

Abstract
Objective
To investigate the relevance of dysfunctional T cells in immune-mediated myopathies. We analyzed T-cell exhaustion and senescence, in the context of programmed cell death protein 1 (PD1)-related immunity in skeletal muscle biopsies from patients with immune-mediated necrotizing myopathy (IMNM), sporadic inclusion body myositis (sIBM), and myositis induced by immune checkpoint inhibitors (irMyositis).
Methods
Skeletal muscle biopsies from 12 patients with IMNM, 7 patients with sIBM, and 8 patients with irMyositis were analyzed by immunostaining and immunofluorescence as well as by quantitative PCR. Eight biopsies from nondisease participants served as controls.
Results
CD3+CD8+ T cells in biopsies from IMNM, sIBM, and irMyositis were largely PD1-positive, while CD68+ macrophages were sparsely positive to the ligand of programmed cell death protein 1 (PD-L1). The sarcolemma of myofibers was PD-L2+ and was colocalized with major histocompatibility complex (MHC) class I. CD68+ macrophages were colocalized with PD-L2. Senescent T cells were strongly enriched in skeletal muscle of sIBM, revealing a distinct immunologic signature. Biopsies from patients with irMyositis showed mild signs of senescence and exhaustion.
Conclusion
Persistent exposure to antigens in IMNMs and sIBM may lead to T-cell exhaustion, a process controlled by the PD1 receptor and its cognate ligands PD-L1/PD-L2. To our knowledge, these data are the first evidence of presence of dysfunctional T cells and relevance of the PD1 pathway in IMNM, sIBM, and irMyositis. These findings may guide the way to a novel understanding of the immune pathogenesis of immune-mediated myopathies.
Autoantibodies against signal recognition particle (SRP) and 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) play a crucial role in the pathogenesis of immune-mediated necrotizing myopathies (IMNMs).1,2 Levels of autoantibody titres are directly linked to disease activity, and treatment response can be monitored by measuring the levels of autoantibodies in both anti-SRP– and anti-HMGCR–associated necrotizing myopathies.3 Antigens to these antibodies persist over a long time in the human body and stimulate the immune system in a specific way. In contrast to sporadic inclusion body myositis (sIBM), which is characterized by the presence of cytotoxic granzyme B+ and/or perforin+ T cells,4 IMNM is a paucilymphocytic disease. We have recently highlighted the presence of T cells in all examined IMNM biopsies; however, we showed that in IMNM, T cells lack robust cytotoxicity.1 As opposed to IMNM, the specificity of an autoantibody (cN1A) in sIBM is less clear.5–7 Hence, persistence of antigens in IMNM may affect the phenotype of T cells in this disease. Indeed, T cells, exposed to persistent antigen and inflammatory molecules, may functionally deteriorate, a process called T-cell exhaustion.8 Exhausted T cells are subject to complex negative regulation involving signaling through multiple receptors that inhibit functional and proliferative responses, which in turn result in ineffective control of the inflammation.9 The CD28 family member programmed cell death protein 1 (PD1) is the most highly expressed inhibitory receptor on CD8+ T cells during chronic inflammation, and this receptor has a major role in regulating T-cell exhaustion. The role of PD1 expression in healthy adult humans to distinguish between effector T cells and exhausted T cells as well as the interpretation of increased expression levels of PD1 in infection as a single marker of exhaustion, however, has been much debated.10,11 Interestingly, PD1 has been shown to be upregulated early after T-cell activation and is not exclusively associated with states of T-cell dysfunction.12 Importantly, blockade of the PD1 signaling pathway can restore appropriate antigen-specific T-cell responses in chronic infection and in the tumor environment.8 Immune checkpoint inhibitors (ICIs) successfully lead to tumor immune rejection in certain cancers by reactivating tumor response in exhausted T cells.13 However, therapy with these agents causes increased autoimmune responses with development of adverse effects including myositis and myocarditis.14 Exhaustion, anergy, and senescence are common concepts to describe dysfunctional states of T cells associated with increased expression of immune checkpoints, which may harbor overlapping, yet distinctive features, one of which is expression of CD57—low in exhaustion, but high in senescence.9,15,16 Senescent T cells enter a terminal differentiation state owing to excessive cell replication, a process that is associated with irreversible cell cycle arrest and telomere shortening. CD57+ T cells have been studied in sIBM.17
In this study, we hypothesized that persistent antigen presentation in immune-mediated myopathies leads to dysfunctional states of invading T cells. This finding would provide further evidence for a direct recognition of muscular antigens by invading T cells, and a role for T cells in the pathogenesis of these diseases. In addition, we analyzed molecules driving T-cell exhaustion and senescence in skeletal muscle biopsies from IMNM with anti-SRP or anti-HMGCR antibodies and sIBM as well as ICI-associated immune toxicity-related myositis induced by immune checkpoint inhibitors (irMyositis).
