

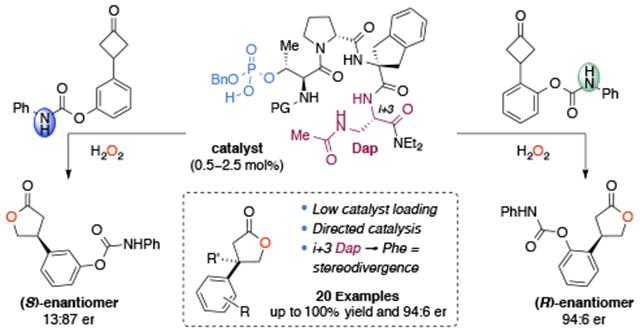
Abstract
Biologically inspired phosphothreonine (pThr)-embedded peptides that function as chiral Brønsted acid catalysts for enantioselective Baeyer–Villiger oxidations (BV) of cyclobutanones with aqueous H2O2 are reported herein. Complementary to traditional BINOL-derived chiral phosphoric acids (CPAs), the functional diversity of the peptidic scaffold provides the opportunity for multiple points of contact with substrates via hydrogen bonding, and the ease of peptide synthesis facilitates rapid diversification of the catalyst structure, such that numerous unique peptide-based CPA catalysts have been prepared. Utilizing a hypothesis-driven design, we identified a pThr-based catalyst that contains an N-acylated diaminopropionic acid (Dap) residue, which achieves high enantioselectivity with catalyst loadings as low as 0.5 mol%. The power of peptide-based multi-site binding is further exemplified through reversal in the absolute stereochemical outcome upon repositioning of the substrate–directing group (ortho- to meta). Modifications to the i+3 residue (LDap to LPhe) lead to an observed enantiodivergence without inversion of any stereogenic center on the peptide catalyst, due to noncovalent interactions. Structure–selectivity and 1H-1H-ROESY studies revealed that the proposed hydrogen bonding interactions are essential for high levels of enantioinduction. The ability for the phosphopeptides to operate as multifunctional oxidation catalysts expands the scope of pThr catalysts and provides a framework for the future selective diversification of more complex substrates, including natural products.
Keywords: asymmetric catalysis, Baeyer–Villiger, chiral phosphoric acid, peptides, phosphothreonine
Graphical Abstract
INTRODUCTION:
Phosphate esters are ubiquitous in biochemistry.1 Phosphothreonine (pThr), among other proteinogenic amino acids (e.g. pSer, pTyr), is the product of site-selective post-translational modifications of proteins, which both induces global conformational changes2,3 and binds signaling molecules, as part of cellular regulation (Figure 1a).4 Yet, no known catalytic site of an enzyme engages pThr as part of the bond-forming or bond-breaking mechanistic machinery. However, chiral phosphoric acids (CPAs) have emerged as powerful catalysts for an ever-expanding list of asymmetric transformations.5 So far, BINOL- or SPINOL-derived CPAs have dominated the expansive literature of CPA-catalyzed reactions (Figure 1b).6 As a result of their C2-symmetry and rigidity, BINOL CPAs have a well-defined binding pocket that facilitate high levels of enantioinduction, predominately arising from the steric bulk on the 3,3’-positions.6e,7 While further modifications to the scaffold can attenuate the electronics and enhance enantioselectivities in select transformations, the ability to construct diverse CPAs with new opportunities for secondary interactions, especially hydrogen bonding, is limited.6,8
Figure 1.
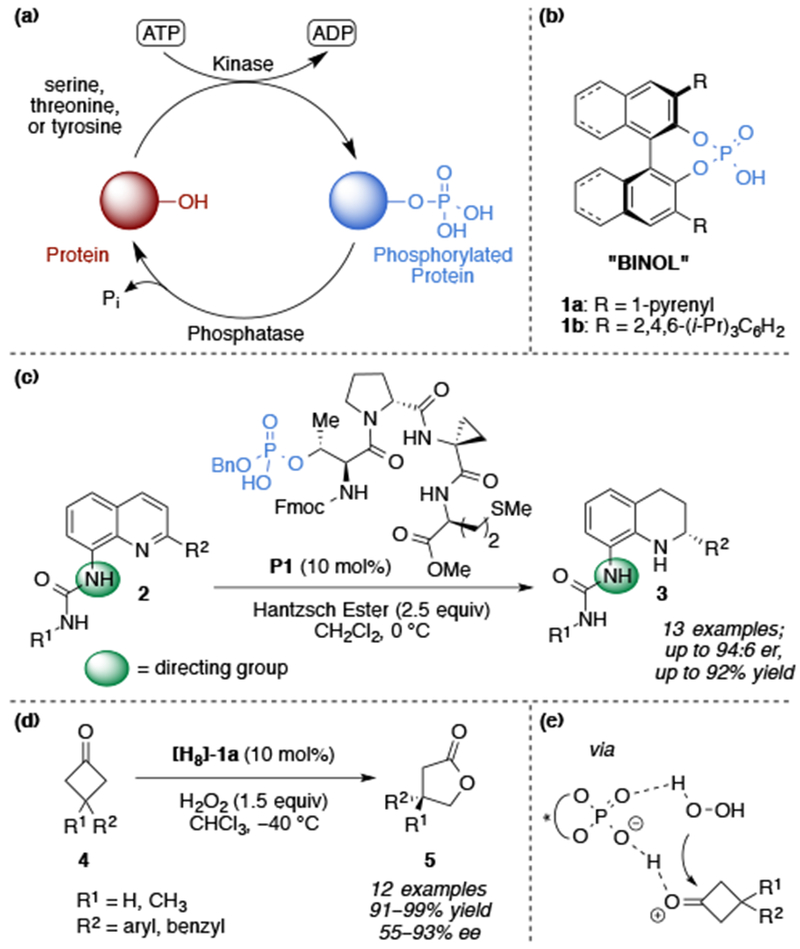
(a) Phosphorylation event as a post-translational modification. (b) BINOL-derived chiral phosphoric acids. (c) Peptide-mediated transfer hydrogenation of 8-aminoquinolines using pThr as a catalytic residue. (d) Ding and co-workers’ pioneering Brønsted acid catalyzed Baeyer–Villiger oxidation of 3-substituted cyclobutanones. (e) Proposed mechanism for the CPA catalyzed BV reaction utilizing H2O2 as the oxidant.
As a primary focus of our group is to functionalize complex molecules, we sought to develop an analogous strategy with added opportunities for non-covalent interactions (NCIs).9 Chiral catalysts that operate on highly functionalized substrates often require less rigid catalysts that possess an increased capacity to interact with substrates through multivalent NCIs, though the explicit, rational design of a suitable catalyst is not always straightforward.10 Additionally, the choice of functionalized model substrates provides an opportunity to examine substrate–catalyst interactions with drug-like molecules.11 Accordingly, we have initiated a program that explores pThr as a biologically inspired catalytic residue in peptide-based Brünsted acid catalysts for a broad array of synthetically interesting reactions. Our first proof-of-principle study established that pThr-based catalysts could indeed mediate enantioselective reactions, including archetypal transfer hydrogenations of functionalized quinolines (Figure 1c) 12 and reductive aminations of functionalized ketones.13 More recently, we have also found that these catalysts facilitate atroposelective cyclodehydrations in complex molecular settings.14 Reported herein are applications of pThr-based catalysts to an entirely orthogonal mode of reactivity, the oxidative ring-expansion of cyclobutanones.
Discovered in 1899, the Baeyer–Villiger (BV) reaction15 remains an indispensable tool for accessing chiral esters and lactones from the corresponding ketone precursors through the use of enzymes,16 organocatalysts,17 and transition metal complexes.18 Inspired by the pioneering work of Ding and co-workers on CPA-catalyzed BV oxidation17b, we endeavored to learn whether pThr-based CPAs could facilitate oxidation chemistry with comparable or complementary selectivities and achieve a benchmark for peptide catalysts for the diversification of more complex substrates (Figure 1d). The enantioselective BV reaction of symmetrical, cyclic ketones is particularly appealing for elucidating catalyst control given the mechanistic ramifications. In general, the migratory aptitude is determined by the substituent best able to stabilize the partial positive charge (e.g. 3° alkyl > 2° alkyl > phenyl > 1° alkyl > methyl).19 However, for symmetrical ketones, the migratory aptitude of the substituents is identical. Furthermore, upon addition of H2O2, two Criegee intermediates may be formed (syn-6 and anti-6), both of which can go on to form either enantiomer of product.20 Therefore the stereochemical outcome is dictated by the ~90° bond rotation of the O-O peroxyl group to obtain the necessary σC–C to σ*O–O orbital overlaps for migration (Scheme 1). The consequence of possible syn- and anti- Criegee intermediates is that the catalysts must posses the ability to control delivery of H2O2, through the differentiation of the diastereotopic faces of the ketone and/or position the dihedral angle of the peroxyl O-O bond with high fidelity. As we show below, the application of pThr-based CPAs in the BV reaction resulted in a remarkable level of catalyst-control through the explicit design of peptide sequences that exploit secondary NCIs, similar to those often associated with enzymes.21 Taken together, the results herein expand the specific capabilities of CPAs for both enantioselective BV and pThr-based catalysts to the previously unreported realm of oxidation catalysis. Yet, it could be that the most exciting ramification of this work is the generalization of CPA catalysis to include scaffolds beyond C2-symmetry, including those that contain catalyst functionality that enables specific interactions with functionalized substrate chemotypes. Shown below are dramatic examples of stereodivergence that highlight this point.
Scheme 1.
