

Abstract
Purpose of review
We review the growing clinical evidence that metabolic acidosis mediates chronic kidney disease (CKD) progression and that treatment to increase the associated low serum bicarbonate (HCO3) in CKD is disease-modifying.
Recent findings
Seven prospective studies of patients with wide ranges of estimated glomerular filtration rates (eGFRs) and serum HCO3 examined the effect on CKD of increasing serum HCO3 using dietary acid reduction with either oral alkali (sodium bicarbonate or sodium citrate), a vegetarian diet very low in acid-producing protein (0.3 g/kg/day) supplemented with ketoanalogues or added base-producing fruits and vegetables. Clinical outcomes included slower kidney function decline (using eGFR measurements) and fewer patients progressing to end-stage kidney disease. Post hoc analyses demonstrated that: treatment of metabolic acidosis for 2 years decreased the number of patients with at least a 40% eGFR decline, a validated surrogate for progression to end-stage kidney disease and across four studies, treatment to increase serum HCO3 by 4–6.8 mEq/l in acidotic patients with CKD was associated with a ∼4 ml/min/1.73 m2 reduction in the rate of eGFR decline over 6–24 months compared with controls.
Summary
Metabolic acidosis appears to enhance CKD progression and its treatment should be studied further as a potential disease-modifying intervention.
Keywords: chronic kidney disease, chronic kidney disease progression, disease-modifying intervention, estimated glomerular filtration rate decline, serum bicarbonate
INTRODUCTION
Metabolic acidosis arises from a fundamental disturbance of acid–base balance and is common in patients with chronic kidney disease (CKD). Optimal cell, tissue and organ function requires the maintenance of hydrogen ion (H+) concentration within a physiologic range, which for serum is 35–45 nM (pH 7.46–7.35). The dynamic process leading to a net gain of H+ in body fluids is called acidosis. When acidosis is due to a gain of ‘fixed acid’ H+ (as opposed to the gain of ‘volatile’ H+ because of accumulation of carbon dioxide gas, causing respiratory acidosis) and this H+ gain decreases the metabolic component of the acid–base equilibrium, bicarbonate (HCO3), it is called metabolic acidosis. If the amount or rate of added H+ exceeds normal excretory capacity or if H+ excretory capacity is reduced as in CKD with reduced glomerular filtration rate (GFR), metabolic acidosis becomes chronic. Epidemiological studies show that the prevalence of metabolic acidosis is approximately 15–19% in CKD stage 3–5 patients, with metabolic acidosis affecting approximately 3 million patients with CKD in the United States [1–4].
The relationship between decreasing serum HCO3 levels and worsening clinical outcomes appears to be a continuum. As serum HCO3 decreases from the normal range in patients with CKD, generally considered as 22–29 mEq/l [5,6], the risk of progression to end-stage kidney disease (ESKD) or mortality increases compared to patients with CKD but without metabolic acidosis [7–11,12▪,13,14]. Indeed, observational studies associate serum HCO3 with kidney disease progression outcomes and show that each 1 mEq/l increase in serum HCO3 from levels below the normal range reduces the risk of kidney outcomes [e.g., initiation of dialysis, 50% reduction in estimated glomerular filtration rate (eGFR)] by 3–8% [7–11,12▪]. Interestingly, risk reduction continues for serum HCO3 increases within the low normal range [9]. Findings from these observational studies are supported by recent prospective interventional studies that show that treatment of metabolic acidosis in CKD stage 3–5 patients can slow the rate of kidney function decline [15,16▪▪,17,18▪▪,19]. In addition, provision of sodium bicarbonate or a diet rich in base-producing fruits and vegetables to CKD stage 3 patients with serum HCO3 in the low normal range (22–24 mEq/l) also slowed the rate of kidney function decline [20]. Post hoc analysis of treatment effect in CKD stage 3–5 patients with overt metabolic acidosis (serum HCO3 <22 mEq/l) [17] showed that fewer patients treated with oral alkali supplementation, in comparison to untreated patients, reached at least 40% eGFR reduction, an endpoint recognized to be a validated surrogate endpoint for ESKD [21–25]. Thus, data from these prospective studies suggest the intriguing possibility that interventions that increase or normalize serum HCO3 in patients with CKD and metabolic acidosis or low normal serum HCO3 may further slow deterioration of the diseased kidney.
Box 1.

no caption available
INCREASING SERUM BICARBONATE SLOWS PROGRESSION OF CHRONIC KIDNEY DISEASE
Five studies have examined the impact of metabolic acidosis treatment with oral alkali or base-producing dietary interventions on the progression of kidney disease in CKD stage 3–5 patients with reduced serum HCO3 (<22 mEq/l) (Table 1) [15,16▪▪,17,18▪▪,19]. The study by de Brito-Ashurst et al. [15] represented an important first step in linking correction of low serum HCO3 and improved outcomes in patients with CKD and metabolic acidosis. In this single-center, open-label, prospective, parallel-group study, 134 CKD stage 4–5 patients with metabolic acidosis (serum HCO3 >16 and <20 mEq/l) were randomized 1:1 to intervention with oral sodium bicarbonate, administered as 600 mg tablets three times per day and increased as necessary to achieve and maintain serum HCO3 at least 23 mEq/l, or standard of care for 2 years. The objectives of the study were to assess the effects of oral sodium bicarbonate therapy on progression of CKD and on nutritional status in these predialysis patients. The study population had an average baseline creatinine clearance of ∼20 ml/min/1.73 m2 and an average baseline serum HCO3 level of ∼20 mEq/l. Study criteria excluded patients with poorly controlled blood pressure (BP; >150/90 mmHg, despite use of four agents), overt congestive heart failure, morbid obesity, and chronic sepsis, all conditions with potential fluid balance complications that could be worsened with ingestion of ∼500 mg of sodium that accompanied the average oral sodium bicarbonate daily dose administered in this study (1.82 ± 0.80 g/day). At the end of the 2-year study, serum HCO3 increased in the treated patients by approximately 4 mEq/l compared with the control group (Fig. 1a); these results were clinically meaningful and statistically significant (P < 0.0001). Compared with the control group, decline in creatinine clearance was slower with sodium bicarbonate supplementation (−1.88 versus −5.93 ml/min/1.73 m2; P < 0.0001), a difference of approximately 4 ml/min/1.73 m2 over the 2-year treatment period (Fig. 1b). Patients who received treatment for their metabolic acidosis were also significantly less likely than controls to experience rapid progression of kidney failure (9 versus 45%; P < 0.0001) or to develop ESKD (6.5 versus 33%; P < 0.001) over the 2-year treatment period (Fig. 2). Thus, this landmark study demonstrated the beneficial effects of increasing serum HCO3 on slowing the progression of kidney disease to an extent that was both statistically significant and clinically meaningful.
