

Supplemental Digital Content is available in the text.
Keywords: acute myocardial infarction; dendritic cells; inflammation; receptors, CXCR4; regulatory; T cells
Abstract
Background:
Acute myocardial infarction (MI) elicits an inflammatory response that drives tissue repair and adverse cardiac remodeling. Inflammatory cell trafficking after MI is controlled by C-X-C motif chemokine ligand 12 (CXCL12) and its receptor, C-X-C motif chemokine receptor 4 (CXCR4). CXCR4 antagonists mobilize inflammatory cells and promote infarct repair, but the cellular mechanisms are unclear.
Methods:
We investigated the therapeutic potential and mode of action of the peptidic macrocycle CXCR4 antagonist POL5551 in mice with reperfused MI. We applied cell depletion and adoptive transfer strategies using lymphocyte-deficient Rag1 knockout mice; DEREG mice, which express a diphtheria toxin receptor–enhanced green fluorescent protein fusion protein under the control of the promoter/enhancer region of the regulatory T (Treg) cell–restricted Foxp3 transcription factor; and dendritic cell-depleted CD11c-Cre iDTR mice. Translational potential was explored in a porcine model of reperfused MI using serial contrast-enhanced magnetic resonance imaging.
Results:
Intraperitoneal POL5551 injections in wild-type mice (8 mg/kg at 2, 4, 6, and 8 days) enhanced angiogenesis in the infarct border zone, reduced scar size, and attenuated left ventricular remodeling and contractile dysfunction at 28 days. Treatment effects were absent in splenectomized wild-type mice, Rag1 knockout mice, and Treg cell–depleted DEREG mice. Conversely, treatment effects could be transferred into infarcted splenectomized wild-type mice by transplanting splenic Treg cells from POL5551-treated infarcted DEREG mice. Instructive cues provided by infarct-primed dendritic cells were required for POL5551 treatment effects. POL5551 injections mobilized Treg cells into the peripheral blood, followed by enhanced Treg cell accumulation in the infarcted region. Neutrophils, monocytes, and lymphocytes displayed similar mobilization kinetics, but their cardiac recruitment was not affected. POL5551, however, attenuated inflammatory gene expression in monocytes and macrophages in the infarcted region via Treg cells. Intravenous infusion of the clinical-stage POL5551 analogue POL6326 (3 mg/kg at 4, 6, 8, and 10 days) decreased infarct volume and improved left ventricular ejection fraction in pigs.
Conclusions:
These data confirm CXCR4 blockade as a promising treatment strategy after MI. We identify dendritic cell–primed splenic Treg cells as the central arbiters of these therapeutic effects and thereby delineate a pharmacological strategy to promote infarct repair by augmenting Treg cell function in vivo.
Clinical Perspective.
What Is New?
Inflammatory cell trafficking between hematopoietic organs and sites of tissue injury is controlled by C-X-C motif chemokine ligand 12 (CXCL12) and its receptor, CXCR4. We show that bolus injections of a highly selective peptidic macrocycle CXCR4 antagonist enhance tissue repair and functional recovery after reperfused acute myocardial infarction in mice.
The therapeutic effects require dendritic cell priming and are specifically mediated by regulatory T cells. Intermittent CXCR4 blockade mobilizes regulatory T cells from their splenic reservoir, leading to their enhanced recruitment to the infarcted region and regulatory T cell–dependent attenuation of inflammatory gene expression in monocytes and macrophages.
Highlighting the translational potential of this strategy, CXCR4 blockade also reduces infarct volume and improves systolic function in a porcine closed-chest model of reperfused acute myocardial infarction.
What Are the Clinical Implications?
This study outlines a new pharmacological approach to augment regulatory T cell function and tissue repair after acute myocardial infarction.
These findings should stimulate further research into the therapeutic potential of CXCR4 blockade after myocardial infarction and in other acute conditions wherein excessive innate or adaptive immune responses cause immunopathology.
Acute myocardial infarction (MI) is caused by thrombotic occlusion of a coronary artery, which leads to progressive cell death in the nonperfused territory. Improved reperfusion strategies have enhanced myocardial salvage in most patients with acute MI,1 but patients with extensive myocardial injury remain at risk of developing chronic heart failure.2 MI triggers an inflammatory response that leads to the necrotic area being replaced with highly vascularized granulation tissue and eventually a collagen-rich scar. The heart can undergo deleterious changes in left ventricular (LV) geometry and function during this vulnerable period before scar formation has stabilized the infarct area. The inflammatory response must therefore be tightly regulated to promote healing and prevent postinfarction heart failure.3,4
Inflammatory cell trafficking from hematopoietic organs to sites of tissue injury is coordinated by chemokine-chemokine receptor networks.5 Several chemokines produced in the infarcted myocardium provide exit cues for circulating leukocytes expressing their cognate receptors.6–8 Therapeutically modulating chemokine-chemokine receptor interactions may promote infarct healing by limiting excessive inflammation-induced tissue damage or by enhancing the recruitment of angiogenic cell populations to the infarct wound.9–11 Pharmacological blockade of the C-X-C motif chemokine receptor 4 (CXCR4) with the small-molecule antagonists plerixafor (AMD3100) and burixafor (TG-0054) has been shown to improve heart function after MI in mice and pigs, respectively.10–12 Plerixafor is clinically used to mobilize CXCR4-expressing hematopoietic stem and progenitor cells from the bone marrow to the peripheral blood for collection and subsequent transplantation in patients with hematologic malignancies.13 Under steady-state conditions, hematopoietic stem and progenitor cells are retained in the bone marrow by interacting with the CXCR4 ligand, C-X-C motif chemokine ligand 12 (CXCL12), which is constitutively produced by bone marrow stromal cells.14 After MI, CXCL12 expression increases in the infarct area and serves as a homing signal for circulating stem and progenitor cells and proangiogenic cell populations referred to as endothelial progenitor cells.15–18 Along these lines, the beneficial effects of plerixafor after MI have been associated with enhanced mobilization and cardiac recruitment of endothelial progenitor cells and increased infarct vascularization.10,11 CXCR4, however, is widely expressed in the hematopoietic system,19 and mobilization of hematopoietic stem and progenitor cells by CXCR4 antagonists is accompanied by marked leukocytosis that affects all hematopoietic lineages in patients and mice.20 Notably, neither hematopoietic stem and progenitor cells, endothelial progenitor cells, nor any specific myeloid or lymphoid cell population has been causally linked to the beneficial effects of CXCR4 blockade after MI.
POL5551 and its close analogue and clinical-stage compound POL6326 are potent, selective, and reversible CXCR4 antagonists that have been developed by protein epitope mimetics technology.21–23 Both compounds are peptidic macrocycles that mimic the β-hairpin structure of the CXCR4-binding motif of CXCL12. Here, we investigated the therapeutic potential and mode of action of POL5551 in a mouse model of reperfused acute MI. We show that POL5551 promotes tissue repair and functional adaptation after MI via Foxp3+ regulatory T (Treg) cells. Supporting the translational potential of this strategy, we find that POL6326 enhances systolic function in a porcine closed-chest model of reperfused acute MI. Our observations define a specific mode of action of CXCR4 blockade after MI and outline a new pharmacological approach to enhance Treg cell function and tissue repair after acute MI.
Methods
On reasonable request, the data, analytic methods, and study materials will be made available to other researchers for the purposes of reproducing the results. Extended methods are provided in the Methods section in the online-only Data Supplement.
Reagents
The CXCR4 antagonists POL5551 and POL6326 were provided by Polyphor.21,22 Mice were treated with POL5551 (8 mg/kg per dose) or POL6326 (8 mg/kg per dose) and pigs with POL6326 (3 mg/kg per dose). For continuous dosing in mice, an osmotic minipump (Alzet, model 1007D) was implanted in a subcutaneous interscapular pocket. Pumps were filled with vehicle only or POL5551 to deliver the compound at a dose of 8 mg/kg/d for up to 7 days.
Mice
C57BL/6NCrl wild-type (WT) mice were purchased from Charles River. DEREG (depletion of regulatory T cell) mice express a simian diphtheria toxin (DT) receptor–enhanced green fluorescent protein (eGFP) fusion protein under the control of the endogenous forkhead box P3 (Foxp3) promoter/enhancer region on a bacterial artificial chromosome transgene. The transcription factor Foxp3 is highly restricted to Treg cells and confers immunosuppressive functions to naïve T cells.24,25 In DEREG mice, DT receptor–eGFP expression is observed in fully functional CD4+ Foxp3+ Treg cells, thereby allowing their DT-induced ablation or fluorescent detection. To deplete Foxp3+ Treg cells, mice were intraperitoneally injected with DT (Merck, catalog number #322326, 25 ng/g body weight) immediately before and 24 hours after MI.26 DT-treated WT littermates served as controls. Recombination activating gene 1 (Rag1) deficient (knockout) mice (which do not produce mature T and B cells), CD11c-Cre mice (which express Cre recombinase under the control of the CD11c promoter/enhancer region), iDTR mice (in which Cre-mediated excision of a STOP cassette renders cells sensitive to DT), and Tie2-GFP mice (which express green fluorescent protein under the control of the murine endothelium-specific Tie2 promoter) were purchased from the Jackson Laboratory. To deplete CD11c+ dendritic cells (DCs), CD11c-Cre iDTR mice were intraperitoneally injected with DT (50 ng/g body weight) 24 hours before and immediately before MI.27
Mouse Model of Reperfused Acute MI
All procedures in mice were approved by the authorities in Hannover, Germany (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit). MI was induced in 8- to 10-week-old male mice by transient left anterior descending coronary artery ligation. In sham-operated control mice, the ligature around the left anterior descending coronary artery was not tied. LV end-diastolic area (LVEDA) and LV end-systolic area (LVESA) were recorded by high-resolution 2-dimensional transthoracic echocardiography. Fractional area change (%) was calculated as [(LVEDA−LVESA)/LVEDA]×100. LV pressure-volume loops were recorded with a micromanometer-tipped conductance catheter.
