs). Journal of medicinal chemistry. 2015;58(4): 1581–1595. [DOI] [PubMed] [Google Scholar]
- 5.D’Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther. 2015;147: 32–54. [DOI] [PubMed] [Google Scholar]
- 6.Eletr ZM, Wilkinson KD. Regulation of proteolysis by human deubiquitinating enzymes. Biochim Biophys Acta. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritorto MS, Ewan R, Perez-Oliva AB, et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat Commun. 2014;5: 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nature reviews Molecular cell biology. 2011;12(7): 439–452. [DOI] [PubMed] [Google Scholar]
- 9.Lee JT, Gu W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010;17(1): 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell biochemistry and biophysics. 2011;60(1-2): 61–68. [DOI] [PubMed] [Google Scholar]
- 11.Torgersen ML, Simonsen A. Autophagy: Friend or foe in the treatment of fusion protein-associated leukemias? Autophagy. 2013;9(12). [DOI] [PubMed] [Google Scholar]
- 12.Sun H, Kapuria V, Peterson LF, et al. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood. 2011;117(11): 3151–3162. [DOI] [PubMed] [Google Scholar]
- 13.Lawson AP, Long MJ, Coffey RT, et al. Naturally Occurring Isothiocyanates Exert Anticancer Effects by Inhibiting Deubiquitinating Enzymes. Cancer research. 2015;75(23): 5130–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mattern MR, Wu J, Nicholson B. Ubiquitin-based anticancer therapy: carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim Biophys Acta. 2012;1823(11): 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colland F The therapeutic potential of deubiquitinating enzyme inhibitors. Biochemical Society transactions. 2010;38(Pt 1): 137–143. [DOI] [PubMed] [Google Scholar]
- 16.Edelmann MJ, Nicholson B, Kessler BM. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev Mol Med. 2011;13: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31(19): 2373–2388. [DOI] [PubMed] [Google Scholar]
- 18.Lamberto I, Liu X, Seo HS, et al. Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem Biol. 2017;24(12): 1490–1500 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550(7677): 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kategaya L, Di Lello P, Rouge L, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550(7677): 534–538. [DOI] [PubMed] [Google Scholar]
- 21.Gavory G, O’Dowd CR, Helm MD, et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nature chemical biology. 2018;14(2): 118–125. [DOI] [PubMed] [Google Scholar]
- 22.O’Dowd CR, Helm MD, Rountree JSS, et al. Identification and Structure-Guided Development of Pyrimidinone Based USP7 Inhibitors. ACS Med Chem Lett. 2018;9(3): 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Omura S, Asami Y, Crump A. Staurosporine: new lease of life for parent compound of today’s novel and highly successful anti-cancer drugs. J Antibiot (Tokyo). 2018. [DOI] [PubMed] [Google Scholar]
- 24.Stone RM, Manley PW, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018;2(4): 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl). 2015;7: 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinstock J, Wu J, Cao P, et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Med Chem Lett. 2012;3(10): 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Arcy P, Brnjic S, Olofsson MH, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17(12): 1636–1640. [DOI] [PubMed] [Google Scholar]
- 28.Lawson AP, Bak DW, Shannon DA, et al. Identification of deubiquitinase targets of isothiocyanates using SILAC-assisted quantitative mass spectrometry. Oncotarget. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasima N, Aggarwal BB. Targeting proteasomal pathways by dietary curcumin for cancer prevention and treatment. Curr Med Chem. 2014;21(14): 1583–1594. [DOI] [PubMed] [Google Scholar]
- 30.Zhou B, Zuo Y, Li B, et al. Deubiquitinase inhibition of 19S regulatory particles by 4-arylidene curcumin analog AC17 causes NF-kappaB inhibition and p53 reactivation in human lung cancer cells. Mol Cancer Ther. 2013;12(8): 1381–1392. [DOI] [PubMed] [Google Scholar]