Table 1.
Studies of oral bicarbonate supplementation demonstrate slowing of chronic kidney disease progression
| Reference | Study population (CKD stage and serum HCO3 level) | Key exclusion criteria | Study treatment(s) | Treatment duration | Clinical evidence of treatment effect |
| [15] | Patients (N = 134) with stage 4/5 CKD and serum HCO3 >16 to <20 mEq/l | Poorly controlled BP (>150/90 mmHg despite use of four agents); overt congestive heart failure; malignant disease; morbid obesity; cognitive impairment; chronic sepsis | Sodium bicarbonate (1.82 ± 0.80 g/daya) or no treatment (standard of care) | 2 years | Sodium bicarbonate slowed the rate of GFR decline and progression to end-stage kidney disease |
| [16▪▪] | Patients (N = 207) with stable stage 4/5 CKD and serum HCO3 ∼16–18 mEq/l | Patients with poorly controlled BP (≥145/85 mmHg), relevant comorbidities (diabetes, heart failure, hepatic disease, digestive diseases), uremic complications (pericarditis, polyneuropathy), or feeding inability (anorexia, nausea) | Very low-protein diet (VLPD; 0.3 g/kg/day, ketoanalogue supplemented) or low-protein diet (LPD; 0.6 g/kg/day) | 18 months (3-month LPD run-in; 15-months treatment after randomization) | The VLPD slowed the rate of GFR decline and progression to end-stage kidney disease |
| [17] | Patients (N = 59) with Stage 3/4 CKD because of hypertensive nephropathy and serum HCO3 <22 mEq/l | Known primary kidney disease, diabetes, or fasting blood glucose >110 mg/dl, malignancy, chronic infection, clinical evidence of cardiovascular disease, peripheral edema, heart/liver failure or nephrotic syndrome, renal artery stenosis or primary hyperaldosteronism | Sodium citrate (1 mEq/kg bicarbonate equivalent per day) or no treatment (standard of care) | 2 years | Sodium citrate slowed the rate of eGFR decline and decreased urine biomarkers of kidney injury (ET-1, NAG, albumin, TGF-β) |
| [18▪▪] | Patients (N = 188) with CKD stage 3/4 and serum bicarbonate <22 mEq/l | Patients with structural or functional anomalies of the gastrointestinal tract; decompensated chronic liver disease; decompensated heart failure; morbid obesity (BMI ≥40 kg/m2); malignancy; chronic infections; prior bicarbonate therapy for a duration of >2 weeks; receiving immunosuppression therapy | Sodium bicarbonate (2.3 g/day) or no treatment (standard of care) | 6 months | Sodium bicarbonate slowed the rate of GFR decline and was associated with preservation of lean body mass and mid-arm muscle circumference |
| [19] | Patients (N = 71) with stage 4 CKD because of hypertensive nephropathy and serum HCO3 <22 mEq/l | Known primary kidney disease, diabetes or fasting blood glucose ≥110 mg/dl, malignancy, chronic infection, clinical evidence of cardiovascular disease, peripheral edema, heart/liver failure or nephrotic syndrome, plasma potassium level >4.6 mEq/l; taking or inability to stop taking drugs (other than ACE inhibitors) that limit potassium excretion | Sodium bicarbonate (1 mEq/kg/day) or fruits/vegetables to reduce dietary acid by 50% | 1 year | Both sodium bicarbonate and dietary fruits and vegetables decreased urine biomarkers of kidney injury (albumin, NAG, and TGF-β) |
| [20] | Patients (N = 108) with stage 3 CKD because of hypertensive nephropathy and serum HCO3 22–24 mEq/l | Known primary kidney disease, diabetes or fasting blood glucose ≥110 mg/dl, malignancy, chronic infection, clinical evidence of cardiovascular disease, peripheral edema, heart/liver failure or nephrotic syndrome, plasma potassium level >4.6 mEq/l; taking or inability to stop taking drugs (other than ACE inhibitors) that limit potassium excretion | Sodium bicarbonate (0.3 mEq/kg/day), or fruits/vegetables to reduce dietary acid by 50%, or no treatment (standard of care) | 3 years | Both sodium bicarbonate and fruits/vegetables slowed the rate of eGFR decline and decreased urine biomarkers of kidney injury (albumin, NAG angiotensinogen) |
| [34] | Patients (N = 120) with stage 2 CKD because of hypertensive nephropathy and serum HCO3 ∼26 mEq/l | Known primary kidney disease, diabetes or fasting blood glucose >110 mg/dl, malignancy, chronic infection, clinical evidence of cardiovascular disease, peripheral edema or heart failure, smoking or oral tobacco use | Sodium bicarbonate (0.5 mEq/kg/day) or sodium chloride (0.5 mEq/kg/day) or placebo | 5 years | Sodium bicarbonate slowed the rate of eGFR decline and decreased urine biomarkers of kidney injury (ET-1, albumin, NAG) |
ACE, angiotensin-converting enzyme; BP, blood pressure; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; ET-1, endothelin-1; HCO3, bicarbonate; LPD, low protein diet; NAG, N-acetyl-β-D-glucosaminidase; RRT, renal replacement therapy; TGF-β, transforming growth factor β; VLPD, very low-protein diet.
aMean (SD).
FIGURE 1.