Splenectomy
Mice were injected with butorphanol 2 mg/kg SC and anesthetized with 2% isoflurane via face mask. The abdominal cavity was entered through a small incision under the left rib cage, splenic vessels were cauterized, and the spleen was removed. In control mice, the abdomen was opened, but the spleen was left in situ (laparotomy only).
Adoptive Splenic Mononuclear Cell Transfer
Spleens were aseptically removed from donor mice and placed in DMEM (Thermo Fisher Scientific). Splenocyte suspensions were prepared by carefully mincing the spleen with the plunger tip of a 1-mL syringe while passing the tissue through a 100-μm cell strainer (BD Biosciences). Erythrocytes were depleted by NH4Cl lysis. Cells were suspended in DMEM, layered onto Ficoll-Paque (GE Healthcare), and centrifuged for 20 minutes at 2000g at room temperature. Splenic mononuclear cells (MNCs) were collected from the buffy coat and washed with PBS. Splenic MNCs from mice belonging to the same experimental group were pooled and suspended in PBS. Cells were injected via a jugular vein catheter into splenectomized recipient mice (1.7×107 MNCs per mouse, corresponding to the average number of MNCs isolated from 1 donor mouse).
Adoptive Splenic Monocyte Transfer
Splenic monocytes were prepared from splenic MNCs with a kit from Miltenyi Biotec (#130-100-629). In brief, highly pure, untouched monocytes were isolated by depleting nontarget cells (T cells, B cells, natural killer cells, DCs, erythroid cells, and granulocytes) using magnetic cell labeling and separation. Monocytes from mice belonging to the same experimental group were pooled, suspended in PBS, and infused via a jugular vein catheter into splenectomized recipient mice (1×106 monocytes per mouse, corresponding to the average number of monocytes isolated from 1 donor mouse).
Inflammatory Cell Isolation
Peripheral blood was drawn from the right ventricle. Splenocyte suspensions were prepared with a gentleMACS dissociator (Miltenyi Biotec). Blood and spleen erythrocytes were depleted by NH4Cl lysis. Inflammatory cells were isolated from the infarcted region of the LV by enzymatic digestion and mechanical dissociation with a gentleMACS dissociator.
Treg Cell Quantification and Isolation
Inflammatory cells were isolated from DEREG mice, incubated with a CD16/CD32 antibody (clone 2.4G2, mouse BD Fc Block, BD Biosciences; dilution 1:100), and stained with a CD4-APC antibody (clone RM4-5, BioLegend; dilution 1:100). CD4+ Foxp3+/eGFP+ Treg cells were counted by flow cytometry. For cell transfer experiments, CD4+ Foxp3+/eGFP+ Treg cells were isolated by fluorescence-activated cell sorting using a FACSAria IIu instrument (Becton Dickinson) and infused via a jugular vein catheter into splenectomized recipient mice (2×105 Treg cells per mouse, corresponding to the average number of Treg cells isolated from 1 donor mouse).
Closed-Chest Model of Reperfused MI and Magnetic Resonance Imaging in Pigs
All procedures in pigs were approved by the Animal Ethics Committee of the Hungarian National Food Chain Safety Office (approval number 23.1./02322/009/2008). Acute MI was induced in domestic (DanBred hybrid) female pigs by percutaneous balloon occlusion of the mid left anterior descending coronary artery as described previously by our group.28 Three days and 6 weeks after MI, LV end-diastolic and end-systolic volumes (LVEDV and LVESV) and infarct volume were determined by contrast-enhanced magnetic resonance imaging, and LV ejection fraction (LVEF; %) was calculated as [(LVEDV−LVESV)/LVEDV]×100. LV myocardium showing late contrast enhancement was quantified to assess infarct volume.
Statistical Analyses
We randomly allocated mouse littermates and pigs to the different experimental groups. Whenever possible, the investigators were blinded to group allocation during the experiment and when assessing the outcome. No animals were excluded from the analyses. On the basis of visual inspection, data were normally distributed, with similar variances in the different groups. With small sample sizes, we did not apply statistical tests for normality or equality of variances. Data are presented as mean±SEM unless otherwise stated. The 2-independent-sample t test was used to compare 2 groups. For comparisons among >2 groups, 1-way ANOVA was used if there was 1 independent variable and 2-way ANOVA if there were 2 independent variables. The Dunnett post hoc test was used for multiple comparisons with a single control group. The Tukey post hoc test was used to adjust for multiple comparisons. In the pig study, we used ANCOVA to compare LVEF changes within (3 days to 6 weeks) and between treatment groups (placebo versus POL6326), with treatment group as the main factor and LVEF at 3 days as a covariate. Differences in least-squares means and corresponding 95% CIs were calculated based on the ANCOVA model. Other end points were analyzed using the same methods. We considered a 2-tailed P value <0.05 to indicate statistical significance. K.C.W. had full access to all the data in the study and takes responsibility for the integrity of the data and the data analysis.
Results
POL5551 Promotes Tissue Repair and Improves Heart Function After MI
To explore the therapeutic potential of POL5551 after MI, we subjected mice to transient coronary artery ligation. This model simulates the situation in patients with acute MI receiving reperfusion therapy and was used throughout the present study. At 2, 4, 6, and 8 days after MI, mice were injected intraperitoneally with POL5551 or vehicle (0.9% NaCl) only (Figure 1A). Each POL5551 bolus injection was followed by a rapid (within 2 hours) mobilization of neutrophils, monocytes, and lymphocytes into the peripheral blood (Figure 1B). Cell numbers returned to baseline levels within 24 hours after each injection (Figure 1B). Ly6Chigh and Ly6Clow monocyte populations showed similar mobilization kinetics (Figure I in the online-only Data Supplement). Compared with vehicle-only–treated control mice, treatment with POL5551 enhanced the number of proliferating Ki67+ IB4+ endothelial cells in the infarct border zone at 3 days after MI (Figure 1C). This angiogenic effect was associated with more perfused capillaries at 7 days (Figure 1D) and an increase in total capillary density in the infarct border zone at 3, 7, and 28 days (Figure 1E). POL5551 is a highly selective CXCR4 antagonist with no C-X-C motif chemokine receptor 7 (CXCR7) agonistic activity (Figure II in the online-only Data Supplement). POL5551 did not directly act on endothelial cells, as has been reported for TC14012,29 a combined CXCR4 antagonist and CXCR7 agonist (Figure 1F and Figure III in the online-only Data Supplement). POL5551-treated mice developed smaller infarct scars (Figure 1G) and less pronounced LV remodeling and systolic and diastolic dysfunction as shown by echocardiography and pressure-volume measurements (Figure 1H and 1I and Table I in the online-only Data Supplement).
Figure 1.
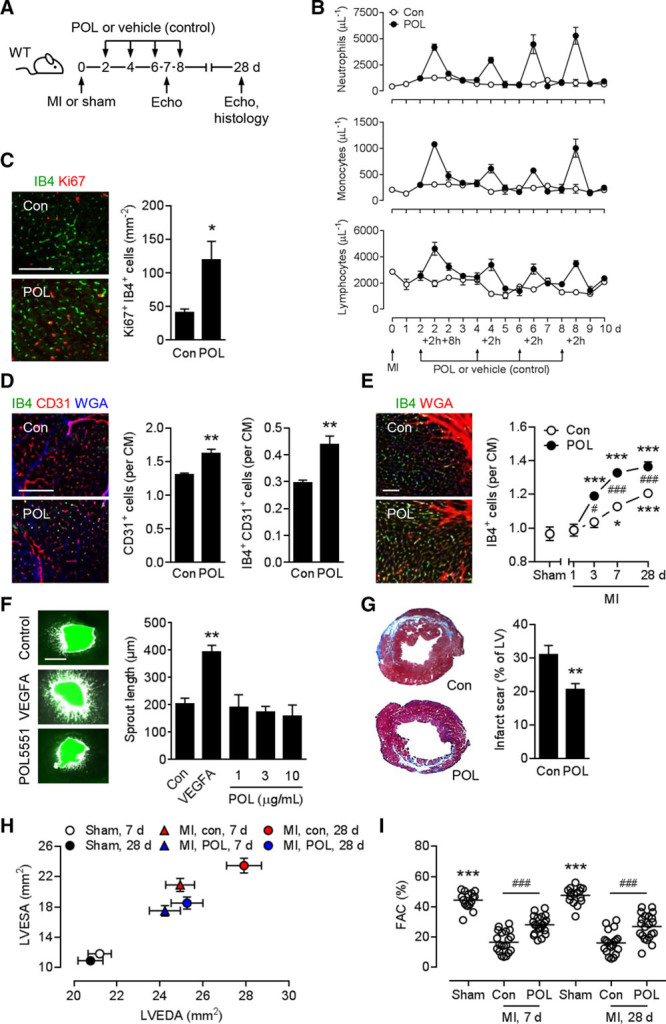
POL5551 mobilizes inflammatory cells and promotes therapeutic effects after MI. A, Experimental setup for all panels except F. Myocardial infarction (MI) was induced in wild-type (WT) mice. Additional mice underwent sham surgery. POL5551 (POL) or vehicle only (control, Con) was intraperitoneally injected 2, 4, 6, and 8 days after MI. Echo indicates echocardiography. B, Neutrophil, monocyte, and lymphocyte numbers in the peripheral blood; 3 to 5 mice per time point. C, Ki67+ isolectin B4 (IB4)+ endothelial cells in the infarct border zone 3 days after MI. Representative fluorescent images (scale bar, 100 μm). Summary data from 5 mice per group. *P<0.05 (2-independent-sample t test). D, CD31+ capillary density and IB4+ CD31+ perfused capillary density in the border zone 7 days after MI. For quantification, cardiomyocyte (CM) borders were visualized by wheat germ agglutinin (WGA) staining. Scale bar, 100 μm. Data from 4 control and 5 POL5551-treated mice. **P<0.01 (2-independent-sample t test). E, Fluorescein-labeled IB4+ capillary density in the border zone 1, 3, 7, and 28 days after MI. Extracellular matrix and CM borders were highlighted by WGA staining. Fluorescent images were taken at 28 days (scale bar, 50 μm). Data from 7 sham-operated mice (28 days) and 6 to 14 infarcted mice per group and time point. *P<0.05, ***P<0.001 vs sham (for each treatment arm, 1-way ANOVA with Dunnett post hoc test). #P<0.05, ###P<0.001, POL5551 vs control (2-independent-sample t test). F, MI was induced in Tie2-GFP mice. After 24 hours, tissue samples from the infarcted region were removed and cultured for 3 days in the absence (Con) or presence of vascular endothelial growth factor A (VEGFA, 50 ng/mL) or POL5551 (concentration range covering mean and maximum plasma concentrations after bolus injection of 8 mg/kg POL5551 in mice).21 Images show green fluorescent protein fluorescence (scale bar, 500 μm). Bar graph depicts average sprout length. Explants from 3 to 6 mice per group. **P<0.01 vs control (1-way ANOVA with Dunnett post hoc test). G, Scar size 28 days after MI. Tissue sections stained with Masson’s trichrome. Data from 12 mice per group. LV indicates left ventricle. **P<0.01 (2-independent-sample t test). H and I, Sixteen sham-operated mice, 20 infarcted control mice, 21 infarcted POL5551-treated mice. H, LV end-diastolic area (LVEDA) and LV end-systolic area (LVESA) as determined by echocardiography 7 and 28 days after sham surgery or MI. LVEDA and LVESA: P<0.001, MI vs sham for both treatment groups at both time points (1-way ANOVA with Dunnett post hoc test). LVEDA: P<0.05, control vs POL5551-treated mice at 28 days; LVESA: P<0.01, control vs POL5551 at 7 days, P<0.001, control vs POL5551 at 28 days (2-independent-sample t tests). I, Fractional area change (FAC). Circles represent individual mice. Horizontal bars are the means. ***P<0.001 vs both MI groups at each time point (1-way ANOVA with Dunnett post hoc test). ###P<0.001 (2-independent-sample t tests).
