

Abstract
α,α’-Trehalose plays roles in the synthesis of several cell wall components involved in pathogenic mycobacteria virulence. Its absence in mammalian biochemistry makes trehalose-related biochemical processes potential targets for chemotherapy. The trehalose-6-phosphate synthase (TPS)/trehalose-6-phosphate phosphatase (TPP) pathway, also referred to as the OtsA/OtsB2 pathway, is the major pathway involved in the production of trehalose in Mycobacterium tuberculosis (Mtb). In addition, TPP is essential for Mtb survival. We describe the synthesis of 6-phosphonic acid 4 (TMP), 6-(methylene)phosphonic acid 5 (TEP), and 6-N-phosphonamide 6 (TNP) derivatives of α,α’-trehalose. These non-hydrolyzable substrate analogs of TPP were examined as inhibitors of Mtb, M. lentiflavum (Mlt), and M. triplex (Mtx) TPP. In all cases the compounds inhibited Mtx TPP most strongly, with TMP (IC50 = 288 ± 32 μM) inhibiting most strongly, followed by TNP (IC50 = 421 ± 24 μM) and TEP (IC50 = 1959 ± 261 μM). The results also indicate significant differences in the analog binding profile when comparing Mtb TPP, Mlx TPP, and Mtx TPP homologs.
Keywords: trehalose, trehalose 6-phosphate phosphatase, enzyme inhibitors, Mycobacterium tuberculosis, carbohydrates
Graphical Abstract
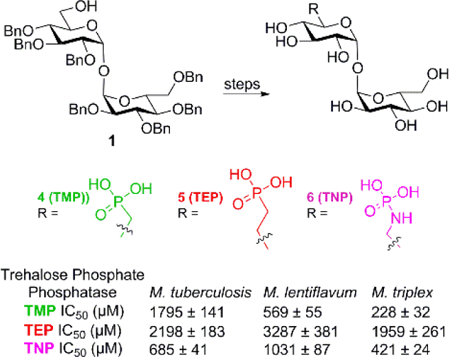
Introduction
α,α’-Trehalose (1) is an essential disaccharide present in plants,[1] some bacteria,[2] fungi,[3] parasitic nematodes,[4] and insects;[5] however, it is absent in mammals. Depending on context, trehalose can serve as an energy source, metabolic regulator, anti-desiccant, and cell wall component.[2] The lack of mammalian trehalose synthesis, import, or processing pathways makes those pathways potential targets for chemotherapy in Mycobacterium tuberculosis (Mtb).[6] In Mtb, three pathways for synthesizing trehalose are known,[6b] they are the TreY-TreZ, TreS and the OtaA-OtsB pathways (Figure 1). The OtsA/OtsB pathway is considered most prevalent in replicating Mtb.[7] OtsA catalyzes the formation of trehalose-6-phosphate (2, T6P) from UDP-glucose or ADP-glucose and glucose-6-phosphate.[8] OtsB has two homologues, OtsB1 and OtsB2, and only OtsB2 has phosphatase activity and is essential for Mtb growth.[7, 9] The phosphate moiety of T6P is removed by OtsB2 to yield trehalose. The OtaA-OtsB pathway is also referred to as the trehalose-6-phosphate synthase (TPS)/trehalose-6-phosphate phosphatase (TPP) pathway and TPP is used in place of OtsB2 hereafter.
Figure 1.
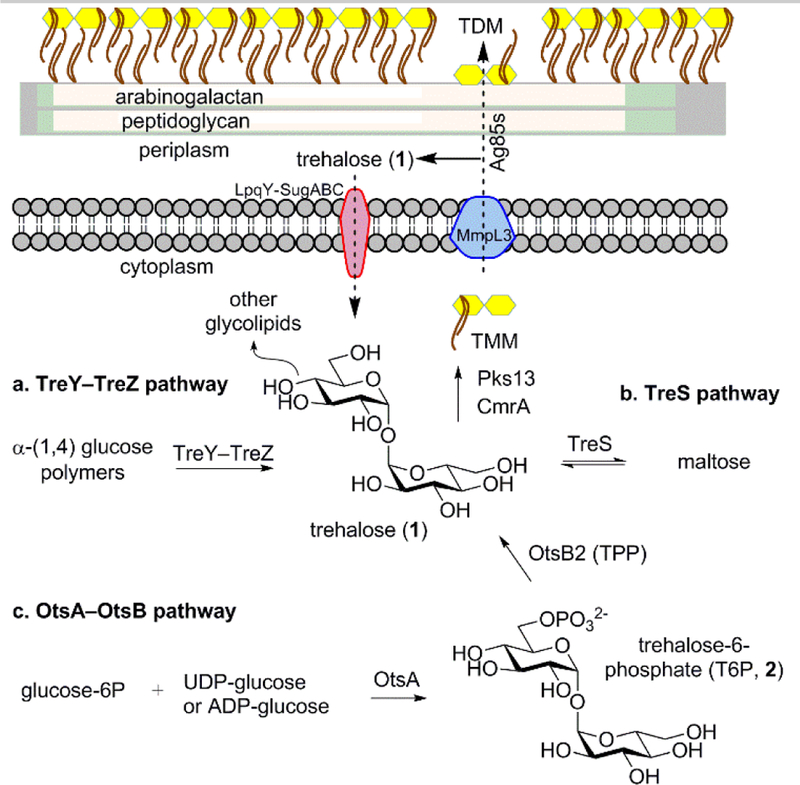
Routes for the synthesis and recycling of trehalose, as well as its conversion to TMM/TDM in Mtb. a. TreY–TreZ pathway; b. TreS pathway; c. OtsA–OtsB pathway.
Trehalose has several fates related to virulence and cell wall synthesis. These include conversion into intracellular α-glucan, sulfolipid-1, diacyltrehalose, and polyacyltrehalose. [10] Trehalose is also modified with mycolic acids by polyketide synthase 13 (Pks13) to form mono-α-alkyl β-ketoacyl trehalose (TMMk).[11] TMMk is reduced to trehalose monomycolate (TMM) by CmrA[12] which is transported to the cell wall by MmpL3 (Figure 1).[13] The Antigen 85 Complex (Ag85) transfers[14] the mycolic acids to the cell wall arabinogalactan (AG) or to a second molecule of TMM to form trehalose dimycolate (TDM) a key virulence factor.[15] Free trehalose produced by the action of Ag85s on TMM is salvaged by the action of the ABC transporter LpqY-SugABC.[6a] It is also known that Mtb can enter an antibiotic resistant state under the stress of low oxygen.[16] Intriguingly, trehalose has been linked to the ability of Mtb to adapt its metabolism under these conditions.[17] Thus, compounds interfering with the various trehalose-producing pathways may offer new approaches for more effective treatments against Mtb.
Recently, 6-sulfate, 6-(methylene)phosphonate, and 6-(fluoromethylene)fluorophosphonate derivatives of α,α’-trehalose were reported active (Ki =130–480 μM) against Mtb TPP[18] while aryl-D-glucopyranoside 6-sulfates were also reported as mimics of T6P with activity against TPP.[19] Based on the activity of these α,α’-trehalose derivatives against Mtb TPP we developed heptabenzyl α,α’-trehalose derivative 3 and synthetic routes to access 6-phosphonic acid 4 (TMP), 6-(methylene)phosphonic acid 5 (TEP), 6-N-phosphonamide 6 (TNP), and a 6-oxirane 7 derivatives of 6-deoxy-α,α’-trehalose (Figure 2). The library of 6-deoxy-α,α’-trehalose derivatives was evaluated for inhibition against TPP homologs encoded by Mtb, Mlt and Mtx. Validamycin A (8) was included due to topological similarity with α,α’-trehalose and for known inhibitory activity against E. coli TPP.[20] The Mlt and Mtx TPP homologs were included in this study because of the high sequence identity with the Mtb TPP, 72% and 71%, respectively, and the 100% identity in catalytic residues. Additionally, the relative paucity of available Mtb TPP structural information suggests protein structural dynamics that hinder crystallization. The Mlt and Mtx TPP homologs lack two large loops indicated in the Mtb TPP sequence and are being pursued as TPP surrogates to afford structural determination of mycobacterial TPP enzymes. Finally, the Mtb, Mlt, and Mtx TPP enzymes in this study exhibit KM values of 640,[7] 130,[21] and 82 μM,[21] respectively.
Figure 2.
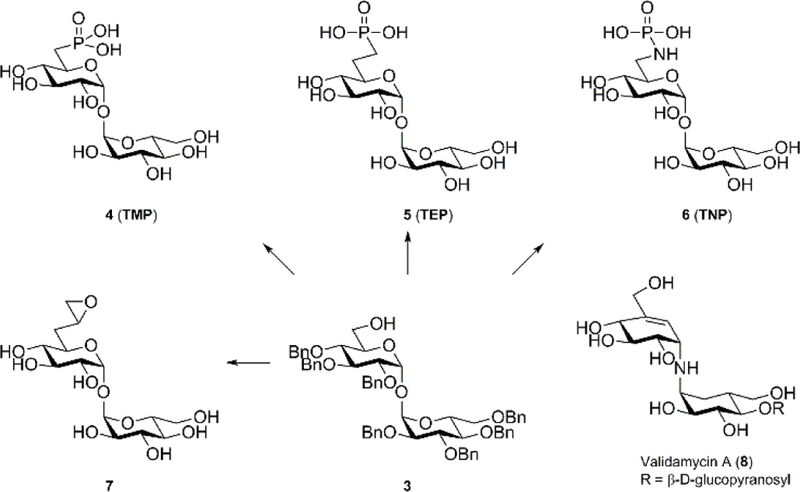
6-deoxy-α,α’-trehalose derivatives prepared from heptabenzyl α,α’-trehalose derivative 3 and Validamycin A (8) which shows topological similarities with trehalose.
