

Abstract
μ opioid receptor (MOR) agonists have been widely applied for treating moderate to severe pain. However, numerous adverse effects have been associated with their application, including opioid-induced constipation (OIC), respiratory depression, and addiction. On the basis of previous work in our laboratory, NAP, a 6β-N-4′-pyridyl substituted naltrexamine derivative, was identified as a peripheral MOR antagonist that may be used to treat OIC. To further explore its structure–activity relationship, a new series of NAP derivatives were designed, synthesized, and biologically evaluated. Among these derivatives, NFP and NYP significantly antagonized the antinociception effect of morphine. Whereas NAP acted mainly peripherally, its derivatives NFP and NYP actually can act centrally. Furthermore, NFP produced significantly lesser withdrawal symptoms than naloxone at similar doses. These results suggest that NFP has the potential to be a lead compound to treat opioid abuse and addiction.
Graphical Abstract
INTRODUCTION
Opioid receptors belong to the class A rhodopsin-like G-protein-coupled receptor (GPCR) family and may be classified into three main subtypes known as the μ opioid receptor (MOR), κ opioid receptor (KOR), δ opioid receptor (DOR).1–4 Among them, MOR is the main pharmacological target for opioid medications, such as morphine.5 After agonist binding, the conformation change of the MOR will lead to activation of the receptor, which may further result in opening of inwardly rectifying K+ (GIRK) channels, inhibition of the voltage-gated Ca2+ (VGCC) channels, and inhibition of adenyl cyclase activity.6–9 These changes may further induce a variety of changes in membrane morphology, neurons excitation, neurotransmitters release, and expression of genes through the second messenger systems.10 The behavioral effects of MOR activation include antinociception and addiction or abuse.11,12 Opioid addiction or abuse has become a global epidemic.13,14 Moreover, opioid abuse increases the prevalence of diseases such as HIV, tuberculosis, and hepatitis, especially for users who inject these drugs.15–19 It is reported that over 33 000 people died due to opioid overdose in the U.S. in 2015,20which has increased over 14 times in the past 20 years.21
Currently, there are two approaches to treat opioid addiction, which are detoxification and maintenance therapy by using opioid receptor agonists,22–24 partial agonists,25–27 or antagonists.28,29 Among them, opioid antagonists have been used as potential opioid addiction treatments for years.28,29 The synthesized opioid antagonists include peptide opioids and non-peptide opioids. CTOP (Figure 1) and CTAP (Figure 1) are cyclic peptides that are highly effective opioid antagonists with high MOR selectivity and metabolic stability, but their poor bioavailability limited their development as clinical therapeutics.30–32 Compared to peptide opioids, non-peptide opioid antagonists may have advantages of high bioavailability, easily crossing the blood–brain barrier (BBB), and good metabolic stability.32 The classical opioid antagonists used in opioid addiction treatments are naltrexone (Figure 1) and naloxone (Figure 1). As a result of their different half-lives, naloxone is a short-acting medication while naltrexone is used for long-term treatment.33,34 As opioid antagonists, they do not carry the side effects related to opioid agonists, such as addiction and respiratory suppression.35 However, high doses of naltrexone and naloxone may induce hepatotoxicity36,37 and cardiovascular and pulmonary problems.38 Besides, the interactions between these compounds and the DOR or KOR may induce mood changes39,40 or block transmission of neurotransmitters,41–43 though more evidence is still needed to further prove them.
Figure 1.

MOR selective antagonists.
The side effects associated with low selectivity MOR antagonists prompt researchers to develop new opioid antagonists. β-Funaltrexamine (β-FNA, Figure 1), a MOR antagonist, has similar potency to the MOR and KOR.44b Cyprodime (Figure 1), another MOR antagonist, has moderate selectivity to the MOR (Ki,δ/μ = 39 and Ki,κ/μ = 10).45,46 Clocinnamox (Figure 1) and methocinamox (Figure 1) are potent MOR antagonists but with similar binding affinity to all three opioid receptors.47 Meanwhile, it has been reported that activation of the KOR and further dynorphin release have a relationship with mood behaviors, such as drug-related withdrawal, aversion, and relapse induced by stress.48 Studies have shown that norBNI (Ki,κ = 1.8 nM) increased the symptoms of opioid withdrawal in rats, while in another study with KOR knockout mice precipitation of opioid withdrawal was not observed.49,50a De Marco et al. studied a new highly selective KOR partial agonist as anodyne with less harmful side effects.50b These studies suggest that KOR probably is a potential target for drug addiction and side effects related to mood behaviors. Collectively, highly selective and potent opioid antagonists are still extremely desirable.
Keeping this in mind, a number of new chemical entities have been designed and synthesized by modifying the structure of naltrexone to achieve highly selective opioid ligands.51–53 One such compound from our lab, 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(4′-pyridylcarboxamido)-morphinan (NAP) (Figure 1), a 6β-N-4′-pyridyl substituted naltrexamine derivative, was identified as a peripherally selective MOR antagonist (Ki,δ/μ ≈ 747, Ki,δ/κ ≈ 163; ED50 = 0.0088 mg/kg).54–56 To avoid diarrhea induced at high doses of NAP, the second generation NAP derivatives were designed and synthesized by modifying the C(6)-pyridyl ring system of NAP which was shown to interact with the MOR “address” region. NMP (17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-{[4′-(3′-methylpyridyl)]carboxamido}morphinan), a second generation NAP derivative, had improved pharmaco logical properties compared to NAP since it not only acted as a peripherally selective MOR antagonist but also improved motility of the gastrointestinal tract in vivo.57a,58 Further molecular modeling studies showed that the methyl group at 3′ position on the pyridyl group could reinforce the interactions between NMP and the “address” region of the MOR.58 On the basis of these previous study results, we sought to further explore the influence on binding affinity of 3′ position on the pyridyl ring of NAP to opioid receptors based on the “message–address” concept.57a,58 Thus, we designed, synthesized, and biologically evaluated a series of new compounds as the third generation NAP derivatives. Two new compounds, NFP and NYP, were characterized in vitro and in vivo as opioid receptor antagonists. Interestingly, these two compounds were found to be centrally acting with fewer withdrawal effects compared to naloxone, which was contrary to the peripherally acting NAP and NMP.
RESULTS AND DISCUSSION
Molecular Design.
Our molecular modeling study on NAP in the three opioid receptors demonstrated that the C(6)-pyridyl ring of NAP may strengthen the selectivity of NAP for the MOR over the DOR and KOR through aromatic stacking and hydrogen-bonding interactions.44 Meanwhile, a molecular dynamics simulation study of NMP in the MOR, DOR, and KOR showed that the 3′-methyl group on the pyridyl ring may reinforce the π–π stacking interaction between the aromatic pyridyl ring of NMP and W3187.35 of the MOR, which resulted in the high selectivity of NMP to the MOR over the DOR and KOR.58 Further modeling study results indicated that the characteristics of the substituents on the 3′ position of pyridyl ring of NAP derivatives, such as steric hindrance, electrostatic, and hydrophobic effects, may have signiffcant effect on the binding affinity and selectivity of NAP derivatives to the three opioid receptors.58 Therefore, in designing the third generation NAP derivatives, substitutions with different electronic, bulky, steric, and hydrophobic properties were introduced at the 3′ position of pyridyl ring (Figure 2).
Figure 2.

Structures of newly designed the third generation NAP derivatives.
Chemistry.
Twelve new NAP derivatives (Table 1) were synthesized. Compounds 1–6 were derivatives with electron-withdrawing groups at the 3′ position on the pyridyl ring with a corresponding increment in size of the substituents. Compounds 7–12 were derivatives with electron-donating groups at the 3′ position on the pyridyl ring with different bulkiness. For compounds 1–3, 9, and 12, the 3′-substituted pyridylcarboxylic acids were from commercial resources. For compounds 4–8, 10, and 11, the 3′-substituted pyridylcarbox ylic acids had to be prepared by ourselves as shown in Scheme 1 (Supporting Information).57b–d In addition, 6β-naltrexamine was synthesized as reported previously with a yield of 58% (Scheme 2).44a Then the 4′-pyridylcarboxylic acids were coupled with 6β-naltrexamine by EDCI/HOBt method.44,57a After that, the reaction crudes were treated with K2CO3 to get compounds 1–12 with reasonable yields (Scheme 2). All compounds were then transferred to hydrochloride salt and used for in vitro and in vivo studies.
Table 1.