Methods
Patients
Clinical data of all patients enrolled in this study are listed in the table. We included patients with clinical, serologic, and morphologic signs and symptoms consistent with IMNM18 and sIBM according to present criteria,19 as well as patients treated with ICIs suffering from ICI-induced myositis.
Table.
Characteristics of all patients included in the study

Standard protocol approvals, registrations, and patient consents
Informed consent was obtained from all patients at each institution involved. The Charité Ethics Committee (EA2/163/17), the French Ministry of Research, as well as the Research Ethics Committee of the Pitié-Salpêtrière Hospital (Paris, France) (AC 2008-87) granted ethical approval.
Skeletal muscle specimens
We analyzed skeletal muscle biopsies from 12 patients diagnosed with IMNM according to the clinico-sero-morphologic European Neuromuscular Centre criteria.18 Also, skeletal muscle biopsies from 7 patients with sIBM were included (clinically and morphologically definite sIBM)19 and 9 patients with myositis as an adverse event after ICI treatment according to recent data.20 In addition, we investigated control skeletal muscle biopsies (n = 8) from patients with nonspecific myalgia, in whom muscle weakness and morphologic abnormalities in skeletal muscle biopsies were absent; creatine kinase levels were normal; no signs of systemic inflammation and no myositis-specific antibodies (MSAs) or myositis-associated antibodies had been detected. All skeletal muscle specimens had been cryopreserved at −80°C prior to analysis.
Morphologic analysis
All stains were performed on 7 μm cryomicrotome sections, according to standard procedures. Immunohistochemical and double immunofluorescence stains were obtained as described previously.21 The following antibodies were used for staining procedures: (primary antibody, dilution, company/clone): mouse anti-human CD3, 1:50, Dako/UCHT1; mouse anti-human CD68, 1:100 Dako/EBM1; MHCI, 1:1.000, Dako/w6/32; PD1, 1:100, Abcam/NAT105; PDL1, 1:100, Cell signalling/E1L3N; PDL2, 1:100, Abcam/Ty25.
RNA extraction and PCR
Total RNA extraction from muscle specimens and quantitative reverse transcription PCR were performed as described previously.21
cDNA was synthesized using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). For quantitative PCR (qPCR) reactions, 2 ng of cDNA were used, and for analysis, the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) was used, running conditions: 95°C 0:20, 95°C 0:01, 60°C 0:20, 45 cycles (values above 40 cycles were defined as not expressed). All genes were run as triplicates, and each run contained the reference gene (PGK1) as internal control, to normalize the expression of the target genes. The qPCR assay identification numbers, TaqMan Gene Exp Assay from Life Technologies/ThermoFisher are listed as follows:
CD28, Hs01007422_m1; CD39, Hs00969556_m1; CD80, Hs01045161_m1; CD244, Hs00175569_m1; EOMES, Hs00172872_m1; IL7R, Hs00902334_m1; KLRG1, Hs00195153_m1; LAG3, Hs00958444_g1; PD1, Hs01550088_m1; PDL1, Hs00204257_m1; PDL2, Hs00228839_m1; PGK1, Hs99999906_m1 TBX21, Hs00894392_m1; TIM3, Hs00958618_m1; VISTA, Hs00735289_m1.
The ΔCT of healthy controls was subtracted from the ΔCT of patients with IMNM to determine the differences (ΔΔCT) and fold change (2^−ΔΔCT) of gene expression. Gene expression was illustrated by the log10 of fold change values compared to the healthy controls.
Statistical analysis
Kruskal-Wallis one-way analysis of variance followed by Bonferroni-Dunn correction of the post hoc tests was used to analyze quantitative differences of messanger ribonuclein acid (mRNA) transcripts. Data are presented as mean ± standard error of the mean. The level of significance was set at p < 0.05. GraphPad Prism 5.02 software (GraphPad Software, Inc, La Jolla, CA) was used for statistical analysis.
Data availability statement
The authors of this manuscript state that they have carefully documented data, methods, and materials used to conduct the research in the article. Data not provided in the article because of space limitations can be made available at the request of other investigators for purposes of replicating procedures and results. To our knowledge, there are no legal or ethical reasons or any embargoes on datasets, which may restrict these data availability policy.