Results and Discussion
Chemistry studies.
Prior research has shown that 6-sulfate, 6-(methylene)phosphonate TEP, and 6-(fluoromethylene)phosphonate derivatives of α,α’-trehalose possessed inhibitory activity against Mtb TPP;[18] however, no microbiological evaluation was reported. That data, in addition to the know growth inhibition data of 6-modified derivatives of α,α’-trehalose against mycobacteria,[14b, 22] prompted us to investigate the synthesis of additional 6-modified derivatives. In the current work we focused on synthesis and study of 6-phosphonate α,α’-trehalose TMP, an alternate route to 6-(methylene)phosphonate analogue TEP, a 6-N-phosphonamide analogue TNP, and an 6-oxirane analogue 7 due to similarity with reported Mtb TPP inhibitors (Figure 2).[18]
The target 6-phosphonate analog TMP was accessible through a heptabenzyl α,α’-trehalose intermediate 3[22c] prepared by our reported route.[22d] The route allows gram scale access to unsymmetrical 6-modified α,α’-trehalose analogues. In order to access 6-phosphonate TMP, heptabenzyl α,α’-trehalose derivative 3 was converted to the 6-iodo-6-deoxy-α,α’-trehalose derivative 9 in 70% yield by treatment with triphenylphosphine in the presence of iodine and imidazole (Scheme 1). Iodide 9 was subjected to Michaelis–Arbuzov conditions by treatment with trimethyl phosphite to afford 6-phosphonate derivative 10 in 58% yield. The phosphonate ester 10 was deprotected with bromotrimethylsilane (TMSBr) to afford the 6-phosphonic acid derivative 11. The latter was debenzylated by hydrogenolysis with 20% Pd(OH)2 on carbon under 1 atm. of H2 to afford 6-phosphonate TMP.
Scheme 1.
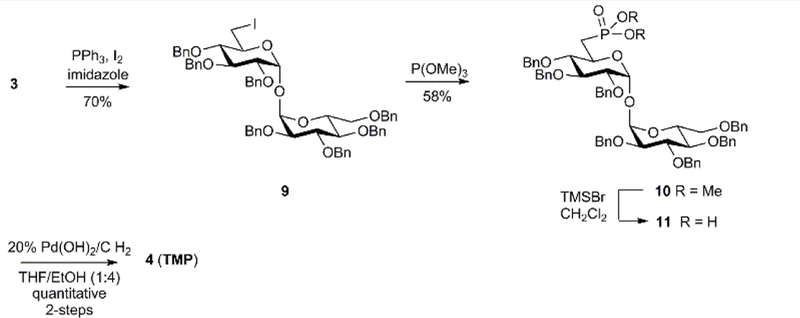
Synthesis of 6-phosphonic acid-α,α’-trehalose derivative TMP.
The target 6-(methylene)phosphonic acid analog TEP was also accessible through intermediate 3. Intermediate 3 was first subjected to a Swern oxidation to afford aldehyde 12 in 62% yield (Scheme 2).[18] Aldehyde 12 was converted to the phosphonate 13 in 49% yield via a Horner–Wadsworth–Emmons reaction utilizing tetramethyl methylenediphosphonate and sodium hydride. The phosphonate ester 13 was deprotected with bromotrimethylsilane (TMSBr) to afford the 6-(vinylphosphonic acid) derivative 14. The latter was debenzylated by hydrogenolysis with 20% Pd(OH)2 on carbon under 1 atm. of H2 to afford 6-(methylene)phosphonic acid TEP. The route is a minor modification of reported work[18] which used tetraethyl methylenediphosphonate in the Horner–Wadsworth–Emmons reaction along with other modifications in reagents and reaction sequence.
Scheme 2.
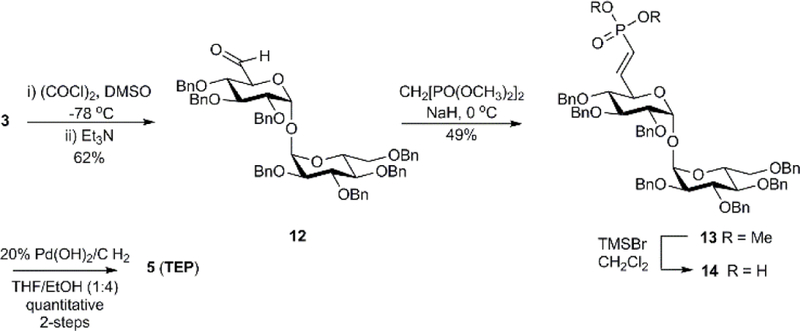
Synthesis of 6-(methylenephosphonic acid)-α,α’-trehalose derivative TEP.
The target 6-phosphoramidic acid analog TNP was accessible through iodide intermediate 9. Iodide 9 was subjected nucleophilic substitution with sodium azide to afford azide 15 in 82% (Scheme 3). Azide 15 was reduced to amine 16 via a Staudinger reaction reduction in good yield. The 6-amino-α,α’-trehalose derivative 16 was treated with dibenzylphosphoryl chloride to afford dibenyzl phosphoramidate 17. Global deprotection of the benzyl groups with 10% Pd on carbon under 1 atm. of H2 yielded 6-phosphoramidic acid analog TNP.
Scheme 3.
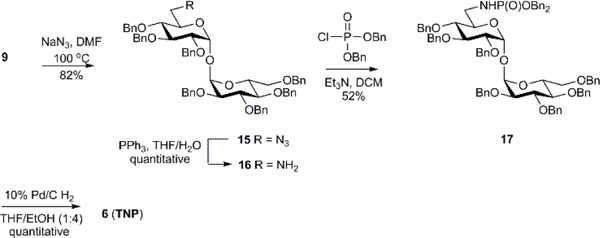
Synthesis of 6-(phosphoramidic acid)-α,α’-trehalose derivative TNP.
The target oxirane analog 7 was likewise accessible through iodide intermediate 9. Iodide 9 was subjected to nucleophilic substitution with trimethylsilylacetylene followed by deprotection of the silyl group with tetrabutylammonium fluoride (TBAF) to afford compound 18 in 48% yield over 2 steps (Scheme 4). Compound 18 was subjected to Birch reduction to reduce the benzyl groups and convert the alkyne to an alkene. The resulting hydroxyl groups were protected by acetylation to afford compound 19 in 40% yield.[23] The alkene 19 was treated with meta-chloroperoxybenzoic acid (mCPBA) to afford epoxide 20 as a mixture of diastereomers in 59% yield. Deprotection of the acetyl groups of 20 was achieved with NEt3/MeOH/H2O (1:3:1) to afford oxirane analog 7.[24]
Scheme 4.
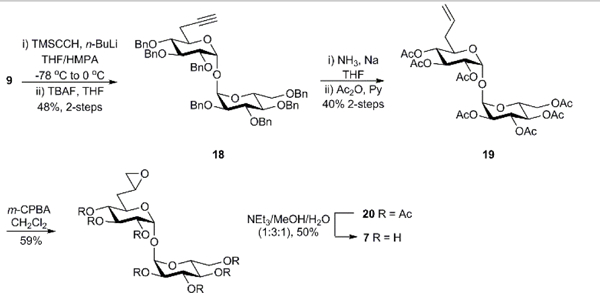
Synthesis of 6-oxiranyl-α,α’-trehalose derivative 7.
Enzyme inhibition studies.
To assess the ability of non-hydrolysable mimics of the natural TPP substrate, T6P, to inhibit TPP, TMP, TEP, TNP, 7 and Validamycin A (8) were evaluated. These non-hydrolysable mimics were tested against the TPP homologs encoded by Mtb, Mlt and Mtx. All of the inhibitory curves and resulting IC50 values are given in Figure 3. Few general trends of inhibition for the three homologs were observed. TEP exhibited the weakest inhibition for each enzyme, which is consistent with the expected enzyme mechanism. The presence of an ethyl moiety between the phosphonate and trehalose moiety is sufficiently large as to prevent appropriate simultaneous coordination of these two components of the inhibitor. The inhibitory activity of TMP and TNP exhibited IC50 values lacking a consistent pattern. TNP was the most potent inhibitor against the Mtb TPP, while TMP exhibited the lowest IC50 values for both the Mlt and Mtx TPP homologs. Compound 7 (data not shown) showed no inhibition. For comparison, Validamycin A (8) was also tested as an inhibitor of the mycobacterial TPPs as it had been previously shown to completely inhibit E. coli TPP at a concentration of 25 μM.[20] Interestingly, Validamycin A exhibited unexpectedly high IC50 values for the mycobacterial TPPs (Figure 3, J, K, and L). Neither the structure of the E. coli TPP structure nor the T6P-bound Mtb TPP structure have been deposited in the PDB, so it is difficult to rationalize the observed difference between the E. coli TPP and mycobacterial TPPs in response to Validamycin A. The structure of an inactive Cryptococcus neoformans TPP with T6P bound shows how both the essential Mg2+ is coordinated by the conserved Aspartate residues and how the phosphate moiety of T6P interacts with the Mg2+ as well as conserved Histidine and Lysine active site residues.[25] However, the residues in both the catalytic domain and the cap domain of C. neoformans TPP that interact directly with the glucose moieties of trehalose are not highly conserved in TPP homologs. It is also shown that structural changes in C. neoformans TPP are stimulated by binding of T6P. Therefore, it is likely that the sequence differences and structural dynamics lead to differential inhibition by Validamycin A.