Radioligand Binding Results at the MOR, KOR, and DOR and [35S]GTPγS Functional Assay at the MOR for Third Generation NAP Derivatives
| Compounds R | Ki (nM) | Selectivity | MOR [35S]GTPγS Binding | |||||
|---|---|---|---|---|---|---|---|---|
| MOR | DOR | KOR | δ/μ | κ/μ | EC50 (nM) | % Emax of DAMGO | ||
| NTXa | N/A | 0.26 ± 0.02 | 117.1 ± 8.9 | 5.15 ± 0.26 | 450 | 20 | ND | 7.75 ± 0.20 |
| NAPa | 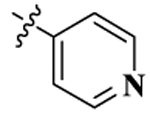 |
0.37 ± 0.07 | 277.5 ± 8.0 | 60.7 ± 5.6 | 747 | 163 | 1.14 ± 0.38 | 22.72 ± 0.84 |
| 1(NFP) | 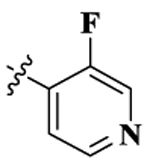 |
0.36 ± 0.02 | 156.93 ± 7.81 | 4.80 ± 0.26 | 435.8 | 13.3 | 1.20 ± 0.19 | 34.97 ± 3.07 |
| 2(NYP) | 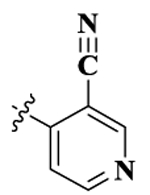 |
0.87 ± 0.09 | 46.29 ± 7.86 | 2.74 ± 0.47 | 53.2 | 3.1 | 1.28 ± 0.20 | 26.05 ± 0.74 |
| 3 | 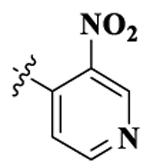 |
0.17 ± 0.02 | 95.61 ± 5.90 | 4.46 ± 0.11 | 562.4 | 26.5 | 0.53 ± 0.01 | 39.10 ± 2.57 |
| 4 | 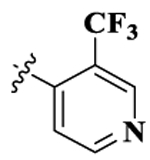 |
0.43 ± 0.01 | 61.79 ± 5.0 | 12.53 ± 0.89 | 143.7 | 29.1 | 1.38 ± 0.20 | 31.88 ± 3.39 |
| 5 | 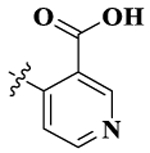 |
0.76 ± 0.08 | 55.58 ± 6.02 | 2.43 ± 0.19 | 73.2 | 3.2 | 1.83 ± 0.18 | 29.27 ± 1.42 |
| 6 | 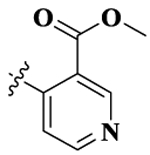 |
4.31 ± 0.29 | 137.31 ± 17.55 | 11.74 ± 1.15 | 31.9 | 2.7 | 13.40 ± 1.36 | 29.19 ± 0.84 |
| NMPb | 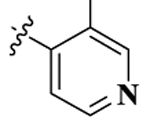 |
0.58 ± 0.25 | 273.6 ± 1.8 | 96.7 ± 12.2 | 470 | 166 | 1.52 ± 0.26 | 30.63 ± 0.55 |
| 7 | 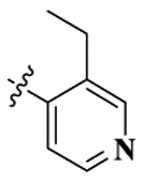 |
0.32 ± 0.05 | 42.86 ± 8.27 | 9.18 ± 1.36 | 134.1 | 28.8 | 2.29 ± 0.28 | 41.65 ± 3.14 |
| 8 | 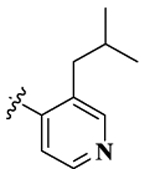 |
7.20 ± 0.81 | 148.75 ± 15.96 | 75.63 ± 3.87 | 20.7 | 10.5 | 19.70 ± 2.37 | 27.30 ± 0.25 |
| 9 | 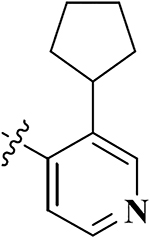 |
0.60 ± 0.06 | 33.99 ± 3.99 | 8.16 ± 1.17 | 56.7 | 13.7 | 1.85 ± 0.16 | 56.67 ± 3.14 |
| 10 | 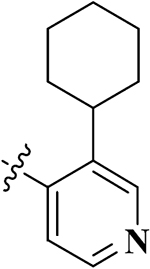 |
0.78 ± 0.09 | 11.24 ± 1.91 | 3.32 ±0.54 | 14.4 | 4.2 | 2.46 ± 0.30 | 51.59 ± 3.73 |
| 11 | 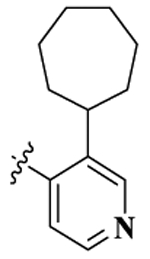 |
0.93 ± 0.13 | 14.97 ± 2.44 | 5.16 ± 0.54 | 16.1 | 5.6 | 5.11 ± 0.16 | 48.32 ± 3.34 |
| 12 | 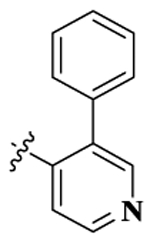 |
0.63 ± 0.05 | 44.83 ± 3.70 | 3.42 ± 0.38 | 71.1 | 5.4 | 1.46 ± 0.18 | 44.42 ± 3.12 |
Scheme 1.

Synthesis of 3′-Substituted Pyridylcarboxylic Acids for the Newly Designed NAP Derivatives
Scheme 2.

Synthesis Route of the Third Generation NAP Derivatives (Compounds 1–12)
In vitro binding and functional studies were first conducted to evaluate the binding affinity, selectivity, and efficacy of the compounds to the opioid receptors. In vivo studies were further conducted to characterize the behavioral profiles of these compounds.
In Vitro Radioligand Binding and MOR[35S]GTPγS Functional Studies.
The competition radioligand binding assay was used to determine the binding affinity and selectivity of the new NAP derivatives for the MOR, KOR, and DOR. Chinese hamster ovary (CHO) cell lines expressing mouse monocloned opioid receptors were used for this assay. The MOR was labeled with [3H]naloxone, while the KOR and DOR were both labeled with [3H]diprenorphine.
As shown in Table 1, all the compounds maintained subnanomolar binding affinity to the MOR except compounds 6 and 8 which showed nanomolar binding affinity. Compared to the first and second generation lead compounds, NAP (Ki,MOR = 0.37 ± 0.07 nM) and NMP (Ki,MOR = 0.58 ± 0.25 nM), most of the third generation derivatives still maintained high binding affinity to the MOR. This indicated that the substituent groups with different electronic properties and sizes at 3′ position on the pyridyl ring did not significantly affect the binding affinity of these derivatives to the MOR. However, it is worth noting that esterification of the carboxylic acid at the 3′ position (compound 6) and introduction of an isobutyl group at the 3′ position (compound 8) resulted in a significant reduction in their MOR binding affinity. As mentioned above, the structural features of substituents on the 3′ position of pyridyl ring, such as the steric hindrance, electrostatic, and hydrophobic effects, would significantly affect the binding affinity and selectivity of NAP derivatives to the three opioid receptors.58Compound 6 was an esterification product of compound 5. After esterification, the original ionic interactions between carboxylic moiety in compound 5 and basic residues in the binding pocket of the MOR would be weakened in compound 6 significantly so that the binding affinity of compound 6 to the MOR was reduced. Furthermore, the isobutyl group on the pyridyl ring of compound 8 may result in steric clashes or unfavorable interactions with hydrophilic residues in the binding pocket that would decrease the binding affinity of compound 8 to the MOR. Interestingly, compared to compound 8, compounds 9–12 carried even bulkier substituents on the pyridyl ring while still maintaining high binding affinities to the MOR at the subnanomolar level. One possible explanation of such an observation could be that the electrostatic and hydrophobic interactions induced by the much larger substituents on the pyridyl ring (compounds 9–11) may facilitate the compounds to interact with the binding pocket favorably in the MOR in a different binding mode than that of compound 8.
The selectivity of these compounds for the MOR over the DOR varied with different substituent groups at 3′ position on the pyridyl ring. In general, derivatives with electron-with drawing groups showed higher selectivity for the MOR over the DOR than derivatives with electron-donating groups. For example, compounds 1–6 showed more than 30-fold selectivity for the MOR over the DOR, especially for compounds 1 (NFP, δ/μ = 435.8) and 3 (δ/μ = 562.4). Both compounds had similar selectivity for the MOR over the DOR as NMP (δ/μ = 470). For the derivatives with electron- donating groups, compounds 7, 9, and 12 showed 50- to 134- fold selectivity for the MOR over the DOR while compounds 8, 10, and 11 showed 14- to 20-fold selectivity for the MOR over the DOR. Thus, it seemed that introduction of bulky and alkyl groups at position 3′ on the pyridyl ring may reduce their selectivity for the MOR over the DOR.
On the KOR, there was a significant improvement in the binding affinity of all the compounds compared to NAP and NMP except for compound 8 which showed similar binding affinity to the KOR as of NAP and NMP. Compounds 1–7 and 9–12 maintained one-digit nanomolar or two-digit nanomolar binding affinity to the KOR. Thus, collectively, with substitution at 3′ position on the pyridyl ring, the third generation NAP derivatives seemed to act as MOR/KOR dual-selective ligands.
The [35S]GTPγS functional assay was then conducted to determine the potency and relative efficacy of the synthesized compounds at the MOR. The potency was determined as the EC50 while the efficacy was determined as the Emax relative to DAMGO, a MOR full agonist. From these results, it seemed that all the compounds acted as partial agonists with low to moderate efficacy. Derivatives with electron-withdrawing groups (compounds 1–6) showed 20–40% MOR stimulation with one-digit nanomolar potency except compound 6. Meanwhile, derivatives with electron-donating groups (compounds 7–12) showed more than 40% efficacy with one- or two-digit nanomolar potencies except compound 8 which had the lowest potency.
In Vivo Tail Immersion Assay.
All the newly synthesized NAP derivatives were further assessed for their acute agonistic and/or antagonistic effects using the tail immersion assay in mice as previously reported.44,57 The baseline latency (control) was determined before the tested compound was injected into the mice. For the study of agonism, the time of tail immersion was 20 min (time that morphine’s antinociceptive effect starts to peak) after the test compound was injected (sc). For the study of antagonism, the test compound was given 5 min before morphine. The tail immersion test was then conducted 20 min after morphine was given. The percent maximum possible effects (%MPE) was calculated for each mouse using at least five mice per drug. Briefly, the derivatives were studied for their ability to produce antinociception or block the antinociception produced by morphine (10 mg/kg). As shown in Figure 3A, none of these new derivatives showed significant antinociception compared to morphine at the same dose (10 mg/kg). However, when they were tested as antagonists at the same dose (Figure 3B, 10 mg/kg), compounds 1 (NFP) and 2 (NYP) antagonized morphine’s antinociception effect significantly. The %MPEs of morphine (10 mg/kg) in the presence of NFP and NYP (10 mg/kg) were only 6.2 ± 2.4% and 7.8 ± 5.4%, respectively. The antagonism of NFP and NYP was shown to be dose-dependent (Figure 4A and Figure 4B,AD50 values of NFP and NYP were 2.82 (1.34–5.94) mg/kg with 95% confidence level (CL) and 1.75 (0.88–3.47) mg/kg with 95% CL, respectively). In contrast to NFP and NYP, other NAP derivatives did not significantly block morphine’s antinociception at the tested dose of 10 mg/kg. These results suggested that NFP and NYP had the ability to cross the blood–brain barrier (BBB) and block the analgesic effect of morphine in CNS.
Figure 3.

Tail immersion assay results for the third generation NAP derivatives as agonist (A) and antagonist (B) in the presence of morphine in mice at a single dose of 10 mg/kg. Morphine (10 mg/kg) and saline were used as positive and negative controls (n = 5, ∗∗∗ indicates P < 0.005).
Figure 4.

Dose–response studies of NFP (A) and NYP (B) as antagonists in mice. Morphine (10 mg/kg) and saline were used as positive and negative controls (n = 5, ∗∗∗ indicates P < 0.005).