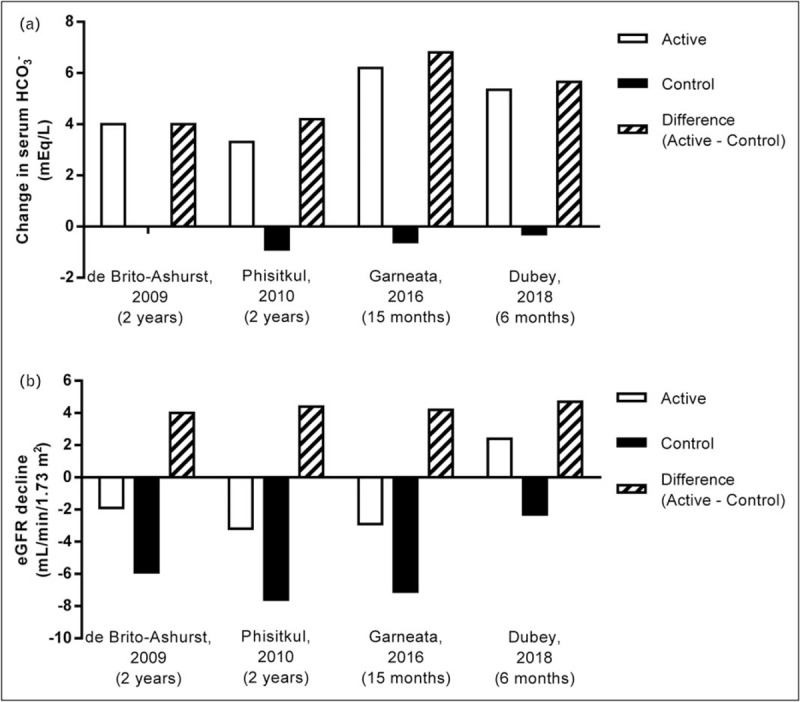
Serum bicarbonate (HCO3) increase and eGFR decline in chronic kidney disease (CKD) patients with metabolic acidosis (serum HCO3 <22 mEq/l) treated with NaHCO3 or sodium citrate supplementation versus control. Summary of changes in serum HCO3 (panel a) and estimated glomerular filtration rate (eGFR) decline (panel b) in CKD patients with metabolic acidosis (serum HCO3 <22 mEq/l) treated with NaHCO3 or sodium citrate supplementation (active) versus control patients. Data were extracted from the respective studies listed on the x-axis and cited in the reference section [15,16▪▪,17,18▪▪]; all values are mean changes from beginning to the end of the treatment period. The mean serum HCO3 values at the end of treatment for the de Brito-Ashurst et al. [15] study were estimated from graphed data. CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; HCO3, bicarbonate.
FIGURE 2.
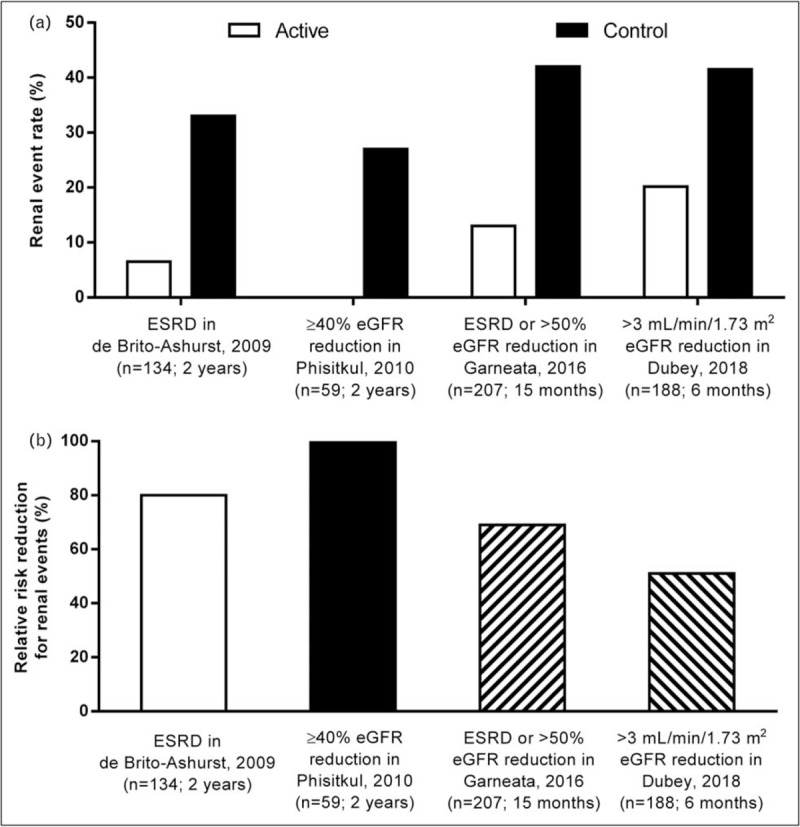
Reduction in kidney events in chronic kidney disease (CKD) patients with metabolic acidosis (serum HCO3 <22 mEq/l) treated with NaHCO3 supplementation versus control. Summary of the reduction in kidney event rate (panel a) and relative risk reduction for kidney events (panel b) in CKD patients with metabolic acidosis (HCO3 <22 mEq/l) treated with NaHCO3 supplementation (active) versus control patients. Data were extracted from the respective studies listed on the x-axis and cited in the Reference section [15,16▪▪,17,18▪▪]. Kidney event rate (panel a) was significantly different for active versus control in the de Brito-Ashurst et al. [15] (P < 0.001), Garneata et al. [16▪▪] (P < 0.001), and Dubey et al. [18▪▪] (P = 0.001) studies; the data presented for Phisitkul et al. [17] were derived from the patient level data analysis described in Table 3. CKD, chronic kidney disease; HCO3, bicarbonate.