Results
Clinical data
For the analysis of exhaustion and senescence markers, we included 12 patients with IMNM (7 male, mean age 47 [18–80]), 7 with sIBM (6 male, mean age 70 [64–75], 9 with irMyositis (7 male, mean age 71 [56–87]), and 8 healthy controls (6 male, mean age 30 [2–49]) (table).
Of all patients with IMNM, 59% (7 patients) harbored anti-SRP autoantibodies, 41% (5 patients) had anti-HMGCR autoantibodies, and for the patients with sIBM, the autoantibody status for NT5C1A was unknown and not obtainable retrospectively. One patient with ICI-related myositis had anti-Sjögren syndrome–related antigen A autoantibodies. No other MSAs were found in this cohort.
Expression and localization of PD1 and its cognate ligands PD-L1 and PD-L2 in IMNM, sIBM, and irMyositis
We were not able to detect any significant difference between biopsies from SRP- and HMGCR-positive patients with IMNM either by qPCR analyses or by immunohistochemical stains (figure 1). We will, therefore, report all results for both SRP- and HMGCR-positive IMNM as one IMNM phenotype.
Figure 1. Log10-fold changes of markers of T-cell exhaustion and senescence in samples from patients with IMNM.

We did not observe statistically significant differences between patients with anti-SRP and anti-HMGCR autoantibodies. Statistical analysis was performed using Kruskal-Wallis one-way analysis of variance with Dunn multiple comparison test. HMGCR = hydroxy-3-methylglutaryl-coenzyme A reductase; SRP = signal recognition particle.
In skeletal muscle biopsies of patients with IMNM, CD3+ T cells were mostly PD1 positive (figure 2, A and D). Ligand of programmed cell death protein 1 (PD-L1) was only expressed on macrophages in the endomysium, within myophagocytosis, and not on the sarcolemma of myofibers (figure 2B). PD-L1+ macrophages did not closely interact with PD1+ T cells. The sarcolemma of myofibers was weakly PD-L2+, the expression of which is colocalized with major histocompatibility complex (MHC) class I (figure 2, C and G). PD1+ T cells are localized adjacent to PD-L2+ sarcolemmal structures—thus may form an immunologic synapse (figure 2F). PD-L2 was also expressed on many CD68+ macrophages (figure 2E).
Figure 2. Immunohistochemical stains of skeletal muscle biopsies from patients with IMNM, sIBM, or irMyositis.

In IMNM biopsies, PD1+ cells are diffusely distributed in the endomysium (A) and most of them correspond to CD3+ T cells (D). Macrophages in myophagocytosis and endomysial macrophages are PD-L1+ (B) but the sarcolemma of myofibers is not (B). CD68 + macrophages coexpress PD-L2 (E), they colocalize with MHC class I+ sarcolemmal membranes (G), and PD1+ lymphocytes interact with PD-L2+ sarcolemmal structures (F) (A and C, original magnification ×200; B, original magnification ×400; D-F, original magnification ×600). In sIBM biopsies, PD1+ cells are CD3+, and they are detectable in the endomysium surrounding myofibers (H, K). PD-L1 is faintly detectable in macrophages within myophagocytosis but not on the sarcolemma and not on endomysial or perimysial macrophages (I). The sarcolemma of myofibers and macrophages are PD-L2+ and colocalize with sarcolemmal MHC class I (J, N). CD68 + macrophages coexpress PD-L2 (L), and PD1+ lymphocytes only occasionally interact with PD-L2+ sarcolemmal structures (M) (H-J, original magnification ×200; K-N, original magnification ×600). In irMyositis, PD1+ cells are detectable in the endomysium and they are CD3+ with variable intensity (O, R). PD-L1 is positive on macrophages but not all CD68 + macrophages express PD-L1 (P). The sarcolemma of myofibers is moderately PD-L2 positive (Q) and faintly coexpresses MHC class I (U). CD68 + macrophages interact with PD-L2+ sarcolemmal structures (S). Single PD1+ T cells interact with PD-L2 sarcolemmal structures (T) (O-Q, original magnification ×200; R-U original magnification ×600). IMNM = immune-mediated necrotizing myopathy; irMyositis = myositis induced by immune checkpoint inhibitors; sIBM = sporadic inclusion body myositis.