Figure 3.
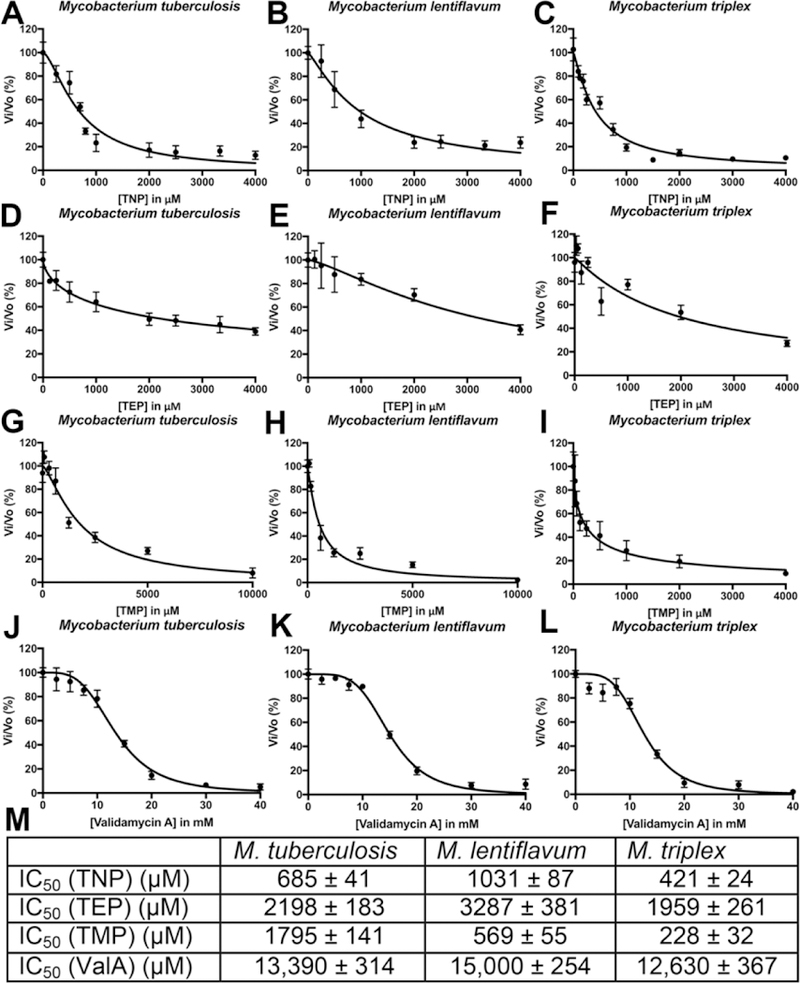
TPP Inhibition studies. A, B and C; TPP inhibition curves for TNP. D. E, F; TPP inhibition curves for TEP. G, H, and I; TPP inhibition curves for TMP. J, K, L; TPP inhibition curves for Validamycin A. M; IC50 values of TNP, TEP, TMP and Validamycin A against Mtb TPP, Mlt TPP and Mtx TPP.
Discussion
TPP is an essential enzyme for survival and virulence of Mtb. It catalyzes the second reaction in the de novo trehalose biosynthesis pathway. Since trehalose is not produced by mammals, they possess no homologues of TPP rendering it an intriguing drug target. Since enzymes evolve to strongly bind their transition-states, initial inhibitory studies on any enzyme typically attempt to mimic the expected transition states. In the case of TPP, formation of trehalose occurs through an associative-two-step mechanism (Figure 4).[26]
Figure 4.
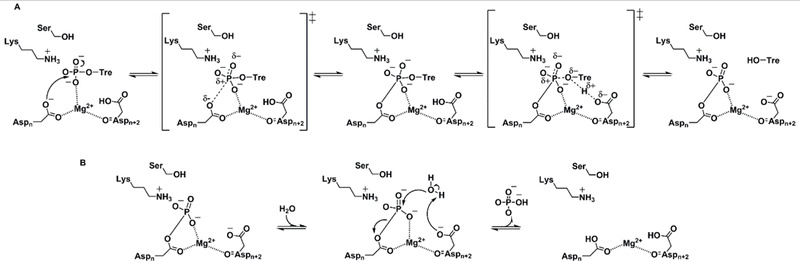
Associative-two-step mechanism of TPP. A. First step of the reaction. B. Second step of the reaction. Aspn corresponds to the nucleophile. Aspn+2 corresponds to the general acid/general base.
During the first step of the reaction, the nucleophilc aspartate, Asp153/120 (numbering corresponds with Mtb TPP/Mlt TPP residue numbering: Aspn in Figure 4), launches an attack on the phosphoryl group of T6P, resulting in the formation of a penta-coordinate trehalose-phosphoaspartyl enzyme intermediate. Then acting as a general acid, Asp155/122 (Aspn+2 in Figure 4) protonates trehalose promoting formation of the phosphoaspartyl intermediate and release of free trehalose. The proposed pentacoordinate phosphorous transition states and intermediates are challenging to emulate in stable organic compounds. Therefore, we have employed phosphonate compounds as substrate analogues for the initial inhibitory studies.
A magnesium co-factor, required for this reaction to occur, affords the correct positioning of the substrate phosphoryl group and for neutralization of the negative charges forming on the transition-state. The three non-hydrolysable phosphorus-based compounds were made to mimic the T6P substrate and assess the impact of length and atom character of the chain connecting trehalose and the phospho-mimic. While the TMP IC50 against the Mtx TPP and Mtb TPP homologs was only 1.8- to 2.8-fold worse than the determined T6P KM, respectively, TEP exhibited an IC50 that was 3.4- to 135-fold worse than the determined T6P KM. The third inhibitor, TNP, was synthesized to test the role for the O-6 oxygen of T6P binding by TPP. The IC50 of TNP suggests binding affinity similar to the Mtb TPP binding of the T6P substrate. When compared to the inhibitory activities of TMP and TEP against Mtb TPP, this suggests enhanced affinity due to the presence of a hydrogen bond acceptor and donor at position 6 which has an important role in enzyme recognition. Compound 7, on the other hand, appeared unable to interact with any of the the enzymes. The moderate inhibition data is consistent with other studies that describe T6P analogs designed to mimic phosphate.[18–19] One potential path forward to improve inhibition may be to mimic phosphate with α-hydrodroxy- or α-amino- phosphonates. These bidentate moieties may have the potential to bind magnesium ions and improve inhibitory potential. In addition, α-hydrodroxy- or α-amino- groups may make up for the loss of the putative H-bond acceptor absent in the current phosphonate series. It is expected that the compounds in the current series could potentially be imported by Mtb by LpqY-SugABC (Figure 1) which recognizes trehalose and some of its derivatives.[14b, 22] For example, it has been shown that Mtb could take-up extracellular trehalose including a fluorescein-containing trehalose probe and incorporate the materials into growing bacilli.[22a] The same investigators showed that 6-fluoro-6-deoxy-α,α’-trehalose and 6-bromo-6-deoxy-α,α’-trehalose possessed activity (MIC 200 μg/mL against Mtb.[22a]
For future studies it would interesting to investigate how these new compounds might effect trehalose production, Mtb growth or recovery from dormancy, Ag85 activity, TMM/TDM production, or biofilm formation. Related compounds, like 6-azido-6-deoxy-α,α’-trehalose, are known to inhibit growth of M. aurum (MIC 200 μg/mL) as well as inhibit Ag85C activity, synthesis of TMM, TDM, and reduced cell wall–bound mycolic acids and TMM export. [14b] While a library of N,N-dialkylamino and 6,6-bis(sulfonamido) analogs of α,α’-trehalose was shown active (MIC 16–128 μg/mL) against Mtb H37Ra.[22b] A second 6,6-bisalkyl library of α,α’-trehalose derivatives was found active against M. smegmatis.[22c] In addition, 6,6-bis(α-ketoesters/amides) analogs of α,α’-trehalose have been reported to be active against Ag85C[22d] and 6-deoxy-, 6-fluoro-6-deoxy-, and 6-azido-6-deoxy-α,α’-trehalose were shown to inhibit (MIC 50–100 μM) M. smegmatis biolfim formation.[22e] Collectively, these studies suggest several possible applications of the target compounds.