- 31.Si X, Wang Y, Wong J, Zhang J, McManus BM, Luo H. Dysregulation of the ubiquitin-proteasome system by curcumin suppresses coxsackievirus B3 replication. J Virol. 2007;81(7): 3142–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Melandri F, Berdo I, et al. Delta12-Prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin-protein aggregation without proteasome inhibition. Biochemical and biophysical research communications. 2004;319(4): 1171–1180. [DOI] [PubMed] [Google Scholar]
- 33.Mullally JE, Moos PJ, Edes K, Fitzpatrick FA. Cyclopentenone prostaglandins of the J series inhibit the ubiquitin isopeptidase activity of the proteasome pathway. The Journal of biological chemistry. 2001;276(32): 30366–30373. [DOI] [PubMed] [Google Scholar]
- 34.Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer research. 2006;66(18): 9235–9244. [DOI] [PubMed] [Google Scholar]
- 35.Eldridge AG, O’Brien T. Therapeutic strategies within the ubiquitin proteasome system. Cell Death Differ. 2010;17(1): 4–13. [DOI] [PubMed] [Google Scholar]
- 36.Kapuria V, Levitzki A, Bornmann WG, et al. A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination. Cell Signal. 2011;23(12): 2076–2085. [DOI] [PubMed] [Google Scholar]
- 37.Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer research. 2010;70(22): 9265–9276. [DOI] [PubMed] [Google Scholar]
- 38.de Jong A, Merkx R, Berlin I, et al. Ubiquitin-based probes prepared by total synthesis to profile the activity of deubiquitinating enzymes. Chembiochem. 2012;13(15): 2251–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Mazurkiewicz M, Hillert EK, et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci Rep. 2016;6: 26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altun M, Kramer HB, Willems LI, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chemistry & biology. 2011;18(11): 1401–1412. [DOI] [PubMed] [Google Scholar]
- 41.Sun HS, Kapuria V, Peterson LF, et al. Inhibition of Usp9x Deubiquitinase Activity by WP1130 Reduces Mcl-1 Levels and Induces Apoptosis In Cells From Patients with Plasma Cell Dyscrasia and Drug-Refractory Multiple Myeloma. Blood. 2010;116(21): 1238–1239. [Google Scholar]
- 42.Baggio R, Shi YQ, Wu YQ, Abeles. From good substrates to good inhibitors: design of inhibitors for serine and thiol proteases. Biochemistry. 1996;35(11): 3351–3353. [DOI] [PubMed] [Google Scholar]
- 43.Mullally JE, Fitzpatrick FA. Pharmacophore model for novel inhibitors of ubiquitin isopeptidases that induce p53-independent cell death. Mol Pharmacol. 2002;62(2): 351–358. [DOI] [PubMed] [Google Scholar]
- 44.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib). Cancer investigation. 2004;22(2): 304–311. [DOI] [PubMed] [Google Scholar]
- 45.Tsvetkov P, Myers N, Eliav R, et al. NADH binds and stabilizes the 26S proteasomes independent of ATP. The Journal of biological chemistry. 2014;289(16): 11272–11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee BH, Lee MJ, Park S, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467(7312): 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. The EMBO journal. 2001;20(18): 5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kramer HB, Nicholson B, Kessler BM, Altun M. Detection of ubiquitin-proteasome enzymatic activities in cells: application of activity-based probes to inhibitor development. Biochim Biophys Acta. 2012;1823(11): 2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qian Y, Martell J, Pace NJ, Ballard TE, Johnson DS, Weerapana E. An Isotopically Tagged Azobenzene-Based Cleavable Linker for Quantitative Proteomics. Chembiochem. 2013;14(12): 1410–1414. [DOI] [PubMed] [Google Scholar]
- 50.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18(5): 538–543. [DOI] [PubMed] [Google Scholar]
- 51.Um JW, Im E, Lee HJ, et al. Parkin directly modulates 26S proteasome activity. J Neurosci. 2010;30(35): 11805–11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.An X, Tiwari AK, Sun Y, Ding PR, Ashby CR Jr., Chen ZS BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34(10): 1255–1268. [DOI] [PubMed] [Google Scholar]
- 53.Xia M, Knezevic D, Vassilev LT. p21 does not protect cancer cells from apoptosis induced by nongenotoxic p53 activation. Oncogene. 2011;30(3): 346–355. [DOI] [PubMed] [Google Scholar]
- 54.Baggio RF. A strategy for designing time-dependent protease inhibitors, implications for drug development. Department of Biochemistry. PhD. Brandeis University; 1996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.