In skeletal muscle biopsies of patients with sIBM, numerous CD3+ T cells were PD1 positive (figure 2, H and K). PD-L1 was weakly expressed on single macrophages in myophagocytosis or in the endomysium, and not on the sarcolemma of myofibers (figure 2I). PD-L1+ macrophages did not show close interaction with PD1+ T cells. The sarcolemma of myofibers was PD-L2+, the expression of which is intensely colocalized with MHC class I (figure 2, J and N). PD1+ T cells only occasionally interacted with PD-L2+ sarcolemmal structures (figure 2M). PD-L2 was also expressed on many CD68+ macrophages and was co-localized with the sarcolemma of MHC class I+ myofibers (figure 2, L and N).
In skeletal muscle biopsies of patients with irMyositis, PD1+ T cells were diffusely distributed and frequently clustered close to myofiber necrosis (figure 2O). PD-L1 was strongly positive on many endomysial CD68+ macrophages and in myophagocytic lesions but not on the sarcolemma of myofibers (figure 2, P,R,T). PD1+ T cells only occasionally interacted with PD-L2+ sarcolemmal structures (figure 2T). PD-L2 was strongly positive on the sarcolemma of myofibers and was colocalized with MHC class I (figure 2, Q, S, U).
Molecular expression of exhaustion and senescence markers in IMNM, sIBM, and irMyositis
Consistent with our findings from immunohistochemical stains, expression levels of PD1 (PDCD1), PDL1 (CD274), and PDL2 (PDCD1LG2) in IMNM samples were overall increased by qPCR analyses. Expression levels showed a large variance among individual patients and differences to healthy controls were nonsignificant in statistical analyses. Patients with high expression of PD1, however, also showed high expression levels of other markers of T-cell exhaustion and vice versa (figure 3A). The T-cell exhaustion marker TIM3 was significantly elevated in IMNM samples (compared to healthy controls). We did not find increased expression levels of markers of T-cell senescence in our IMNM samples (figure 3, B and C). Conversely, we observed clear signs of both T-cell senescence and T-cell exhaustion in sIBM samples. mRNA levels for characteristic T-cell exhaustion markers PD1, EOMES, TBX21, LAG3, CD244,8 and KLRG1 were significantly increased (figure 3, B and C). The PD1 ligand PDL2 was also significantly increased (figure 3A). Despite significant elevation of KLRG1 as a marker of T-cell senescence, CD28 and CD80 were also significantly elevated in the sIBM samples. IL7R, which has been shown to be downregulated in T-cell exhaustion, was significantly upregulated (figure 3C). In irMyositis, we observed 2 distinct subsets of expression pattern. In samples of 2 patients with irMyositis, we found clear signs of T-cell senescence, but not of T-cell exhaustion. In these cases, surface expression of CD57 on immunohistochemical stains was high while gene expression of PD1 and CD28 were suppressed. These 2 patients had a severe clinical phenotype, leading to death of an associated myocarditis in one case. In all other cases, CD57 surface expression was undetectable, while TIM3 and LAG3 as well as PDL1 and PDL2 expression were significantly increased (figure 3, A–C).
Figure 3. Markers of T-cell exhaustion and senescence in patient skeletal muscle samples.

mRNA levels of genes related to T-cell exhaustion (A and B) and T-cell senescence (C) from IMNM (n = 12), sIBM (n = 7), and irMyositis (n = 9) samples. Statistical analysis was performed using Kruskal-Wallis one-way analysis of variance with Dunn multiple comparison test. *p < 0.05; **p < 0.01; ***p < 0.001—difference between displayed groups in log fold changes. #p < 0.05; ##p < 0.01; ###p < 0.001—differences between dCT of patient groups and normal control. IMNM = immune-mediated necrotizing myopathy; irMyositis = myositis induced by immune checkpoint inhibitors; sIBM = sporadic inclusion body myositis.
Expression of inflammatory markers in sIBM and IMNM
To further characterize the nature and the effect of the observed T-cell subsets in sIBM and IMNM, we investigated markers of inflammation and T-cell activation in muscle biopsies of patients with IMNM and sIBM. In sIBM samples, we found strong upregulation of gene expression of gamma interferon (IFNγ), IL6, and IL12. These markers were also increased in IMNM samples regardless of the antibody subtype (HMGCR or SRP) but to a lower degree (figure 4).
Figure 4. Markers of inflammation in IMNM (n = 42) and sIBM (n = 7) skeletal muscle samples.