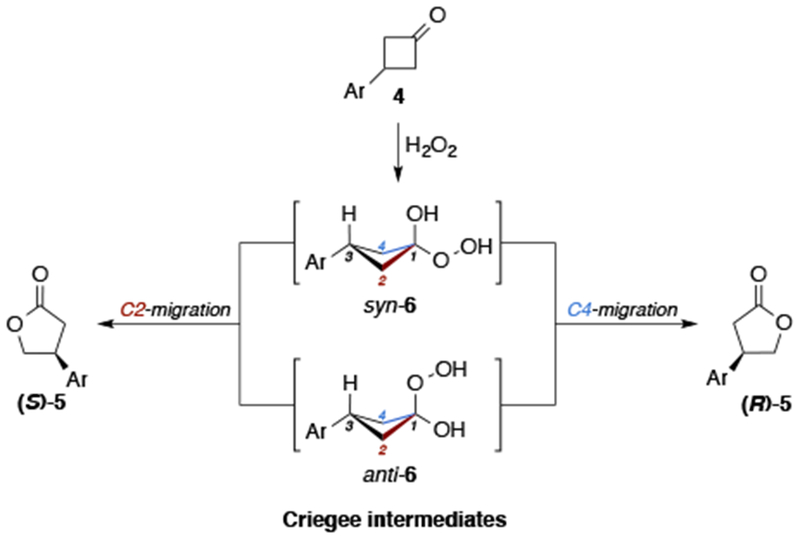
Syn/anti Criegee intermediates.
Results and Discussion
Preliminary studies in our laboratory on pThr-based peptide catalysts for the enantioselective BV oxidation commenced with 3-phenylcyclobutanone (4a) and peptide catalyst P2 (Scheme 2). Employing similarly established reaction conditions17b, the Baeyer–Villiger oxidation of 4a proceeded to give 5a with high conversion, albeit with low enantioselectivity (55:45 er). With 4b, bearing a prochiral quaternary carbon, γ-butyrolactone 5b was obtained in 95% conversion with a slight increase in enantioselectivity (57:43 er). Given the modularity of peptide catalysts, we focused on sequential residue optimization first. We chose peptide sequences that are predicted to adopt well-defined secondary structures, namely β-turns, and identified aspects of the peptide catalyst responsible for enantioselectivity (Table 1).22
Scheme 2.
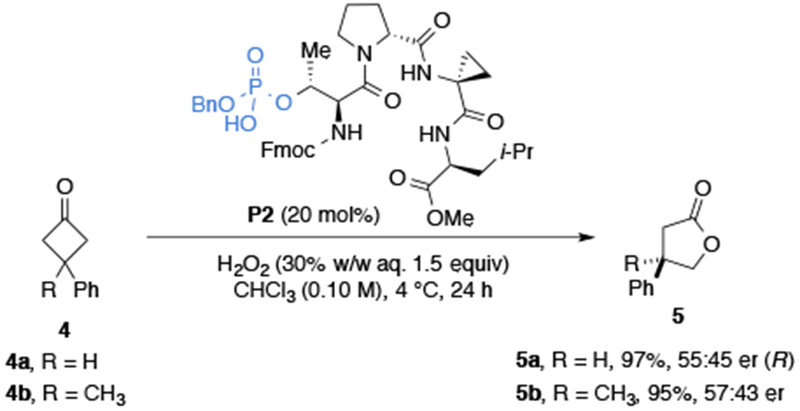
Preliminary pThr-catalyzed Baeyer–Villiger oxidation of 4a–b.
Table 1.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | Catalyst |
conv (%)c | erd | |||||
| PG | i | i + 1 | i + 2 | i + 3 | ||||
| 1 | P3 | Fmoc | pThr(Bn) | d-Pro | Acpc | Leu-NMe2 | 87 | 64:36 |
| 2 | P4 | Fmoc | pThr(Bn) | d-Pro | Acpc | Leu-d-Phe-NMe2 | 67 | 66:34 |
| 3 | P5 | Fmoc | pThr(Bn) | d-Pro | Acpc | Phe-NMe2 | 88 | 64:36 |
| 4 | P6 | Fmoc | pThr(Bn) | d-Pro | Acpc | Ala-NMe2 | 85 | 62:38 |
| 5 | P7 | Fmoc | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 92 | 69:31 |
| 6 | P8 | Fmoc | pThr(Bn) | d-Pro | Aib | d-Phe-NMe2 | 69 | 52:48 |
| 7 | P9 | Fmoc | pThr(Bn) | Pro | Aib | Phe-NMe2 | 54 | 44:56 |
| 8e | P8 | Fmoc | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 19 | 51:49 |
| 9 | P10 | Fmoc | Thr(Bn) | d-Pro | Aib | Phe-NMe2 | <5 | n/a |
| 10 | P11 | Fmoc | pThr(H) | d-Pro | Aib | Phe-NMe2 | 44 | 55:45 |
| 11 | P12 | Fmoc | allo-pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 47 | 51:49 |
| 12 | P13 | Fmoc | pPhSer(Bn) | d-Pro | Aib | Phe-NMe2 | 98 | 67:33 |
| 13 | P14 | Fmoc | pSer(Bn) | d-Pro | Aib | Phe-NMe2 | 73 | 58:42 |
| 14 | P15 | Fmoc-Phe | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 96 | 70:30 |
| 15 | P16 | Fmoc-d-Phe | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 88 | 67:33 |
| 16 | P17 | Fmoc-Pro | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 88 | 72:28 |
| 17 | P18 | Fmoc-d-Pro | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 93 | 73:27 |
| 18f | P18 | Fmoc-d-Pro | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 73 (75)g | 78:22 |
| 19h | P18 | Fmoc-d-Pro | pThr(Bn) | d-Pro | Aib | Phe-NMe2 | 50 | 77:23 |
| 20h | (R)-1b | 97 | 64:36 | |||||
Reaction conditions: Cyclobutanone 4b (0.05 mmol, 1.0 equiv) at 0 → 4 °C (ambient temperature) with [4b] = 0.10 M CHCl3.
Reported results are the average of two trials.
Determined by 1H NMR relative integrations.
Enantiomeric ratios were determined by CSP-HPLC analysis (Chiralpak IA, 210 nm). Absolute configuration was not determined.
Reaction performed with f-BuOOH (70% w/w aq. 1.5 equiv).
Reaction was carried out at −15 °C.
Yield in parentheses determined by 1H NMR internal standard (1,3,5-trimethoxybenzene).
Reaction was carried out at −25 °C.
Abbreviations: pThr, phosphothreonine; Bn, benzyl; Acpc, 1-aminocyclopropane carboxylic acid; Aib, 2-aminoisobutryic acid; pPhSer, β-threo-phosphophenylserine; pSer, phosphoserine, Bz, benzoyl.
Given the significant influence of the C-terminal group on enantioselectivity in prior studies, the dimethyl amide analog P3 was prepared first, which afforded 5b in 87% conversion and 64:36 er (Table 1, entry 1) with 10 mol% catalyst loading.23 While other reactions investigated from our group have demonstrated that the i+2 residue has a pronounced effect on the enantioselectivity, modifications to the i+2 or i+3 positions (P4–P7) provided little influence on the enantioselectivity in the BV oxidation of 4b (entries 2–5) (see Table 1 for “i+n” nomenclature).23 Investigation into the diastereomeric series of the tetrameric peptides revealed the l-stereochemical configuration of the i+3, and d-stereochemical configuration of Pro i+1, was crucial (entry 6–7). While a number of reaction parameters were evaluated, permutations from the aforementioned conditions did not offer any advantages (see SI). However, when H2O2 is substituted with t-BuOOH, 5b is obtained in only 19% conversion and poor enantioselectivity (entry 8). This observation suggests that a similar mechanism proposed by Ding and co-workers is operative, which invokes the phosphoric acid as a bifunctional catalyst.24 Furthermore, when the phosphate moiety is removed, as in P10, catalysis is lost, highlighting the catalytic role of the phosphorylated amino acid (entry 9).
Given the relatively subtle effects on enantioselectivity observed thus far, we sought to make alterations to the catalytically active residue, as well as modifications nearest to pThr, that might enhance enantioselectivity. In contrast to rigid C2-symmetric CPAs, non-equivalent rotamers about the β-C–OpThr bond, in addition to the stereogenic P(V) and rapid tautomerization of the acidic proton, provide numerous, chemically distinct catalyst conformers.25 When the phosphate benzyl group is removed, thereby producing a non-stereogenic phosphorous atom, the enantioselectivity is diminished (55:45 er, entry 10). Inversion of the Thr β-stereochemistry to allo-pThr ablates catalyst activity and selectivity, resulting in 47% conversion and 51:49 er (entry 11). Subsitution of the β-methyl group with a more bulky β-phenyl substituent (P13) affords the product in 97% conversion, although surprisingly the enantioselectivity remains unaffected (67:33, entry 12) given the close proximity of the β-substituent to the phosphate moiety. The importance of the β-substituent is further highlighted as replacement of pThr with pSer resulted in 78% conversion and 58:42 er (entry 13).26 While elongation of the C-terminus did not attenuate enantioselectivity (entry 2), an additional d-Pro at the N-terminus resulted in increased selectivity (73:27 er), (entries 14–17). At low temperatures (−15, −25 °C) 5b could be obtained in up to 78:22 er with a slight decrease in yield (entries 18–19). Notably, P18 provided higher enantioselectivity than commercially available BINOL derived CPA, (R)-TRIP 1b (64:36 er), and comparable selectivities (81:19 er) to those previously reported.17b
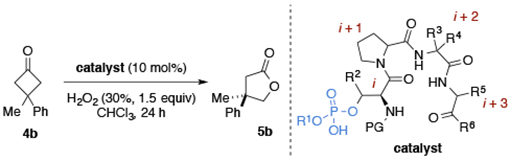
In order to gain additional information about the catalyst structure and develop hypotheses about NCIs, we determined the solid-state structure of P4 using X-ray crystallography (Figure 2a). Interestingly, there were three intramolecular hydrogen bonds, including the predicted 10-membered β-turn hydrogen-bond between N–H(i+3)⋯O(i) and N–H(i)⋯O(i+4).22 Recently, the structures of various dimethylaminoalanine (Dmaa)-embedded β-turn catalysts were extensively studied, and we wondered whether the principles learned would be applicable in ongoing catalyst design.27 In order to make a direct comparison, we obtained a crystal structure of the Dmaa analog (7) (Figure 2b).28 Indeed, both catalysts showed nearly identical secondary structures in the solid-state, despite the rather large change to the catalytically active residue (Fmoc-pThr(Bn) vs. Boc-Dmaa) as depicted by the similar hydrogen bond lengths and structural overlays (see SI for details). Moreover, both peptides exhibit similar φ and ψ dihedral angles that are consistent with a type II’ β-turn secondary structure, and only show a significant difference in the conformation of the i+3 Phe side chain orientation.22 One observation from the solid-state structure of P4 was the relatively exposed nature of the phosphate moiety, which may position substrates in such a way as to prevent favorable NCIs needed to achieve a highly enantioselective BV reaction.