It was intriguing to us that two of the third generation NAP derivatives, NFP and NYP, crossed the BBB and antagonized morphine’s antinociception, while NAP was identified as a P-glycoprotein substrate and failed to cross the BBB effectively. Apparently introduction of fluoro and nitrile groups on the pyridyl ring may play an important role in improving their CNS penetration. To be noted, about 5–15% of drugs on the market are fluorinated compounds,59,60 and it has been shown that these compounds have improved metabolic stability and physicochemical properties. Meanwhile fluorination seems to enhance the CNS penetration ability of non-CNS drugs and the efflux function of P-glycoprotein.61,55 This may help explain why NFP crossed the BBB while NAP did not.61 In addition, it has been reported that introducing nitrile groups may also enhance metabolic stability and improve hydrogen bonding interactions with residues in the orthosteric binding site of protein targets.62 Currently, there are at least 30 drugs containing nitrile groups on the market and more than 20 drug candidates containing nitrile groups under clinical trials.62 An example of such a drug is piritramide, an opioid analgesic that crosses the BBB and is used for postoperation pain.63
In Vivo Withdrawal Study.
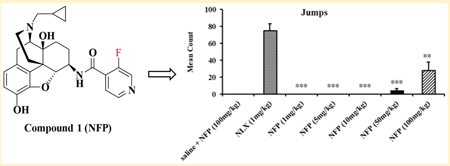
Opioid antagonists such as naloxone and naltrexone have been applied to reverse the effects of opioid agonists in cases of opioid overdose and in opioid addiction treatments. However, it has been shown that naloxone and naltrexone produce significant withdrawal symptoms when administered, which limits their use in opioid addiction treatments.64 Since NFP and NYP were identified as opioid antagonists in vivo, a withdrawal study was conducted using morphine-pelleted mice to determine whether these two compounds would produce withdrawal effects similar to naloxone. In this study, somatic symptoms of opioid withdrawal (shakes, jumps, and paw tremors) were quantified over a period of 20 min.55a As shown in Figure 5, NFP produced significantly fewer wet dog shakes, jumps, and paw tremors at 10 mg/kg than naloxone at 1 mg/kg. Meanwhile, at much higher doses (50 and 100 mg/kg) NFP produced wet dog shakes (Figure 5A) and paw tremors (Figure 5C) but not significantly different from those of naloxone at 1 mg/kg. More interestingly, at a high dose of 50 mg/kg NFP still produced significantly fewer jumps (Figure 5B) than naloxone at 1 mg/kg. NYP, on the other hand, produced significantly fewer wet dog shakes and jumps at 1 mg/kg than naloxone at 1 mg/kg, but at doses 5 mg/kg and higher it produced wet dog shakes (Figure 6A) and jumps (Figure 6B) similar to naloxone at 1 mg/kg. Also, NYP produced similar paw tremors (Figure 6C) at 5 mg/kg similar to naloxone at 1 mg/kg.
Figure 5.
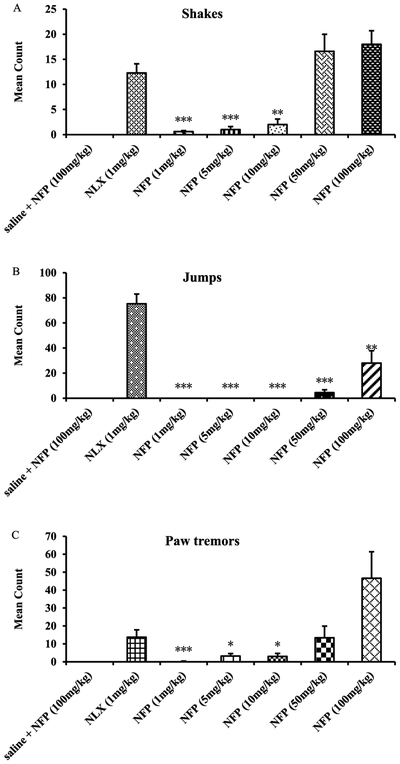
In vivo withdrawal study of NFP in morphine-pelleted mice (n = 5). The first column in each figure represents the withdrawal effects of NFP in placebo-pelleted mice, while the second to the seventh show the withdrawal effects of naloxone (NLX, 1 mg/kg) and NYP in morphine-pelleted mice (n = 5, ∗ means P < 0.05, ∗∗ means P < 0.005, ∗∗∗ indicates P < 0.0005).
Figure 6.
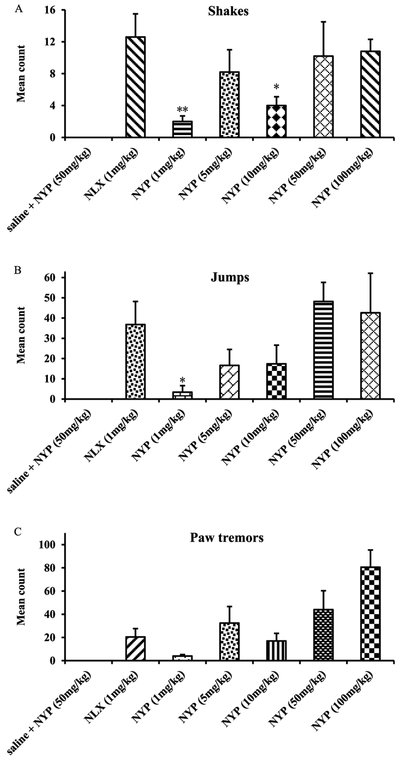
In vivo withdrawal study of NYP in morphine-pelleted mice (n = 5). The first column in each figure represents the withdrawal effects of NYP in placebo-pelleted mice, while the second to the seventh show the withdrawal effects of naloxone (NLX, 1 mg/kg) and NYP in morphine-pelleted mice (n = 5, ∗ indicates P < 0.05, ∗∗ indicates P < 0.005, ∗∗∗ indicates P < 0.0005).
It has been reported that naloxone can antagonize morphine’s antinociception effect potently (AD50 at 3.1 mg/kg) in a tail flick assay55b while it may induce significant withdraw symptom (e.g., Figures 5 and 6, at 1 mg/kg). Compared to naloxone, NFP can effectively across BBB entering into brain and reverse morphine’s antinociception effect at a similar potency level but showed much lesser withdrawal symptoms. These results are very encouraging since they suggest that NFP may have potential application in opioid addiction treatments with more compatible patient compliance profiles compared to naloxone.
We then further characterized NFP’s partial agonism and antagonism via concentration–effect curves for stimulation of [35S]GTPγS binding in mMOR-CHO cells by NFP and DAMGO alone (Figure 7A) in comparison to NFP curves in the presence of an EC90 DAMGO concentration (Figure 7B). Naltrexone was examined as a positive control for MOR antagonism (Figure 7B). Relative to DAMGO (normalized Emax = 100 ± 2.6% and EC50 = 32.5 ± 1.1 nM), NFP was a partial agonist with an Emax value of 34.9 ± 1.5% and EC50 value of 1.21 ± 0.19 nM, in agreement with our initial screening results (Table 1). In the presence of 0.3 μM DAMGO, NFP produced a concentration-dependent inhibition of DAMGO-stimulated [35S]GTPγS binding, achieving maximal inhibition (minimum) at 37.7 ± 1.5% of the maximal stimulation produced by DAMGO. Similarly, naltrexone produced concentration-dependent inhibition, with maximal inhibition at 12.8 ± 0.4% of maxima DMAGO stimulation. Functional Ki values were then calculated from these data. NFP and naltrexone inhibited DAMGO-stimulated [35S]GTPγS binding with Ki values of 0.86 ± 0.09 and 0.67 ± 0.05 nM, similar to their Ki values for MOR binding (Table 1). NFP had a relative efficacy that was approximately 35% of DAMGO and 3-fold that of naltrexone under these experimental conditions.
Figure 7.

MOR partial agonism and antagonism by NFP in mMORCHO cells. Ligand concentration–effect for stimulation of [35S]GTPγS binding in membranes from mMOR-CHO cells was determined alone (A) or in the presence of 0.3 μM DAMGO (B). Data are the mean ± SEM (n = 4–5) of the percentage of maximal stimulation (produced by a maximally effective DAMGO concentration of 0.3 μM).
Modeling Study.
As described above, NFP (Figure 8) showed dual-selective binding affinity to the MOR and KOR and displayed significant selectivity over the DOR (Ki,δ/μ = 435.8). Meanwhile, NFP acted as an MOR partial agonist with moderate efficacy (Table 1) and a KOR partial agonist with low efficacy (EC50 = 13.62 ± 1.57 nM, 26.71 ± 1.94% max effect of U50,488H) and a DOR antagonist (EC50 not determined, 7.22 ± 1.17% max effect of SNC80). However, the results of its in vivo studies displayed that NFP acted as an effective opioid antagonist in the CNS. To understand its selectivity profile to the opioid receptors, the antagonist-bound crystal structures of MOR (PDB code 4DKL),64 KOR (PDB code 4DJH),65 and DOR (PDB code 4EJ4)66 were adopted from Protein Data Bank, http://www.rcsb.org, and NFP was docked into the three receptors by GOLD 5.4.67,68 The binding poses of NFP with the highest CHEM-PLP scores from the docking studies were chosen as the optimal binding poses of the ligand in the MOR, KOR, and DOR.
Figure 8.

Chemical structure of NFP with atom notations from the docking studies.
Similar to the lead compound NAP, NFP was designed and synthesized according to the “message–address” concept where the “message” moiety (epoxymorphinan moiety) was assumed to determine its efficacy and the “address” moiety (pyridyl ring) to contribute to its selectivity (Figure 8).58 Comparing the binding poses of NFP in the MOR, KOR, and DOR with those of NAP, we found that the identical “message” moiety of NFP and NAP bound with the same domain of the MOR, KOR, and DOR through similar interactions with the conserved residues in the three receptors.58 Basically, the epoxymorphinan moiety formed hydrophobic interactions with the conserved residues M3.36, W6.48, and H6.52 (superscript numbers follow the Ballesteros–Weinstein numbering method for GPCRs69) and hydrogen bonding interactions with Y3.33. Meanwhile, an ionic interaction was formed between the conserved D3.32 and the nitrogen atom at 17-position of the epoxymorphinan moiety of NFP (Figure 9). Collectively, the interactions between the “message” moiety (epoxymorphinan moiety) of NFP and the conserved residues seemed to have no significant influence on the selectivity of NFP to the MOR, KOR, and DOR.
Figure 9.