Conclusions
The inhibitory results show that the TEP analog with the longer linker and lacking a hydrogen bond acceptor significantly weakens affinity by any TPP homolog. In contrast, the lone pair of electrons on the nitrogen atom of the aminophosphonate moiety in the TNP analog may interact with the magnesium co-factor to enhance binding. In contrast, the Mlx TPP and Mtx TPP exhibited a different pattern of response to the inhibitors, particularly the inhibition by TMP. While this lack of correlation is troubling, it is well-documented that TPP enzyme active sites undergo a conformational change upon binding substrate and structural dynamics can have a profound effect on substrate or inhibitor affinity.[25] In this context, the otherwise minor differences in active site residues of the three tested homologs may be the source of the inconsistent response to these inhibitors. This suggests that structural changes in Mtb TPP due to substrate-analog binding may be distinct from changes occurring in the Mlx TPP and Mtx TPP homologs, particularly in the case of TMP binding. Resolution of this discrepancy will follow structural determination of TPP/inhibitor complexes and complementary molecular dynamic studies.
Experimental Section
General Methods
All chemicals and solvents were purchased from Fisher Scientific, Acros Organics, Alfa Aesar or Sigma-Aldrich. Solvents were dried by distillation, other standard procedures and through a solvent purification system by passing HPLC grade solvent through activated alumina and copper columns. All reactions were carried out at room temperature under nitrogen atmosphere using a nitrogen balloon unless mentioned in procedure. Reactions were monitored by TLC (silica gel, f254) under UV light or by charring (5% sulfuric acid-methanol) and the purification was performed by flash column chromatography on silica gel (230–400 mesh) using the solvent system specified, solvents were used without purification for chromatography. 1H NMR and 13C NMR were recorded on a Bruker Avance III 600 MHz spectrometer using CDCl3 or D2O as internal references. 31P NMR were recorded on a VXRS 400 MHz using CDCl3 or D2O as solvents. High resolution mass spectrometry was performed on a Waters SYNAPT HRMS nano ESI-MS instrument and low-resolution mass spectrometry was performed on an ESquire-LC-MS.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-iodo-6-deoxy-α-D-glucopyranosyl-(1→1)-α-D-glucopyranoside (9).
To a solution of 3 (0.950 g, 0.977 mmol, 1eq.) in tetrahydrofuran (10 mL) at 0 °C was added triphenylphosphine (0.307 g, 1.17 mmol, 1.2 eq.), imidazole (0.166 g, 2.44 mmol, 2.5 eq.) and iodine (0.297 g, 1.17 mmol, 1.2 eq.). The reaction was refluxed for 3 h. After completion, water (10 mL) was added to the reaction flask and organic layer was extracted with ethyl acetate (15 mL). The organic layer was washed successively with Na2S2O3 (10 mL X 3) and water (10 mL X 3). The ethyl acetate was dried over anhydrous Na2SO4 and filtered. Purification was performed by flash column chromatography on silica gel (5% ethyl acetate-hexanes) to afford a colorless viscous liquid (0.768 g, 70% yield): Rf = 0.35 (20% ethyl acetate-hexanes); 1H NMR (600 MHz, CDCl3): δ 7.38–7.24 (m, 33H, aromatic), 7.13 (dd, 2H, J = 7.4, 1.9 Hz, aromatic), 5.27 (d, 1H, J = 3.5 Hz, H-1), 5.23 (d, 1H, J = 3.5 Hz, H-1’), 4.98 (m, 3H, benzylic), 4.85 (m, 3H, benzylic), 4.72 (m, 5H, benzylic), 4.56 (d, 1H, J = 12.1 Hz, benzylic), 4.47 (d, 1H, J = 10.8 Hz, benzylic), 4.40 (d, 1H, J = 12.1 Hz, benzylic), 4.14 (m, 1H, H-5’), 4.09 (t, 1H, J = 9.3 Hz, H-3), 4.03 (t, 1H, J = 9.4 Hz, H-3’), 3.69 (m, 2H, H-4’, H-5), 3.62 (dd, 1H, J = 9.6, 3.6 Hz, H-2’), 3.52 (m, 2H, H-6a’, H-4), 3.41 (m, 2H, H-6b’, H-4), 3.25 (dd, 1H, J = 10.9, 4.5 Hz, H-6a), 3.12 (dd, 1H, J = 10.8, 2.9 Hz, H-6b); 13C NMR (150 MHz, CDCl3): δ 139.03, 138.81, 138.52, 138.44, 138.35, 138.30, 138.02, 128.74, 128.68, 128.63, 128.62, 128.62, 128.60, 128.22, 128.20, 128.14, 128.14, 128.11, 127.95, 127.89, 127.86, 127.80, 127.60, 94.61, 94.24, 82.06, 81.89, 81.40, 79.81, 79.68,77.92, 75.90, 75.85, 75.68, 75.35, 73.74, 73.34, 73.01, 70.97, 68.92, 68.30, 66.12, 9.59; HRMS (ESI): calculated for C61H63IO10, 1105.3364 [M+Na]+; observed, m/z = 1105.3331 [M+Na]+.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-methylphosphonate-6-deoxy-α-D-glucopyranosyl-(1→1)-α-D-glucopyranoside (10).
Trimethyl phosphite (10 mL) was added to compound 9 (0.500 g, 2.16 mmol) and the reaction was refluxed for 36 h. After the completion, excess trimethyl phosphite was evaporated under reduced pressure. The product was purified by flash column chromatography on silica gel (5% ethanol-choroform) to afford the product as a colorless solid (0.202 g, yield 58%): Rf = 0.30 (8% ethanol-chloroform); 1H NMR (600 MHz, CDCl3): δ 7.38–7.14 (m, 35H, aromatic), 5.47 (d, 1H, J = 3.5 Hz, H-1), 5.26 (d, 1H, J = 3.5 Hz, H-1’), 4.97 (m, 4H, benzylic), 4.84 (d, 3H, J = 10.8 Hz, benzylic), 4.71 (m, 3H, benzylic), 4.65 (d, 1H, J = 11.2 Hz, benzylic), 4.55 (d, 1H, J = 12.3 Hz, benzylic), 4.49 (d, 1H, J = 11 Hz, benzylic), 4.43 (d, 1H, J = 12.1 Hz, benzylic), 4.30 (m, 1H, H-5), 4.11 (m, 3H, H-3, H-3’, H-5’), 3.66 (m, 2H, H-2, H-2’), 3.61 (m, 6H, 2X-OCH3), 3.55 (m, 1H, H-4’), 3.42 (t, 1H, H-4), 2.12 (m, 1H, H-6a), 1.88 (m 1H, H-6b); 13C NMR (150 MHz, CDCl3): δ 139.11, 138.87, 138.84, 138.68, 138.58, 138.33, 138.14, 128.62, 128.61, 128.58, 128.55, 12853, 128.48, 128.17, 128.10, 128.01, 127.76, 127.73, 92.55, 92.14, 82.18, 82.09,82.03, 81.50, 81.49, 79.81, 79.73, 78.01, 75.82, 75.71, 75.17, 75.15, 73.70, 73.27, 72.92, 70.83, 68.66, 66.92, 66.88, 66.11, 52.46, 52.41, 52.32, 52.28, 15.52; 31P NMR (162 MHz, CDCl3): δ 32.13 (s, 1-P); LRMS (ESI): calculated for C63H69PO13, 1087.4 [M+Na]+; observed, m/z = 1087.5 [M+Na]+.
6-Deoxy-6-(phosphonic acid)-α,α’-trehalose (4, TMP).
To a solution of 10 (0.080 g, 0.075 mmol) in dichloromethane (4 mL) was added bromotrimethylsilane (129 μL, 0.977 mmol, 13 eq.) dropwise and the resulting solution stirred for 2 h at room temperature. A solution of methanol-water (1.1:1.8 mL) was added followed by concentration to dryness to afford phosphonicacid 11. The residue was dissolved in 3 mL of tetrahydrofuran-ethanol (1:4) and 20% Pd(OH)2/C (80 mg) was added. The suspension was stirred overnight at room temperature under 1 atm. of hydrogen. The catalyst was filtered away through a plug of Celite® 545 that was washed with 20% methanol-dichloromethane. The filtrate and washings were concentrated to dryness to afford product TMP as a colorless solid (0.030 g, quantitative yeild): 1H NMR (600 MHz, D2O): δ 5.24 (d, 1H, J = 3.5 Hz, H-1), 5.02 (d, 1H, J = 3.8 Hz, H-1’), 3.94 (m, 1H, H-5), 3.75–3.53 (m, 7H, H-2, H-2’, H-4, H-4’, H-5’, H-6a’, H-6b’), 3.33 (t, 1H, J = 9.4 Hz, H-3’), 3.15 (t, 1H, J = 9.4 Hz, H-3), 2.12 (m, 1H, H-6a), 1.75 (m, 1H, H-6b); 13C NMR (150 MHz, D2O): δ 92.93, 92.88, 74.32, 74.22, 72.25, 72.05, 71.11, 70.91, 69.59, 68.03, 34.82; 31P NMR (162 MHz, D2O): δ 30.78 (s, 1-P); HRMS (ESI): calculated for C12H23PO13, 429.0774 [M+Na]+; observed, m/z = 429.0774 [M+Na]+.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-carbaldehyde-α-D-glucopyranosyl-(1→1)-α-D-glucopyranoside (12).