Figure 2.
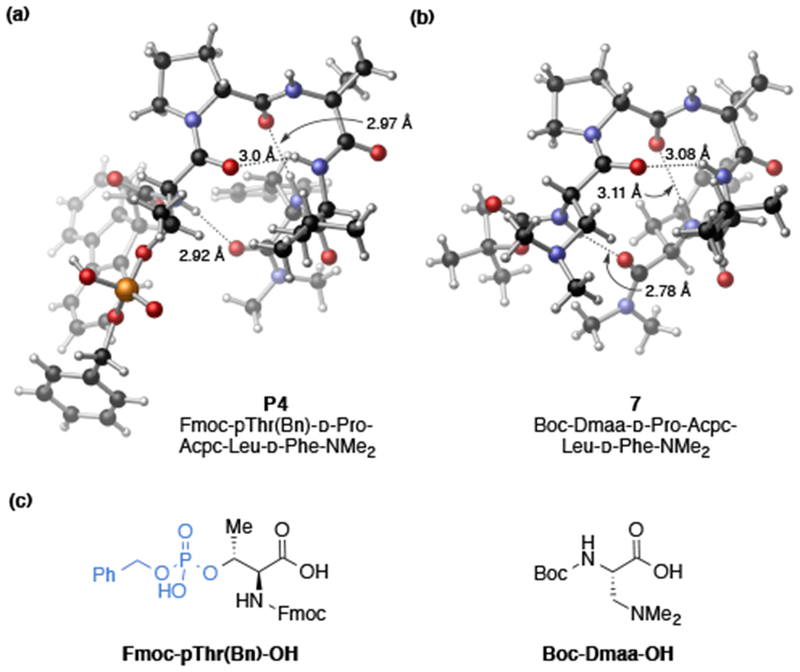
Comparison of X-ray crystal structures of pThr- and Dmaa-embedded peptides. (a) X-ray structure of P4. Two 1,4-dioxane solvent molecules omitted for clarity (see SI for details). (b) X-ray structure of 7. Two distinct packing polymorphs were observed in the unit cell (7a, 7b), only one is shown for clarity (See SI for details). (c) Catalytically active residues (pThr and Dmaa) embedded in i position of conserved peptide sequence.
Given the numerous examples of peptide catalyst-substrate interactions involving hydrogen bonding networks and the diversity of functionalization in drug-like molecules, we hypothesized an additional hydrogen bond donor or acceptor on the substrate might engage in further secondary contacts with the catalyst and perturb enantioselectivity.23,29 Using substrate 4c, bearing a carbamate ortho-substituent and 10 mol% of P7, product 5c was obtained in 97% yield and 66:34 er (Scheme 3). Initially, this result suggested additional functionality on the substrate had no influence on the enantioselectivity of the reaction, and that perhaps the selectivities observed thus far are due largely in part to steric hindrance. Based on the relative positions of the i pThr and i+3 Leu side chains in the solid state of P4, we postulated that an i+3 side chain-directing group (e.g. amide functionality) would afford the opportunity for substrate 4c to engage in attractive secondary interactions with the amino acid side chains synergistically.
Scheme 3.
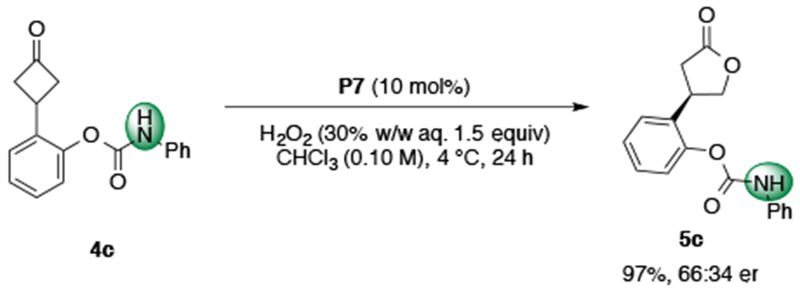
Baeyer–Villiger oxidation of carbamate 4c.
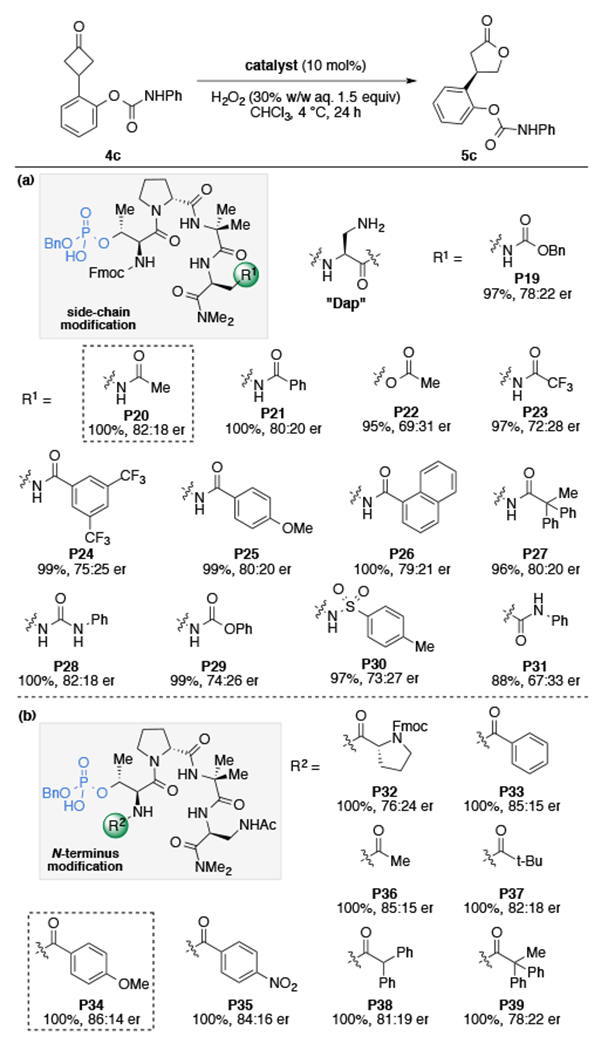
While more than half of proteinogenic amino acids possess side-chain functionality that can engage in hydrogen bonding interactions, for our purposes, we envisioned an ideal amino acid residue to be neutral, easily diversifiable, and sufficiently short as to reduce the rotational degrees of freedom. We imagined that the readily available 2,3-diaminoproprionic acid (Dap) residue, a biologically significant non-proteinogenic amino acid, would fulfill all these requirements.30 Indeed, when Dap(Cbz) (P19) was incorporated in the peptidic scaffold, the reaction delivered product 5c in 97% yield and 78:22 er, lending credence to our hypothesis (Table 2a). Encouraged by these results, we began to further investigate the role of the i+3 Dap residue on the enantioselectivity of the BV reaction.
Table 2.
 |
Reaction conditions: Cyclobutanone 4c (0.025 mmol, 1.0 equiv), catalyst (10 mol%), and H2O2 (30% w/w aq. 1.5 equiv) at 0 → 4 °C (ambient temperature) with [4c] = 0.10 M CHCl3.
Conversion determined by 1H NMR relative integrations and enantiomeric ratios determined by CSP-HPLC analysis (Chiralpak IA, 230 nm).
Reported results are the average of two trials.
Hydrogenation of the Cbz-protecting group allowed access to a variety of side chain analogs, following amino functionalization, and in comparing catalysts P19–P31, a number of differences were realized (Table 2a). Initially, acetamide P20 and benzamide P21 were prepared, which both resulted in increased enantioselectivity (82:18 er and 80:20 er, respectively), whereas the serine analog (P22) afforded 5c in decreased selectivity (69:31 er). Replacement of the acetamide (P20) with electron-deficient amides (P23–P24) resulted in generally lower selectivity, while the electron-rich amide P25 gave similar results (80:20 er). There was little difference observed in enantioselectivity among bulkier amides (P26, P27), signifying that the increased selectivity of the catalyst is likely not attributable to steric factors alone. Catalysts containing a phenyl urea performed equally well (P28), although carbamate P29 and sulfonamide P30 afforded the product with reduced enantioselectivity. The directionality of the amide group was shown to be crucial, as the Asn analog P31 resulted in a substantial decrease in selectivity (67:33 er). Together, these results suggest that while the N–H of the side chain might not be directly involved in the putative secondary interactions, an amido group on the side chain is necessary for achieving high selectivities.31 The enantioselectivity could then be further increased through N-terminal modification (Table 2b, P32–P39). It was found that an electron-rich N-terminal amide was advantageous (P34, 86:14 er), while bulkier amides afforded decreased selectivities. Although a steric effect is possible, the increased enantioselectivity is thought to be associated with changes in the electronic nature of the N-terminal amide that alters the intramolecular hydrogen bonding of the peptide.13
With P34 identified as a more selective catalyst, we set out to perform an optimization of the reaction conditions. A catalyst loading screen with 10, 5, and 2.5 mol% peptide P34 resulted in a slight increase in enantioselectivity with no loss in conversion at 4 °C in 24 hours (Table 3, entries 1–3). By reducing the reaction concentration from 0.10 M to 0.05 M in CHCl3, 5c was obtained in 99% yield and 88:12 er (entry 4). The enantioselectivity was further increased by lowering the reaction temperature to −15 °C (89:11 er), although at −25 °C the selectivity was not improved (entries 5–6). We chose to briefly re-examine the i+2 position and found that when Aib was replaced with Aic (P40), the product was obtained in 99% conversion and 92:8 er under the optimized reaction conditions. Additionally, by switching the dimethylamide C-terminal group with a diethylamide (P41), the lactone could be obtained with 99% yield and 94:6 er. With 0.5 mol% of P41, 5c could be obtained in 88% yield and nearly the same enantioselectivity (92:8 er) with prolonged reaction time, which demonstrates both the robustness and efficiency of the pThr-embedded catalysts.32 It should be noted that when (R)-1b is used as the catalyst under the reaction conditions optimized for the peptide catalyst, 5c is obtained in 74% yield and 77:23 er (entry 10). Although the mode of catalysis is likely the same, the ability for the peptide catalyst to engage in additional hydrogen bonding interactions offers an advantage in achieving highly selective transformations of more functionalized substrates. With the optimized conditions in hand, peptide P41 was used to evaluate the substrate scope of the enantioselective BV of cyclobutanones.