Splenic MNCs Are Required and Sufficient for the Treatment Effects of POL5551
After MI, the spleen continuously supplies neutrophils and monocytes via the bloodstream to satisfy the high demand for these cells at the site of inflammation.30,31 To delineate the importance of specific cell subsets for the beneficial effects of intermittent CXCR4 blockade, we reassessed the therapeutic potential of POL5551 in splenectomized mice. Mice underwent splenectomy 2 days after MI, before the first injection of either POL5551 or vehicle only (Figure 2A). POL5551 did not enhance capillary density in the infarct border zone (Figure 2B), did not reduce infarct scar size (Figure 2C), and did not attenuate LV remodeling and systolic or diastolic dysfunction in splenectomized mice (Figure 2D and 2E and Table II in the online-only Data Supplement), thereby indicating that the spleen is required for its therapeutic effects. The treatment effects of POL5551 were not affected in infarcted mice that had undergone laparotomy only 2 days after MI (data not shown).
Figure 2.
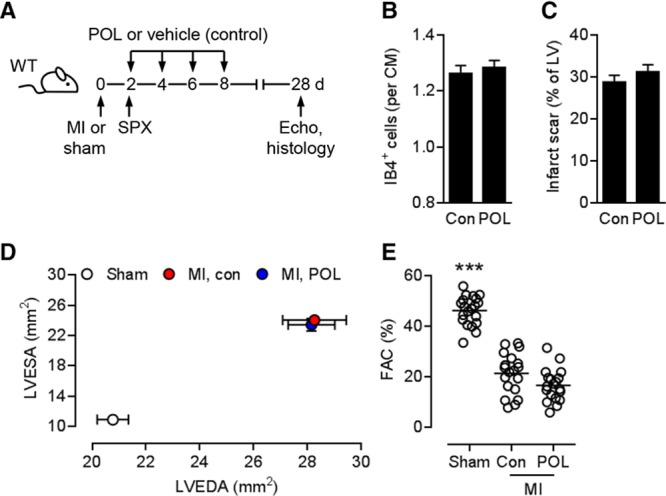
Splenectomy abolishes the therapeutic effects of POL5551. A, Experimental setup. Myocardial infarction (MI) was induced in wild-type (WT) mice. Additional mice underwent sham surgery. POL5551 (POL) or vehicle only (control) was intraperitoneally injected 2, 4, 6, and 8 days after MI. Infarcted mice underwent splenectomy (SPX) immediately before the first injection. B, Fluorescein-labeled isolectin B4 (IB4)+ capillary density in infarct border zone 28 days after MI; 6 mice per group. CM indicates cardiomyocyte. C, Scar size 28 days after MI; 14 mice per group. D and E, 20 sham-operated mice, 19 control mice, 19 POL5551-treated mice. D, LVEDA and LVESA as determined by echocardiography 28 days after sham surgery or MI. LVEDA and LVESA: P<0.001, MI vs sham for both treatment groups (2-way ANOVA with Tukey post hoc test). E, Fractional area change (FAC). ***P<0.001 vs both MI groups (2-way ANOVA with Tukey post hoc test). Con indicates control; Echo, echocardiography; LV, left ventricular; LVEDA, left ventricular end-diastolic area; and LVESA, left ventricular end-systolic area.
We next explored whether splenic MNCs from POL5551-treated mice can transfer the therapeutic effects into untreated mice. Sham or MI surgeries were performed in donor mice (Figure 3A). After 2 days, donor mice were treated with POL5551 or vehicle only, and splenic MNCs, consisting mainly of lymphocytes and monocytes (Figure IV in the online-only Data Supplement), were prepared 24 hours later. Two days after undergoing MI surgery, recipient mice were splenectomized and then infused with donor MNCs. Capillary density in the infarct border zone was greater in mice that had received MNCs from POL5551-treated infarcted donors than in mice that had received MNCs from vehicle-only–treated infarcted donors (Figure 3B). MNCs from POL5551-treated infarcted donors also reduced infarct scar size (Figure 3C) and attenuated LV remodeling and systolic and diastolic dysfunction in recipient mice (Figure 3D and 3E and Table III in the online-only Data Supplement). MNCs from sham-operated donors did not exert any therapeutic effects regardless of whether the donors had been treated with POL5551 or vehicle only (Figure 3B through 3E).
Figure 3.
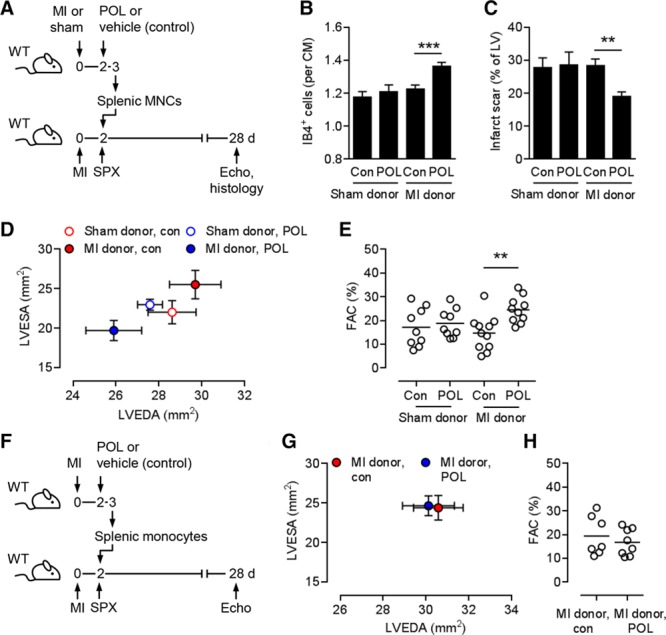
Importance of splenic MNCs for the therapeutic effects of POL5551. A, Experimental setup for B through E. Sham or myocardial infarction (MI) surgeries were performed in wild-type (WT) donor mice. Two days after MI, donor mice were intraperitoneally injected with POL5551 (POL) or vehicle only (control), and splenic mononuclear cells (MNCs) were isolated 24 hours later. Recipient WT mice underwent MI surgery, were splenectomized after 2 days, and then were infused intravenously with donor MNCs. Echo indicates echocardiography; and SPX, splenectomy. B through E, 9 to 11 mice per group. B, Fluorescein-labeled isolectin B4 (IB4)+ capillary density in the infarct border zone 28 days after MI. CM indicates cardiomyocyte; and Con, control. C, Scar size 28 days after MI. LV indicates left ventricle. D, LV end-diastolic area (LVEDA) and LV end-systolic area (LVESA) as determined by echocardiography 28 days after MI. LVESA: P<0.05, recipients receiving MNCs from infarcted POL5551-treated vs infarcted vehicle-only–treated donors (2-independent-sample t test). E, Fractional area change (FAC). F, Experimental setup for G and H. MI was induced in WT donor and recipient mice. Two days after MI, donor mice were intraperitoneally injected with POL5551 or vehicle only (con), and splenic monocytes were isolated 24 hours later. Two days after MI, recipient mice were splenectomized and then infused intravenously with donor monocytes. Seven to 8 mice per group. G, LVEDA and LVESA 28 days after MI. H, FAC. **P<0.01, ***P<0.001 (2-independent-sample t tests).