Docking studies results of NFP in the MOR (a, PDB code 4DKL), KOR (b, PDB code 4DJH), and DOR (c, PDB code 4EJ4), respectively, with the highest CHEM-PLP scores: NFP atoms, carbon (magenta); key residue atoms in MOR, carbon (green); key residue atoms in KOR, carbon (orange); key residue atoms in DOR, carbon (light blue); oxygen (red); nitrogen (blue). The red dashed lines represent the electrostatic interactions. The yellow dashed lines represent the hydrogen bonds.
The fluorine atom on the pyridyl ring (“address” moiety) of NFP is a strong electron withdrawing group that would weaken consequence, the nitrogen atom on the pyridyl ring of NFP to keep a proton compared to the case of NAP and NMP.58 In consequence, the nitrogen atom on the pyridyl ring of NFP might form electrostatic interactions with the conserved residue E2295.35 in the MOR, D2235.35 in the KOR, and D2105.35 in the DOR. In addition, the pyridyl ring of NFP could also form hydrophobic interactions with the conserved hydrophobic residue LECL2 and FECL2 (Table S1). Thus, the “address” moiety (pyridyl ring) of NFP seemed to bind to the same domain (termed “address” domain) in the MOR, KOR, and DOR. However, several nonconserved residues located at this “address” domain (T218ECL2, T2255.31, and L2325.38 in the MOR, S211ECL2, Y2195.31, and M2265.38 in the KOR, and M199ECL2, S2065.31, and T2135.38 in the DOR, Figure 9) could also form direct interactions with the “address” moiety (pyridyl ring) of NFP.
As the distances shown in Figure 9, the electrostatic interaction between the nitrogen atom on the pyridyl ring of NFP and E2295.35 in the MOR was much stronger than those in the NFP/KOR and NFP/DOR complexes, which may help to explain the highest affinity of NFP to the MOR. For the NFP/KOR complex, the nonconserved Y2195.31 and S211ECL2 could also form hydrogen bond with the nitrogen atom on the pyridyl ring of NFP, which may facilitate the binding of NFP to the KOR. However, for the NFP/DOR complex, no additional interaction being observed between the “address” moiety of NFP and the residues located in the “address” domain of DOR except the possibly weakest electrostatic one from D2105.35 (Table S1). Overall, the different interactions between the “address” moiety of NFP and the “address” domain of the MOR, KOR, and DOR helped explain the binding affinity profile observed for NFP in the three receptors.
CONCLUSIONS
On the basis of previous studies from our group, the third generation 6β-N-4′-pyridyl substituted naltrexamine derivatives were designed, synthesized, and evaluated in both in vitro and in vivo assays. The in vitro competition assays showed that the third generation compounds bearing various substitutions at the 3′ position of pyridyl ring carried a different selectivity profile than NAP. In that, the third generation compounds were typically MOR/KOR dual-selective while NAP was highly MOR selective. Most of the new compounds retained their binding affinity to the MOR with subnanomolar level and one-digit nanomolar binding affinity to the KOR. Meanwhile, most of the compounds had low to moderate efficacies at the MOR with one-digit nanomolar potencies. Among these derivatives, NFP and NYP significantly antagonized the antinociception produced by morphine in a dose dependent fashion. The in vivo studies showed that NFP produced significantly fewer withdrawal symptoms than naloxone at similar doses. These findings suggest that NFP may serve as a lead compound to develop novel MOR/KOR dual selective ligand for treating opioid addiction and abuse.
EXPERIMENTAL SECTION
Chemistry.
General Methods.
Reagents were purchased from either Sigma-Aldrich or Alfa Aesar. TLC analyses were carried out on the Analtech Uniplate F254 plates. Chromatographic purification was conducted on silica gel column (230–400 mesh, Merck). 1H (400 MHz) and 13C (100 MHz) nuclear magnetic resonance (NMR) spectra were recorded at ambient temperature with tetramethylsilane as the internal standard on a Varian Mercury 400 MHz NMR spectrometer.
Melting points were recorded with OptiMelt automated melting point system (Fisher Scientific). IR spectra were obtained with Nicolet iS10 instrument (Thermo Scientific). Applied Bio Systems 3200 Q trap with a turbo V source for TurbolonSpray was used for MS analysis. HPLC analysis was performed by a Varian ProStar 210 system on Agilent Microsorb-MV 100–5 C18 (250 mm × 4.6 mm) at 210 nm with water/acetonitrile (25/75, 30/70 or 35/65, 0.01% TFA in water) at 0.8 mL/min over 30 min. The purity of the third generation NAP derivatives was determined by above analytical methods, and their purity was identified as ≥95%.
General Procedure.
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (2.5 equiv), hydrobenzotriazole (2.5 equiv), 4 Å molecular sieves, and trimethylamine (5 equiv) were added to a solution of the carboxylic acid (2.5 equiv) in DMF on an ice–water bath under N2 protection. After 30 min, a solution of 6β-naltrexamine (1 equiv) in DMF was added dropwise. The mixture was kept stirring overnight at room temperature and filtered the next day. The filtrate was then concentrated under reduced pressure to remove the solvent. The residue was dissolved in MeOH, and potassium carbonate (2 equiv) was added to the mixture. The reaction mixture was stirred overnight at room temperature. Next day, the mixture was concentrated and the residue was purified with silica gel column using methanol/dichloromethane (1% NH3H2O) as the solvent system to obtain the target compounds.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-fluoro-4′-pyridyl)acetamido]morphinan (compound 1, NFP).
Compound 1 was synthesized as shown in the general procedure with a yield of 48%. 1H NMR (400 MHz, CDCl3) δ 8.94 (s, 1 H), 8.74 (d, J = 8.08 Hz, 1 H), 8.58 (m, 1 H), 8.43 (m, 1 H), 7.46 (t, J = 5.6 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 6.45 (d, J = 8.1 Hz, 1H), 4.77 (s, 1 H), 4.45 (d, J = 8.1 Hz, 1 H), 3.52 (m, 1H), 2.90 (m, 2 H), 2.47 (m, 2 H), 2.21 (m, 2 H), 2.02 (m, 1 H), 1.87 (m, 1 H), 1.73 (m, 1 H), 1.48 (m, 1 H), 1.37 (m, 1 H), 1.21 (m, 1 H), 1.15 (m, 1 H), 0.73 (m, 1 H), 0.35 (m, 2 H), 0.01 (m, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 161.54, 156.46, 153.90, 146.23, 146.18, 142.06, 140.46, 139.10, 138.86, 131.24, 131.05, 130.93, 123.52, 123.23, 118.47, 117.12, 90.39, 69.52, 61.75, 58.38, 51.90, 47.06, 43.66, 30.32, 29.97, 24.33, 22.19, 9.22, 3.65, 3.52. 19F NMR (75 MHz, DMSO-d6) δ –192.29. IR (diamond, cm–1) Vmax 2942.19, 2829.33, 1655.64, 1529.02, 1487.78, 1454.25, 1416.41, 1322.93, 1238.29, 1204.29, 1186.88, 1152.58, 1131.78, 1100.14, 1034.82, 1017.27, 985.87, 942.11, 919.01, 898.28, 882.36, 799.14, 785.35, 763.19, 746.38, 667.00. Mass spectrum C26H28FN3O4 m/z calcd 465.2064, obsd 466.2159 [M + H]+. Mp 277.6–280.6 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-cyano-4′-pyridyl)acetamido]morphinan (Compound 2, NYP).
Compound 2 was synthesized as shown in the general procedure with a yield of 48%. 1H NMR (400 MHz, DMSO-d6) δ 9.15–9.13 (m, 2 H), 8.94 (s, 1 H), 7.92 (d, J = 3.6 Hz, 1 H), 7.44 (s, 1 H), 7.31 (s, 1 H), 7.18 (s, 1 H), 6.77 (d, J = 8.08 Hz, 1 H), 6.69 (d, J = 8.08 Hz, 1 H), 5.06 (d, J = 8.24 Hz, 1 H), 3.38–3.33 (m, 3 H), 3.11–3.06 (m, 3 H), 2.9 (m, 1 H), 1.93–1.90 (m, 1 H), 1.51–1.43 (m, 3 H), 1.10–1.09 (m, 1 H), 0.67 (m, 1 H), 0.60 (m, 1 H), 0.51 (m, 1 H), 0.42 (m, 1 H). 13C NMR (100 MHz, DMSO-d6) δ 167.45, 167.09, 156.63, 144.66, 142.37, 142.15, 139.45, 129.84, 125.95, 121.26, 120.30, 118.68, 117.50, 87.36, 70.14, 62.25, 57.32, 52.30, 47.03, 46.66, 30.59, 27.74, 23.65, 21.51, 6.32, 5.70, 3.24. IR (diamond, cm–1) Vmax 3111.05, 2087.54, 1721.23, 1626.30, 1501.34, 1467.64, 1371.61, 1322.77, 1079.34, 1031.03, 858.34, 731.40. Mass spectrum C27H28N4O4 m/z calcd 472.2111, obsd 474.2049 [M + 2H]+. Mp 222.8–224.5 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-nitro-4′-pyridyl)acetamido]morphinan (Compound 3).
Compound 3 was synthesized as shown in the general procedure with a yield of 52%. 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1 H), 8.94 (m, 1 H), 8.87 (m, 1 H), 7.53 (m, 1 H), 6.50 (d, J = 8.1 Hz, 1H), 6.44(d, J = 8.1 Hz, 1H), 4.35(d, J = 7.8 Hz, 1H), 3.42 (m, 1H), 2.90 (m, 2 H), 2.49 (m, 2 H), 2.22 (m, 2 H), 2.01 (m, 1 H), 1.88 (m, 1 H), 1.70 (m, 1 H), 1.57 (m, 1 H), 1.39 (m,1 H), 1.19 (m, 2 H), 0.73 (m, 1 H), 0.35 (m, 2 H), 0.01 (m, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 163.07, 154.58, 145.17, 142.56, 142.01, 140.49, 139.18, 131.16, 123.46, 122.82, 118.53, 117.14, 90.25, 69.52, 61.69, 58.32, 54.85, 52.11, 47.04, 43.66, 30.28, 29.84, 23.57, 22.18, 9.11, 3.68, 3.48. IR (diamond, cm–1) Vmax 3072.08, 2952.94, 2820.03, 1681.02, 1639.45, 1615.13, 1567.61, 1550.22, 1531.35, 1451.63, 1393.77, 1357.98, 1348.34, 1329.88, 1277.14, 1262.58, 1253.30, 1241.33, 1227.99, 1189.63, 1147.14, 1121.29, 1035.17, 984.65, 945.33, 931.99, 922.82, 915.19, 896.65, 882.54, 858,32, 828.95, 806.17, 763.88, 740.60, 732.83, 721.00, 672.69. Mass spectrum C26H28N4O4 m/z calcd 492.2009, obsd 493.2087 [M + H]+. Mp 266.46–269 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-trifluorometyl-4′-pyridyl)acetamido]morphinan (Compound 4).