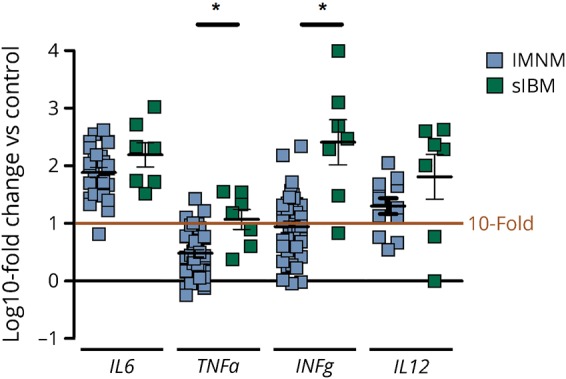
Markers for interstitial inflammation were upregulated in samples of IMNM and sIBM. All changes were significant at p < 0.01 compared to control samples (n = 8). TNFα and IFNγ levels differed significantly between IMNM and sIBM samples. Statistical analysis was performed using Kruskal-Wallis one-way analysis of variance with Dunn multiple comparison test. *p < 0.05—difference between displayed groups in log fold changes. IMNM = immune-mediated necrotizing myopathy; sIBM = sporadic inclusion body myositis.
Discussion
In this study, we provide systematic evidence of the presence of dysfunctional T cells and activation of the PD1 pathway in IMNM, inclusion body myositis, and immune toxicity-related myopathy. This finding has implications for the understanding of the role of T cells in the pathogenesis of these diseases. States of T-cell exhaustion and senescence have been studied extensively in chronic viral infections and cancer, and are associated with prolonged antigen exposure and subsequent adverse effects on T-cell function.9,22,23 The role of T-cell exhaustion and T-cell senescence in autoimmune disease has gathered increased interest in recent years,24,25 and co-inhibitory receptors have been shown to play a role in regulating severity of autoimmune disease.26 Despite a heterogeneously used nomenclature, T-cell exhaustion and senescence are actively regulated and, at least in part, reversible T-cell states associated with defective T-cell function.8
In this study, we demonstrate that T-cell exhaustion and senescence can be detected in sIBM and IMNM. We were also able to demonstrate that in irMyositis, T cells with an exhausted phenotype invade the skeletal muscle tissue, and that myofibers upregulate ligands of the inhibitory coreceptor PD1. Upregulation of PD-L1 and PD-L2 has been demonstrated in several autoimmune diseases,27–29 and PD-L1/PD-1 and PD-L2/PD-1 interaction has been reported to control the severity of autoimmune disease.29 Furthermore, PD-L1 has been reported to be expressed on muscle fibers in sIBM, polymyositis, and dermatomyositis in vivo and to inhibit T-cell activation in vitro.30 Using double immunohistochemistry/fluorescence, we were able to detect expression of PD1 ligands PD-L1 on macrophages and PD-L2 on myofibers in sIBM, IMNM, and irMyositis. PD-L2 staining on myofibers was partially overlapping PD1 staining on CD3+ lymphocytes, implicating the formation of so called “immunologic synapses” and a potential role of PD-L2/PD1 interaction in modulating T-cell activation.31 These findings were also reflected by significantly increased PDL2 mRNA levels in sIBM and irMyositis and increased PDL1 mRNA levels in irMyositis. PDL1 and PDL2 expression have been shown to be driven by IFNγ.31 Consistent with this report, IFNγ mRNA levels were significantly elevated in sIBM and IMNM samples compared to control, with significantly higher levels in sIBM than in muscle tissues of IMNM cases. IFNγ is a key regulatory molecule of cytotoxic T cells (CTLs),32,33 and CTLs have been reported to invade skeletal muscle in sIBM and to a much lesser degree also in IMNM.1,34 Thus far, there are no studies on the role of T cells in irMyositis in the literature. In this study, we now provided evidence that a subset of the invading T cells exhibit markers of severe T-cell exhaustion in sIBM, IMNM, and irMyositis.
Using immunohistochemistry and mRNA qPCR, we were able to detect high levels of gene expression for PD1 and several other inhibitory coreceptors (KLRG1, LAG3, TIM3, EOMES, TBET) associated with a severely exhausted phenotype23 in sIBM skeletal muscle samples. In qPCR analysis of IMNM skeletal muscle biopsies, TIM3 mRNA levels were significantly increased. TIM3 is an important marker for T-cell exhaustion8 and is a negative regulator of T-cell activation in infections35,36 and cancer.37 In addition, TIM-3 is a key modulator of T-cell activation and disease severity in autoimmune disease.26,38 T-cell exhaustion predicts favorable outcome in several immune-mediated diseases.25 Correlating clinical outcome with expression levels of exhaustion markers was beyond the scope of this study, but might explain in parts the heterogeneous findings in this group. Hence, we demonstrate that T cells in skeletal muscles of patients with IMNM can show an “exhausted” phenotype, which might be the consequence of prolonged exposure of these cells to high levels of autoantigen. Levels of autoantigens in sera of IMNM patients are actually directly associated with disease activity and creatine kinase levels.3 This also indirectly fosters the hypothesis that anti-SRP and anti-HMGCR autoantibodies play decisive roles in IMNM.1 We did not find any significant difference in expression levels between anti-HMGCR+, and anti-SRP+ IMNM, and interindividual variance was high among IMNM samples.