Oxalyl chloride (0.07 mL, 0.80 mmol, 4 eq.) was slowly added to a solution of dimethyl sulfoxide (0.11 mL, 1.60 mmol, 8 eq.) in dichloromethane (5 mL) maintained at −78 °C. The solution was allowed to stir at that temperature for 20 min. To this solution was added 3 (0.20 g, 0.020 mmol, 1eq.) dissolved in 2 mL of dichloromethane. This solution was allowed to stir for 1 hour at −78 °C. Triethylamine (0.45 mL, 3.2 mmol, 16 eq.) was added and the solution was stirred and additional 20 min. The reaction was allowed to warm to room temperature over 30 min. dichloromethane (5 mL) was added to the solution followed by successive washings with saturated NH4Cl solution (10 mL X 3) and saturated NaCl solution (10 mL X 3). The organic layer was dried over anhydrous Na2SO4 and concentrating under reduced pressure. The residue was purified by flash column chromatography on silica gel (40% ethyl acetate-hexanes) to afford the product as a viscous liquid (0.12 g, 62% yeild): Rf = 0.37 (30% ethyl acetate-hexanes); 1H NMR (600 MHz, CDCl3): δ 9.36 (s, 1H, H-6), 7.40–7.13 (m, 35H, aromatic), 5.23 (d, 1H, J = 3.5 Hz, H-1), 5.15 (d, 1H, J = 3.7 Hz, H’−1), 5.02 (dd, 2H, J = 10.9, 5.6 Hz, benzylic), 4.87 (m, 5H, benzylic), 4.73 (d, 1H, J = 11.7 Hz, benzylic), 4.67 (m, 3, H-5, benzylic), 4.57 (m, 3H, benzylic), 4.48 (d, 1H, J = 10.6 Hz, benzylic), 4.41 (d, 1H, J = 12.1 Hz, benzylic), 4.15 (m, 1H, H-5’), 4.10 (t, 1H, J = 9.6 Hz, H-3), 4.02 (t, 1H, J = 9.4 Hz, H-3’), 3.70 (t, 1H, J = 9.6 Hz, H-4’), 3.60 (dd, 1H, J = 9.7, 3.7 Hz, H-2’), 3.57–3.51 (m, 3H, H-2, H-4, H-6a’), 3.40 (dd, 1H, J = 10.6, 1.8 Hz, H-6b’); 13C NMR (150 MHz, CDCl3): δ 197.98, 138.93, 138.60, 138.35, 137.99, 137.95, 137.91, 137.55, 128.71, 128.64, 128.61, 128.58, 128.54, 128.51, 128.38, 128.32, 138.23, 128.20, 128.10, 128.05, 127.92, 127.87, 127.83, 127.78, 127.66, 95.29, 94.66, 81.97, 81.73, 79.50, 78.85, 78.51, 77.78, 76.00, 75.81, 75.38, 74.85, 73.70, 73.40, 72.90, 71.05, 68.20; LRMS (ESI): calculated for C61H62O11, 993.4 [M+Na]+; observed, m/z = 993.4 [M+Na]+.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-deoxy-6E-(dimethyl vinylphosphonate)-α,α’-trehalose (13).
Sodium hydride, 60% dispersion in oil, (7 mg, 0.30 mmol, 3 eq.) was added to a solution of tetramethyl methylenediphosphonate (0.03 mL, 0.15 mM, 1.5 eq.) in tetrahydrofuran (3 mL) maintained at 0 °C. The mixture was stirred for 30 min. A solution of aldehyde 12 (0.10 g, 0.10 mmol, 1eq.) in 1 mL of tetrahydrofuran was added dropwise to the anion. The solution was allowed to warm to room temperature and stirred for another 2 h. The solvent was evaporated and the residue purified by flash chromatography on silica gel (5% methanol-dichloromethane) to afford 13 as a colorless viscous liquid (0.054 g, 49% yeild): Rf = 0.48 (10% methanol-dichloromethane); 1H NMR (600 MHz, CDCl3): δ 7.38–7.23 (m, 33H, aromatic), 7.24–7.13 (m, 2H, aromatic), 6.96–6.88 (m, 1H, H-6), 5.94 (m, 1H, H-7), 5.23 (d, 1H, J = 3.7 Hz, H-1), 5.1 (d, 1H, J = 3.7 Hz, H-1’), 5.0 (dd, 2H, J = 10.8, 2.2 Hz, benzylic), 4.86 (m, 4H, benzylic), 4.75 (m, 1H, H-5), 4.72–4.76 (m, 2H, benzylic), 4.57 (m, 4H, benzylic), 4.47 (d, 1H, J = 10.8 Hz, benzylic), 4.39 (d, 1H, J = 12.1 Hz, benzylic), 4.15 (m, 1H, H-5’), 4.09 (t, 1H, J = 9.06 Hz, H-3), 4.03 (t, 1H, J = 9.06 Hz, H-3’), 3.66 (m, 6H, H-4’, 2X-OCH3), 4.56 (m, 2H, H-2 H-2’), 3.51 (m, 1H, H-6a’), 3.38 (dd, 1H, J = 10.6, 1.8 Hz, H-6b’), 3.25 (dd, 1H, J = 10.1, 9.2 Hz, H-4); 13C NMR (150 MHz, CDCl3): δ 149.17, 138.57, 138.35, 138.01, 137.76, 137.67, 137.50, 137.36, 128.28, 128.18, 127.75, 127.39, 127.15, 116.68, 115.42, 94.30, 93.63, 81.64, 81.58, 81.19, 78.98, 78.60, 77.41, 75.48, 75.40, 75.31, 74.87, 73.27, 72.71, 72.59, 70.45, 70.31, 70.17, 67.86, 52.10; 31P NMR (162 MHz, CDCl3): δ 32.42 (s, 1-P); HRMS (ESI): calculated for C64H69PO13, 1099.4373 [M+Na]+; observed, m/z = 1099.4370 [M+Na]+.
6-Deoxy-6-(methylenephosphonic acid)-α,α’-trehalose (5, TEP).
Bromotrimethylsilane (83.4 μL, 0.652 mmol, 13 eq.) was added dropwise to a solution of 13 (0.09 g, 0.05 mmol) in dichloromethane (4 mL) and the resulting solution was stirred for 2 h at room temperature. The solution was quenched with 2 mL of methanol-water (1.1:1.8) followed by concentration to dryness. To the solution of benzylated disaccharide 14 was taken up in 3 mL of tetrahydrofuran-ethanol (1:4) and 20% Pd(OH)2/C (50 mg) was added. The mixture was stirred overnight under hydrogen (1 atm.). The catalyst was filtered off through Celite® 545 and washed with 20% methanol-dichloromethane. The combined filtrate and washings were concentrated to dryness to afford TEP as viscous syrup (0.019 g, quantitative yield): 1H NMR (600 MHz, D2O): δ 5.04 (s, 2H, H-1, H-1’), 3.74–3.64 (m, 6H, H-6a’, H-6b’, H-5, H-5’, H-3, H-3’), 3.55–3.50 (m, 2H, H-2, H-2’), 3.32 (t, 1H, J = 9.6 Hz, H-4’), 3.16 (t, 1H, J = 9.4 Hz, H-4), 1.98 (m, 1H, H-6a), 1.84 (m, 1H, H-6b), 1.59 (m, 2H, H-7a, H-7b); 13C NMR (600 MHz, D2O): δ 93.39, 93.11, 73.14, 72.43, 72.36, 72.08, 71.61, 71.50, 71.11, 70.88, 69.60, 60.41, 24.33; 31P NMR (162 MHz, D2O): δ 27.29 (s, 1-P); HRMS (ESI): calculated for C13H29PO13, 443.0930 [M+Na]+; observed, m/z = 443.0946 [M+Na]+.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-deoxy-6-azido-α,α’-trehalose (15).