Dietary interventions that severely limit the amount of acid-producing animal protein or increase the consumption of base-producing fruits and vegetables ameliorate metabolic acidosis, the rate of eGFR decline, and markers of kidney injury [16▪▪,19,20]. A recent study by Garneata et al. [16▪▪] explored the potential contributions of very low-protein, plant-based diets and ketoanalogue supplementation on nephropathy progression. In this single-center, open-label, prospective, parallel-group study, 207 patients with CKD stage 4–5 and metabolic acidosis were treated for 15 months. One hundred four (104) patients were randomized to receive a vegetarian, ketoanalogue-supplemented, very low-protein diet (VLPD) (0.3 g/kg/day) and the remaining 103 patients served as the control group and received a typical low-protein (from plant and animal sources) diet (LPD) (0.6 g/kg/day). The patient population had a baseline eGFR of ∼18 ml/min/1.73 m2 and a serum HCO3 of ∼17 mEq/l. Patients with poorly controlled arterial BP (≥145/85 mmHg), diabetes, or heart failure were excluded and only 14% of the screened patients met all the eligibility criteria, agreed to adhere to the dietary requirements, and were randomized in this study. Baseline serum HCO3 was similar in the two treatment groups, with median values of 16.7 and 16.8 mEq/l in the VLPD and LPD groups, respectively. However, at the end of study, serum HCO3 significantly increased (+6.2 mEq/l) in the VLPD group but slightly decreased (−0.6 mEq/l) in the LPD group, a difference between groups of 6.8 mEq/l (Fig. 1a). The rate of kidney function decline, as measured by eGFR, was slower in patients on the VLPD diet compared to controls on the LPD diet. At the end of the 15-month treatment period, the patients on the VLPD regimen had a −2.9 ml/min/1.73 m2 decrease of eGFR relative to their baseline value, whereas patients on the LPD regimen had a −7.1 ml/min/1.73 m2 decrease relative to their baseline value, a difference between groups of 4.2 ml/min/1.73 m2 (Fig. 1b). The mean eGFR at end of study was significantly higher (P < 0.01) in the VLPD group (15.1 ml/min/1.73 m2, 95% confidence interval 13.2–17.4) than in the LPD group (10.8 ml/min/1.73 m2; 95% confidence interval 9.0–12.2). Patients eating the VLPD diet were significantly less likely (P < 0.001) than LPD controls to meet the primary endpoint of kidney replacement therapy or a decrease in eGFR of more than 50% (13 versus 42%; VLPD and LPD, respectively) over the 15-month intervention period (Fig. 2).
The study by Phisitkul et al.[17] was designed to examine the treatment effect of oral sodium citrate on the rate of eGFR decline in CKD stage 3–4 patients with metabolic acidosis, in addition to determining the effects on four biomarkers of kidney injury, including urine endothelin-1 (a surrogate of kidney endothelin production that mediates progressive kidney injury), urine N-acetyl−β-D-glucosaminidase (NAG; a marker of kidney tubulointerstitial injury), urine albumin (a general marker of progressive kidney injury), and urine transforming growth factor β (TGF-β; a general marker of progressive kidney injury) [25–30]. The study was a single-center, open-label, prospective, parallel-group study of 59 patients with hypertension-associated nephropathy and metabolic acidosis. Patients were treated for 6 months with antihypertensive drug regimens including angiotensin-converting enzyme inhibitors to control their BP, and then were assigned to treatment for metabolic acidosis with oral sodium citrate (1 mEq/kg bicarbonate equivalent daily in three divided doses; N = 30) or no treatment (N = 29) for 2 years. The 29 patients serving as controls were unable or unwilling to take sodium citrate. The population of study patients had a baseline mean eGFR value of ∼33 ml/min/1.73 m2 and mean serum HCO3 of ∼20 mEq/l. At the end of the 2-year treatment period, mean (± SE) serum HCO3 levels had increased significantly (+3.3 mEq/l) in sodium citrate-treated patients but had decreased significantly (−0.9 mEq/l) in untreated patients; the difference between the treated and untreated groups (4.2 mEq/l) was also significantly different (P < 0.0001) at the end of the treatment period (Fig. 1a). Comparisons using linear mixed models demonstrated that the yearly rate of eGFR decline was significantly slower in the sodium citrate-treated group compared with the no treatment group. The comparison between treatment and no treatment was −1.60 ± 0.13 versus −3.79 ± 0.30 ml/min/year (P < 0.0001), respectively, when eGFR was calculated using plasma creatinine values, and −1.82 ± 0.08 versus −4.38 ± 0.98 ml/min/year (P < 0.0001), respectively, when eGFR was calculated using plasma cystatin C values. In addition to slowing kidney decline, treatment of metabolic acidosis with sodium citrate was also associated with significant decreases in the urine biomarkers of kidney injury (endothelin-1, NAG, albumin, and TGF-β).
The benefits of oral alkali treatment in the control of metabolic acidosis may be realized in a short course of therapy. A recent study by Dubey et al.[18▪▪] examined the effect of sodium bicarbonate administration over a 6-month period in acidotic patients with stage 3–4 CKD, with respect to changes from baseline in lean body mass (LBM), mid-arm muscle circumference (MAMC), and eGFR. The study was a single-center, open-label, prospective, parallel-group study of 188 patients with a stable eGFR, defined as less than 5% fluctuations during a 4-week preenrollment observation period. Patients with CKD of unidentified cause (CKDu) were included in the study, as previously defined [31], and CKDu was the most common cause of CKD in the enrolled patients (52.1%). Patients with decompensated chronic liver disease, decompensated heart failure, and morbid obesity were excluded from the study. The patients were assigned to standard of care and oral sodium bicarbonate (2.3 g/day; intervention arm; N = 94;) or standard of care only (control arm; N = 94). The population of study patients had a baseline mean eGFR value of ∼30 ml/min/1.73 m2 and mean serum HCO3 of ∼18 mEq/l. At the end of the 6-month treatment period, mean serum HCO3 levels increased significantly (+5.4 mEq/l) in treated patients but decreased (−0.3 mEq/l) in untreated patients, a difference between groups of 5.7 mEq/l (Fig. 1a). The intervention arm showed an increase in eGFR by 2.4 ml/min/1.73 m2, whereas the eGFR in the control arm decreased by −2.3 ml/min/1.73 m2 (Fig. 1b). A rapid decline in eGFR (defined as >3 ml/min/1.73 m2) was seen in 41.5% of the control patients versus 20.2% of the treatment patients (P = 0.001) (Fig. 2). In addition, the intervention group showed significantly increased LBM index and MAMC versus the control group. It is important to note that the intervention group had a significantly increased requirement for diuretics (35.1%) versus the control group (18.1%) (P = 0.008), along with worsening hypertension and edema, but this latter trend did not reach significance. Overall, adverse effects (excluding increases in diuretic prescriptions and hospitalizations; all hospitalizations were due to acute kidney injury) were higher in the treatment group (77.7%) versus the control group (41.4%) (P = 0.01). This short-term (6 month) study using oral alkali to treat metabolic acidosis in patients with CKD suggested that the course of treatment could preserve LBM, MAMC, and kidney function, but came at the expense of increased sodium intake and the increased requirement for diuretics in a population of patients already prone to sodium-sensitive comorbidities.