Considering the key role of splenic monocytes in orchestrating inflammation and repair after their relocation to the infarcted myocardium,30,31 we repeated the previous experiment and transplanted only magnetic cell labeling and separation–enriched splenic monocytes from POL5551-treated infarcted donor mice into infarcted and splenectomized recipient mice (Figure 3F and Figure IV in the online-only Data Supplement). To our surprise, adoptively transferring splenic monocytes from POL5551-treated infarcted donors did not attenuate LV remodeling and systolic dysfunction in recipient mice (Figure 3G and 3H).
Splenic Treg Cells Mediate the Treatment Effects of POL5551
The above adoptive cell transfer experiments pointed to splenic lymphocytes as potential mediators of the treatment effects of POL5551. To address this hypothesis, we induced MI in Rag1 knockout mice and their WT littermates and treated the animals with POL5551 or vehicle only. POL5551 attenuated adverse LV remodeling and systolic dysfunction in Rag1 WT but not Rag1 knockout mice, which indicates that T cells or B cells are required for the treatment effects (Figure V in the online-only Data Supplement).
Recent studies have implicated Treg cells in promoting tissue repair and functional recovery after MI.32–35 We therefore repeated the experiment in Treg cell–depleted DEREG mice. DEREG mice were injected with DT or saline immediately before and 24 hours after MI. Treatment with POL5551 or vehicle only was started 2 days after MI (Figure 4A). DT efficiently depleted CD4+ Foxp3+/eGFP+ Treg cells in DEREG mice (Figure 4B). POL5551 enhanced the capillary density in the infarct border zone (Figure 4C), reduced infarct scar size (Figure 4D), and ameliorated adverse remodeling and systolic dysfunction (Figure 4E and 4F and Table IV in the online-only Data Supplement) in saline-injected, non–Treg cell–depleted DEREG mice. Treg cell depletion, however, completely abolished the treatment effects of POL5551 (Figure 4C through 4F and Table IV in the online-only Data Supplement). The treatment effects of POL5551 were not affected in DT-treated WT mice (Figure VI in the online-only Data Supplement).
Figure 4.
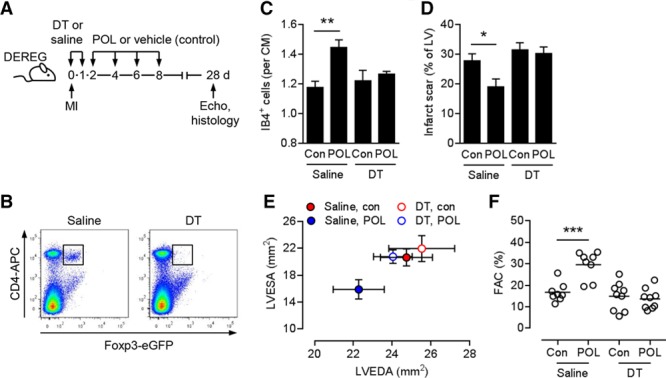
Importance of regulatory T cells for the therapeutic effects of POL5551. A, Experimental setup. Myocardial infarction (MI) was induced in DEREG mice. Diphtheria toxin (DT) or saline was intraperitoneally injected immediately before and 24 hours after MI. POL5551 (POL) or vehicle only (control) was intraperitoneally injected 2, 4, 6, and 8 days after MI. Echo indicates echocardiography. B, Representative flow cytometry panels confirming CD4+ Foxp3+/eGFP+ regulatory T cell depletion in splenocytes 24 hours after the second DT injection. C through F, Eight to 9 mice per group. C, Fluorescein-labeled isolectin B4 (IB4)+ capillary density in the infarct border zone 28 days after MI. CM indicates cardiomyocyte; and Con, control. D, Scar size 28 days after MI. LV indicates left ventricle. E, LV end-diastolic area (LVEDA) and LV end-systolic area (LVESA) as determined by echocardiography 28 days after MI. LVESA: P<0.05, POL5551 vs control in saline-injected mice (2-independent-sample t test). F, Fractional area change (FAC). *P<0.05, **P<0.01, ***P<0.001 (2-independent-sample t tests).
To further examine the importance of Treg cells for the therapeutic effects of CXCR4 blockade, we performed adoptive splenic MNC transfer experiments using Treg cell–depleted or non–Treg cell–depleted DEREG mice as donors and WT mice as recipients (Figure 5A). MI was induced in donor and recipient mice. Donor mice were injected with DT or saline immediately before and 24 hours after MI. Two days after MI, donor mice were treated with POL5551, and splenic MNCs were prepared 24 hours later. Two days after MI, recipient mice were splenectomized and then infused with donor MNCs (Figure 5A). Capillary density in the infarct border zone was greater (Figure 5B), infarct scar size was smaller (Figure 5C), and postinfarction LV remodeling (Figure 5D) and systolic dysfunction (Figure 5E) were less severe in recipient mice transplanted with non–Treg cell–depleted MNCs than in mice that were transplanted with Treg cell–depleted MNCs. These findings further supported our conclusion that splenic Treg cells are required for the treatment effects of POL5551.
Figure 5.
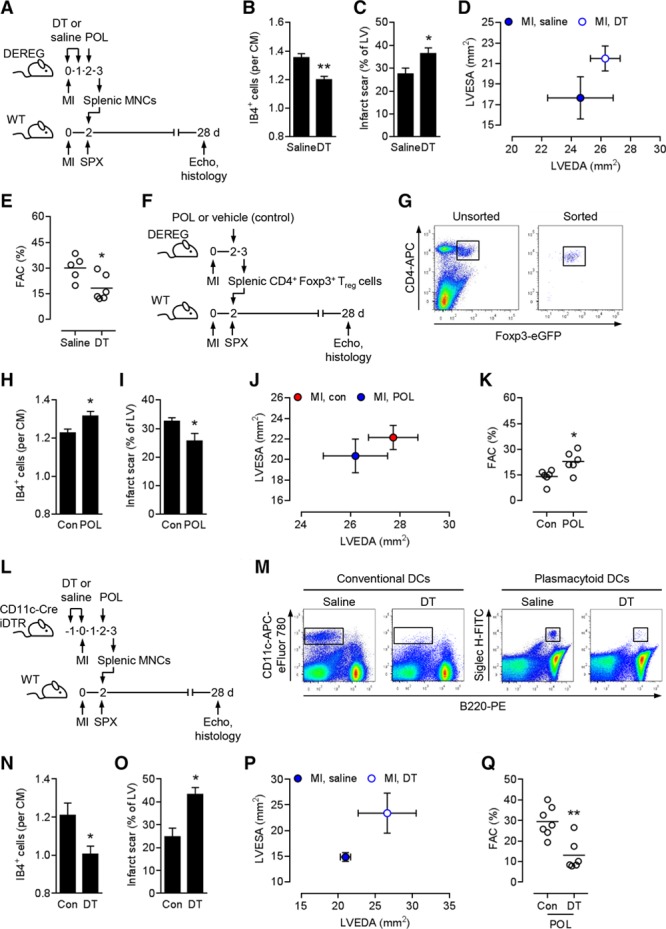
Splenic regulatory T cells are required and sufficient for the therapeutic effects of POL5551. A, Experimental setup for B through E. Myocardial infarction (MI) was induced in DEREG donor and wild-type (WT) recipient mice. Donor mice were intraperitoneally injected with diphtheria toxin (DT) or saline immediately before and 24 hours after MI. Two days after MI, donor mice were intraperitoneally injected with POL5551 (POL), and splenic mononuclear cells (MNCs) were isolated 24 hours later. Two days after MI, recipient mice were splenectomized (SPX) and then infused intravenously with donor MNCs. Echo indicates echocardiography. B through E, Five mice treated with MNCs from saline-injected donors, 6 mice treated with MNCs from DT-injected donors. B, Fluorescein-labeled isolectin B4 (IB4)+ capillary density in the infarct border zone 28 days after MI. CM indicates cardiomyocyte. C, Scar size 28 days after MI. LV indicates left ventricle. D, LV end-diastolic area (LVEDA) and LV end-systolic area (LVESA) as determined by echocardiography 28 days after MI. E, Fractional area change (FAC). F, Experimental setup for H through K. MI was induced in DEREG donor and WT recipient mice. Two days after MI, donor mice were intraperitoneally injected with POL5551 or vehicle only (Con), and splenic CD4+ Foxp3+/eGFP+ regulatory T (Treg) cells were isolated 24 hours later. Two days after MI, recipient mice were splenectomized and then infused intravenously with donor Treg cells. G, Representative flow cytometry panels showing unsorted splenic MNCs (left) and sorted splenic Treg cells (right) that were used for transplantation. CD4+ Foxp3+/eGFP+ Treg cells are highlighted in both panels. H through K, 6 mice that received Treg cells from vehicle-only–treated donors, 6 mice that received Treg cells from POL5551-treated donors. H, Capillary density in the border zone 28 days after MI. I, Scar size 28 days after MI. J, LVEDA and LVESA 28 days after MI. K, FAC. L, Experimental setup for N through Q. MI was induced in CD11c-Cre iDTR donor and WT recipient mice. Donor mice were intraperitoneally injected with DT or saline 24 hours before and immediately before MI. Two days after MI, donor mice were intraperitoneally injected with POL5551, and splenic MNCs were isolated 24 hours later. Two days after MI, recipient mice were splenectomized and then infused intravenously with donor MNCs. M, Representative flow cytometry panels showing non–dendritic cell (DC)–depleted (saline) and DC-depleted (DT) splenic MNCs that were used for transplantation. DCs are highlighted in all panels. N through Q, Seven mice treated with MNCs from saline-injected donors, 6 mice treated with MNCs from DT-injected donors. N, Capillary density in the border zone 28 days after MI. O, Scar size 28 days after MI. P, LVEDA and LVESA 28 days after MI. LVESA: P<0.05 (2-independent-sample t test). Q, FAC. *P<0.05, **P<0.01 (2-independent-sample t tests).