Sodium azide (0.114 g, 1.76 mol, 5 eq.) was added to a solution of iodide 9 (0.380 g, 351 mmol, 1eq.) in DMF (8 mL). The mixture was heated to reflux for 24 h. The solution was diluted with ethyl acetate and washed with saturated NaCl solution (10 mL X 3). The organic layer was dried over anhydrous Na2SO4, filtered, and the solvent evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel (8% ethyl acetate-hexanes) to afford 15 as a colorless viscous syrup (0.300 g, 86% yield): Rf = 0.33 (20% ethyl acetate:hexanes); 1H NMR (600 MHz, CDCl3): δ 1H NMR (600 MHz, CDCl3): 7.32 (m, 33H, aromatic), 7.12 (m, 2H, aromatic), 5.22 (d, 2H, J = 3.5 Hz, H-1, H-1’), 4.99 (dd, J = 10.8, 7.3 Hz, 2H, benzylic), 4.85 (m, 4H, benzylic), 4.71 (m, 4H, benzylic), 4.62 (s, 1H, benzylic), 4.55 (m, 2H, benzylic), 4.46 (d, 1H, J = 10.6 Hz,, benzylic), 4.38 (d, 1H, J = 12.1 Hz, benzylic), 4.16 (m, 1H, H-5’, H-5), 4.02 (m, 2H, H-3’, H-3), 3.68 (m, 1H, H-4’), 3.61 (m, 1H, H-2), 3.56 (m, 1H, H-2’), 3.49 (m, 2H, H-6a’, H-6b’), 3.37 (m, 1H, H-4), 3.18 (m, 2H, H-6a, H-6b); 13C NMR (150 MHz, CDCl3): δ 138.83, 138.66, 138.29, 138.16, 138.06, 137.79, 128.49, 128.43, 128.38, 128.37, 128.03, 128.01, 127.93, 127.90, 127.89, 127.73, 127.66, 127.62, 127.57, 127.47, 127.38, 94.50, 94.01, 81.82, 81.46, 79.50, 79.36, 78.30, 77.69, 77.26, 77.04, 76.83, 75.62, 75.21, 75.13, 73.53, 72.96, 72.78, 70.74, 70.35, 68.10, 51.08; HRMS (ESI): calculated for C61H63IO10, 1020.4411 [M+Na]+; observed, m/z = 1020.4424 [M+Na]+.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-deoxy-6-amino-α,α’-trehalose (16).
Triphenylphosphine (0.236 g, 0.900 mmoL, 3 eq.) and water (0.027 g, 1.50 mmoL, 5 eq.) were added to compound 15 (0.300 g, 0.300 mmol, 1 eq.) dissolved in tetrahydrofuran (8 mL). The solution was stirred for 12 h and concentrated under reduced pressure. Ethyl acetate (10 mL) and water (10 mL) were added to the residue and the organic layer separated and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue subjected to flash column chromatography on silica gel (10% acetone-hexanes) to afford 16 as a white solid (0.290 g, quantitative yield): Rf = 0.21 (30% acetone:hexanes); 1H NMR (600 MHz, CDCl3): δ 7.67 (m, 9H, aromatic), 7.52 (m, 4H, aromatic), 7.44 (m, 9H, aromatic), 7.37–7.29 (12H, aromatic), 7.14 (dd, 2H, J = 7.7, 1.5 Hz, aromatic), 5.22 (d, 1H, J = 3.7 Hz, H-1), 5.20 (d, 1H, J = 3.7 Hz, H-1’), 5.01 (t, 2H, J = 10 Hz, benzylic), 4.88 (m, 3H, benzylic), 4.83 (d, 1H, J = 10.8 Hz, benzylic), 4.72 (m, 5H, benzylic), 4.64 (m, 1H, J = 11.2 Hz, benzylic), 4.55 (d, 1H, J = 12.3 Hz, benzylic), 4.48 (d, 1H, J = 10.8 Hz, benzylic), 4.39 (d, 1H, J = 12.1 Hz, benzylic), 4.18 (dd, 1H, J = 12.1, 2.2 Hz, H-5’), 4.08 (td, 2H, J = 9.3, 7.1 Hz, H-3, H-3’), 4.00 (m, 1H, H-5), 3.7 (t, 1H, J = 9.7 Hz, H-4’), 3.63 (dd, 1H, J = 9.7,3.7 Hz, H-2’), 3.53 (m, 2H, H-6a’, H-4), 3.41 (m, 2H, H-6b’, H-4), 2.81 (dd, 1H, J = 13.7, 2.8 Hz, H-6a), 2.66 (dd, 1H, J = 13.8, 5.3 Hz, H-6b); 13C NMR (150 MHz, CDCl3): δ 138.66, 138.63, 138.16, 138.04, 138.01, 137.62, 132.67, 131.98, 131.93, 131.87, 131.79, 131.76, 128.37, 128.29, 128. 21, 128.16, 127.97, 127.81, 127.77, 127.77, 127.70, 127.51, 127.47, 127.36, 127.33, 127.29, 127.27, 127.19, 93.94, 93.57, 81.67, 81.56, 79.54, 79.28, 78.08, 77.58, 75.41, 75.34, 74.87, 74.70, 73.32, 72.81, 72.60, 71.71, 70.46, 67.97, 42.34; LRMS (ESI): calculated for C61H63IO10, 972.5 [M+H]+; observed, m/z = 972.9 [M+H]+.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-deoxy-6-(dibenyzl phosphoramidate)-α,α’-trehalose (17).
Dibenzylphosphoryl chloride (0.045 g, 0.154 mmol, 3 eq.) dissolved in 2 mL of dichloromethane was added to compound 16 (0.050 g, 0.051 mmoL, 1eq.) dissolved in 5 mL of dichloromethane. Triethylamine (0.051 g, 0.51 mmoL, 10 eq.) was added and the solution stirred for 6 h. The solution was concentrated under reduced pressure and the residue subjected to flash column chromatography on silica gel (8% acetone-hexanes) to afford a 17 as aviscous syrup (0.033 g, 52% yield): Rf =0.32 (30% acetone:hexanes); 1H NMR (600 MHz, CDCl3): δ 7.3 (m, 43H, aromatic), 7.14 (m, 2H, aromatic), 5.12 (d, 1H, J = 3.7 Hz, H-1), 5.10 (d, 1H, J = 3.7 Hz, H-1’), 4.97 (m, 6H, benzylic), 4.81 (m, 5H, benzylic), 4.63 (m, 6H, benzylic), 4.53 (m, 1H, benzylic), 4.45 (m, 1H, benzylic), 4.38 (m, 1H, benzylic), 4.12 (m, 1H, H-5’), 4.01 (m, 3H, H-3, H-3’, H-5’), 3.65 (m, 1H, H-4’), 3.56 (m, 1H, H-2’), 3.50 (m, 2H, H-5, H-6a’), 3.44 (m, 2H, H-2), 3.37 (m, 1H, H-6b’), 3.08 (m, 1H, H-4), 2.93 (m, 1H, H-6a), 2.70 (m, 1H, H-6b); 13C NMR (150 MHz, CDCl3): δ 138.96, 138.91, 138.46, 138.35, 138.23, 138.21, 137.96, 128.70, 128.65, 128.62, 128.56, 128.54, 128.51, 128.43, 128.40, 128.23, 128.14, 128.06, 128, 127.96, 127.91, 127.86, 127.72, 127.59, 127.56, 127.52, 94.38, 93.91, 79.55, 79.32, 77.87, 77.83, 75.73, 75.66, 75.25, 75.03, 73.66, 72.97, 72.93, 70.83, 68.30, 68.24, 68.20, 68.14, 68.11, 41.53; 31P NMR (162 MHz, D2O): δ 10.89 (s, 1-P); HRMS (ESI): calculated for C61H63IO10, 1232.5289 [M+H]+; observed, m/z = 1232.5276 [M+H]+.
6-(phosphoramidic acid)-α,α’-trehalose (6, TNP).
A catalytic amount of 20% Pd(OH)2/C was added to a solution of benzylated disaccharide derivative 17 (0.020 g, 0.016 mmol) dissolved in 3 mL of tetrahydrofuran-ethanol (1:4). The mixture was stirred overnight under hydrogen (1 atm.). The catalyst was filtered away through a plug of Celite® 545 and washed with 20% methanol-dichloromethane. The combined filtrate and washings were concentrated to dryness afford TNP as a viscous syrup (0.006 g, quantitative yield): 1H NMR (600 MHz, D2O): δ 5.12 (d, 1H, J = 3.5 Hz, H-1), 5.07 (d, 1H, J = 3.5 Hz, H-1’), 3.87 (m, 1H, H-5), 3.82 (m, 1H, H-6a’), 3.75 (m, 4H, H-3, H-3’, H-5’, H-6b’), 3.64 (m, 1H, H-4’), 3.55 (m, 2H, H-2, H-2’), 3.33 (m, 1H, H-4), 3.25 (m, 1H, H-6a), 3.03 (m, 1H, H-6b); 13C NMR (150 MHz, D2O): δ 93 .52, 93.35, 72.46, 72.14, 71.40, 70.99, 70.66, 69.53, 68.09, 60.36, 40.44; 31P NMR (162 MHz, D2O): δ 4.62 (s, 1-P); HRMS (ESI): calculated for C12H23PO13, 422.1064 [M+H]+; observed, m/z = 422.1052 [M+H]+.