A series of studies compared the effects of dietary base-producing fruits and vegetables with those of oral sodium bicarbonate in the control of serum HCO3 and rate of eGFR decline in CKD patients with metabolic acidosis (serum HCO3 <22 mEq/l) or low normal (22–24 mEq/l) serum HCO3. In the first of these studies by Goraya et al.[19], patients with stage 4 CKD because of hypertension-associated nephropathy and serum HCO3 <22 mEq/l were assigned to either a diet with added base-producing fruits and vegetables or 1 mEq/kg/day sodium bicarbonate. The fruits and vegetables were provided in sufficient quantity to reduce dietary acid by 50%, calculated from 3-day diaries of daily food intake. The primary outcome measure was the change in eGFR, with secondary outcomes related to plasma and urine acid–base measures and markers of kidney injury. Estimated GFR was not different between the sodium bicarbonate and fruits and vegetables groups after the 1-year treatment. However, metabolic acidosis, as measured by an increase in serum HCO3, was significantly improved in both groups (P < 0.01). Similarly, 8-h net acid excretion was significantly lower in both treatment groups (P < 0.01), with a larger effect observed in the oral sodium bicarbonate group compared with the fruits and vegetables group (P < 0.01). Both interventions were associated with lower-than-baseline urine indices of kidney injury, including albumin, NAG, and TGF-β.
A more recent study [20] enrolled CKD stage 3 patients with hypertension-associated nephropathy (eGFR 30–59 ml/min/1.73 m2) and low normal serum HCO3 (22–24 mEq/l), a level higher than that for which kidney disease outcomes quality initiative (KDOQI) and kidney disease improving global outcomes (KDIGO) recommend treatment [5,6]. The primary outcome measure was the change from baseline in eGFR after 3 years of treatment with oral sodium bicarbonate or an added fruits and vegetables diet that was designed to reduce dietary acid by 50% (achieved with an average dose of 25.2 mEq/day or 2.1 g/day of sodium bicarbonate, or with the necessary amounts of fruits and vegetables, guided by 3-day food intake diaries to calculate potential dietary acid load); these interventions were compared with a usual care group which received no additional bicarbonate supplementation. At the end of the 3-year treatment period, mean serum HCO3 was significantly higher in the patients receiving oral sodium bicarbonate (24.0 ± 0.6 versus 23.1 ± 0.6 mEq/l baseline; P < 0.01) or fruits and vegetables (23.9 ± 0.6 versus 23.0 ± 0.6 mEq/l baseline; P < 0.01) and significantly lower in patients who received usual care (22.4 ± 0.6 versus 23.0 ± 0.5 mEq/l baseline; P < 0.01). Mean creatinine eGFR and cystatin C eGFR decreased over the 3-year treatment period in all three treatment groups, but the decrease from baseline to year 2 and from baseline to year 3 was significantly less in the oral sodium bicarbonate and fruits and vegetables groups than in the usual care group (P < 0.05). In addition, treatments with oral sodium bicarbonate and fruits and vegetables were both associated with significant decreases (P < 0.05) in three markers of kidney damage over the 3-year treatment period, including urinary albumin, NAG, and urine angiotensinogen, a surrogate of kidney angiotensin II levels that has been associated with progressive kidney injury. These results suggest that dietary acid reduction is kidney protective in CKD patients with metabolic acidosis and serum HCO3 in the low normal range and higher than that for which current treatment guidelines recommend alkali therapy.
As shown above, treatments that increase serum HCO3 in CKD stage 3–5 patients with metabolic acidosis or low-normal serum HCO3 demonstrate significant slowing of CKD progression. Early stage CKD patients (e.g., CKD stage 2) without apparent decreases in serum HCO3 may nonetheless have compensatory responses to nonvolatile acid accumulation that might promote kidney injury [32▪,33]. To test the potential benefit of therapy to reduce H+ retention in these early stage CKD patients, hypertension-associated nephropathy patients with reduced but relatively preserved eGFR (∼75 ml/min/1.73 m2) and serum HCO3 in the normal range (∼26 mEq/l) were studied by Mahajan et al.[34] to evaluate whether sodium bicarbonate could reduce the rate of eGFR decline in this patient population receiving adequate BP control. Comparisons using linear mixed models demonstrated that the rate of eGFR decline was significantly lower in the oral sodium bicarbonate treatment group (−1.47 ± 0.19 ml/min/year) compared to the sodium chloride (−2.05 ± 0.19 ml/min/year; P = 0.029) treatment group and the placebo group (−2.13 ± 0.19 ml/min/year; P = 0.014). Overall, this study demonstrated that oral alkali therapy slowed the rate of eGFR decline in early-stage hypertensive nephropathy patients with reduced but relatively preserved eGFR.
The combined findings of the studies summarized above indicate that alkali supplementation, in the form of oral bicarbonate or dietary modification, in patients with CKD and metabolic acidosis (serum HCO3 <22 mEq/l) or low normal serum HCO3 (22–24 mEq/l) can preserve kidney function. A tabular summary of the studies is provided in Table 1. It is important to note that in these studies, patients with CKD and common sodium-sensitive comorbidities, such as heart failure, edema, and uncontrolled hypertension, were excluded as a precaution considering the added sodium load of oral alkali supplementation and increased potassium intake accompanying a fruits and vegetable, vegetarian-based diet.