Next, we explored whether POL5551-primed splenic Treg cells are also sufficient to promote tissue repair and functional recovery after MI. To this end, MI was induced in DEREG donor and WT recipient mice. Two days after MI, donor mice were treated with POL5551 or vehicle only. Splenic CD4+ Foxp3+/eGFP+ Treg cells were prepared 24 hours later (Figure 5F and 5G). Two days after MI, recipient mice were splenectomized and then infused with donor Treg cells (Figure 5F). Capillary density in the infarct border zone was greater (Figure 5H), infarct scar size was smaller (Figure 5I), and postinfarction LV remodeling (Figure 5J) and systolic dysfunction (Figure 5K) were less severe in recipient mice transplanted with Treg cells from POL5551-treated donors than in mice that were transplanted with Treg cells from vehicle-only–treated donors.
DCs Are Required for POL5551 Treatment Effects
The MNC transfer experiments shown in Figure 3A through 3E indicated that instructive cues provided by the infarct are required for POL5551 treatment effects. MI has recently been shown to prime DCs, which in turn promote Treg cell activation.36 We therefore performed adoptive splenic MNC transfer experiments using DC-depleted or non–DC-depleted CD11c-Cre iDTR infarcted mice as donors and infarcted WT mice as recipients (Figure 5L). Donor mice were injected with DT or saline 24 hours before and immediately before MI. Two days after MI, donor mice were treated with POL5551, and splenic MNCs were prepared 24 hours later (Figure 5M). Two days after MI, recipient mice were splenectomized and then infused with donor MNCs (Figure 5L). Capillary density in the infarct border zone was greater (Figure 5N), infarct scar size was smaller (Figure 5O), and postinfarction LV remodeling (Figure 5P) and systolic dysfunction (Figure 5Q) were less severe in recipient mice transplanted with MNCs from non–DC-depleted donors than in mice that were transplanted with MNCs from DC-depleted donors.
POL5551 Enhances Treg Cell Mobilization, Cardiac Recruitment, and Immune-Regulatory Function
We treated infarcted DEREG mice with POL5551 or vehicle only and quantified CD4+ Foxp3+/eGFP+ Treg cells in the circulation and the infarcted LV by flow cytometry. Each POL5551 injection was followed by rapid Treg cell mobilization into the peripheral blood (Figure 6A). Treg cell mobilization displayed similar kinetics to other inflammatory cell populations (Figure 1B) and was paralleled by enhanced Treg cell accumulation in the infarcted LV (Figure 6B). Enhanced Treg cell recruitment was confined to the infarcted region (Figure 6C). In both vehicle-only– and POL5551-treated mice, between 87% and 96% of CD4+ Foxp3+/eGFP+ Treg cells in the infarcted LV (both in the infarcted and noninfarcted regions) expressed the activation marker CD69 antigen (5 mice per group, P=NS). POL5551 did not increase neutrophil, monocyte, or macrophage (Ly6Chigh and Ly6Clow) or total lymphocyte accumulation in the infarcted region (Figure VII in the online-only Data Supplement). Circulating Treg cell numbers were much lower in infarcted mice that had undergone splenectomy before POL5551 injection (Figure 6D), which points to the spleen as the main Treg cell reservoir. Indeed, POL5551 did not enhance Treg cell accumulation in the infarcted region in splenectomized mice (Figure 6E).
Figure 6.
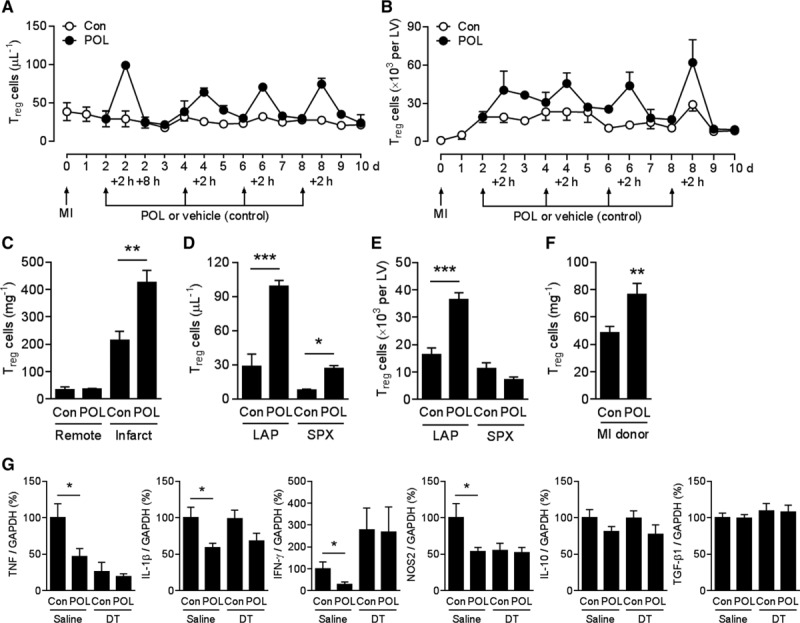
POL5551 enhances Treg cell mobilization, cardiac recruitment, and immune-modulatory function. A and B, Myocardial infarction (MI) was induced in DEREG mice, and POL5551 (POL) or vehicle only (control [Con]) was intraperitoneally injected after 2, 4, 6, and 8 days. CD4+ Foxp3+/eGFP+ regulatory T (Treg) cell numbers (A) in the peripheral blood and (B) in the infarcted left ventricle (LV); 3 to 9 mice per time point. C, CD4+ Foxp3+/eGFP+ Treg cell numbers in infarcted and noninfarcted (remote) region 2 hours after first vehicle only (Con) or POL5551 injection; 5 mice per group. D and E, Two days after MI, DEREG mice underwent splenectomy (SPX) or sham surgery (laparotomy only [LAP]) and were then intraperitoneally injected with POL5551 or vehicle only (Con). CD4+ Foxp3+/eGFP+ Treg cell numbers (D) in the peripheral blood 2 hours later and (E) in the infarcted LV 24 hours later; 4 to 9 mice per group. F, MI was induced in DEREG donor and wild-type recipient mice. Two days after MI, donor mice were injected with POL5551 or vehicle only (Con), and splenic mononuclear cells were isolated 24 hours later. Two days after MI, recipient mice were splenectomized and then infused intravenously with 17×106 donor mononuclear cells (as shown in a pilot experiment, mononuclear cells from POL5551- and vehicle-only–treated donors contained comparable numbers of CD4+ Foxp3+/eGFP+ Treg cells). Two hours later, Treg cells were quantified in the infarcted region in recipient mice; 11 mice per group. G, DEREG mice were intraperitoneally injected with diphtheria toxin (DT) or saline immediately before and 24 hours after MI. Two days after MI, mice were intraperitoneally injected with POL5551 or vehicle only (Con). Two days later, Ly6Clow/high monocytes and macrophages were isolated from the infarcted region, and the expression levels of tumor necrosis factor (TNF), interleukin 1β (IL-1β), interferon-γ (IFN-γ), nitric oxide synthase 2 (NOS2), IL-10, and transforming growth factor β1 (TGF-β1) were determined by reverse-transcription quantitative polymerase chain reaction. Eight to 10 mice per group. *P<0.05, **P<0.01, ***P<0.001 (2-independent-sample t tests).
The cell transfer experiments shown in Figure 5F through 5K, where we had transplanted equal numbers of splenic CD4+ Foxp3+/eGFP+ Treg cells from POL5551- and vehicle-only–treated donors, indicated that POL5551 also enhances Treg cell functionality. We therefore explored whether POL5551 enhances the capacity of Treg cells to home to the infarct. Indeed, Treg cell recruitment to the infarcted region was greater in mice that had been transplanted with Treg cells from POL5551-treated donors than in mice that had been transplanted with equal numbers of Treg cells from vehicle-only–treated donors (Figure 6F). Treg cells from POL5551-treated donors also showed a somewhat greater activity in Treg cell suppression assays (Figure VIII in the online-only Data Supplement). Expression of T cell activation markers on Treg cells from POL5551- or vehicle-only–treated donors did not differ significantly (Table V in the online-only Data Supplement).
We next explored whether POL5551 modulates gene expression in myeloid cells and whether Treg cells may be involved. DEREG mice were injected with either DT or saline immediately before and 1 day after MI. Two days after MI, mice were treated with POL5551 or vehicle only. Ly6Clow/high monocytes and macrophages were isolated from the infarcted region 2 days later and used for reverse-transcription quantitative polymerase chain reaction analyses. POL5551 attenuated the expression of tumor necrosis factor, interleukin 1β, interferon-γ, and nitric oxide synthase 2 in Ly6Clow/high monocytes and macrophages in non–Treg cell–depleted but not in Treg cell–depleted DEREG mice (Figure 6G). Expression levels of interleukin 10 and transforming growth factor β1 were not altered by POL5551 (Figure 6G).