2,2’,3,3’,4,4’,6’-Hepta-O-benzyl-6-deoxy-6-ethynyl-α,α’-trehalose (18).
n-Butyllithium (0.415 mL, 2 M) was slowly added to a solution of trimethylsilylacetylene (118 μL, 0.831 mmol, 3 eq.) in tetrahydrofuran (3 mL) at −78 °C. The solution was allowed to warm to 0 °C and was strirred for 30 min. Afterward, the solution was allowed to warm to room temperature over 50 min. The solution of anion was added to another round bottom flask containing iodide 9 (0.300 g, 0.277 mmol, 1 eq). To the resulting solution was added hexamethylphosphoramide (0.768 mL, 4.43 mmol, 16 eq.). The solution changed to brown in color and was stirred for 12 h at room temperature. The solution was concentrated under reduced pressure and the residue subjected to flash column chromatography on silica gel (5% ethyl acetate-hexanes) to afford 18 as a colorless viscous syrup (0.095 g, 35% yield): Rf = 0.73 (30% ethyl acetate-hexanes); 1H NMR (600 MHz, CDCl3): δ 7.26–7.15 (m, 33H, aromatic), 7.06 (dd, 2H, J = 7.5 Hz, 1.8, aromatic), 5.17 (dd, 2H, J = 8.9, 3.6 Hz, H-1, H-1’), 4.93 (dd, 2H, J = 10.8, 6.2 Hz, benzylic), 4.8 (m, 4H, benzylic), 4.63 (m, 5H, benzylic), 4.48 (d, 1H, J = 12.1 Hz, benzylic), 4.39 (d, 1H, J = 10.6 Hz, benzylic), 4.32 (d, 1H, J = 12.1 Hz, benzylic), 4.09 (m, 2H, H-5, H-5’), 3.97 (td, 2H, J = 9.3, 7.7 Hz, H-3, H-3’), 3.62 (t, 1H, J = 9.7 Hz, H-3), 3.53 (ddd, 3H, J = 9.5, 7.6, 3.6 Hz, H-4’, H-2, H-2’), 3.45 (dd, 1H, J = 10.7, 3.2 Hz, H-6a’), 3.31 (dd, 1H, J = 10.6, 1.8 Hz, H-6b’), 2.37 (m, 1H, H-6a), 2.25 (dt, 1H, J = 17, 3.3 Hz, H-6b), 1.95 (t, 1H, J = 2.6 Hz, H-7 alkyne); 13C NMR (150 MHz, CDCl3): δ 139.03, 138.90, 138.49, 138.45, 138.30, 137.97, 128.59, 128.53, 128.17, 128.13, 127.59, 127.54, 94.54 (anomeric), 94.46 (anomeric), 81.96, 81.67, 80.46, 80.20, 79.72, 79.52, 77.82, 75.83, 75.80, 75.59, 75.28, 73.66, 72.95, 72.87, 71.05, 70.82, 68.63, 68.24, 21.24; LRMS (ESI): calculated for C63H64O10, 1003.4 [M+Na]+; observed, m/z = 1003.9 [M+Na]+.
2,2’,3,3’,4,4’,6’-Hepta-O-acetyl-6-deoxy-6-vinyl-α,α’-trehalose (19).
Compound 18 (0.245 g, 0.250 mmol) was dissolved in 2 mL of tetrahydrofuran in a three-neck flask equipped with a dry ice condenser. The flask wwas then set in a dry ice-acetone bath. Ammonia gas at was passed through the condenser until several milliliters of ammonia collected. A small piece of sodium metal was added to the solution, which immediately turned turquois in color. The ammonia was refluxed at −33 °C for 15 minutes. The reaction was quenched with 5 mL of methanol. The ammonia was evaporated and the remaining liquids removed under reduced pressure and the residue further dried under high vacuum for 12 h. To the dried residue was added pyridine (3 mL), acetic anhydride (3 mL) and a catalytic amount of DMAP. The solution was stirred for 12 h and then the solvent was concentrated under reduced pressure. The residue was subjected to flash column chromatography on silica gel (5% ethyl acetate-hexanes) to afford 19 as an amorphous solid (0.095 g, 59% yield): Rf = 0.33 (30% ethyl acetate:hexanes); 1H NMR (600 MHz, CDCl3): δ 5.72 (m, 1H, H-7), 5.48 (m, 2H, H-3, H-3’), 5.27 (d, 2H, J = 3.7 Hz, H-1, H-1’), 5.01 (m, 7H, H-2, H-2’, H-4, H6a,b, H8a,b), 4.23 (m, 1H, H-6a’), 4.04 (m, 3H, H-4’, H-5’, H-6b’), 3.38 (m, 1H, H-5), 2.06 (m, 21H, 7XCH3); 13C NMR (150 MHz, CDCl3): δ 170.67, 170.06, 169.95, 169.70, 169.69, 169.65, 169.61, 132.39, 118.48, 91.94, 71.78, 70.10, 69.74, 69.10, 68.56, 68.07, 61.78, 35.45, 29.72, 20.75, 20.64; HRMS (ESI): calculated for C61H63IO10, 647.2187 [M+H]+; observed, m/z = 647.2193 [M+H]+.
2,2’,3,3’,4,4’,6’-Hepta-O-acetyl-6-deoxy-6-oxiranyl-α,α’-trehalose (20).
Compound 19 (0.050 g, 0.077 mmol, 1 eq.) was dissolved in dichloromethane (3 mL) and 3-chloroperoxybenzoic acid (0.066 g, 0.387 mmol, 5 eq.) was added. The solution was stirred for an hour at room temperature and the solvent was was evaporated under reduced pressure. The reside was subjected to flash column chromatography on silica gel (10% ethyl acetate-hexanes) to afford 20 as an amorphous solid (0.030 g, 59% yield): Rf = 0.42 (30% ethyl acetate:hexanes); 1H NMR (600 MHz, CDCl3): δ 5.51 (m, 3H), 5.32 (m, 4H), 5.08 (m, 7H), 4.92 (t, 1H, J = 9.7 Hz), 4.23 (dt, 2H, J = 12.6, 6.3 Hz), 4.06 (m, 5H), 2.98 (m, 1H), 2.78 (m, 2H) 2.43 (td, 2H, J = 5.5, 2.7 Hz), 2.17 (m, 6H, 2XCH3), 2.08 (m, 36H, 12XCH3); 13C NMR (150 MHz, CDCl3): δ 170.69, 170.11, 170.10, 170.02, 169.98, 169.95, 169.94, 169.87, 169.87, 169.79, 196.73, 169.65, 169.64, 169.62, 92.29 (anomeric), 92.11 (anomeric), 91.75 (anomeric), 91.34 (anomeric), 72.20, 71.85, 70.24, 70.13, 70.05, 70.00, 69.95, 69.92, 69.40, 69.22, 68.62, 68.17,68.09, 67.49, 67.38, 61.85, 48.27, 48.18, 20.65; LRMS (ESI): calculated for C61H63IO10, 685.2 [M+Na]+; observed, m/z = 685.3 [M+Na]+.
6-deoxy-6-oxiranyl-α,α’-trehalose (7)
Compound 20 (0.044 g, 0.066 mmol) was dissolved in 9 mL of a mixture of triethylamine-methanol-water (2:6:1). The solution was stirred for an hour in dark. The solvent was evaporated under reduced pressure and subjected to flash column chromatography on silica gel using ethyl acetate-isopropanol-water (6:3:1) as an eluent to afford 7 as an viscous syrup (0.012 g, 50% yield): Rf = 0.45 (6:3:1 ethyl acetate-isopropanol-water); 1H NMR (600 MHz, D2O): δ 5.05 (m, 2H, anomeric), 3.87 (m, 1H), 3.72 (m, 5H), 3.54 (m, 2H), 3.33 (m, 2H), 3.19 (m, 1H), 3.09 (m, 2H), 1.79 (m, 2H). 13C NMR (150 MHz, D2O): δ 93.54, 93.38, 93.21, 93.04, 73.24, 73.19, 72.53, 72.50, 72.41, 72.11, 71.97, 71.38, 71.07, 71.05, 70.92, 70.29, 69.62, 69.59, 69.52, 69.16, 60.42, 51.44, 50.44, 48.28, 46.71, 46.59, 33.63, 23.19, 20.40, 8.15; HRMS (ESI): calculated for C61H63IO10, 369.1397 [M+H]+; observed, m/z = 369.1219 [M+H]+.
Protein expression and purification.
The otsB2 gene, encoding for the Mtb (UniProt code: P9WFZ5) trehalose-6-phosphate phosphatase, TPP, was amplified using Mtb genomic DNA and inserted into a modified pET-28 plasmid. Codon optimized genes encoding the TPP orthologs from Mlt (UniProt code: A0A0E4GVV3) and Mtx (UniProt code: A0A024JUE5) were purchased from IDT Inc., amplified and inserted into modified pET-32 plasmids. The resulting pDR28-otsB2, pDR32-Mlt-otsB2 and pDR32-Mtx-otsB2 plasmids were used in this study to encode and produce recombinant TPP possessing N-terminal polyhistidine tags. The sequences of the expression plasmids mentioned above were confirmed by DNA sequencing (MWG Operon). These plasmids were used to transform E. coli T7 Rosetta. The cells were then cultured at 37 °C in Luria Broth to an O.D.600 nm of 0.6. Gene expression was then induced by adding IPTG to a final concentration of 1 mM followed by a 24-hour incubation at 16 °C. The bacteria were harvested by centrifugation and resuspended with buffer A (20 mM Tris pH 7.5, 150 mM NaCl, 5 mM MgCl2 and 5 mM β-mercaptoethanol). Following bacterial resuspension, 10 μM of lysozyme and 100 μM of DNase I were added to the resuspended cells, which were then incubated for 30 minutes on ice before sonication. The lysate was then centrifuged at 15,000 × g for 45 min and the resulting supernatant applied to a 5 mL HiTrap Talon Crude column (GE Healthcare) previously equilibrated with buffer A. Proteins were eluted from the column by applying a step gradient. First, one column volume of 12% buffer B (150 mM imidazole, 150 mM NaCl, 5 mM MgCl2 and 5 mM β-mercaptoethanol) was applied followed by eight column volumes of 100% buffer B. The purity of the protein was assessed using SDS-PAGE. The purified proteins were then dialyzed into 20 mM Tris pH 8.0 for enzymatic assays.