Table 3.
In depth optimization of peptide and reaction conditions.a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | cat. | i + 2 | X | cat. (mol%) | conc. [M] | T (°C) | conv. (%)b | erc |
| 1 | P34 | Aib | NMe2 | 10 | 0.10 | 4 | 100 | 85:15 |
| 2 | P34 | Aib | NMe2 | 5 | 0.10 | 4 | 100 | 86:14 |
| 3 | P34 | Aib | NMe2 | 2.5 | 0.10 | 4 | 100 | 86:14 |
| 4 | P34 | Aib | NMe2 | 2.5 | 0.05 | 4 | 100 | 88:12 |
| 5 | P34 | Aib | NMe2 | 2.5 | 0.05 | −15 | 99 | 89:11 |
| 6d | P34 | Aib | NMe2 | 2.5 | 0.05 | −25 | 100 | 88:12 |
| 7 | P40 | Aic | NMe2 | 2.5 | 0.05 | −15 | 99 | 92:8 |
| 8 | P41 | Aic | NEt2 | 2.5 | 0.05 | −15 | 99 | 94:6 |
| 9e | P41 | Aic | NEt2 | 0.5 | 0.05 | −15 | 88 | 92:8 |
| 10 | (R)-1b | 2.5 | 0.05 | −15 | 74 | 77:23 | ||
Reaction conditions: Cyclobutanone 4c (0.05 mmol, 1.0 equiv) and H2O2 (30% w/w aq. 1.5 equiv). Reported results are the average of two trials (0.05 mmol).
Conversion determined by 1H NMR relative integrations.
Enantiomeric ratios were determined by CSP-HPLC analysis (Chiralpak IA, 230 nm).
Reaction performed for 48 hours.
Reaction performed for 96 hours.
Abbreviations: Aic, 2-aminoindane-2-carboxylic acid.
As shown in Table 4, P41 provides a number of N-substituted substrates with high enantioselectivities. In general, N-aryl carbamates are processed with high enantioselectivity, and electron-donating or electron-withdrawing groups in the para-position do not lead to significant perturbations in the enantioselectivity, such that substrates 4d–4g were smoothly converted to the corresponding lactones in nearly quantitative yield and good enantioselectivities within 24 hours (93:7 – 94:6 er) (Table 4, entries 2–5). When the reaction was carried out with a cyclobutanone containing a p-t-butyl phenyl carbamate, the product was obtained in 97% yield and 94:6 er (entry 6). Additionally, the dioxolane product can be isolated in 96% yield and 93:7 er (entry 7). Benzyl or alkyl substituted carbamates are generally less well tolerated, requiring longer reaction times (72–84 hours) and decreased enantioselectivity (82:18 – 85:15 er) (entries 8–9). Using the carbamate-functionalized analog of 4b, which contains a methyl group at quaternary position C3, lactone 5l can be isolated in 94% yield and 80:20 er, albeit with increased reaction time (entry 10). Lastly, we were able to establish the absolute configuration of the lactone products 5e and 5f by X-ray diffraction (see SI for details).
Table 4.
Carbamate substrate scope.a
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | time (h) | yield (%)b | erc |
| 1 | H | C6H5 (c) | 24 | 99 | 94:6 |
| 2 | H | 4-OMeC6H4 (d) | 24 | 99 | 93:7 |
| 3 | H | 4-CF3C6H4 (e) | 24 | 98 | 93:7 |
| 4 | H | 4-BrC6H4 (f) | 24 | 94 | 94:6 |
| 5 | H | 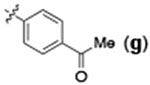 |
24 | 95 | 92:8 |
| 6 | H | 4-f-BuC6H4 (h) | 24 | 97 | 94:6 |
| 7 | H | 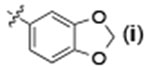 |
24 | 96 | 93:7 |
| 8 | H | CH3CH2 (j) | 84 | 96 | 85:15 |
| 9 | H | C6H5CH2 (k) | 72 | 95 | 82:18 |
| 10 | CH3 | C6H5 (l) | 72 | 94 | 80:20 |
Reaction conditions: Cyclobutanone 4 (0.10 mmol, 1.0 equiv), P41 (2.5 mol%), and H2O2 (30% w/w aq. 1.5 equiv) at −15 °C with [4c] = 0.05 M CHCl3. Reported results are the average of two trials.
Isolated yield after chromatography.
Enantiomeric ratios were determined by CSP-HPLC analysis.
See supporting information for details on assignment of absolute configuration. The configuration of products 5c-d, 5g–l were assigned in analogy to 5e and 5f.
Further studies were then undertaken to gain a better understanding of the apparent directing group effects and key substrate-catalyst interactions that are required to achieve a highly enantioselective transformation. To probe the putative hydrogen bonding with the i+3 Dap residue, the phenylalanine analog P42 was prepared, containing only a single-point mutation (Phe) at the i+3 position (Figure 3a). Various substrates were then subjected to the optimized reaction conditions using 2.5 mol% of P41 or P42 for 24 hours. In agreement with our earlier observations, catalyst P42 (Phe) afforded product 5c in 78% yield and 79:21 er, which corresponds to a decrease in Δ(ΔΔG‡) of 0.73 kcal·mol−1 compared to P41 (Dap) (Figure 3). When the carbamate directionality is inverted, 5m was obtained with excellent enantioselectivity using P41 (Dap, 91:9 er) and again diminished yield and enantioselectivity with catalyst P42 (Phe). The same trend is observed for products 5n–5p, wherein urea32 and amide groups are better matched for the Dap-containing peptide P41, which contains more functionality for attractive secondary interactions, than for P42 containing Phe. The presence of a hydrogen bond donor is necessary for achieving high selectivities, which we believe arises in part to essential hydrogen bonding interactions occurring in the transition structure of the reaction.29
Figure 3.
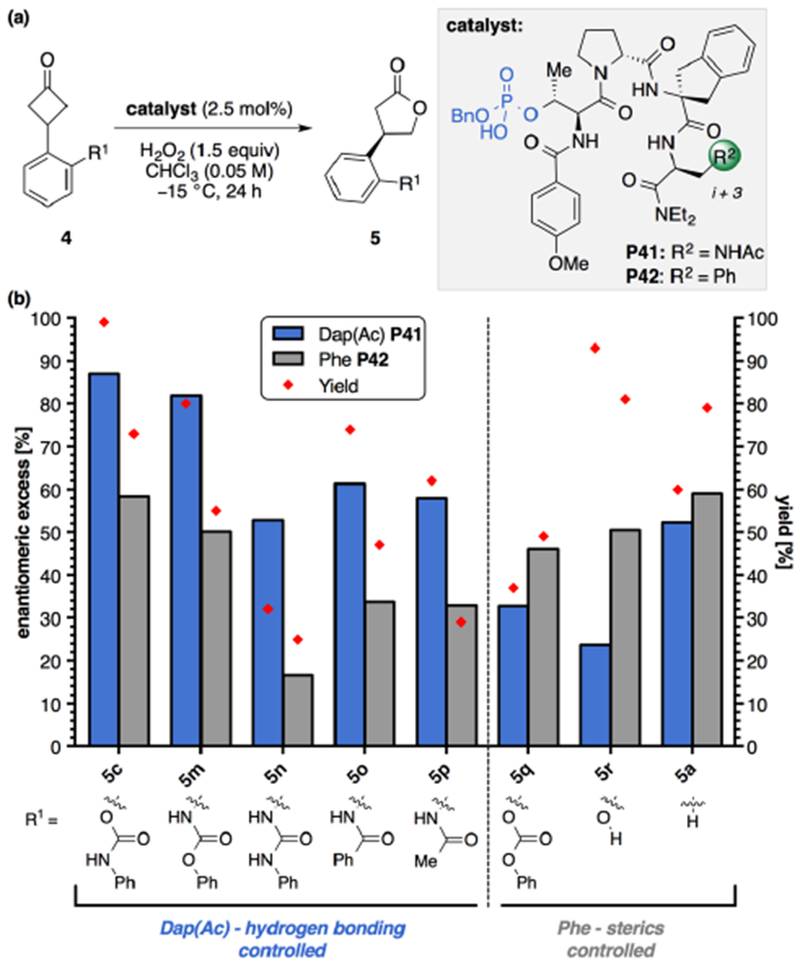
(a) General reaction scheme for BV oxidation with peptide catalyst and (b) structure–selectivity study with P41 and Phe analog (P42). Reaction conditions: Cyclobutanone 4 (0.05 mmol, 1.0 equiv), catalyst (2.5 mol%), and H2O2 (30% w/w aq. 1.5 equiv) at −15 °C with [4c] = 0.05 M CHCl3. Yield was determined by 1H NMR analysis of the crude reaction mixture using an internal standard (1,3,5-trimethoxybenzene). Enantiomeric ratios were by CSP-HPLC analysis. Reported results are the average of two trials. The absolute configuration of products 5m–r were assigned in analogy to 5e and 5f. The absolute configuration of 5a was assigned as (R)-(−)-5a by comparison of the measured optical rotation to literature values.17a
Altogether, these results demonstrate a synergistic effect between the nature of the appended substrate directing group and the multifunctional nature of the peptide catalysts. It is notable that in all of the substrates tested thus far, the yields are consistently better with P41 (Dap), which suggests associative interactions that lead to a rate acceleration for oxidations compared with P42 (Phe). Using catalyst P41 (Dap), with carbonate substrate 4q lacking an N–H, 5q was obtained in only 37% yield and 66:34 er. In contrast, catalyst P42 (Phe) performs better, generating the product in 49% yield and 78:22 er. The difference in selectivities achieved with catalyst P41 when the N–H of 4c is replaced with an oxygen atom (4q) corresponds to a Δ(ΔΔG‡) of 1.07 kcal·mol−1 (94:6 er for 5c and 66:34 er for 5q). Substrates 4r and 4a are again obtained in better enantioselectivity with the Phe catalyst P42, although the phenol is obtained in higher yield with P41 (Dap). Tuning of the i+3 position provides a complementary pair of catalysts that can obtain good-to-high enantioselectivities for a range of substrates through secondary NCIs.