Compound 4 was synthesized as shown in the general procedure with a yield of 51%. 1H NMR (400 MHz, DMSO-d6) δ 9.38 (s, 1 H), 9.04(s, 1 H), 9.02 (m, 1 H), 8.98 (m, 1 H), 7.56 (m, 1 H), 6.75 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 4.62(d, J = 8.1 Hz, 1H), 3.38 (m, 1H), 3.04 (m, 2 H), 2.83 (m, 2 H), 2.43 (m, 2 H), 2.16 (m, 1 H), 1.82 (m, 2H), 1.84 (m, 1 H), 1.62 (m, 1 H), 1.41 (m, 1 H), 0.86 (m, 1 H), 0.76 (m, 2 H), 0.53 (m, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 164.68, 154.10, 147.20, 143.25, 142.09, 141.39,129.55, 123.01, 122.22, 121.83, 121.17, 119.33, 117.98, 89.34, 69.64, 61.60, 60.67, 51.54, 45.67, 32.18, 29.30, 27.69, 22.97, 21.96, 13.95, 5.71, 5.05. 19F NMR (75 MHz, DMSO-d6) δ –57.80. IR (diamond, cm–1) Vmax: 3216.30, 2935.48, 2831.91, 1676.72, 1607.94, 1557.26, 1502.39, 1471.37, 1452.47, 1422.76, 1377.05, 1328.16, 1281.32, 1194.14, 1132.27, 1081.31, 1054.16, 1038.57, 1027.12, 999.69, 982.16, 927.80, 890.56, 858.47, 821.56, 785.26, 720.13, 691.36, 675.16. Mass spectrum C27H28F3N3O4 m/z calcd 515.2032, obsd 516.2089 [M + H]+. Mp 224.9–227.5 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-carboxyl-4′-pyridyl)acetamido]morphinan (Compound 5).
Compound 5 was synthesized as shown in the general procedure with a yield of 23%. 1H NMR (400 MHz, DMSO-d6) δ 9.14 (d, J = 0.84 Hz, 1 H), 9.12 (d, J = 4.76 Hz, 1 H), 8.99 (s, 1 H), 7.91 (d, J = 1.08 Hz, 1 H), 7.90 (d, J = 0.92 Hz, 1 H), 6.62–6.52 (m, 2 H), 4.99 (d, J = 8.08 Hz, 1 H),4.92 (s, 1 H), 3.91–3.82 (m, 1 H), 3.16 (d, J = 5.24 Hz, 1 H), 3.03 (t, 2 H), 2.97 (s, 1 H), 2.63–2.60 (m, 1 H), 2.57–2.53 (m, 1 H), 2.40–2.30 (m, 2 H), 2.24–2.18 (m, 1 H), 2.02–1.95 (m, 1 H), 1.59–1.55 (m, 1 H), 1.18–1.44 (m, 1 H), 1.41–1.34 (m, 1 H), 1.26–1.23 (m, 1 H), 0.87–0.82 (m, 1 H), 0.48–0.46 (m, 2 H), 0.13–0.11 (m, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 167.65, 167.25, 156.53, 144.44, 141.89, 141.13, 139.43, 125.90, 119.61, 117.53, 88.10, 70.04, 62.06, 58.80, 52.80, 49.03, 48.08, 44.14, 31.23, 22.66, 4.26, 4.00. IR (diamond, cm–1) Vmax 3345.73, 3039.98, 2478.91, 2079.72, 1782.60, 1725.96, 1651.70, 1630.27, 1614.51, 1556.21, 1505.72, 1455.55, 1398.02, 1361.26, 1263.68, 728.87, 708.62. Mass spectrum C27H29N3O6 m/z calcd 491.2056, obsd 474.2033 [M-CH3]+. Mp 271.2–273.7 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-methylformate-4′-pyridyl)acetamido]morphinan (Compound 6).
Compound 6 was synthesized as shown in the general procedure with a yield of 32%. 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1 H), 9.12 (s, 1 H), 9.08 (s, 1 H), 9.00 (d, J = 3.68 Hz, 1 H),7.91 (d, J = 3.68 Hz, 1 H), 7.76 (d, J = 3.52 Hz, 1 H), 7.31 (s, 1 H), 7.26 (d, d, J = 8.24 Hz, 1 H), 7.00 (d, J = 8.24 Hz, 1 H), 5.16 (7.26 (d, J = 8.24 Hz, 1 H)), 3.83 (s, 3H), 3.55 (s, 1 H), 3.50 (s, 1H), 3.36–3.35 (m, 1 H), 3.27–3.24 (s, 1 H), 3.23 (s, 2 H), 3.16–3.12 (m, 1 H), 2.95 (m, 1 H), 2.66–2.63 (m, 1 H), 1.99–1.95 (m, 1 H), 1.57–1.44 (m, 3 H), 1.23–1.19 (m, 1 H), 1.13 (m, 1 H), 0.69 (m, 1 H), 0.62 (m, 1 H), 0.54 (m, 1 H), 0.45 (m, 1 H). 13C NMR (100 MHz, DMSO-d6) δ 167.04, 165.51, 163.83, 156.57, 154.31, 150.66, 146.23, 144.61, 139.14, 134.01, 131.22, 130.14, 123.75, 122.67, 120.93, 117.47, 89.86, 70.03, 61.97, 57.38, 53.57, 52.41, 49.14, 46.62, 30.53, 27.58, 24.12, 21.11, 6.29, 5.70, 3.29. IR (diamond, cm–1) Vmax 3554.62, 2953.39, 2088.05, 1716.47, 1633.27, 1557.57, 1489.97, 1446.28, 1367.76, 1290.11, 1263.40, 1234.61, 1193.08, 1152.84, 1128.11, 1101.15, 1076.56, 1032.71, 995.14, 960.71, 938.56, 914.77, 892.11, 855.03, 834.94, 797.66, 780.76, 732.58, 685.21, 662.07. Mass spectrum m/z calcd C28H31N3O6 505.2213, obsd 506.2227 [M + H]+. Mp 210.5–212.3 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-ethyl-4′-pyridyl)acetamido]morphinan (Compound 7).
Compound 7 was synthesized as shown in the general procedure with a yield of 61%. 1H NMR (400 MHz, DMSO-d6) δ 9.00(s, 1 H), 8.50 (m, 2 H), 7.25 (m, 1 H), 6.70 (d, J = 8.1 Hz, 1H), 6.54(d, J = 8.1 Hz, 1H), 4.53(d, J = 7.8 Hz, 1H), 3.61 (m, 1H), 3.01 (m, 2 H), 2.74 (q, J = 7.6 Hz, 2H), 2.56 (m, 2 H), 2.29 (m, 2 H), 2.12 (m, 1H), 2.00 (m, 1H), 1.82 (m, 1 H), 1.62 (m, 1 H), 1.48 (m, 1 H), 1.34 (m,1 H), 1.25 (m, 1 H), 1.16 (t, J = 7.6 Hz, 3H), 0.84 (m, 1 H), 0.47 (m, 2 H), 0.12 (m, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 166.80, 150.49, 147.32, 143.38, 142.17, 140.54, 135.81, 131.33, 123.50, 120.89, 118.39, 117.10, 90.38, 69.58, 61.74, 58.37, 54.86, 51.53, 47.05, 43.68, 30.09, 24.32, 23.14, 22.19, 15.54, 9.22, 3.65, 3.51. IR (diamond, cm–1) Vmax 3307.45, 3084.08, 2946.81, 2931.57, 2822.35, 1659.73, 1592.75, 1530.90, 1503.18, 1471.36, 1454.36, 1431.17, 1404.97, 1368.65, 1322.47, 1260.16, 1239.39, 1227.39, 1190.27, 1154.54, 1128.18, 1114.19, 1096.93, 1082.03, 1059.66, 1036.40, 1028.24, 974.96, 932.47, 922.72, 915.97, 899.73, 877.40, 855.23, 818.18, 799.14, 784.30, 761.82, 744.37, 709.36, 675.07. Mass spectrum m/z calcd C28H33N3O4 475.2471, obsd 476.2539 [M + H]+. Mp 238.6–241.3 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-isobutyl-4′-pyridyl)acetamido]morphinan (Compound 8).
Compound 8 was synthesized as shown in the general procedure with a yield of 48%. 1H NMR (400 MHz, CDCl3) δ 8.61 (d, J = 5.04 Hz, 1 H), 8.58 (s, 1 H), 8.48–8.46 (m, 2 H), 7.86 (d, J = 5.04 Hz, 1H), 7.23 (d, J = 4.92 Hz, 1 H), 6.96 (d, J = 8.16 Hz, 1 H), 6.74 (d, J = 8.20, 1H), 4.56 (d, J = 4.80 Hz, 1 H), 4.27–4.21(m, 1H), 3.15 (s, 1 H), 3.11 (s, 1 H), 2.90–2.86 (m, 1 H), 2.68–2.66 (m, 2 H), 2.41–2.39 (m, 1 H), 2.27–2.25 (m, 1 H), 1.96–1.85 (m, 1 H), 1.65–1.63 (m, 1 H), 1.26 (s, 1 H), 0.93 (d, J = 2.20 Hz, 2 H), 0.91 (d, J = 2.16 Hz, 2 H), 0.84 (d, J = 7.96 Hz, 6 H), 0.57–0.55 (m, 2 H), 0.16–0.15 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 167.37, 164.01, 153.50, 152.53, 148.14, 143.75, 137.28, 136.07, 134.23, 132.66, 132.14, 123.90, 122.76, 120.83, 119.34, 92.94, 69.91, 62.25, 59.63, 49.70, 47.07, 43.86, 40.21, 39.46, 30.39, 30.21, 28.70, 23.25, 22.55, 4.19, 3.94, 0.13. IR (diamond, cm–1) Vmax 3212.54, 3055.22, 2957.95, 2869.52, 2580.83, 2089.08, 1756.35, 1659.98, 1633.88, 1446.95, 1257.27, 1234.91, 1057.99, 851.95, 772.04, 666.71. Mass spectrum m/z calcd C30H37N3O4 503.2784, obsd 504.2653 [M + H]+. Mp 195.5–197.8 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-cyclopentanyl-4′-pyridyl)acetamido]morphinan (Compound 9).