ADDITIONAL ANALYSIS RELATING SERUM BICARBONATE INCREASE AND REDUCTION OF ESTIMATED GLOMERULAR FILTRATION RATE DECLINE IN PATIENTS TREATED WITH ORAL BICARBONATE SUPPLEMENTATION
The findings of the above prospective studies and the association of better kidney outcomes with treatment of metabolic acidosis in CKD suggest a promising new approach to slowing disease progression, apart from current renin-angiotensin-aldosterone system inhibition therapy [35,36]. There is a clear medical need for new therapies for CKD. In view of this need, the Food and Drug Administration (FDA) has considered new recommendations for kidney outcome measures used in the evaluation of new therapies [21]. The FDA accepts a 57% decline in eGFR, corresponding to a doubling of serum creatinine, as an endpoint for clinical trials of kidney disease progression because it represents a marked loss of kidney function that is predictive of eventual kidney failure. Recent initiatives coordinated by the National Kidney Foundation and the FDA are evaluating alternative endpoints to shorten the duration of clinical trials, reduce sample size, and include patients with earlier stages of CKD. Based on a series of meta-analyses of cohorts and clinical trials, as well as simulations of trial designs and analytic methods, these efforts have shown that an eGFR decline of 40% is an acceptable surrogate endpoint to assess treatment effects of new therapies designed to slow CKD progression [22–24]. It is instructive then to see how this endpoint might be used in light of the prospective studies reported above.
Post hoc analyses of the reported data in selected studies show a relationship between changes in serum HCO3 and eGFR decline. For example, subject-level data on changes in serum HCO3 and eGFR from the study by Phisitkul et al.[17] were evaluated (Tables 2 and 3). Analysis of these data showed that during the 2-year treatment period, patients in the sodium citrate-treated group (N = 30) had a mean (± SD) increase in serum HCO3 of 3.27 ± 1.00 mEq/l, whereas those untreated (N = 29) had a decrease in serum HCO3 of −0.90 ± 0.64 mEq/l (Table 2), with the difference between groups reaching a high level of significance at the end of the treatment period (P < 0.0001). Within the sodium citrate group, 10.0, 63.3, and 26.67% of the patients had serum HCO3 values that increased by less than 2 mEq/l, 2 to less than 4 mEq/l, and at least 4 mEq/l, respectively, during the 2-year treatment period. In contrast, all patients without sodium citrate treatment had serum HCO3 increases less than 2 mEq/l during the 2-year treatment period. In the evaluation of eGFR change within the sodium citrate and no sodium citrate treatment groups, significant benefit was seen with sodium citrate over the 2-year treatment period. The mean (± SD) change in eGFR was −3.2 ± 1.45 ml/min/1.73 m2 for the sodium citrate treatment group, whereas the control group had a significantly larger (P < 0.0001) mean (± SD) decline in eGFR of −7.59 ± 3.26 ml/min/1.73 m2 (Table 3). Notably, 27.59% (N = 8) of the patients in the control group had at least a 40% decline in eGFR over the 2-year treatment period, whereas none of the patients in the sodium citrate treatment group had an eGFR decline of this magnitude. Thus, this post hoc analysis of patient-level data from the Phisitkul et al.[17] study demonstrated that significant increases in serum HCO3 in CKD patients with metabolic acidosis reduced the likelihood of reaching the clinical endpoint of at least 40% eGFR decline.
Table 2.
Mean serum bicarbonate (mEq/l) change from baseline over 24-month treatment period comparing sodium citrate and no sodium citrate groups
| Sodium citrate group (N = 30) | No sodium citrate (N = 29) | P value | |
| Baselinea | 20.78 (1.17) 17.2–23.7 |
20.55 (0.79) 18.6–21.6 |
0.3745 |
| Month 6a | 20.53 (1.14) 17–22.2 |
20.50 (0.80) 19–21.8 |
0.9175 |
| Month 30a | 23.80 (0.96) 20.8–25.4 |
19.60 (1.17) 17.3–21.2 |
<0.0001 |
| Change (month 30–month 6)a | 3.27 (1.00) 1.4–5.1 |
−0.90 (0.64) −2.2–0.3 |
<0.0001 |
| % change (month 30–month 6)a | 16.16% (5.47%) 6.45–25.63% |
−4.45% (3.21%) −10.63–1.49% |
<0.0001 |
| <2 mEq/l changeb (month 6 to month 30) | 3 (10%) | 29 (100%) | |
| 2–<4 mEq/l changeb (month 6 to month 30) | 19 (63.33%) | 0 | |
| ≥4 mEq/l changeb (month 6 to month 30) | 8 (26.67%) | 0 |
aValues are mean (SD); minimum to maximum.
bValues are number (percentage).
Adapted with permission from [17].
Table 3.
Mean eGFR (ml/min/1.73 m2) change from baseline over 24-month treatment period comparing sodium citrate and no sodium citrate groups
| Sodium citrate group (N = 30) | No sodium citrate (N = 29) | P value | |
| Baselinea | 33.02 (8.48) 18–57.6 |
33.38 (8.39) 19–49 |
0.8706 |
| Month 6a | 32.67 (8.21) 18–56 |
32.52 (8.33) 18–47 |
0.9449 |
| Month 30a | 29.47 (8.77) 14–52 |
24.93 (9.74) 10–44 |
0.0656 |
| Change (month 30–month 6)a | −3.2 (1.45) −6 to 0 |
−7.59 (3.26) −17 to −2 |
<0.0001 |
| % Change (month 30–month 6)a | −10.82% (6.44%) −24 to 0% |
−25.72% (13.84%) −53.13 to −4.35% |
<0.0001 |
| ≥40% decrease eGFRb (month 6 to month 30) | 0 | 8 (27.59%) |
eGFR, estimated glomerular filtration rate.
aValues are mean (SD); minimum to maximum.
bValues are number (percentage).
Adapted with permission from [17].
Based on data provided in four of the prospective studies that enrolled CKD stage 3–5 patients with overt metabolic acidosis (serum HCO3 <22 mEq/l) [15,16▪▪,17,18▪▪], it is possible to calculate an event rate corresponding to initiation of dialysis or kidney transplant or at least 40% reduction of eGFR. In this analysis, the difference between the treatment and control groups in the increase in serum HCO3 from the beginning to the end of the treatment period ranged from 4 to 6.8 mEq/l (Fig. 1a). The corresponding difference in eGFR decline between the treatment and control groups was between 4.0 and 4.2 ml/min/1.73 m2 over the 6–24-month treatment durations (Fig. 1b). Further, the change in serum HCO3 of 4–6.8 mEq/l corresponded with a reduction in the observed kidney event rate of between 51 and 100% in the treated groups as compared to the control groups over the four studies (Fig. 2). Taken together, these four prospective studies provide strong evidence that raising serum HCO3 in CKD patients with metabolic acidosis significantly improves clinical outcomes.