Expression of PD1 and other markers for T-cell exhaustion and T-cell senescence in tumor infiltrating T cells, as well as the expression of PD1 ligands on tumor cells have been well studied in several cancers. Inducing and maintaining T-cell inhibition through activation of inhibitory molecules on tumor-specific T cells have been implicated as one of the driving mechanisms for tumor immune escape.23 Blocking these inhibitory signals leads to robust beneficial anticancer immune responses.39,40 In this study, we provide the first evidence that T cells in irMyositis acquire an exhausted phenotype with high expression of PD-1, LAG-3, and TIM-3, reflecting a similar pattern found on tumor infiltrating T cells.23 Interestingly, in 2 severe cases of irMyositis with associated immune myocarditis, we observed no signs of T-cell exhaustion in our samples but distinct markers of T-cell senescence. These samples exhibited high surface expression levels of CD57 and low expression of CD28 and PD1.41 There was no association with the delay from symptom onset to biopsy or specific immunosuppressive treatment before biopsy. Further studies with larger sample sizes are needed to confirm the association of symptom severity and T-cell senescence. Differences in the distribution of weakness and frequency of a concomitant autoimmune myocarditis between cases showing predominantly T-cell exhaustion or T-cell senescence would be of particular interest. Consistent with previous reports addressing CD57 expression on invading T cells in sIBM,17 we also found CD57 expression in samples of sIBM. Furthermore, expression levels of KLRG1 and CD244 were significantly increased in sIBM samples as markers of exhaustion as well as T cell differentiation.12,23 However, IL7R, a receptor typically downregulated in exhausted T cells and involved in a sustained effective T-cell response,8 was also significantly upregulated in our sIBM samples. These findings suggest a heterogenous composition of senescent, exhausted, as well as differentiated cytotoxic invading T cells in sIBM. Analyzing T-cell subsets in the peripheral blood and muscle infiltrates of sIBM patients has previously revealed an oligoclonal expansion of CD8+ T cells and a deficiency of regulatory T cells,42 and the PDL1-PD1 axis has been shown to convert Th1 cells into T regs.43 Our finding of pronounced T-cell exhaustion and senescence in sIBM samples corroborates the notion of sIBM as a primarily inflammatory myopathy.
In this study, we report retrospective analyses of skeletal muscle biopsies with confirmed sIBM, IMNM, and irMyositis. We were therefore unable to analyze expression patterns on a single-cell level to further disentangle different invading T cell subsets, which might lead to a misinterpretation of the prevailing T-cell population. Nevertheless, in this study, we present clear evidence of T-cell exhaustion, and T-cell senescence to varying degrees in all 3 studied myositis entities: In IMNM samples, we found evidence of exhaustion, but no signs of senescence. Clearer signs of T-cell exhaustion were visible in irMyositis with a subset of patients showing markers of T-cell senescence, but not of exhaustion. In sIBM, we found high expression of markers for T-cell exhaustion and T-cell senescence. Differences in the duration of disease might interfere with the immune phenotype of the entities studied herein. Taken together, our findings implicate the involvement of muscle-specific T cells with specific phenotypes in the development of sIBM, IMNM, and irMyositis. Activation of coinhibitory receptors could therefore be a viable treatment target in sporadic immune-mediated myositis entities mandating further prospective studies correlating markers of T-cell exhaustion with clinical outcome.
Glossary
- CTL
cytotoxic T cell
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- ICI
immune checkpoint inhibitors
- IMNM
immune mediated necrotizing myopathy
- IFNγ
gamma interferon
- irMyositis
myositis induced by immune checkpoint inhibitors
- MHC
major histocompatibility complex
- mRNA
messanger ribonuclein acid
- MSA
myositis-specific antibody
- PD1
programmed cell death protein 1
- PD-L1/2
ligand of programmed cell death protein 1/2
- qPCR
quantitative PCR
- sIBM
sporadic Inclusion body myositis
- SRP
signal recognition particle
Authors contributions
S. Knauss, C. Preusse, W. Stenzel, H. Radbruch, R. Mothes: study concept and design, acquisition, analysis and interpretation of data. Y. Allenbach, S. Leonard-Louis, M. Touat, N. Fischer, V. Matyash, W. Böhmerle, M. Endres, H.-H. Goebel, O. Benveniste: analysis and interpretation of data.