Continuous POL5551 Infusion Mobilizes Treg Cells but Prevents Their Cardiac Recruitment and Therapeutic Effects
Consistent with previous reports,15,16 CXCL12 expression was significantly increased in the infarcted region, peaking at 7 days and returning to baseline levels at 28 days (Figure 7A). To assess the importance of CXCL12 for Treg cell recruitment to the infarcted region, we treated infarcted DEREG mice with POL5551 bolus injections as before or with a continuous POL5551 infusion starting 2 days after MI (Figure 7B). Bolus injections and continuous POL5551 infusion both mobilized CD4+ Foxp3+/eGFP+ Treg cells into the circulation (Figure 7C). Continuous POL5551 infusion, however, prevented Treg cell recruitment to the infarcted region (Figure 7D). In a related experiment, we treated infarcted WT mice with POL5551 bolus injections or with a continuous POL5551 infusion (Figure 7E). Unlike the bolus injections, continuous POL5551 infusion did not enhance infarct border-zone capillarization (Figure 7F), did not reduce infarct scar size (Figure 7G), and did not attenuate LV remodeling and systolic dysfunction (Figure 7H and 7I).
Figure 7.
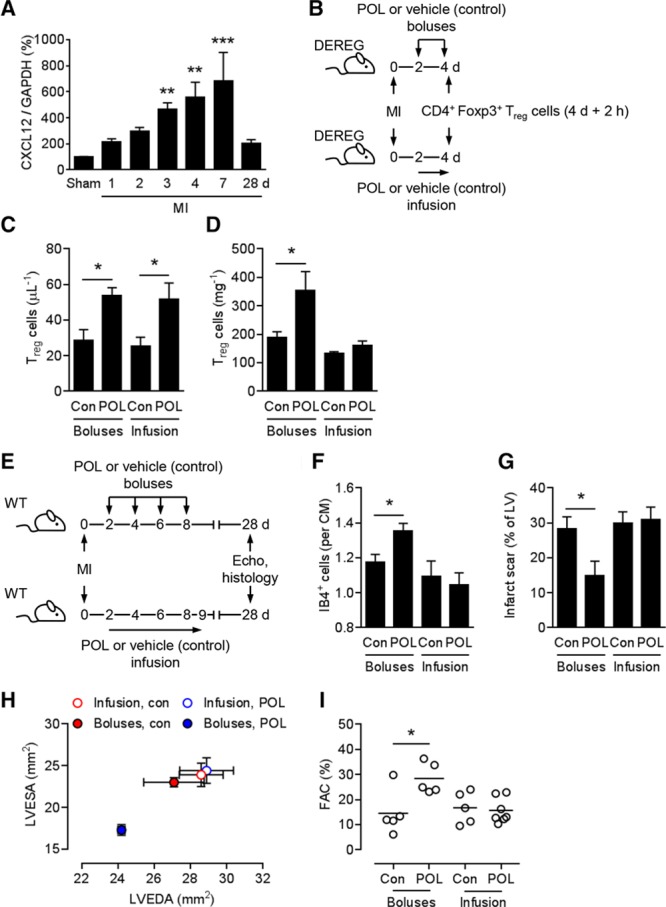
Continuous POL5551 infusion mobilizes regulatory T cells but prevents their cardiac recruitment and therapeutic effects. A, Expression levels of C-X-C motif chemokine ligand 12 (CXCL12) in the infarcted region at various time points after myocardial infarction (MI) or sham operation (day 7) as determined by reverse-transcription quantitative polymerase chain reaction. Five to 9 mice per group. **P<0.01, ***P<0.001 vs sham (1-way ANOVA with Dunnett post hoc test). B, Experimental setup for C and D. MI was induced in DEREG mice. POL5551 (POL) or vehicle only (control [Con]) was injected intraperitoneally 2 and 4 days after MI or infused subcutaneously starting 2 days after MI. C, CD4+ Foxp3+/eGFP+ regulatory T (Treg) cell numbers in the peripheral blood and (D) in the infarcted region at 4 days + 2 hours after MI. Four to 5 mice per group. E, Experimental setup for F through I. MI was induced in wild-type (WT) mice. POL5551 or vehicle only (Con) was injected intraperitoneally 2, 4, 6, and 8 days after MI or infused subcutaneously for 7 days starting 2 days after MI. 5 to 7 mice per group. Echo indicates echocardiography. F, Fluorescein-labeled isolectin B4 (IB4)+ capillary density in the infarct border zone 28 days after MI. CM indicates cardiomyocyte. G, Scar size 28 days after MI. LV indicates left ventricle. H, LV end-diastolic area (LVEDA) and LV end-systolic area (LVESA) as determined by echocardiography 28 days after MI. LVEDA: P<0.05, POL5551 vs vehicle-only boluses; LVESA: P<0.001, POL5551 vs vehicle-only boluses (2-independent-sample t tests). I, Fractional area change (FAC). *P<0.05 (2-independent-sample t tests).
POL6326 Treatment Effects in Pigs With Reperfused Acute MI
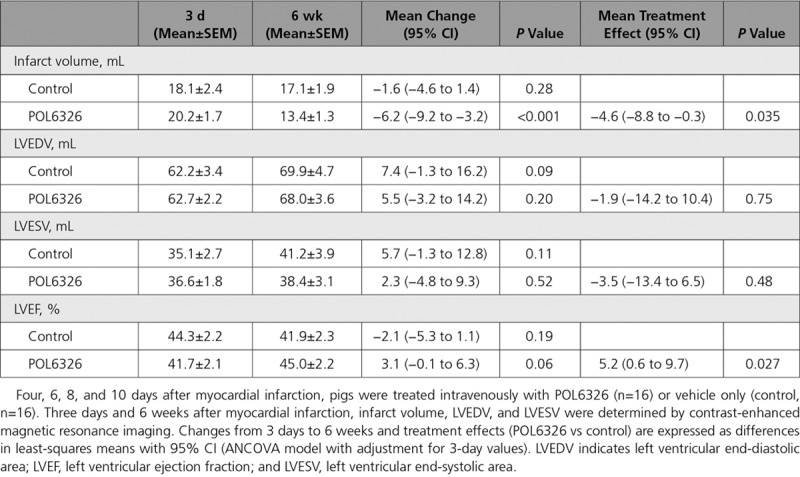
We treated pigs with reperfused acute MI with POL6326 to explore the translational potential of CXCR4 blockade with a peptidic macrocycle antagonist. Like POL5551, POL6326 mobilized Treg cells, enhanced Treg cell recruitment to the infarct, attenuated tumor necrosis factor expression in monocytes and macrophages in the infarcted region, enhanced infarct border-zone capillarization, reduced infarct scar size, and attenuated LV remodeling and systolic dysfunction in mice (Figure IX in the online-only Data Supplement). At 4, 6, 8, and 10 days after MI, pigs were infused intravenously with the clinical stage compound POL6326 or vehicle only. Contrast-enhanced cardiac magnetic resonance imaging was performed 3 days and 6 weeks after MI (Table). Infarct volume decreased, on average, by 1.6 mL in the control group (P=0.28) and by 6.2 mL in the POL6326 group (P<0.001), which corresponds to a POL6326 treatment effect of −4.6 mL (P=0.035). Increases in LV end-diastolic and end-systolic volumes over time tended to be more pronounced in the control group than in the POL6326 group. Mean LVEF decreased by 2.1 percentage points in the control group (P=0.19) and increased by 3.1 percentage points in the POL6326 group (P=0.06), which corresponds to a POL6326 treatment effect of 5.2 percentage points (P=0.027).
Table.
POL6326 Treatment Effects in Pigs With Reperfused Acute Myocardial Infarction
Discussion
Patients who develop heart failure after MI remain at high risk of death and frequent hospitalizations.37 Available strategies to prevent postinfarction heart failure aim at limiting ischemic injury and chronic LV remodeling.38 Infarct healing may afford an additional time window of therapeutic opportunity wherein transient interventions can have a lasting impact on cardiac function.4 In the present study, repeated bolus application of POL5551, a highly selective peptidic macrocycle CXCR4 antagonist, promoted angiogenesis and tissue repair and attenuated LV remodeling and systolic and diastolic dysfunction in a mouse model of reperfused acute MI. As shown by a series of cell depletion and transfer experiments, splenic CD4+ Foxp3+ Treg cells mediated these therapeutic effects.
Treg cells are a subpopulation of CD4+ T cells that maintain immune homeostasis by controlling adaptive and innate immune responses at multiple levels. Dysregulated Treg cell generation or function has been implicated in autoimmunity, cancer, and cardiovascular disease.39 CXCR4 is expressed on Treg cells and other lymphocyte populations.40 Of note, CXCR4 was originally discovered as a coreceptor for the entry of human immunodeficiency virus into CD4+ T cells, and the CXCR4 antagonist plerixafor was first developed as an antiviral agent.41 During the initial pharmacodynamic studies, marked increases in peripheral white blood cell counts were observed after systemic injections of plerixafor, with lymphocytes representing one component of this generalized leukocytosis.42 In fact, plerixafor mobilizes T cells (including Treg cells) and B cells more efficiently than granulocyte-colony stimulating factor.40 Consistent with this earlier report, POL5551 led to a rapid mobilization of Foxp3+ Treg cells in our study. Within 24 hours after each POL5551 injection, Foxp3+ Treg cell numbers in the circulation returned to baseline levels, consistent with Treg cell redistribution into tissues after the interruption of the CXCR4/CXCL12 interaction had ended because of rapid clearance of the drug.21 POL5551 enhanced cardiac recruitment of Foxp3+ Treg cells, which indicates that some of the mobilized Foxp3+ Treg cells homed to the infarcted myocardium. Neutrophils, Ly6Chigh and Ly6Clow monocytes, and lymphocytes displayed similar mobilization kinetics after POL5551 injections, but cardiac recruitment of these cells did not increase. Notably, continuous infusion of POL5551 prevented mobilized Treg cell homing to the CXCL12-expressing infarcted region and did not promote tissue repair and functional recovery. This indicates that Treg cells are recruited to the infarcted region via the CXCR4/CXCL12 axis (not excluding the involvement of additional chemokines)7 and reconciles our findings with reports from the cancer field that show that continuous CXCR4 blockade inhibits Treg cell homing to CXCL12-expressing tumors.43,44
CXCL12/CXCR4 signals are critical for Treg cell trafficking between the bone marrow and the periphery.45 Other anatomic niches harboring Treg cells via CXCR4/CXCL12 interactions remain poorly defined. In our study, Foxp3+ Treg cell mobilization was strongly reduced in splenectomized mice, which points to the spleen as the main Foxp3+ Treg cell reservoir amenable to POL5551-mediated mobilization. Splenectomy also abolished POL5551 effects on Treg cell recruitment to the infarct, infarct border-zone angiogenesis, scar size, and LV remodeling. Likewise, depleting mature T and B cells in Rag1 knockout mice or specifically depleting Foxp3+ Treg cells in DEREG mice eliminated the therapeutic effects of POL5551. Adoptive cell transfer experiments using splenic MNCs, Foxp3+ Treg cell–depleted splenic MNCs, or splenic Foxp3+ Treg cells from POL5551-treated infarcted donor mice confirmed splenic Foxp3+ Treg cells as the prime mediators of the POL5551 treatment effects.