CONCLUSION
The importance of normalizing serum HCO3 in CKD patients with metabolic acidosis is reflected in the leading treatment guidelines for CKD. The US-based National Kidney Foundation's KDOQI guidelines state that serum HCO3 levels should be maintained at 22 mEq/l or greater and recommend the use of oral alkali supplements to attain this goal, whereas maintaining an awareness of the complications related to the use of such supplements [6]. Similarly, guidelines from KDIGO suggest that in patients with CKD and serum bicarbonate less than 22 mEq/l treatment with oral bicarbonate supplementation can be given to maintain serum bicarbonate within the normal range unless contraindicated [5]. Several large retrospective database analyses [7–10] show that clinical outcomes for CKD patients with serum HCO3 levels that are below normal (i.e., <22 mEq/l) are significantly worse compared with patients with normal serum HCO3 (i.e., 22–29 mEq/l). In addition, several single-center, prospective studies examining the effects of sodium bicarbonate, sodium citrate, or dietary interventions have also elucidated the beneficial effects of treating chronic metabolic acidosis [15,16▪▪,17,18▪▪,19,20,34]. In four of the seven key interventional studies discussed here, the provision of oral bicarbonate supplementation increased serum HCO3 in enrolled CKD patients with baseline serum HCO3 less than 22 mEq/l, often within 6 months of treatment initiation. Further, long-term treatment of metabolic acidosis (1–3 years) or treatment of hypertension-associated nephropathy patients with normal serum HCO3 belying an increased acid load (5 years), showed direct benefit on reducing eGFR decline and improving kidney outcomes. Importantly, these improvements were accompanied by a reduction in the expression of urine biomarkers that reflect continued injury to kidneys exposed to chronic acid loading. A post hoc analysis of data from the Phisitkul et al.[17] study showed that the provision of oral alkali to CKD patients with metabolic acidosis reduced the number of patients who experienced at least 40% decline in eGFR, an accepted surrogate endpoint of kidney disease progression to ESKD [21–24]. Although 27.6% of control patients in this study showed at least 40% decline in eGFR over the 2-year treatment interval, no patients in the sodium citrate treatment group experienced eGFR declines of this magnitude (Table 3; Fig. 2). A limitation of the studies reviewed here is in the enrollment of patients who did not have severe comorbidities commonly found in CKD (e.g., edema, heart failure, poorly-controlled hypertension), as treatment with sodium-based oral alkali might aggravate fluid homeostasis in such patients [37▪▪]. Based on these findings, it seems likely that nutritional and pharmacologic management of metabolic acidosis preserves kidney function, and larger scale, multicenter testing of this hypothesis is warranted.
Acknowledgements
The authors wish to thank Jerry Buysse, Jun Shao, and Dawn Parsell (employees of Tricida, Inc., South San Francisco, CA) for editorial assistance, assistance with figures, and review of the manuscript.
Financial support and sponsorship
D.E.W. received financial support and sponsorship from Tricida, Inc., South San Francisco, CA during this work; N.G. has no financial support or sponsorship to report related to this work. The authors also report grant support and sponsorship from NIH grant R21DK113440 (Wesson, Principle Investigator), outside of the subject matter of this work.
D.E.W. receives support for his salary paid through Baylor Scott and White Health from Tricida, Inc.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Eustace JA, Astor B, Muntner PM, et al. Prevalence of acidosis and inflammation and their association with low serum albumin in chronic kidney disease. Kidney Int 2004; 65:1031–1040. [DOI] [PubMed] [Google Scholar]
- 2.Moranne O, Froissart M, Rossert J, et al. Timing of onset of CKD-related metabolic complications. J Am Soc Nephrol 2009; 20:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raphael K, Zhang Y, Ying J, Greene T. Prevalence of and risk factors for reduced serum bicarbonate in chronic kidney disease. Nephrology 2014; 19:648–654. [DOI] [PubMed] [Google Scholar]
- 4.Inker LA, Coresh J, Levey AS, et al. Estimated GFR, albuminuria, and complications of chronic kidney disease. J Am Soc Nephrol 2011; 22:2322–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int Suppl 2013; 3:1–150. [Google Scholar]
- 6.National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42 Suppl 3:S1–S201. [PubMed] [Google Scholar]
- 7.Shah SN, Abramowitz M, Hostetter TH, Melamed ML. Serum bicarbonate levels and the progression of kidney disease: a cohort study. Am J Kidney Dis 2009; 54:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobre M, Yang W, Chen J, et al. Association of serum bicarbonate with risk of renal and cardiovascular outcomes in CKD: a report from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis 2013; 62:670–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raphael KL, Wei G, Baird BC, et al. Higher serum bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kidney Int 2011; 79:356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tangri N, Stevens LA, Griffith J, et al. A predictive model for progression of chronic kidney disease to kidney failure. JAMA 2011; 305:1553–1559. [DOI] [PubMed] [Google Scholar]
- 11.Menon V, Tighiouart H, Vaughn NS, et al. Serum bicarbonate and long-term outcomes in CKD. Am J Kidney Dis 2010; 56:907–914. [DOI] [PubMed] [Google Scholar]
- 12▪.Harambat J, Kunzmann K, Azukaitis K, et al. Metabolic acidosis is common and associates with disease progression in children with chronic kidney disease. Kidney Int 2017; 92:1507–1514. [DOI] [PubMed] [Google Scholar]; This study looked at the prevalence and determinants of metabolic acidosis in children and examined the associations between various baseline serum bicarbonate cohorts and outcomes in pediatric CKD patients comprising the Cardiovascular Comorbidity in Children with CKD Study. During a median follow-up of 3.3 years, metabolic acidosis prevalence was 43, 60, and 45% in CKD stages 3, 4, and 5, respectively. The risk of CKD progression was significantly higher for those patients with a time-varying serum bicarbonate <18 mmol/l, compared with those having serum bicarbonate levels above 22 mEq/l.