Compound 9 was synthesized as shown in the general procedure with a yield of 58%. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1 H), 8.43 (m, 1 H), 7.05 (m, 1 H), 7.03 (m, 1H), 6.74 (d, J = 8.1 Hz, 1H), 6.58 (2 d, J = 8.1 Hz, 1H), 4.45 (d, J = 7.8 Hz, 1H), 3.99 (m, 1H), 3.33 (m, 1 H), 3.06 (m, 2H), 2.63 (m, 2 H), 2.36 (m, 2 H), 2.18 (m, 2H), 2.08 (m, 2H), 1.83 (m, 3H), 1.65 (m, 16 H), 1.49 (m, 2 H), 0.82 (m, 1 H), 0.52 (m, 2 H), 0.12 (m, 2 H). 13C NMR (100 MHz, MeOH-d4) δ 170.58, 149.17, 147.62, 146.33, 143.88, 142.80, 140.26, 122.25, 120.57, 119.18, 92.20, 71.59, 64.10, 59.53, 53.32, 41.77, 35.59, 35.51, 31.29, 26.59, 25.02, 24.07, 5.35, 3.80. IR (diamond, cm–1) Vmax 3135.82, 3042.97, 2956.45, 2868.49, 1635.34, 1543.77, 1504.22, 1451.51, 1406.10, 1324.84, 1242.99, 1156.26, 1124.74, 1060.06, 1032.41, 1012.61, 986.31, 919.54, 855.36, 799.44, 781.28, 747.15, 677.37. Mass spectrum C31H37N3O4 m/z calcd 515.2784, obsd 516.2896 [M + H]+. Mp 231.1–232.8 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-cyclohexanyl-4′-pyridyl)acetamido]morphinan (Compound 10).
Compound 10 was synthesized as shown in the general procedure with a yield of 51%. 1H NMR (400 MHz, CDCl3) δ 8.66(s, 1 H), 8.46 (m, 1 H), 7.11 (m, 1 H), 6.89 (m, 1H), 6.74 (d, J = 8.1 Hz, 1H), 6.58(d, J = 8.1 Hz, 1H), 4.43(d, J = 7.8 Hz, 1H), 4.06 (m, 1H), 3.19 (m, 1 H), 2.91(m, 2H), 2.63 (m, 2 H), 2.36 (m, 2 H), 2.18 (m, 2H), 1.85 (m, 5H), 1.74 (m, 1 H), 1.66 (m, 2 H), 1.45 (m, 5H), 1.26 (m,3 H), 0.82 (m, 1 H), 0.52 (m, 2 H), 0.12 (m, 2 H). 13C NMR (100 MHz, MeOH-d4) δ 172.30, 149.20, 147.66, 146.05, 143.84, 142.16, 141.31, 132.55, 125.43, 122.40, 120.11, 118.63, 92.74, 71.79, 63.78, 60.27, 53.44, 45.34, 40.59, 35.16, 34.94, 31.83, 31.36, 27.84, 27.73, 27.05, 25.56, 23.59, 10.25, 4.48, 4.20. IR (diamond, cm–1) Vmax 3269.93, 2925.57, 2850.60, 1644.45, 1537.65, 1504.66, 1450.26, 1411.24, 1374.41, 1324.67, 1240.14, 1188.89, 1149.13, 1130.35, 1095.73, 1035.49, 984.59, 918.19, 881.92, 856.04, 828.64, 798.39, 785.52, 762.60, 745.35, 728.43, 702.73, 677.85. Mass spectrum C32H39N3O4 m/z calcd 529.2941, obsd 530.3025 [M +H]+. Mp 248.6–249.7 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-cyclohentanyl-4′-pyridyl)acetamido]morphinan (Compound 11).
Compound 11 was synthesized as shown in the general procedure with a yield of 65%. 1H NMR (400 MHz, CDCl3) δ 8.58(s, 1 H), 8.43 (m, 1 H), 7.07 (m, 1 H), 6.89 (m, 1H), 6.74 (d, J = 8.1 Hz, 1H), 6.58(d, J = 8.1 Hz, 1H), 4.44 (d, J = 7.8 Hz, 1H), 4.04 (m, 1H), 3.06(m, 3H), 2.63 (m, 2 H), 2.36 (m, 2 H), 2.18 (m, 2H), 1.47–1.92 (m, 17H), 0.83 (m, 1 H), 0.52 (m, 2 H), 0.13 (m, 2 H). 13C NMR (100 MHz, MeOH-d4) δ 170.34, 149.43, 147.35, 145.13, 143.83, 143.39, 142.18, 132.54, 125.41, 122.28, 120.11, 118.62, 92.74, 71.79, 63.78, 60.27, 53.43, 45,35, 42.38, 37.53, 37.42, 31.82, 31.36, 28.74, 28.73, 28.63, 28.47, 25.64, 23.59, 10.23, 4,48, 4.19. IR (diamond, cm–1) Vmax 3269.95, 3074.45, 2923.19, 2853.17, 1644.22, 1536.48, 1504.37, 1453.20, 1411.13, 1374.26, 1324.43, 1239.86, 1186.87, 1154.29, 1129.40, 1095.41, 1035.39, 984.37, 918.44, 896.79, 882.17, 856.61, 829.74, 798.64, 785.78, 762.42, 745.58, 728.45, 703.20, 676.77. Mass spectrum C33H41N3O4 m/z calcd 543.3097, obsd 544.3253 [M + H]+. Mp 240.1–242.5 °C.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-phenyl-4′-pyridyl)acetamido]morphinan (Compound 12).
Compound 12 was synthesized as shown in the general procedure with a yield of 60%. 1H NMR (400 MHz, DMSO-d6) δ 9.06(s, 1 H), 8.66(s, 1 H), 8.65 (m, 2 H), 7.48 (m, 4 H), 7.37 (m, 2H), 6.60 (d, J = 8.1 Hz, 1H), 6.51(d, J = 8.1 Hz, 1H), 4.43(d, J = 7.8 Hz, 1H), 3.48 (m, 1H), 3.17 (m, 1 H), 2.96 (m, 2H), 2.56 (m, 2 H), 2.30 (m, 2 H), 2.07 (m, 1H), 1.96 (m, 1H), 1.82 (m, 1 H), 1.54 (m, 1 H), 1.39 (m, 1 H), 1.23 (m,3 H), 0.83 (m, 1 H), 0.46 (m, 2 H), 0.11 (m, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 166.73, 150.11, 148.48, 143.55, 142.12, 140.48, 136.30, 133.45, 131.25, 128.25, 128.49, 127.80, 123.45, 121.48, 118.56, 117.07, 90.18, 69.51, 61.69, 58.33, 51.59, 48.57, 46.95, 43.67, 29.87, 23.71, 22.16, 9.18, 3.66, 3.49. IR (diamond, cm–1) Vmax 3267.19, 2926.62, 1759.45, 1655.72, 1537.27, 1490.98, 1477.79, 1445.55, 1399.94, 1329.53, 1277.45, 1236.21, 1200.83, 1154.28, 1131.52, 1095.64, 1068.52, 1037.44, 1006.16, 984.61, 934.19, 914.70, 880.93, 852.37, 793.70, 758.91, 699.07, 671.71. Mass spectrum C32H33N3O4 m/z calcd 523.2471, obsd 524.2552 [M + H]+. Mp 232.4–235.2 °C.
Biological Evaluation.
Drugs and Chemicals.
Morphine (morphine sulfate pentahydrate salt) and naloxone were procured from the National Institute of Drug Abuse (NIDA), Bethesda, MD, and then made into a 10 μM stock solution by dissolving in distilled water, which was further diluted to the desired concentrations. All other reagents were purchased from either Sigma-Aldrich or Thermo Fisher.
Animals.
Male Swiss Webster mice (25–30 g, 7–8 weeks, Harlan Laboratories, Indianapolis, IN) were raised in animal care quarters and maintained at room temperature on light–dark cycle. Food and water were available ad libitum. Protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Commonwealth University and complied with the recommendations of the IASP (International Association for the Study of Pain).
Competitive Radioligand Binding and [35S]GTPγS Functional Studies.
In the competition binding assay, 30 μg of membrane protein was incubated with the corresponding radioligand in the presence of different concentrations of test compounds in TME buffer (50 mM Tris, 3 mM MgCl2, and 0.2 mM EGTA, pH 7.4). The bound radioligand was separated by filtration using the Brandel harvester. Specific (i.e., opioid receptor-related) binding at the MOR, KOR, and DOR was determined as the difference in binding obtained in the absence and presence of 5 μM naltrexone, U50,488, and SNC80, respectively. The IC50 values were determined and converted to Ki values using the Cheng–Prusoff equation.
In the [35S]GTPγS functional assay, 10 μg of MOR-CHO membrane protein was incubated with 10 μM GDP, 0.1 nM [35S]GTPγS, assay buffer (TME + 100 mM NaCl), and varying concentrations of the compounds under investigation for 90 min in a 30 °C water bath. Nonspecific binding was determined with 20 μM unlabeled GTPγS. 3 μM DAMGO was included in the assay as maximally effective concentration of a full agonist for the MOR. All assays were determined in duplicate and repeated at least 4 times. Percent DAMGO-stimulated [35S]GTPγS binding was defined as (net-stimulated binding by ligand/net-stimulated binding by 3 μM DAMGO) × 100.
Data Analysis of [35S]GTPγS Binding Assays.
All samples were assayed in duplicate and repeated at least four times for a total of ≥4 independent determinations. Results were reported as mean values ± SEM. Concentration–effect curves were fit by nonlinear regression to a one-site binding model, using GraphPad Prism software, to determine EC50 and Emax values. IC50 values were obtained from Hill plots, analyzed by linear regression using GraphPad Prism software. Binding Ki values were determined from IC50 values using the Cheng–Prusoff equation: Ki = IC50/[1 + ([L]/KD)], where [L] is the concentration of competitor and KD is the KD of the radioligand.