Splenic MNC transfer experiments comparing infarcted versus noninfarcted and DC-depleted versus non–DC-depleted donor mice revealed that instructive cues provided by the infarct (specifically by DCs) are required for POL5551 treatment effects. Considering that Foxp3+ Treg cells are the splenic MNC population that mediates POL5551 treatment effects and that infarcted myocardium–primed DCs promote Treg cell expansion and activation,36 we conclude that MI-primed DCs are required to render Treg cells responsive to POL5551.
Expanding the circulating Treg cell pool by adoptive Treg cell transfer or treatment with a superagonistic anti-CD28 antibody enhances Treg cell recruitment to the infarcted region, improves wound healing, and attenuates LV remodeling in mice and rats with acute MI.32–34 The therapeutic effects of POL5551 may therefore relate in part to increased Treg cell availability. Adoptive Treg cell transfer experiments indicated that POL5551 additionally enhances Treg cell functionality. Specifically, POL5551 increased the capacity of Treg cells to home to the infarct and (slightly) promoted Treg cell immunosuppressive function.
Acute MI activates autoreactive CD4+ T cells,46,47 which could trigger autoimmune damage to the myocardium in mice and possibly patients with type 1 diabetes mellitus.48 Postinfarction autoimmune injury, however, is usually not observed in nondiabetic mice.47,48 Suppression of autoimmune injury, therefore, might not be the prime mechanism whereby Treg cell mobilization or Treg cell expansion32–34 promotes therapeutic effects after MI. Instead, Treg cells have been proposed to modulate innate immune responses after MI at multiple levels.34,35 Treg cell depletion before the infarct could lead to higher monocyte and macrophage accumulation in the infarcted region.34,35 Still, treatment with POL5551, starting 2 days after MI, did not affect monocyte and macrophage numbers in the infarcted myocardium. Similarly, treatment with an anti-CD28 antibody 2 days after MI did not affect monocyte and macrophage accumulation,34 which indicates that timing could be important or that endogenous Treg cell effects on innate immune cell recruitment cannot be further enhanced by these interventions. Treg cells have also been shown to modulate inflammatory gene expression in monocytes and macrophages in the infarcted myocardium.34 Consistently, we found that POL5551, via Foxp3+ Treg cells, attenuated the expression of tumor necrosis factor, interleukin 1β, interferon-γ, and nitric oxide synthase 2 in monocytes and macrophages in the infarcted region.
In conclusion, our data confirm CXCR4 blockade as a promising treatment strategy to enhance tissue repair and functional recovery after acute MI.10–12 We identify CD4+ Foxp3+ Treg cells as the central arbiters of these therapeutic effects and thereby delineate a new pharmacological strategy to acutely and transiently augment Treg cell function in vivo.49 Highlighting the translational potential of this approach, the clinical-stage compound POL6326 reduced infarct volume and promoted LVEF recovery in a porcine closed-chest model of reperfused acute MI.50 These findings should stimulate further research into the therapeutic potential of CXCR4 antagonists after MI and in other acute conditions wherein excessive innate or adaptive immune responses cause immunopathology.
Acknowledgments
We are grateful to Dr Matthias Ballmaier from the fluorescence-activated cell sorting core facility and Dr Christine S. Falk from the Institute of Transplant Immunology at Hannover Medical School for support with cell sorting.
Sources of Funding
This study was supported by grants from the German Research Foundation (WO 552/9-2, WO 552/10-1, and Excellence Cluster Von Regenerativer Biologie zu Rekonstruktiver Therapie 2 [REBIRTH-2] to Dr Wollert). Polyphor (Allschwil, Switzerland) provided financial support.
Disclosures
Drs Chevalier and Dembowsky were employees of Polyphor during experiments and manuscript preparation. The other authors report no conflicts.
Supplementary Material
Footnotes
Sources of Funding, see page 1810
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.118.036053.
References
- 1.Bagai A, Dangas GD, Stone GW, Granger CB. Reperfusion strategies in acute coronary syndromes. Circ Res. 2014;114:1918–1928. doi: 10.1161/CIRCRESAHA.114.302744. doi: 10.1161/CIRCRESAHA.114.302744. [DOI] [PubMed] [Google Scholar]
- 2.Stone GW, Selker HP, Thiele H, Patel MR, Udelson JE, Ohman EM, Maehara A, Eitel I, Granger CB, Jenkins PL, Nichols M, Ben-Yehuda O. Relationship between infarct size and outcomes following primary PCI: patient-level analysis from 10 randomized trials. J Am Coll Cardiol. 2016;67:1674–1683. doi: 10.1016/j.jacc.2016.01.069. doi: 10.1016/j.jacc.2016.01.069. [DOI] [PubMed] [Google Scholar]
- 3.Westman PC, Lipinski MJ, Luger D, Waksman R, Bonow RO, Wu E, Epstein SE. Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. 2016;67:2050–2060. doi: 10.1016/j.jacc.2016.01.073. doi: 10.1016/j.jacc.2016.01.073. [DOI] [PubMed] [Google Scholar]
- 4.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 7.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–2187. doi: 10.2353/ajpath.2010.090759. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, McAlpine CS, Mindur JE, Chan CT, Iwamoto Y, Tricot B, Wojtkiewicz GR, Weissleder R, Libby P, Nahrendorf M, Stone JR, Becher B, Swirski FK. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med. 2017;214:3293–3310. doi: 10.1084/jem.20170689. doi: 10.1084/jem.20170689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, Gorbatov R, Iwamoto Y, Dutta P, Wojtkiewicz G, Courties G, Sebas M, Borodovsky A, Fitzgerald K, Nolte MW, Dickneite G, Chen JW, Anderson DG, Swirski FK, Weissleder R, Nahrendorf M. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation. 2013;127:2038–2046. doi: 10.1161/CIRCULATIONAHA.112.000116. doi: 10.1161/CIRCULATIONAHA.112.000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jujo K, Hamada H, Iwakura A, Thorne T, Sekiguchi H, Clarke T, Ito A, Misener S, Tanaka T, Klyachko E, Kobayashi K, Tongers J, Roncalli J, Tsurumi Y, Hagiwara N, Losordo DW. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc Natl Acad Sci U S A. 2010;107:11008–11013. doi: 10.1073/pnas.0914248107. doi: 10.1073/pnas.0914248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jujo K, Ii M, Sekiguchi H, Klyachko E, Misener S, Tanaka T, Tongers J, Roncalli J, Renault MA, Thorne T, Ito A, Clarke T, Kamide C, Tsurumi Y, Hagiwara N, Qin G, Asahi M, Losordo DW. CXC-chemokine receptor 4 antagonist AMD3100 promotes cardiac functional recovery after ischemia/reperfusion injury via endothelial nitric oxide synthase-dependent mechanism. Circulation. 2013;127:63–73. doi: 10.1161/CIRCULATIONAHA.112.099242. doi: 10.1161/CIRCULATIONAHA.112.099242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu WT, Jui HY, Huang YH, Su MY, Wu YW, Tseng WY, Hsu MC, Chiang BL, Wu KK, Lee CM. CXCR4 antagonist TG-0054 mobilizes mesenchymal stem cells, attenuates inflammation, and preserves cardiac systolic function in a porcine model of myocardial infarction. Cell Transplant. 2015;24:1313–1328. doi: 10.3727/096368914X681739. doi: 10.3727/096368914X681739. [DOI] [PubMed] [Google Scholar]
- 13.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, Calandra G, Bridger G, Dale DC, Srour EF. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karpova D, Bonig H. Concise review: CXCR4/CXCL12 signaling in immature hematopoiesis: lessons from pharmacological and genetic models. Stem Cells. 2015;33:2391–2399. doi: 10.1002/stem.2054. doi: 10.1002/stem.2054. [DOI] [PubMed] [Google Scholar]
- 15.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, Topol EJ, Penn MS. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003;362:697–703. doi: 10.1016/S0140-6736(03)14232-8. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 16.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal cell-derived factor-1α plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110:3300–3305. doi: 10.1161/01.CIR.0000147780.30124.CF. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- 17.Segers VF, Tokunou T, Higgins LJ, MacGillivray C, Gannon J, Lee RT. Local delivery of protease-resistant stromal cell derived factor-1 for stem cell recruitment after myocardial infarction. Circulation. 2007;116:1683–1692. doi: 10.1161/CIRCULATIONAHA.107.718718. doi: 10.1161/CIRCULATIONAHA.107.718718. [DOI] [PubMed] [Google Scholar]
- 18.Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res. 2012;110:624–637. doi: 10.1161/CIRCRESAHA.111.243386. doi: 10.1161/CIRCRESAHA.111.243386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loetscher M, Geiser T, O’Reilly T, Zwahlen R, Baggiolini M, Moser B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J Biol Chem. 1994;269:232–237. [PubMed] [Google Scholar]