Having demonstrated that ortho- directing groups lead to high enantioselectivity, we were interested in the question, whether alternative placement of the identical directing group could allow for switching of catalysis trajectories. To examine whether the peptides could recognize more distal functionality we prepared substrate 4s, which repositions the N-aryl carbamate from the ortho- to the meta- position. Under the optimized conditions, lactone 5s was obtained in 97% yield and 13:87 er with 2.5 mol% P41 (Scheme 4). Moreover, the X-ray structure of 5s revealed that the opposite absolute configuration was obtained, compared to substrates containing an ortho- directing group, now favoring the S-enantiomer (see SI for X-ray analysis).34 The results of the divergence in the absolute stereochemistry can be explained by either (1) delivery of H2O2 to the diastereotopic face of the ketone with the same C–C σ-bond migration or (2) the substrate–catalyst complex repositioning the peroxyl group in the Criegee intermediate ~90° to achieve the opposite, but equivalent C–C bond migration (Scheme 1). It is possible that both explanations can account for the divergence, as the addition of H2O2 to either face of the prochiral ketone leads to syn and anti isomers, which can both afford either enantiomer of product. The ability for P41 to recognize both substrates in a manner that achieves the opposite absolute stereochemistry of the lactone products (R-5c and S-5s), under otherwise identical reaction conditions, is notable.
Scheme 4.
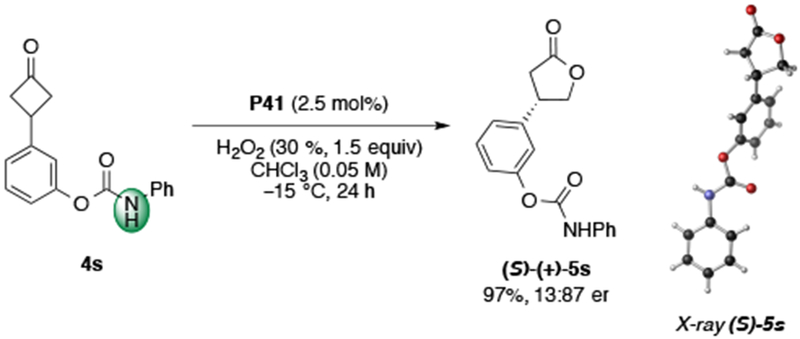
Observed reversal in absolute stereochemistry in the BV of meta-4s.
In continuing our analysis of the effect of Dap(Ac) and Phe substitution at the i+3 position on enantioselectivity, we applied catalyst P42, bearing the Phe i+3 residue, to the oxidation of substrate 4s (Figure 4a). With 2.5 mol% P42, the opposite enantiomer lactone (R)-5s is obtained in 74% yield and 66:34 er under the optimal reaction conditions, relative to P41 (Dap).35 However, it should be noted that the absolute configuration of product (R)-5s is the same as the γ-butyrolactone products 5 bearing an ortho- directing group, previously obtained from the BV reaction with P42 (Figure 3). This observed inversion in enantioselectivity is all the more impressive given the constituent amino acids in both peptides (P41 and P42) have the same stereochemical configuration, yet afford opposite enantiomers of product 5s.23d The Δ(ΔΔG‡) of 1.3 kcal·mol−1 is a consequence of how the two catalysts are able to recognize the substrate upon binding to form different substrate–catalyst complexes. Moreover, when the reaction is carried out with (R)-1b as the catalyst, (R)-5s is obtained in 86% yield and 73:27 er, which matches the absolute stereochemistry and selectivities obtained in the oxidation of 4c with (R)-1b (Table 3, entry 10). This divergence in enantioselectivity could stem from either (1) a conformational change in the catalyst structure upon mutation of the i+3 position or (2) a perturbation in the secondary, non-covalent interactions between the side-chains of the peptide and the functionalized substrate. Once again, it appears that the catalyst P42 with an i+3 Phe residue and (R)-TRIP (1b) achieve selectivity through steric hindrance, whereas P41 bearing the Dap residue operates through multivalent NCIs, resulting in the products bearing opposite absolute configurations.
Figure 4.
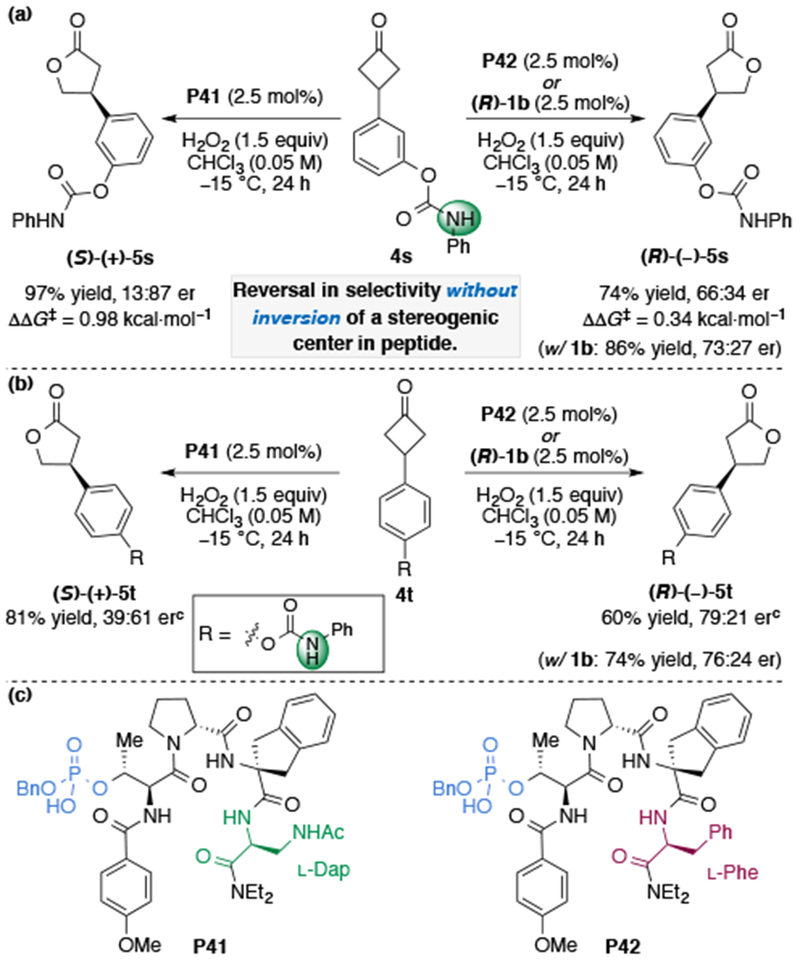
Divergences in enantioselectivity in meta- and para-N-aryl carbamate without inversion of a catalyst stereogenic center.a,b
a Conditions: Cyclobutanone 4 (0.05 mmol, 1.0 equiv), H2O2 (30% w/w aq. 1.5 equiv), catalyst (2.5 mol%) at −15 °C with [4] = 0.05 M CHCl3. b Yield was determined by 1H NMR analysis of the crude reaction mixture using an internal standard (1,3,5-trimethoxybenzene), and enantiomeric ratios determined by CSP-HPLC analysis. Reported results are the average of two trials. c Absolute configuration of 5t assigned in analogy to 5s.
After realizing this intriguing divergence in the absolute sense of enantioinduction by catalysts P41 and P42 with 4s, we prepared substrate 4t, which contains a remote para-N-aryl carbamate directing group (Figure 4b). When we applied optimized catalyst P41 bearing the i+3 Dap(Ac) residue to the oxidation of 4t, lactone 5t was obtained in 81% yield and diminished selectivity (39:61 er). Alternatively, phenylalanine analog P42 afforded the product in 56% yield and 79:21 er, similar to the results obtained with (R)-1b (74% yield, 76:24 er). Once again, P42 and (R)-1b afford the opposite absolute stereochemistry of lactone product relative to P41. Based on the results presented hitherto, catalyst P42 appears to provide enantioinduction through steric repulsive interactions, whereas catalyst P41, containing an i+3 Dap residue can engage the substrate in additional hydrogen-bonding interactions. These differences in mechanism can lead to a divergence in the absolute stereochemistry of product without inversion of a catalyst stereocenter, and seem to suggest that even a remote directing group can engage in NCIs with the peptidic scaffold and perturb the enantioselectivity.
To explain the emerging trends from the structure–selectivity studies and intriguing divergence in observed enantiomeric products 5s–t, we sought to understand the effect of i+3 substitution on peptide secondary structure using 2D NMR spectroscopy (Figure 5a) (see SI for details).36 In the case of P41, a number of nOe contacts were present between the i+3 Dap residue and the i+1 d-Pro residue that indicated the Dap side chain may be orientated on the top face of the peptide turn. However, for P42 no nOe contacts were observed between the i+3 Phe side chain and the peptide backbone, which suggests the phenyl ring is positioned away from the peptide turn. Yet, nOe contacts are still observed between the βC–H of the i pThr residue and the βC–H of the i+3 residues for both peptides, indicating some similarity between the positioning of the Phe and Dap residues in the peptide secondary structure. Furthermore, the orientation of the pThr(Bn) residue is likely the same in each catalyst as indicated by five additional conserved interresidue contacts present at the i catalyst position. DMSO titration studies were then conducted to elucidate the hydrogen bonding environments of each N-H proton, which further showed structural similarities between P41 and P42 (Figure 5b).37 While the N–H(i) exhibits a subtle downfield shift, the N–H(i+2) and N–H(i+3) peaks remain constant and shift upfield respectively, suggesting both N–H(i+2) and N–H(i+3) being previously engaged in intramolecular hydrogen bonding. Together with the observed nOe contacts, these observations are suggestive of both peptides adopting a type I’ double β-turn.18,22 It is worth noting that P41 and P42 appear to adopt a type I’ β-turn secondary structure in solution, whereas previously P4 adopted a type II’ β-turn in the solid-state.38
Figure 5.