Tail Immersion Test.
Swiss Webster mice (5 male mice for each group, 7–8 weeks, ENVIGO) were used for this experiment. Water bath temperature was maintained at 56 ± 0.1 °C. The baseline latency (control) was determined before the test compound was injected subcutaneously (sc) into the mice. The average baseline latency obtained for this experiment was 3.0 ± 0.1 s, and only mice with a baseline latency of 2–4 s were used. For the agonism study, tail immersion was conducted 20 min (time that morphine’s anti-nociceptive effect starts to peak) after the test compound was injected. To prevent tissue damage, a 10 s maximum cutoff time was imposed. Antinociception response was calculated as the percentage maximum possible effect (%MPE), where %MPE = [(test – control)/(10 – control)] × 100. For the antagonism study, the test compound was given 5 min before morphine. Tail immersion was then conducted 20 min after giving morphine. %MPE was calculated for each mouse using at least five mice per drug. AD50 values were calculated using the least-squares linear regression analysis followed by calculation of 95% confidence interval by Bliss method.
Withdrawal Assay.
Swiss Webster mice (5 male mice for each group, 7–8 weeks, ENVIGO) were used for the withdrawal study. A 75 mg morphine pellet was implanted into the base of the neck of the mice, and the mice were then given time to recover in their home cages. Before the test began, 30 min was given to the mice for habituation to an open-topped, square, clear Plexiglas observation chamber (26 × 26 × 26 cm3) with lines partitioning the bottom into quadrants. All drugs and test compounds were administered by subcutaneous injection (sc). The withdrawal was precipitated 72 h from pellet implantation with naloxone (1 mg/kg, sc) and the test compounds at different doses. Withdrawal commenced within 3 min after antagonist administration. Escape jumps, paw tremors, and wet dog shakes were quantified by counting their occurrences over 20 min for each mouse (five mice per group). The data is shown as the mean ± SEM.
Statistical Analysis.
One-way ANOVA followed by the post hoc Dunnett test was performed to assess significance using Prism 6.0 software (GraphPad software, San Diego, CA).
Docking Study.
The crystal structures of the MOR (PDB code 4DKL), KOR (PDB code 4DJH), and DOR (PDB code 4EJ4) were obtained from the Protein Data Bank at http://www.rcsb.org. Sybyl-X 2.058 was used to add hydrogen atoms to each receptor and to sketch NFP (Tripos Inc., St. Louis, MO). NFP was then assigned Gasteiger–Hückel charges and energy minimized to a gradient of 0.05 under the TAFF in Sybyl-X 2.0.58
GOLD 5.4 with default settings was used to conduct the docking study. The atoms within 10 Å of the γ-carbon atom of D3.32 of the MOR, KOR, and DOR were defined as the binding site in the three receptors. Automated docking was conducted with a distance constraint of 4 Å between the nitrogen atom at the 17′ position of the epoxymorphinan skeleton and the conserved D3.32. In addition, a hydrogen bond constraint was applied between NFP’s dihydrofuran oxygen and the phenolic oxygen of the conserved Y3.33. The highest scored solutions (CHEM-PLP) were selected and merged into the receptor to obtain the optimal binding poses of NFP in the MOR, KOR, and DOR. After docking, the binding poses were energy minimized in Sybyl-X 2.0.7 with 5000 iterations under TAFF to remove the clashes and strain energy between NFP and the three receptors.70
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to NIDA Drug Supply Program for providing the free base of naltrexone. The work is partially supported by NIH/NIDA Grants DA024022 and DA044855. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
ABBREVIATIONS USED
- OIC
opioid-induced constipation
- GPCR
G-protein-coupled receptor
- MOR
μ opioid receptor
- KOR
κ opioid receptor
- DOR
δ opioid receptor
- NOP
nociception opioid peptide
- GIRK
inwardly rectifying K+
- VGCC
voltage-gated Ca2+
- BBB
blood–brain barrier
- CHO
Chinese hamster ovary
- NFP
compound 1
- NYP
compound 2
- NMR
nuclear magnetic resonance
- sc
subcutaneous injection
- NIDA
National Institute of Drug Abuse
- IACUC
Institutional Animal Care and Use Committee
- NLX
naloxone
- CL
confidence level
- %MPE
percentage maximum possible effect
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01158.
Molecular formula strings and some data (CSV)
Coordinates information for structure representation (PDB)
Coordinates information for structure representation (PDB)
Coordinates information for structure representation (PDB)
NMR and IR spectra for 4′-pyridylcarboxylic acids prepared in our lab, HPLC spectra for all newly synthesized NAP derivatives, and the shortest distances between atoms on critical amino acid residues and atoms on the ligands of NFP in the MOR, KOR, and DOR (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Pasternak GW Opioids and their receptors: are we there yet? Neuropharmacology 2014, 76 (Part B), 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McNicol E; Horowicz-Mehler N; Fisk RA; Bennett K; Gialeli-Goudas M; Chew PW; Lau J; Carr D Americal Pain Society. Management of opioid side effects in cancer-related and chronic noncancer pain: a systematic review. J. Pain 2003, 4, 231–256. [DOI] [PubMed] [Google Scholar]
- (3).Shang Y; Filizola M Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur. J. Pharmacol 2015, 763 (Part B), 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nutt DJ The role of the opioid system in alcohol dependence. J. Psychopharmacol 2014, 28, 8–22. [DOI] [PubMed] [Google Scholar]
- (5).Remesic M; Hruby VJ; Porreca F; Lee YS Recent advances in the realm of allosteric modulators for opioid receptors for future therapeutics. ACS Chem. Neurosci 2017, 8, 1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chakrabarti S; Prather PL; Yu L; Law PY; Loh HH Expression of the mu-opioid receptor in CHO cells: ability of mu opioid ligands to promote alpha-azidoanilido[32P]GTP labeling of multiple G protein alpha subunits. J. Neurochem 1995, 64, 2534–2543. [DOI] [PubMed] [Google Scholar]
- (7).Miyake M; Christie MJ; North RA Single potassium channels opened by opioids in rat locus ceruleus neurons. Proc. Natl. Acad. Sci. U. S. A 1989, 86, 3419–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ortiz-Miranda SI; Dayanithi G; Coccia V; Custer EE; Alphandery S; Mazuc E; Treistman S; Lemos JR mu-Opioid receptor modulates peptide release from rat neurohypophysial terminals by inhibiting Ca2+ influx. J. Neuroendocrinol 2003, 15, 888–894. [DOI] [PubMed] [Google Scholar]
- (9).Barchfeld CC; Maassen ZF; Medzihradsky F Receptor- related interactions of opiates with PGE-induced adenylate cyclase in brain. Life Sci. 1982, 31, 1661–1665. [DOI] [PubMed] [Google Scholar]
- (10).Thorsell A The μ-opioid receptor and treatment response to naltrexone. Alcohol Alcohol 2013, 48, 402–408. [DOI] [PubMed] [Google Scholar]
- (11).Herz A Multiple opiate receptors and their functional significance. J. Neural Transm. Suppl 1983, 18, 227–233. [PubMed] [Google Scholar]
- (12).Sadée W; Wang Z Agonist induced constitutive receptor activation as a novel regulatory mechanism. Mu receptor regulation. Adv. Exp. Med. Biol 1995, 373, 85–90. [DOI] [PubMed] [Google Scholar]
- (13).Degenhardt L; Bucello C; Calabria B; Nelson P; Roberts A; Hall W; Lynskey M; Wiessing L; Mora ME; Clark N; Thomas J; Briegleb C; McLaren J What data are available on the extent of illicit drug use and dependence globally? Results of four systematic reviews. Drug Alcohol Depend. 2011, 117, 85–101. [DOI] [PubMed] [Google Scholar]
- (14).Rehm J; Mathers C; Popova S; Thavorncharoensap M; Teerawattananon Y; Patra J Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [DOI] [PubMed] [Google Scholar]
- (15).Salomon N; Perlman DC; Friedmann P; Ziluck V; Des Jarlais DC Prevalence and risk factors for positive tuberculin skin tests among active drug users at a syringe exchange program. Int. J. Tuberc. Lung Dis 2000, 4, 47–54. [PubMed] [Google Scholar]
- (16).Nelson PK; Mathers BM; Cowie B; Hagan H; Des Jarlais D; Horyniak D; Degenhardt L Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet 2011, 378, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mathers BM; Degenhardt L; Phillips B; Wiessing L; Hickman M; Strathdee SA; Wodak A; Panda S; Tyndall M; Toufik A; Mattick RP 2007 Reference group to the UN on HIV and injecting drug use. Global epidemiology of injecting drug use and HIV among people who inject drugs: a systematic review. Lancet 2008, 372, 1733–1745. [DOI] [PubMed] [Google Scholar]
- (18).Paulozzi LJ; Budnitz DS; Xi Y Increasing deaths from opioid analgesics in the United States. Pharmacoepidemiol. Pharmacoepidemiol. Drug Saf 2006, 15, 618–627. [DOI] [PubMed] [Google Scholar]
- (19).Degenhardt L; Bucello C; Mathers B; Briegleb C; Ali H; Hickman M; McLaren J Mortality among regular or dependent users of heroin and other opioids: a systematic review and meta- analysis of cohort studies. Addiction 2011, 106, 32–51. [DOI] [PubMed] [Google Scholar]
- (20).Rudd RA; Seth P; David F; Scholl L Increases in drug and opioid-involved overdose deaths - United States, 2010–2015. MMWR. Morb. Mortal. Wkly. Rep 2016, 65, 1445–1452. [DOI] [PubMed] [Google Scholar]