Study funding
Deutsche Gesellschaft für Muskelkranke e.V. (DGM) to W. Stenzel.
Disclosure
S. Knauss, C. Preusse, Y. Allenbach, and S. Leonard-Louis report no disclosures. M. Touat served on the scientific advisory board for Agios, Taiho Oncology, received travel funding from Agios, Merck Sharp and Dohme, received research support from Fondation pour la Recherche Medicale. N. Fischer reports no disclosures. H. Radbruch received travel funding and/or speaker honoraria from Novartis, Sanofi, received research support from Novartis, Sanofi, Deutsche Forschungsgemeinschaft. R. Mothes and V. Matyash report no disclosures. W. Boehmerle received speaker honaria from NOGGO e.V., BMS, received research support from NeuroCure, QUEST. M. Endres served on the scientific advisory board for Center of Stroke Research Berlin, Boehringer Ingelheim, Bayer, Amgen, Covidien, received speaker honoraria paid to institution from Boehringer Ingelheim, Pfizer, Sanofi, Bayer, BMS, GSK, Amgen, Novartis, served on the editorial board for Stroke, Journal of Molecular Medicine, Neuropsychopharmacology, Aktuelle Neurologie, Journal of the European Stroke Organization, has been employed by German Center for Neurodegenerative Diseases, received research support from Roche, Bayer, German Ministry of Education and Science, Deutsche Forschungsgemeinschaft, European Union, German Center for Cardiovascular Research, German Center for Neurodegerative Diseases, Berlin Institute of Health, VolkswagenFoundatoin, CoronaFoundation, Fondation Leducq Transatlantic Network. H.-H. Goebel served on the editorial board for Neuromuscular Disorders, Neuropathology and Applied Neurobiology, Bran Pathology, Clinical Neuropathology. C. Benveniste served on the scientific advisory board for Csl Behring, Novartis, LFB, received travel funding from CSL Behring, LFB, consulted for Novartis, CSL Behring, Neovacs, received research support from LFB, Novartis, Shire, CSL Behring, Neovacs, DGOS/INSERM, PHRC, AFM, They Myositis Association. W. Stenzel served on the editorial board for Neuromuscular Disorders, Neuropathology and Applied Neurobiology. Disclosures available: Neurology.org/NN.
References
- 1.Allenbach Y, Arouche-Delaperche L, Preusse C, et al. Necrosis in anti-SRP+and anti-HMGCR+myopathies: role of autoantibodies and complement. Neurology 2018;90:e507–e517. [DOI] [PubMed] [Google Scholar]
- 2.Arouche-Delaperche L, Allenbach Y, Amelin D, et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol 2017;81:538–548. [DOI] [PubMed] [Google Scholar]
- 3.Allenbach Y, Drouot L, Rigolet A, et al. Anti-hmgcr autoantibodies in European patients with autoimmune necrotizing myopathies. Medicine 2014;93:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cherin P, Herson S, Crevon MC, et al. Mechanisms of lysis by activated cytotoxic cells expressing perforin and granzyme-B genes and the protein TIA-1 in muscle biopsies of myositis. J Rheumatol 1996;23:1135–1142. [PubMed] [Google Scholar]
- 5.Benjamin Larman H, Salajegheh M, Nazareno R, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 2013;73:408–418. [DOI] [PubMed] [Google Scholar]
- 6.Pluk H, van Hoeve BJA, van Dooren SHJ, et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann Neurol 2013;73:397–407. [DOI] [PubMed] [Google Scholar]
- 7.Goyal NA, Cash TM, Alam U, et al. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J Neurol Neurosurg Psychiatr 2016;87:373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry EJ. T cell exhaustion. Nat Immunol 2011;12:492–499. [DOI] [PubMed] [Google Scholar]
- 10.Duraiswamy J, Ibegbu CC, Masopust D, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol 2011;186:4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Re-evaluation of PD-1 expression by T cells as a marker for immune exhaustion during SIV infection. PLoS One; 2013;8:e60186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “exhaustion” of human CD8 T cells. Front Immunol Front 2013;4:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei SC, Levine JH, Cogdill AP, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017;170:1120–1133.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kao JC, Liao B, Markovic SN, et al. Neurological complications associated with anti–programmed death 1 (PD-1) antibodies. JAMA Neurol 2017;74:1216–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014;35:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol 2011;11:289–295. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM. Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain 2016;139:1348–1360. [DOI] [PubMed] [Google Scholar]