Steady-state kinetics and inhibition studies.
The EnzChek® Phosphate Assay Kit (E-6646) (Molecular Probes) was used to monitor phosphate production from the hydrolysis of T6P by TPP in a continuous coupled assay. The phosphate released in solution is then used by purine nucleoside phosphorylase (PNP) to convert 2-amino-6-mercapto-7-methylpurine riboside (MESG) into ribose-1-phosphate and 2-amino-6-mercapto-7-methylpurine shifting the absorbance from 330 nm for MESG to 360 nm.
All kinetic and inhibitory studies were performed using the EnzChek® Phosphate Assay Kit (Molecular Probes) at 37 °C and the absorbance was measured at 360 nm using a Synergy™ H4 Hybrid Multi-Mode Microplate Reader (BioTek). The inhibition data were measured using 15 nM Mtb TPP-WT, 5 nM Mlt TPP-WT and 20 nM Mtx TPP-WT varying the concentration of TNP (31 μM to 4 mM), TEP (31 μM to 4 mM), TMP (78 μM to 10 mM), and Validamycin A (3.75 mM to 40 mM) in 25 μL reactions. Normalized data were fitted using the equation Y=100/(1+(XHillSlope)/(IC50HillSlope)) as implemented in Prism 7.0 (GraphPad). All measurements were performed in quadruplicate.
Supplementary Material
Acknowledgements
This work was supported in part by a grant from the National Institutes of Health (Grant No. AI105084) to S.J.S. and D.R.R.
Dedicated to Professor Chi-Huey Wong on his 70th birthday
Notes and references
- [1].Figueroa CM, Lunn JE, Plant Physiol. 2016, 172, 7–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arguelles JC, Arch. Microbiol. 2000, 174, 217–224. [DOI] [PubMed] [Google Scholar]
- [3].Miao Y, Tenor JL, Toffaletti DL, Maskarinec SA, Liu JY, Lee RE, Perfect JR, Brennan RG, Mbio 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cross M, Rajan S, Chekaiban J, Saunders J, Hamilton C, Kim JS, Coster MJ, Gasser RB, Hofmann A, Sci. Rep. 2017, 7, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Becker A, Schloder P, Steele JE, Wegener G, Experientia 1996, 52, 433–439. [DOI] [PubMed] [Google Scholar]
- [6].a) Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR, Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 21761–21766 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) De Smet KAL, Weston A, Brown IN, Young DB, Robertson BD, Microbiol. 2000, 146, 199–208. [DOI] [PubMed] [Google Scholar]
- [7].Murphy HN, Stewart GR, Mischenko VV, Apt AS, Harris R, McAlister MSB, Driscoll PC, Young DB, Robertson BD, J. Biol. Chem. 2005, 280, 14524–14529. [DOI] [PubMed] [Google Scholar]
- [8].Cabib E, Leloir LF, J. Biol. Chem. 1958, 231, 259–275. [PubMed] [Google Scholar]
- [9].Edavana VK, Pastuszak T, Carroll JD, Thampi P, Abraham EC, Elbein AD, Arch. Biochem. Biophys. 2004, 426, 250–257. [DOI] [PubMed] [Google Scholar]
- [10].a) Kalscheuer R, Syson K, Veeraraghavan U, Weinrick B, Biermann KE, Liu Z, Sacchettini JC, Besra G, Bornemann S, Jacobs WR, Nat. Chem. Biol. 2010, 6, 376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fraga J, Maranha A, Mendes V, Pereira PJB, Empadinhas N, Macedo-Ribeiro S, Sci. Rep. 2015, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Middlebrook G, Coleman CM, Schaefer WB, Proc. Natl. Acad. Sci. U.S.A. 1959, 45, 1801–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hatzios SK, Schelle MW, Holsclaw CM, Behrens CR, Botyanszki Z, Lin FL, Carlson BL, Kumar P, Leary JA, Bertozzi CR, J. Biol. Chem. 2009, 284, 12745–12751 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Touchette MH, Holsclaw CM, Previti ML, Solomon VC, Leary JA, Bertozzi CR, Seeliger JC, Bacteriol J. 2015, 197, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gavalda S, Bardou F, Laval F, Bon C, Malaga W, Chalut C, Guilhot C, Mourey L, Daffe M, Quemard A, Chem. Biol. 2014, 21, 1660–1669. [DOI] [PubMed] [Google Scholar]
- [12].a) Lea-Smith DJ, Pyke JS, Tull D, McConville MJ, Coppel RL, Crellin PK, J. Biol. Chem. 2007, 282, 11000–11008 [DOI] [PubMed] [Google Scholar]; b) Bhatt A, Brown AK, Singh A, Minnikin DE, Besra GS, Chem. Biol. 2008, 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grzegorzewicz AE, Pham H, Gundi VAKB, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SEM, Kordulakova J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M, Nat. Chem. Biol. 2012, 8, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Jackson M, Raynaud C, Laneelle MA, Guilhot C, Laurent-Winter C, Ensergueix D, Gicquel B, Daffe M, Mol. Microbiol. 1999, 31 1573–1587 [DOI] [PubMed] [Google Scholar]; b) Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS, Science 1997, 276, 1420–1422. [DOI] [PubMed] [Google Scholar]
- [15].Kacem R, De Sousa-D’Auria C, Tropis M, Chami M, Gounon P, Leblon R, Houssin C, Daffe M, Microbiol. 2004, 150, 73–84. [DOI] [PubMed] [Google Scholar]
- [16].Warner DF, Mizrahi V, Clin. Microbiol. Rev. 2006, 19, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Eoh H, Wang Z, Layre E, Rath P, Morris R, Moody DB, Rhee KY, Nat. Microbiol. 2017, 2: 17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu CL, Dunaway-Mariano D, Mariano PS, Tetrahedron 2017, 73, 1324–1330. [Google Scholar]
- [19].Liu CL, Dunaway-Mariano D, Mariano PS, Eur. J. Med. Chem. 2017, 128, 274–286. [DOI] [PubMed] [Google Scholar]
- [20].Li H, Su H, Kim SB, Chang YK, Hong SK, Seo YG, Kim CJ, J. Biosci. Bioeng. 2012, 113, 224–232. [DOI] [PubMed] [Google Scholar]
- [21].Petit C Structural and inhibitory studies of three essential phosphate utilizing enzymes of Mycobacterium tuberculosis. 2018, pp 158. [Google Scholar]
- [22].a) Backus KM, Boshoff HL, Barry CS, Boutureira O, Patel MK, D’Hooge F, Lee SS, Via LE, Tahlan K, Barry CE, Davis BG, Nat. Chem. Biol. 2011, 7, 228–235; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rose JD, Maddry JA, Comber RN, Suling WJ, Wilson LN, Reynolds RC, Carbohyd. Res. 2002, 337, 105–120 [DOI] [PubMed] [Google Scholar]; c) Wang J, Elchert B, Hui Y, Takemoto JY, Bensaci M, Wennergren J, Chang H, Rai R, Chang CWT, Bioorgan Med. Chem. 2004, 12, 6397–6413 [DOI] [PubMed] [Google Scholar]; d) Sanki AK, Boucau J, Umesiri FE, Ronning DR, Sucheck SJ, Mol. Biosyst .2009, 5, 945–956 [DOI] [PubMed] [Google Scholar]; e) Wolber JM, Urbanek BL, Meints LM, Piligian BF, Lopez-Casillas IC, Zochowski KM, Woodruff PJ, Swarts BM, Carbohydr. Res. 2017, 450, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mikkelsen LM, Krintel SL, Jimenez-Barbero J, Skrydstrup T, J. Org. Chem. 2002, 67, 6297–6308. [DOI] [PubMed] [Google Scholar]
- [24].Lee HH, Hodgson PG, Bernacki RJ, Korytnyk W, Sharma M, Carbohyd. Res. 1988, 176, 59–72. [DOI] [PubMed] [Google Scholar]
- [25].Miao Y, Tenor JL, Toffaletti DL, Washington EJ, Liu JY, Shadrick WR, Schumacher MA, Lee RE, Perfect JR, Brennan RG, Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 7148–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Allen KN, Dunaway-Mariano D, Trends Biochem. Sci. 2004, 29, 495–503 [DOI] [PubMed] [Google Scholar]; b) Lu Z, Dunaway-Mariano D, Allen KN, Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5687–5692 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lahiri SD, Zhang GF, Dunaway-Mariano D, Allen KN, Science 2003, 299, 2067–2071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.