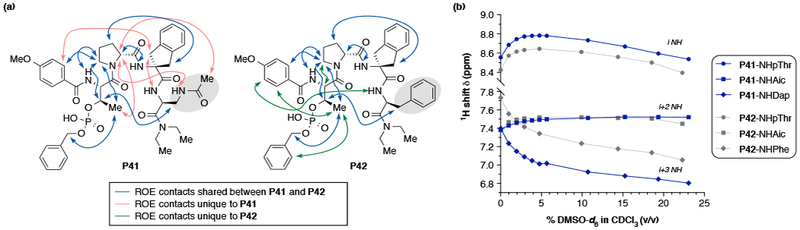
(a) 1H–1H ROESY NMR of peptide catalysts P41 and P42 show both unique and conserved interresidue nOe correlations (800 MHz, 5.0 mM in CDCl3 at 25 °C). (b) 1H NMR solvent titration curve of DMSO-d6 to identify solvent exposed and hydrogen bonded amides for peptides P41 and P42 (5.0 mM concentration in CDCl3 (referenced to 7.26 ppm) at 25 °C).
With these considerations in mind, a mechanistic hypothesis for the formation of the enantioenriched products could be assembled (Figure 6a). In our thinking, we preserve the type I’ β-turn secondary structure in accord with our experimental observations. Given the similarity between the two structures, as determined by 2D NMR and DMSO titration studies, we believe that the incorporation of the Dap(Ac) residue does not result in a different conformation of the peptide. Rather, the i+3 Dap side chain of P41 is orientated on the top face of the catalyst with the C=O of the i+3 side chain acetamide free to engage in a hydrogen bond with the substrate, whereas the i+3 Phe side chain in P42 does not provide additional hydrogen bonding. We envision that the bifunctional phosphate moiety of pThr activates the ketone for addition of H2O2.24 Although speculative, the peroxyl group of the Criegee intermediate could engage in further hydrogen bonding with the peptide backbone amides. In the instances of switching in stereoselection, it is plausible to consider a similar binding model (Figure 6b). However, upon substrate binding with the catalyst side-chain amide, a slight change in the conformation of the peroxyl group results in the opposite stereochemical outcome. Since the i+3 Phe side chain in P42 is unable to engage in further hydrogen bonding, steric hindrance is the predominating factor in stereoselectivity, and thus the absolute stereochemistry of product is unchanged when the carbamate directing group is placed in the o-, m-, or p- positions. The multi-point binding of the substrate-catalyst complex allows for stabilization of the Criegee intermediate and facilitates a highly selective rearrangement, akin to those interactions found in BVO-monoxygenases.16c
Figure 6.
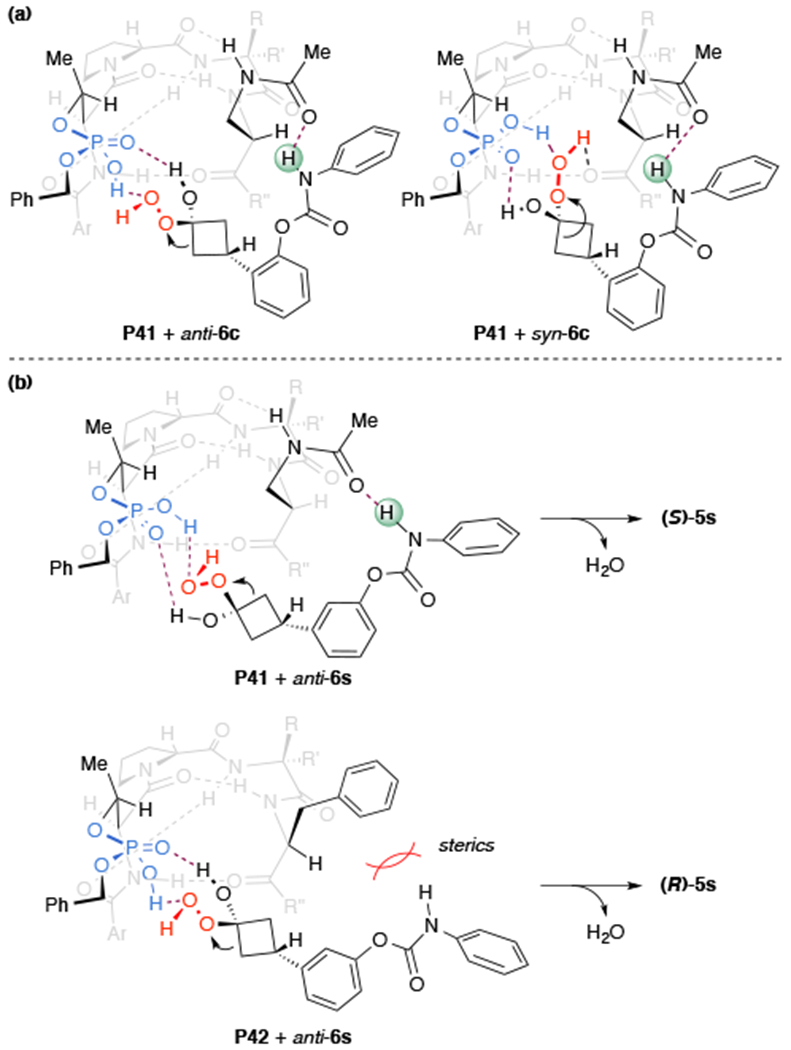
(a) Speculative transition structures showing putative substrate-catalyst hydrogen bonding interactions with an antiperiplanar orientation of the peroxyl group in the Criegee intermediate, which is consistent with the absolute configuration of the product. (b) Speculative transition structures for the switching in stereoselection in substrate 4s, which is meta-substituted.
Conclusion:
In summary, we have reported a multifunctional Brønsted-acid pThr-based peptide catalyst for the enantioselective BV reaction of functionalized cyclobutanones with low catalyst loadings (0.5–2.5 mol%). By utilizing hypothesis-driven design, aided by detailed NMR and crystallographic studies, we have identified the inclusion of an i+3 Dap residue to be advantageous for achieving high selectivities through secondary non-covalent interactions with a variety of directing groups. Moreover, the ability of the peptide to interact with a carbamate directing group in multiple positions of an aryl ring leads to an observed divergence in the absolute stereochemistry (94:6 er to 13:87 er R/S) of the γ-butyrolactone products (5) with a pThr-embedded peptide catalyst containing Dap. Permutations to the i+3 residue provide a complementary pair of catalysts for a further range of cyclobutanone substrates, which affords the products 5 in high yield and stereoselectivities, and in select cases opposite absolute configuration.39 This work provides a framework for the further development of pThr-based catalysts for applications in asymmetric catalysis and modifications of complex molecules, where multiple sites of engagement are necessary to achieve selective functionalization.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health (NIH R01-GM096403). C.R.S. gratefully acknowledges the National Science Foundation Graduate Research Fellowship program for funding (DGE1122492). We also acknowledge Newlyn N. Joseph for the preparation of intermediates towards 4t. The crystallographic data for 4d was acquired at the Advanced Light Source at Lawrence Berkeley National Laboratory. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
The authors declare no competing financial interest.
Crystallographic data are deposited with the Cambridge Crystallographic Data Center under the accession numbers CCDC 1865894 (4d), 1867752 (5c), 1865895 (5e), 1865896 (5f), 1865897 (5s), 1865899 (P4), and 1865898 (7).
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Additional figures, synthetic procedures, characterization data for all compounds, and crystallographic information (PDF).
X-ray crystallographic data for compounds 4d, 5c, 5e, 5f, 5s, P4, 7 (CIF).