- 13.Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Association of serum bicarbonate levels with mortality in patients with nondialysis-dependent CKD. Nephrol Dial Transplant 2009; 24:1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raphael KL, Zhang Y, Wei G, et al. Serum bicarbonate and mortality in adults in NHANES III. Nephrol Dial Transplant 2013; 28:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Brito-Ashurst I, Varagunum M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 2009; 20:2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16▪▪.Garneata L, Stancu A, Dragomir D, et al. Ketoanalogue-supplemented vegetarian very low-protein diet and CKD progression. J Am Soc Nephrol 2016; 27:2164–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that increasing serum HCO3 with a low acid, low protein vegetarian diet supplemented with ketoanalogues slowed nephropathy progression.
- 17.Phisitkul S, Khanna A, Simoni J, et al. Amelioration of metabolic acidosis in patients with low GFR reduced kidney endothelin production and kidney injury, and better preserved GFR. Kidney Int 2010; 77:617–623. [DOI] [PubMed] [Google Scholar]
- 18▪▪.Dubey AK, Sahoo J, Vairappan B, et al. Correction of metabolic acidosis improves muscle mass and renal function in chronic kidney disease stages 3 and 4: a randomized controlled trial. Nephrol Dial Transplant 2018; Jul 24. doi:10.1093/ndt/gfy214 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]; This study demonstrated that treatment of acidotic CKD patients with sodium bicarbonate for as little as 6 months might have a beneficial effect on LBM, MAMC, and reduction of eGFR decline; however, the additional sodium necessitated increased diuretic use and the patient population was primarily CKDu, which by definition excludes patients with severe or uncontrolled hypertension.
- 19.Goraya N, Simoni J, Jo CH, Wesson DE. A comparison of treating metabolic acidosis in CKD stage 4 hypertensive kidney disease with fruits and vegetables or sodium bicarbonate. Clin J Am Soc Nephrol 2013; 8:371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goraya N, Simoni J, Jo CH, Wesson DE. Treatment of metabolic acidosis in patients with stage 3 chronic kidney disease with fruits and vegetables or oral bicarbonate reduces urine angiotensinogen and preserves glomerular filtration rate. Kidney Int 2014; 86:1031–1038. [DOI] [PubMed] [Google Scholar]
- 21.Thompson A, Lawrence J, Stockbridge N. GFR decline as an end point in trials of CKD: a viewpoint from the FDA. Am J Kidney Dis 2014; 64:836–837. [DOI] [PubMed] [Google Scholar]
- 22.Levey AS, Inker LA, Matsushita K, et al. GFR decline as an end point for clinical trials in CKD: a scientific workshop sponsored by the National Kidney Foundation and the US Food and Drug Administration. Am J Kidney Dis 2014; 64:821–835. [DOI] [PubMed] [Google Scholar]
- 23.Inker LA, Heerspink HJ, Mondal H, et al. GFR decline as an alternative end point to kidney failure in clinical trials: a meta-analysis of treatment effects from 37 randomized trials. Am J Kidney Dis 2014; 64:848–859. [DOI] [PubMed] [Google Scholar]
- 24.Greene T, Teng CC, Inker LA, et al. Utility and validity of estimated GFR-based surrogate time-to-event end points in CKD: a simulation study. Am J Kidney Dis 2014; 64:867–879. [DOI] [PubMed] [Google Scholar]
- 25.Loniewski I, Wesson DE. Bicarbonate therapy for prevention of chronic kidney disease progression. Kidney Int 2014; 85:529–535. [DOI] [PubMed] [Google Scholar]
- 26.Kohan DE, Inscho EW, Wesson DE, Pollock DM. Physiology of endothelin and the kidney. Compr Physiol 2011; 1:883–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khanna A, Simoni J, Wesson DE. Endothelin-induced increased aldosterone activity mediates augmented distal nephron acidification as a result of dietary protein. J Am Soc Nephrol 2005; 16:1929–1935. [DOI] [PubMed] [Google Scholar]
- 28.Kohan DE, Barton M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 2014; 86:896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wesson DE, Jo CH, Simoni J. Angiotensin II-mediated GFR decline in subtotal nephrectomy is due to acid retention associated with reduced GFR. Nephrol Dial Transplant 2015; 30:762–770. [DOI] [PubMed] [Google Scholar]
- 30.Wesson DE, Jo CH, Simoni J. Angiotensin II receptors mediate increased distal nephron acidification caused by acid retention. Kidney Int 2012; 82:1184–1194. [DOI] [PubMed] [Google Scholar]
- 31.Jayatilake N, Mendis S, Maheepala P, et al. Chronic kidney disease of uncertain etiology: prevalence and causative factors in a developing country. BMC Nephrol 2013; 14:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32▪.Raphael K. Metabolic acidosis and subclinical metabolic acidosis in CKD. J Am Soc Nephrol 2017; 29:376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]; An important review pointing out the features of eubicarbonatemic metabolic acidosis, where an increasing acid load is held in check by adaptive mechanisms but remains a troubling precursor to full-blown acidosis in the CKD patient.
- 33.Alpern RJ, Sakhaee K. The clinical spectrum of chronic metabolic acidosis: homeostatic mechanisms produce significant morbidity. Am J Kidney Dis 1997; 29:291–302. [DOI] [PubMed] [Google Scholar]
- 34.Mahajan A, Simoni J, Sheather SJ, et al. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int 2010; 78:303–309. [DOI] [PubMed] [Google Scholar]
- 35.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869. [DOI] [PubMed] [Google Scholar]
- 36.Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist Irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860. [DOI] [PubMed] [Google Scholar]
- 37▪▪.Bushinsky DA. Tolerance to sodium in patients with CKD-induced metabolic acidosis: does the accompanying anion matter? Am J Kidney Dis 2018; Dec 3. pii: S0272-6386(18)31009-6. doi: 10.1053/j.ajkd.2018.09.004. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]; An important new review that examines the effect of the anion-accompanying sodium on BP and sodium retention, particularly in treatment of CKD patients with oral alkali. Sodium bicarbonate does not increase BP or sodium retention when administered to patients with CKD under strict dietary sodium chloride limitation (∼10 mEq/day), however, without concurrent sodium chloride restriction, additional sodium, regardless of the accompanying anion, may increase BP and sodium retention in such patients.