- 20.Rettig MP, Ansstas G, DiPersio JF. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia. 2012;26:34–53. doi: 10.1038/leu.2011.197. doi: 10.1038/leu.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karpova D, Dauber K, Spohn G, Chudziak D, Wiercinska E, Schulz M, Pettit AR, Levesque JP, Romagnoli B, Patel K, Chevalier E, Dembowsky K, Bonig H. The novel CXCR4 antagonist POL5551 mobilizes hematopoietic stem and progenitor cells with greater efficiency than Plerixafor. Leukemia. 2013;27:2322–2331. doi: 10.1038/leu.2013.266. doi: 10.1038/leu.2013.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karpova D, Bräuninger S, Wiercinska E, Krämer A, Stock B, Graff J, Martin H, Wach A, Escot C, Douglas G, Romagnoli B, Chevalier E, Dembowski K, Hooftman L, Bonig H. Mobilization of hematopoietic stem cells with the novel CXCR4 antagonist POL6326 (balixafortide) in healthy volunteers: results of a dose escalation trial. J Transl Med. 2017;15:2. doi: 10.1186/s12967-016-1107-2. doi: 10.1186/s12967-016-1107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luther A, Moehle K, Chevalier E, Dale G, Obrecht D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr Opin Chem Biol. 2017;38:45–51. doi: 10.1016/j.cbpa.2017.02.004. doi: 10.1016/j.cbpa.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [PubMed] [Google Scholar]
- 26.Lahl K, Sparwasser T. In vivo depletion of FoxP3+ Tregs using the DEREG mouse model. Methods Mol Biol. 2011;707:157–172. doi: 10.1007/978-1-61737-979-6_10. doi: 10.1007/978-1-61737-979-6_10. [DOI] [PubMed] [Google Scholar]
- 27.Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, von Stebut E, Probst HC, van den Broek M, Riethmacher D, Birnberg T, Blank T, Reizis B, Korn T, Wiendl H, Jung S, Prinz M, Kurschus FC, Waisman A. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor+ regulatory T cells. Immunity. 2012;37:264–275. doi: 10.1016/j.immuni.2012.05.025. doi: 10.1016/j.immuni.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Baranyai T, Giricz Z, Varga ZV, Koncsos G, Lukovic D, Makkos A, Sárközy M, Pávó N, Jakab A, Czimbalmos C, Vágó H, Ruzsa Z, Tóth L, Garamvölgyi R, Merkely B, Schulz R, Gyöngyösi M, Ferdinandy P. In vivo MRI and ex vivo histological assessment of the cardioprotection induced by ischemic preconditioning, postconditioning and remote conditioning in a closed-chest porcine model of reperfused acute myocardial infarction: importance of microvasculature. J Transl Med. 2017;15:67. doi: 10.1186/s12967-017-1166-z. doi: 10.1186/s12967-017-1166-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hao H, Hu S, Chen H, Bu D, Zhu L, Xu C, Chu F, Huo X, Tang Y, Sun X, Ding BS, Liu DP, Hu S, Wang M. Loss of endothelial CXCR7 impairs vascular homeostasis and cardiac remodeling after myocardial infarction: implications for cardiovascular drug discovery. Circulation. 2017;135:1253–1264. doi: 10.1161/CIRCULATIONAHA.116.023027. doi: 10.1161/CIRCULATIONAHA.116.023027. [DOI] [PubMed] [Google Scholar]
- 30.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–137. doi: 10.1084/jem.20111009. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, Yan XX, Nie SF, Liu J, Zhou SF, Li JJ, Yao R, Liao MY, Tu X, Liao YH, Cheng X. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol. 2012;107:232. doi: 10.1007/s00395-011-0232-6. doi: 10.1007/s00395-011-0232-6. [DOI] [PubMed] [Google Scholar]
- 33.Sharir R, Semo J, Shimoni S, Ben-Mordechai T, Landa-Rouben N, Maysel-Auslender S, Shaish A, Entin-Meer M, Keren G, George J. Experimental myocardial infarction induces altered regulatory T cell hemostasis, and adoptive transfer attenuates subsequent remodeling. PLoS One. 2014;9:e113653. doi: 10.1371/journal.pone.0113653. doi: 10.1371/journal.pone.0113653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. doi: 10.1161/CIRCRESAHA.115.303895. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 35.Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, Frangogiannis NG. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol. 2014;307:H1233–H1242. doi: 10.1152/ajpheart.00328.2014. doi: 10.1152/ajpheart.00328.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choo EH, Lee JH, Park EH, Park HE, Jung NC, Kim TH, Koh YS, Kim E, Seung KB, Park C, Hong KS, Kang K, Song JY, Seo HG, Lim DS, Chang K. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation. 2017;135:1444–1457. doi: 10.1161/CIRCULATIONAHA.116.023106. doi: 10.1161/CIRCULATIONAHA.116.023106. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Hsieh AF, Dharmarajan K, Masoudi FA, Krumholz HM. National trends in heart failure hospitalization after acute myocardial infarction for Medicare beneficiaries: 1998–2010. Circulation. 2013;128:2577–2584. doi: 10.1161/CIRCULATIONAHA.113.003668. doi: 10.1161/CIRCULATIONAHA.113.003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szummer K, Wallentin L, Lindhagen L, Alfredsson J, Erlinge D, Held C, James S, Kellerth T, Lindahl B, Ravn-Fischer A, Rydberg E, Yndigegn T, Jernberg T. Improved outcomes in patients with ST-elevation myocardial infarction during the last 20 years are related to implementation of evidence-based treatments: experiences from the SWEDEHEART registry 1995–2014. Eur Heart J. 2017;38:3056–3065. doi: 10.1093/eurheartj/ehx515. doi: 10.1093/eurheartj/ehx515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. 2016;13:167–179. doi: 10.1038/nrcardio.2015.169. doi: 10.1038/nrcardio.2015.169. [DOI] [PubMed] [Google Scholar]
- 40.Kean LS, Sen S, Onabajo O, Singh K, Robertson J, Stempora L, Bonifacino AC, Metzger ME, Promislow DE, Mattapallil JJ, Donahue RE. Significant mobilization of both conventional and regulatory T cells with AMD3100. Blood. 2011;118:6580–6590. doi: 10.1182/blood-2011-06-359331. doi: 10.1182/blood-2011-06-359331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donzella GA, Schols D, Lin SW, Esté JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, Moore JP. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998;4:72–77. doi: 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- 42.De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2:581–587. doi: 10.1038/nrd1134. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 43.Righi E, Kashiwagi S, Yuan J, Santosuosso M, Leblanc P, Ingraham R, Forbes B, Edelblute B, Collette B, Xing D, Kowalski M, Mingari MC, Vianello F, Birrer M, Orsulic S, Dranoff G, Poznansky MC. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 2011;71:5522–5534. doi: 10.1158/0008-5472.CAN-10-3143. doi: 10.1158/0008-5472.CAN-10-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, Fan C, Huang P, Bardeesy N, Zhu AX, Jain RK, Duda DG. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591–1602. doi: 10.1002/hep.27665. doi: 10.1002/hep.27665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, Wei S, David O, Curiel TJ, Zou W. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004;64:8451–8455. doi: 10.1158/0008-5472.CAN-04-1987. doi: 10.1158/0008-5472.CAN-04-1987. [DOI] [PubMed] [Google Scholar]
- 46.Van der Borght K, Scott CL, Nindl V, Bouché A, Martens L, Sichien D, Van Moorleghem J, Vanheerswynghels M, De Prijck S, Saeys Y, Ludewig B, Gillebert T, Guilliams M, Carmeliet P, Lambrecht BN. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep. 2017;18:3005–3017. doi: 10.1016/j.celrep.2017.02.079. doi: 10.1016/j.celrep.2017.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, Frantz S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652–1663. doi: 10.1161/CIRCULATIONAHA.111.044164. doi: 10.1161/CIRCULATIONAHA.111.044164. [DOI] [PubMed] [Google Scholar]
- 48.Gottumukkala RV, Lv H, Cornivelli L, Wagers AJ, Kwong RY, Bronson R, Stewart GC, Schulze PC, Chutkow W, Wolpert HA, Lee RT, Lipes MA. Myocardial infarction triggers chronic cardiac autoimmunity in type 1 diabetes. Sci Transl Med. 2012;4:138ra80. doi: 10.1126/scitranslmed.3003551. doi: 10.1126/scitranslmed.3003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, ten Brinke A, Bushell A, Cools N, Geissler EK, Gregori S, Marieke van Ham S, Hilkens C, Hutchinson JA, Lombardi G, Madrigal JA, Marek-Trzonkowska N, Martinez-Caceres EM, Roncarolo MG, Sanchez-Ramon S, Saudemont A, Sawitzki B. Hurdles in therapy with regulatory T cells. Sci Transl Med. 2015;7:304ps18. doi: 10.1126/scitranslmed.aaa7721. doi: 10.1126/scitranslmed.aaa7721. [DOI] [PubMed] [Google Scholar]
- 50.van der Spoel TI, Jansen of Lorkeers SJ, Agostoni P, van Belle E, Gyöngyösi M, Sluijter JP, Cramer MJ, Doevendans PA, Chamuleau SA. Human relevance of pre-clinical studies in stem cell therapy: systematic review and meta-analysis of large animal models of ischaemic heart disease. Cardiovasc Res. 2011;91:649–658. doi: 10.1093/cvr/cvr113. doi: 10.1093/cvr/cvr113. [DOI] [PubMed] [Google Scholar]