- 18.Allenbach Y, Mammen AL, Stenzel W, Benveniste O. Immune-Mediated Necrotizing Myopathies Working G. 224th ENMC International Workshop: clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscul Disord 2017;28:87–99. [DOI] [PubMed] [Google Scholar]
- 19.Rose MR; ENMC IBM Working Group. 188th ENMC International Workshop: inclusion body myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 2013;23:1044–1055. [DOI] [PubMed] [Google Scholar]
- 20.Touat M, Maisonobe T, Knauss S, et al. Immune checkpoint Inhibitor–related myositis and myocarditis in patients with cancer. Neurology 2018;91:e985–e994. [DOI] [PubMed] [Google Scholar]
- 21.Preusse C, Allenbach Y, Hoffmann O, et al. Differential roles of hypoxia and innate immunity in juvenile and adult dermatomyositis. Acta Neuropathologica Commun 2016;4:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zajac AJ, Blattman JN, Murali-Krishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 1998;188:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol 2013;25:214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinney EF, Smith KG. T-cell exhaustion: understanding the interface of chronic viral and autoinflammatory diseases. Immunol Cell Biol 2016;94:935–942. [DOI] [PubMed] [Google Scholar]
- 25.McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015;523:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monney L, Sabatos CA, Gaglia JL, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nat News 2002;415:536–541. [DOI] [PubMed] [Google Scholar]
- 27.Liang SC, Latchman YE, Buhlmann JE, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol 2003;33:2706–2716. [DOI] [PubMed] [Google Scholar]
- 28.Mataki N, Kikuchi K, Kawai T, et al. Expression of PD-1, PD-L1, and PD-L2 in the liver in autoimmune liver diseases. Am J Gastroenterol 2007;102:302–312. [DOI] [PubMed] [Google Scholar]
- 29.Menke J, Lucas JA, Zeller GC, et al. Programmed death 1 ligand (PD-L) 1 and PD-L2 limit autoimmune kidney disease: distinct roles. J Immunol 2007;179:7466–7477. [DOI] [PubMed] [Google Scholar]
- 30.Wiendl H, Mitsdoerffer M, Schneider D, et al. Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: a novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J 2003;17:1892–1894. [DOI] [PubMed] [Google Scholar]
- 31.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001;2:261–268. [DOI] [PubMed] [Google Scholar]
- 32.Ghanekar SA, Nomura LE, Suni MA, Picker LJ, Maecker HT, Maino VC. Gamma interferon expression in CD8(+) T cells is a marker for circulating cytotoxic T lymphocytes that recognize an HLA A2-restricted epitope of human cytomegalovirus phosphoprotein pp65. Clin Diagn Lab Immunol 2001;8:628–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 2007;96:41–101. [DOI] [PubMed] [Google Scholar]
- 34.Preuße C, Goebel HH, Held J, et al. Immune-mediated necrotizing myopathy is characterized by a specific Th1-m1 polarized immune profile. Am J Pathol 20122012;181:2161–2171. [DOI] [PubMed] [Google Scholar]
- 35.Jones RB, Ndhlovu LC, Barbour JD, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med 2008;205:2763–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin HT, Anderson AC, Tan WG, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA 2010;107:14733–14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8 +T cell dysfunction in melanoma patients. J Exp Med 2010;207:2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rangachari M, Zhu C, Sakuishi K, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat Med 2012;18:1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin-Liberal J, Ochoa de Olza M, Hierro C, Gros A, Rodon J, Tabernero J. The expanding role of immunotherapy. Cancer Treat Rev 2017;54:74–86. [DOI] [PubMed] [Google Scholar]
- 40.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brenchley JM. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2002;101:2711–2720. [DOI] [PubMed] [Google Scholar]
- 42.Benveniste O, Stenzel W, Hilton-Jones D, Sandri M, Boyer O, van Engelen BGM. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken. Acta Neuropathol 2015;129:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amarnath S, Mangus CW, Wang JC, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med 2011;3:111ra120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors of this manuscript state that they have carefully documented data, methods, and materials used to conduct the research in the article. Data not provided in the article because of space limitations can be made available at the request of other investigators for purposes of replicating procedures and results. To our knowledge, there are no legal or ethical reasons or any embargoes on datasets, which may restrict these data availability policy.