REFERENCES:
- (1).Westheimer FH Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [DOI] [PubMed] [Google Scholar]
- (2).(a) For selected examples, see: Pawson T; Scott DJ Signaling Through Scaffold, Anchoring, and Adaptor Proteins. Science 1997, 278, 2075–2080. [DOI] [PubMed] [Google Scholar]; (b) Lu PJ; Zhou XZ; Shen MH; Lu KP Function of WW Domains as Phosphoserine- or Phosphothreonine-Binding Modules. Science 1999, 283, 1325–1328. [DOI] [PubMed] [Google Scholar]
- (3).(a) Selected reviews: Barford D; Das AK; Egloff M-P The Structure and Mechanism of Protein Phosphatases: Insights Into Catalysis and Regulation. Annu. Rev. Biophys. Biomol. Struct 1998, 27, 133–164. [DOI] [PubMed] [Google Scholar]; (b) Hunter T Signaling–2000 and Beyond. Cell 2000, 100, 113–127. [DOI] [PubMed] [Google Scholar]; (c) Cohen P The Regulation of Protein Function by Multisite Phosphorylation–A 25 Year Update. Trends Biochem. Sci 2000, 25, 596–601. [DOI] [PubMed] [Google Scholar]
- (4).(a) Yaffe MB; Elia AEH Phosphoserine/Threonine-Binding Domains. Curr. Opin. Cell Biol 2001, 13, 131–138. [DOI] [PubMed] [Google Scholar]; (b) Yaffe MB; Rittinger K; Volinia S; Caron PR; Aitken A; Leffers H; Gamblin SJ; Smerdon SJ; Cantley LC The Structural Basis for 14–3-3:Phosphopeptide Binding Specificity. Cell 1997, 91, 961–971. [DOI] [PubMed] [Google Scholar]; (c) Verdecia MA; Bowman ME; Lu KP; Hunter T; Noel JP Structural Basis for Phosphoserine-Proline Recognition by Group IV WW Domains. Nat. Struct. Mol. Biol 2000, 7, 639–643. [DOI] [PubMed] [Google Scholar]
- (5).(a) Seminal reports Uraguchi D; Terada M Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation. J. Am. Chem. Soc 2004, 126, 5356–5357. [DOI] [PubMed] [Google Scholar]; (b) Akiyama T; Itoh J; Yokota K; Fuchibe K Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid. Angew. Chem. Int. Ed 2004, 43, 1566–1568. [DOI] [PubMed] [Google Scholar]
- (6).(a) Selected reviews: Akiyama T Stronger Brønsted Acids. Chem. Rev 2007, 107, 5744–5758. [DOI] [PubMed] [Google Scholar]; (b) Terada M Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations. Synthesis 2010, 1929–1982. [Google Scholar]; (c) Zamfir A; Schenker S; Freund M; Tsogoeva BS Chiral BINOL-Derived Phosphoric Acids: Privileged Brønsted Acid Organocatalysts for C-C Bond Formation Reactions. Org. Biomol. Chem 2010, 8, 5262–5276. [DOI] [PubMed] [Google Scholar]; (d) Rueping M; Kuenkel A; Atodiresei I Chiral Brønsted Acids in Enantioselective Carbonylactivations – Activation Modes and Applications. Chem. Soc. Rev 2011, 40, 4539–4549. [DOI] [PubMed] [Google Scholar]; (e) Parmar D; Sugiono E; Raja S; Rueping M Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Bransted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev 2014, 114, 9047–9153. [DOI] [PubMed] [Google Scholar]; (f) Parmar D; Sugiono E; Raja S; Rueping M Addition and Correction to Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev 2017, 117, 10608–10620. [DOI] [PubMed] [Google Scholar]
- (7).Yoon TP; Jacobsen EN Privileged Chiral Catalysts. Science 2003, 299, 1691–1693. [DOI] [PubMed] [Google Scholar]
- (8).(a) Select Examples: Li G; Liang Y; Antilla JC A Vaulted Biaryl Phosphoric Acid-Catalyzed Reduction of α-Imino Esters: The Highly Enantioselective Preparation of α-Amino Esters. J. Am. Chem. Soc 2007, 129, 5830–5831. [DOI] [PubMed] [Google Scholar]; (b) Milo A, Neel AJ; Toste FD; Sigman MS A Data-Intensive Approach to Mechanistic Elucidation Applied to Chiral Anion Catalysis. Science, 2015, 347, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Momiyama N; Tabuse H; Noda H; Yamanaka M; Fujinami T; Yamanishi K; Izumiseki A; Funayama K; Egawa F; Okada S; Adachi H; Terada M Molecular Design of a Chiral Brønsted Acid with Two Different Acidic Sites: Regio-, Diastereo-, and Enantioselective Hetero-Diels–Alder Reaction of Azopyridinecarboxylate with Amidodienes Catalyzed by Chiral Carboxylic Acid-Monophosphoric Acid. J. Am. Chem. Soc 2016, 138, 11353–11359. [DOI] [PubMed] [Google Scholar]; (d) Orlandi M; Coelho JAS; Hilton MJ; Toste FD; Sigman MS Parametrization of Non-covalent Interactions for Transition State Interrogation Applied to Asymmetric Catalysis. J. Am. Chem. Soc 2017, 139, 6803–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kaib PSJ; Schreyer L; Lee S; Properzi R; List B Extremely Active Organocatalysts Enable a Highly Enantioselective Addition of Allyltrimethylsilane to Aldehydes. Angew. Chem. Int. Ed 2016, 55, 13200–13203. [DOI] [PubMed] [Google Scholar]
- (9).(a) Clardy J; Walsh C Lessons from Natural Molecules. Nature 2004, 432, 829–837. [DOI] [PubMed] [Google Scholar]; (b) Han S; Miller SJ Asymmetric Catalysis at a Distance: Catalytic, Site-Selective Phosphorylation of Teicoplanin. J. Am. Chem. Soc 2013, 135, 12414–12421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Giuliano MW; Miller SJ Site-Selective Reactions with Peptide-Based Catalysts. Top. Curr. Chem 2016, 372, 157–201. [DOI] [PubMed] [Google Scholar]
- (10).(a) Newman M; Strzelecka T; Dorner LF; Schildkraut I; Aggarwal AK Structure of Bam HI Endonuclease Bound to DNA: Partial Folding and Unfolding on DNA Binding. Science, 1995, 269, 656–663. [DOI] [PubMed] [Google Scholar]; (b) Miller SJ In Search of Peptide-Based Catalysts for Asymmetric Organic Synthesis. Acc. Chem. Res 2004, 37, 601–610. [DOI] [PubMed] [Google Scholar]; (c) Wennemers H Asymmetric Catalysis with Peptides. Chem. Commun 2011, 47, 12036–12041. [DOI] [PubMed] [Google Scholar]; (d) Lewandowski B; Wennemers H Asymmetric Catalysis with Short-Chain Peptides. Curr. Opin. Chem. Biol 2014, 22, 40–46. [DOI] [PubMed] [Google Scholar]; (e) Davis JJ; Phipps RJ Harnessing Non-Covalent Interactions to Exert Control Over Regioselectivity and Site-Selectivity in Catalytic Reactions. Chem. Sci 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Toste FD; Sigman MS; Miller SJ Pursuit of Noncovalent Interactions for Strategic Site-Selective Catalysis. Acc. Chem. Res 2017, 50, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau LC; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry Informer Libraries: A Chemoinformatics Enabled Approach to Evaluate and Advance Synthetic Methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shugrue CR; Miller SJ Phosphothreonine as a Catalytic Residue in Peptide-Mediated Asymmetric Transfer Hydrogenations of 8-Aminoquinolines. Angew. Chem. Int. Ed 2015, 54, 11173–11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shugrue CR; Featherston AL; Lackner RM; Lin A; Miller SJ Divergent Stereoselectivity in Phosphothreonine (pThr)-Catalyzed Reductive Aminations of 3-Amidocyclohexanones. J. Org. Chem 2018, 83, 4491–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kwon Y; Chinn A; Kim B; Miller SJ Divergent Control of Point and Axial Stereogenicity: Catalytic Enantioselective C–N Bond-Forming Cross-Coupling and Catalyst-Controlled Atroposelective Cyclodehydration. Angew. Chem. Int. Ed 2018, 57, 6251–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bayer AV; Villiger V Einwirkung des Caro’schen Reagens auf Ketone. Ber. Dtsch. Chem. Ges 1899, 32, 3625–3633. [Google Scholar]
- (16).(a) Taschner MJ; Black DJ The Enzymatic Baeyer–Villiger Oxidation: Enantioselective Synthesis of Lactones from Mesomeric Cyclohexanones. J. Am. Chem. Soc 1988, 110, 6892–6893. [Google Scholar]; (b) Reetz MT; Wu S Laboratory Evolution of Robust and Enantioselective Baeyer–Villiger Monooxygenases for Asymmetric Catalysis. J. Am. Chem. Soc 2009, 131, 15424–15432. [DOI] [PubMed] [Google Scholar]; (c) Leisch H; Morley K; Lau PC Baeyer–Villiger Monooxygenases: More Than Just Green Chemistry. Chem. Rev 2011, 111, 4165–4222. [DOI] [PubMed] [Google Scholar]; (d) Li G; Marc G-B; Furst MJLJ; Ilie A; Fraaije MW; Houk KN; Reetz MT Overriding Traditional Electronic Effects in Biocatalytic Baeyer–Villiger Reactions by Directed Evolution. J. Am. Chem. Soc 2018, 140, 10464–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Murahashi S; Ono S; Imada Y Asymmetric Baeyer–Villiger Reaction with Hydrogen Peroxide Catalyzed by a Novel Planar-Chiral Bisflavin. Angew. Chem., Int. Ed 2002, 41, 2366–2368. [DOI] [PubMed] [Google Scholar]; (b) Xu S; Wang Z; Zhang X; Zhang X; Ding K Chiral Brønsted Acid Catalyzed Asymmetric Baeyer–Villiger Reaction of 3-Substituted Cyclobutanones by Using Aqueous H2O2. Angew. Chem., Int. Ed 2008, 47, 2840–2843. [DOI] [PubMed] [Google Scholar]; (c) Xu S; Wang Z; Zhang X; Ding K Asymmetric Baeyer–Villiger Oxidation of 2,3- and 2,3,4-Substituted Cyclobutanones Catalyzed by Chiral Phosphoric Acids with Aqueous H2O2 as the Oxidant. Eur. J. Org. Chem 2011, 1, 110–116. [Google Scholar]; (d) Romney DK; Colvin SM; Miller SJ Catalyst Control over Regio- and Enantioselectivity in Baeyer–Villiger Oxidations of Functionalized Ketones. J. Am. Chem. Soc 2014, 136, 14019–14022. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Giuliano MW; Lin C-Y; Romney DK; Miller SJ; Anslyn EV A Synergistic Combinatorial and Chiroptical Study of Peptide Catalysts for Asymmetric Baeyer–Villiger Oxidation. Adv. Synth. Catal 2015, 357, 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Poudel PP; Arimitsu K; Yamamoto K Self-Assembled Ion-Pair Organocatalysis–Asymmetric Baeyer–Villiger Oxidation Mediated by Flavinium–Cinchona Alkaloid Dimer. Chem. Commun 2016, 52, 4163–4166. [DOI] [PubMed] [Google Scholar]
- (18).(a) Bolm C; Schlingloff G; Weickhardt K Optically Active Lactones from a Baeyer–Villiger-Type Metal-Catalyzed Oxidation with Molecular Oxygen. Angew. Chem. Int. Ed 1994, 33, 1848–1849. [Google Scholar]; (b) Strukul G Transition Metal Catalysis in the Baeyer–Villiger Oxidation of Ketones. Angew. Chem. Int. Ed 1998, 37, 1198–1209. [DOI] [PubMed] [Google Scholar]; (c) Paneghetti C; Gavagnin R; Pinna F; Strukul G New Chiral Complexes of Platinum(II) as Catalysts for the Enantioselective Baeyer–Villiger Oxidation of Ketones with Hydrogen Peroxide: Dissymmetrization of meso-Cyclohexanones. Organometallics 1999, 18, 5057–5065. [Google Scholar]; (d) Watanabe A; Uchida T; Irie R; Katsuki T Zr[bis(salicylidene)ethylenediaminatol-Mediated Baeyer–Villiger Oxidation: Stereospecific Synthesis of Abnormal and Normal lactones. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 5737–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhou L; Liu X; Ji J; Zhang Y; Hu X; Lin L; Feng X Enantioselective Baeyer–Villiger Oxidation: Desymmetrization of Meso Cyclic Ketones and Kinetic Resolution of Racemic 2-Arylcyclohexanones. J. Am. Chem. Soc 2012, 134, 17023–17026. [DOI] [PubMed] [Google Scholar]; (f) Zhou L; Liu X; Ji J; Zhang Y; Wu W; Liu Y; Lin L; Feng X Regio- and Enantioselective Baeyer–Villiger Oxidation: Kinetic Resolution of Racemic 2-Substituted Cyclopentanones. Org. Lett 2014, 16, 3938–3941. [DOI] [PubMed] [Google Scholar]
- (19).(a) For selected reviews: Krow GR Baeyer–Villiger Oxidation of Ketones and Aldehydes. Org. React 1993, 43, 251–798. [Google Scholar]; (b) Renz M; Meunier B 100 Years of Baeyer–Villiger Oxidations. Eur. J. Org. Chem 1999, 737–750. [Google Scholar]; (c) ten Brink G-J; Arends IWCE; Sheldon RA The Baeyer–Villiger Reaction: New Developments toward Greener Procedures. Chem. Rev 2004, 104, 4105–4124. [DOI] [PubMed] [Google Scholar]; (d) Uyanik M; Ishihara K Baeyer–Villiger Oxidation Using Hydrogen Peroxide. ACS Catal. 2013, 3, 513–520. [Google Scholar]
- (20).Bolm C; Luong TKK; Schlingloff G Enantioselective Metal-Catalyzed Baeyer–Villiger Oxidation of Cyclobutanones. Synlett 1997, 1151–1152. [Google Scholar]
- (21).(a) Doyle AG; Jacobsen EN Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]; (b) Knowles RR; Jacobsen EN Attractive Noncovalent Interactions in Asymmetric Catalysis: Links Between Enzymes and Small Molecule Catalysts. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Jarvo ER; Miller SJ Amino Acids and Peptides as Asymmetric Organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar]; (b) Haque TS; Little JC; Gellman SH Stereochemical Requirements for β-Hairpin Formation: Model Studies with Four-Residue Peptides and Depsipeptides. J. Am. Chem. Soc 1996, 118, 6975–6985. [Google Scholar]; (c) Wilmot CM; Thornton JM Analysis and Prediction of the Different Types of β-turn in Proteins. J. Mol. Biol 1988, 203, 221–232. [DOI] [PubMed] [Google Scholar]
- (23).(a) Select examples: Copeland GT; Jarvo ER; Miller SJ Minimal Acylase-Like Peptides. Conformational Control of Absolute Stereospecificity. J. Org. Chem 1998, 63, 6784–6785. [DOI] [PubMed] [Google Scholar]; (b) Mbofana CT; Miller SJ Diastereo- and Enantioselective Addition of Anilide-Functionalized Allenoates to N-Acylimines Catalyzed by a Pyridylalanine-Based Peptide. J. Am. Chem. Soc 2014, 136, 3285–3292. [DOI] [PubMed] [Google Scholar]; (c) Diener ME; Metrano AJ; Kusano S; Miller SJ Enantioselective Synthesis of 3-Arylquinazolin-4(3H)-ones via Peptide-Catalyzed Atroposelective Bromination. J. Am. Chem. Soc 2015, 137, 12369–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hurtley AE; Stone EA; Metrano AJ; Miller SJ Desymmetrization of Diarylmethylamido Bis(phenols) through Peptide-Catalyzed Bromination: Enantiodivergence as a Consequence of a 2 amu Alteration at an Achiral Residue within the Catalyst. J. Org. Chem 2017, 82, 11326–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xu S; Wang Z; Li Y; Zhang X; Wang H; Ding K Mechanistic Investigation of Chiral Phosphoric Acid Catalyzed Asymmetric Baeyer–Villiger Reaction of 3-Substituted Cyclobutanones with H2O2 as the Oxidant. Chem. - Eur. J 2010, 16, 3021–3035. [DOI] [PubMed] [Google Scholar]
- (25).Angione M; Miller SJ Dihedral Angle Restriction Within a Peptide-Based Tertiary Alcohol Kinetic Resolution Catalyst. Tetrahedron 2006, 62, 5254–5261. [Google Scholar]
- (26).(a) Hruby VJ Conformational Restrictions of Biologically Active Peptides via Amino Acid Side Chain Groups. Life Sci. 1982, 31, 189–199. [DOI] [PubMed] [Google Scholar]; (b) Kover KE; Jiao D; Fang S; Hruby VJ Conformational Properties of the Unnatural Amino Acid .beta.-Methylphenylalanine in a Linear Octapeptide System; Correlations of 13C-NMR Chemical Shifts with the Side-Chain Stereochemistry of These Amino Acid Residues. J. Org. Chem 1994, 59, 991–998. [Google Scholar]
- (27).Metrano AJ; Abascal NC; Mercado BQ; Paulson EK; Hurtley AE; Miller SJ Diversity of Secondary Structure in Catalytic Peptides with β-Turn-Biased Sequences. J. Am. Chem. Soc 2017, 139, 492–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Peptide 7 exhibited two packing polymorphs in the solid state that were similar to one another. Only one polymorph is shown for clarity as a point of comparison to P4. See SI for detailed analysis of the polymorphic structures.
- (29).(a) Hoveyda AH; Evans DA; Fu GC Substrate-Directable Chemical Reactions. Chem. Rev 1993, 93, 1307–1370. [Google Scholar]; (b) Bernardi L; Fochi M; Franchini M. Comes; Ricci A Bioinspired Organocatalytic Asymmetric Reactions. Org. Biomol. Chem 2012, 10, 2911–2922. [DOI] [PubMed] [Google Scholar]; (c) Peris G; Jakobsche CE; Miller SJ Aspartate-Catalyzed Asymmetric Epoxidation Reactions. J. Am. Chem. Soc 2007, 129, 8710–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) Viso A; Fernandez de la Pradilla R; Garcia A; Flores A. α,β-Diamino Acids: Biological Significance and Synthetic Approaches. Chem. Rev 2005, 105, 3167–3196. [DOI] [PubMed] [Google Scholar]; (b) Viso A; Fernandez de la Pradilla, R.; Tortosa, M.; Garcia, A.; Flores, A. Update 1 of: α,β-Diamino Acids: Biological Significance and Synthetic Approaches. Chem. Rev 2011, 111, PR1–PR42. [DOI] [PubMed] [Google Scholar]
- (31).Gilli P; Pretto L; Bertolasi V; Gilli G Predicting Hydrogen-Bond Strengths from Acid-Base Molecular Properties. The pKa Slide Rule: Toward the Solution of a Long-Lasting Problem. Acc. Chem. Res 2009, 42, 33–44. [DOI] [PubMed] [Google Scholar]
- (32).Giacalone F; Gruttadauria M; Agrigento P; Noto R Low-loading Asymmetric Organocatalysis. Chem. Soc. Rev 2012, 41, 2406–2447. [DOI] [PubMed] [Google Scholar]
- (33).Due to poor solubility of 4n in CHCl3, the conversion of 4n to 5n was generally low, and therefore the reaction with 4n was conducted for 48 hours.
- (34).Lactone (S)-5s crystallized as two packing polymorphs with a minor component of the minor enantiomer present in the unit cell. Only one polymorph is shown for clarity. See SI for detailed analysis of the absolute stereochemical assignment and full details of the crystal structure.
- (35).Sibi MP; Liu M Reversal of Stereochemistry in Enantioselective Transformations. Can they be Planned or are they Just Accidental? Curr. Org. Chem 2001, 5, 719–755. [Google Scholar]
- (36).(a) Procházková E; Kolmer A; Ilgen J; Schwab M; Kaltschnee L; Fredersdorf M; Schmidts V; Wende RC; Schreiner PR; Thiele CM Uncovering Key Structural Features of an Enantioselective Peptide-Catalyzed Acylation Utilizing Advanced NMR Techniques. Angew. Chem. Int. Ed 2016, 55, 15754–15759. [DOI] [PubMed] [Google Scholar]; (b) Rigling C; Kisunzu JK; Duschmale J; Haussinger D; Wiesner M; Ebert M-C; Wennemers H Conformational Properties of a Peptidic Catalyst: Insights from NMR Spectroscopic Studies. J. Am. Chem. Soc 2018, 140, 10829–10838. [DOI] [PubMed] [Google Scholar]
- (37).(a) For effects of increasing Lewis base concentration: Venkatachalapathi YV; Prasad B. V. Venkataram; Balaram P Conformational Analysis of Small Disulfide Loops. Spectroscopic and Theoretical Studies on a Synthetic Cyclic Tetrapeptide Containing Cysteine. Biochemistry 1982, 21, 5502–5509. [DOI] [PubMed] [Google Scholar]; (b) Raj PA; Balaram P Conformational Effects on Peptide Aggregation in Organic Solvents: Spectroscopic Studies of Two Chemotactic Tripeptide Analogs. Biopolymers 1985, 24, 1131–1146. [DOI] [PubMed] [Google Scholar]; (c) Rai R; Raghothama S; Balaram P Design of a Peptide Hairpin Containing a Central Three-Residue Loop. J. Am. Chem. Soc 2006, 128, 2675–2681. [DOI] [PubMed] [Google Scholar]
- (38).Attempts to obtain crystal structures of either P41 or P42 or analogs thereof, were unsuccessful.
- (39).Tripp AE; Burdette DS; Zeikus JG; Phillips RS Mutation of Serine-39 to Threonine in Thermostable Secondary Alcohol Dehydrogenase from Thermoanaerobacter ethanolicus Changes Enantiospecificity. J. Am. Chem. Soc 1998, 120, 5137–5141. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.