- (21).Manchikanti L; Helm S, 2nd.; Fellows B; Janata JW; Pampati V; Grider JS; Boswell MV Opioid epidemic in the United States.Pain Physician 2012, 15, ES9–E38. [PubMed] [Google Scholar]
- (22).Bart G Maintenance medication for opiate addiction: the foundation of recovery. J. Addict. Dis 2012, 31, 207–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Stotts AL; Dodrill CL; Kosten TR Opioid dependence treatment: options in pharmacotherapy. Expert Opin. Pharmacother 2009, 10, 1727–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).O’Connor PG Methods of detoxification and their role in treating patients with opioid dependence. J. A. M. A 2005, 294, 961–963. [DOI] [PubMed] [Google Scholar]
- (25).Martell BA; Arnsten JH; Krantz MJ; Gourevitch MN Impact of methadone treatment on cardiac repolarization and conduction in opioid users. Am. J. Cardiol 2005, 95, 915–918. [DOI] [PubMed] [Google Scholar]
- (26).Bart G CSAT’s QT interval screening in methadone report: outrageous fortune or sea of troubles? J. Addict. Dis 2011, 30, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ducharme S; Fraser R; Gill K Update on the clinical use of buprenorphine in opioid-related disorders. Can. Fam. Physician 2012, 588, 37–41. [PMC free article] [PubMed] [Google Scholar]
- (28).Saidak Z; Blake-Palmer K; Hay DL; Northup JK; Glass M Differential activation of G-proteins by mu-opioid receptor agonists. Br. J. Pharmacol 2006, 147, 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Helm S; Trescot AM; Colson J; Sehgal N; Silverman S Opioid antagonists, partial agonists, and agonists/antagonists: the role of office-based detoxification. Pain Physician 2008, 11, 225–235. [PubMed] [Google Scholar]
- (30).Pelton JT; Kazmierski W; Gulya K; Yamamura HI; Hruby VJ Design and synthesis of conformationally constrained somatostatin analogues with high potency and specificity for mu opioid receptors. J. Med. Chem 1986, 29, 2370–2375. [DOI] [PubMed] [Google Scholar]
- (31).Hawkins KN; Knapp RJ; Lui GK; Gulya K; Kazmierski W; Wan YP; Pelton JT; Hruby VJ; Yamamura HI [3H]-[H-D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2] ([3H]CTOP), a potent and highly selective peptide for mu opioid receptors in rat brain. J. Pharmacol. Exp. Ther 1989, 248, 73–80. [PubMed] [Google Scholar]
- (32).Abbruscato TJ; Thomas SA; Hruby VJ; Davis TP Blood-brain barrier permeability and bioavailability of a highly potent and mu-selective opioid receptor antagonist, CTAP: comparison with morphine. J. Pharmacol. Exp. Ther 1997, 280, 402–409. [PubMed] [Google Scholar]
- (33).Darke S; Hall W Heroin overdose: research and evidence- based intervention. J. Urban Health 2003, 80, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).White JM; Irvine RJ Mechanisms of fatal opioid overdose. Addiction 1999, 94, 961–972. [PubMed] [Google Scholar]
- (35).Ngo HT; Tait RJ; Hulse GK Comparing drug-related hospital morbidity following heroin dependence treatment with methadone maintenance or naltrexone implantation. Arch. Gen. Psychiatry 2008, 65, 457–465. [DOI] [PubMed] [Google Scholar]
- (36).Dhopesh VP; Taylor KR; Burke WM Survey of hepatitis B and C in addiction treatment unit. Am. J. Drug Alcohol Abuse 2000, 26, 703–707. [DOI] [PubMed] [Google Scholar]
- (37).Hallinan R; Byrne A; Amin J; Dore GJ Hepatitis C virus prevalence and outcomes among injecting drug users on opioid replacement therapy. J. Gastroenterol. Hepatol 2005, 20, 1082–1086. [DOI] [PubMed] [Google Scholar]
- (38).van Dorp E; Yassen A; Dahan A Naloxone treatment in opioid addiction: the risks and benefits. Expert Opin. Drug Saf 2007, 6, 125–132. [DOI] [PubMed] [Google Scholar]
- (39).Filliol D; Ghozland S; Chluba J; Martin M; Matthes HW; Simonin F; Befort K; Gaveriaux-Ruff C; Dierich A; LeMeur M; Valverde O; Maldonado R; Kieffer BL Mice deficient for delta- and mu opioid receptors exhibit opposing alterations of emotional responses. Nat. Genet 2000, 25, 195–200. [DOI] [PubMed] [Google Scholar]
- (40).Jutkiewicz EM The antidepressant-like effects of delta-opioid receptor agonists. Mol. Interventions 2006, 6, 162–169. [DOI] [PubMed] [Google Scholar]
- (41).Wang X; Dergacheva O; Griffioen KJ; Huang ZG; Evans C; Gold A; Bouairi E; Mendelowitz D Action of kappa and delta opioid agonists on premotor cardiac vagal neurons in the nucleus ambiguus. Neuroscience 2004, 129, 235–241. [DOI] [PubMed] [Google Scholar]
- (42).Lishmanov AI; Lasukova TV; Maslov LN; Platonov AA Role of kappa-opioid receptors in the regulation of cardiac resistance to arrhythmogenic effects of ischemia and reperfusion. Ross. Fiziol. Zh. im. I. M. Sechenova 2006, 92, 1419–1428. [PubMed] [Google Scholar]
- (43).Valtchanova-Matchouganska A; Missankov A; Ojewole JA Evaluation of the antidysrhythmic effects of delta- and kappa-opioid receptor agonists and antagonists on calcium chloride-, adrenaline- and ischemia/reperfusion-induced arrhythmias in rats. Methods Find. Exp. Clin. Pharmacol 2004, 26, 31–38. [DOI] [PubMed] [Google Scholar]
- (44).(a) Li G; Aschenbach LC.; Chen J-Y; Cassidy MP; Stevens DL; Gabra BH; Selley DE; Dewey WL; Westkaemper RB; Zhang Y Design, synthesis, and biological evaluation of 6α- and 6β-N-heterocyclic substituted naltrexamine derivatives as μ opioid receptor selective antagonists. J. Med. Chem 2009, 52, 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ward SJ; Portoghese PS; Takemori AE Pharmacological profiles of β-funaltrexamine (β-FNA) and β-chlornaltrexamine (β-CNA) on the mouse vas deferens preparation. Eur. J. Pharmacol 1982, 80, 377–384. [DOI] [PubMed] [Google Scholar]
- (45).Schmidhammer H; Burkard WP; Eggstein-Aeppli L; Smith CF Synthesis and biological evaluation of 14-alkoxymorphinans. 2. (−)-N-(cyclopropylmethyl)-4,14-dimethoxymorphinan-6-one, a se lective mu opioid receptor antagonist. J. Med. Chem 1989, 32, 418–421. [DOI] [PubMed] [Google Scholar]
- (46).Spetea M; Schüllner F; Moisa RC; Berzetei-Gurske IP; Schraml B; Dörfler C; Aceto MD; Harris LS; Coop A; Schmidhammer H Synthesis and biological evaluation of 14-alkoxymorphinans. 21. Novel 4-alkoxy and 14-phenylpropoxy derivatives of the mu opioid receptor antagonist cyprodime. J. Med. Chem 2004, 47, 3242–3247. [DOI] [PubMed] [Google Scholar]
- (47).Broadbear JH; Sumpter TL; Burke TF; Husbands SM; Lewis JW; Woods JH; Traynor JR Methocinnamox is a potent, long-lasting, and selective antagonist of morphine-mediated anti-nociception in the mouse: comparison with clocinnamox, beta-funaltrexamine, and beta-chlornaltrexamine. J. Pharmacol. Exp. Ther 2000, 294, 933–940. [PubMed] [Google Scholar]
- (48).Butelman ER; Yuferov V; Kreek MJ κ-Opioid receptor/dynorphin system: Genetic and pharmacotherapeutic implications for addiction. Trends Neurosci. 2012, 35, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Spanagel R; Almeida OF; Bartl C; Shippenberg TS Endogenous kappa-opioid systems in opiate withdrawal: Role in aversion and accompanying changes in mesolimbic dopamine release. Psychopharmacology 1994, 115, 121–127. [DOI] [PubMed] [Google Scholar]
- (50).(a) Simonin F; Valverde O; Smadja C; Slowe; Kitchen I; Dierich; Le Meur M; Roques BP; Maldonado R; Kieffer BL Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50, 488H and attenuates morphine withdrawal. E. M. B. O. J 1998, 17, 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De Marco R; Bedini A; Spampinato S; Comellini L; Zhao J; Artali R; Gentilucci L Constraining endomorphin-1 by β,α-hybrid dipeptide/heterocycle scaffolds: identification of a novel κ-opioid receptor selective partial agonist. J. Med. Chem 2018, 61, 5751–5757. [DOI] [PubMed] [Google Scholar]
- (51).Pelotte AL; Smith RM; Ayestas M; Dersch CM; Bilsky EJ; Rothman RB; Deveau AM Design, synthesis, and characterization of 6β-naltrexol analogs, and their selectivity for in vitro opioid receptor subtypes. Bioorg. Med. Chem. Lett 2009, 19, 2811–2814. [DOI] [PubMed] [Google Scholar]
- (52).Ward SJ; Portoghese PS; Takemori AE Pharmacological characterization in vivo of the novel opiate, beta-funaltrexamine. J. Pharmacol. Exp. Ther 1982, 220, 494–498. [PubMed] [Google Scholar]
- (53).Yekkirala AS; Lunzer MM; McCurdy CR; Powers MD; Kalyuzhny AE; Roerig SC; Portoghese PS N-naphthoyl-beta-naltrexamine (NNTA), a highly selective and potent activator of μ/kappa-opioid heteromers. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 5098–5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Yuan Y; Li G; He H -J; Stevens, D. L.; Kozak, P.; Scoggins, K. L.; Mitra, P.; Gerk, P. M.; Selley, D. E.; Dewey, W. L.; Zhang, Y. Characterization of 6α- and 6β-N-heterocyclic substituted naltrex amine derivatives as novel leads to development of mu opioid receptor selective antagonists. ACS Chem. Neurosci 2011, 2, 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).(a) Mitra P; Venitz J; Yuan Y; Zhang Y; Gerk PM Preclinical disposition (in vitro) of novel μ-opioid receptor selective antagonists. Drug Metab. Dispos 2011, 39, 1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hernández-Delgadillo GP; Cruz SL Endogenous opioids are involved in morphine and dipyrone analgesic potentiation in the tail flick test in rats. Eur. J. Pharmacol 2006, 546, 54–59. [DOI] [PubMed] [Google Scholar]
- (56).Yuan Y; Stevens DL; Braithwaite A; Scoggins KL; Bilsky E; Akbarali HI; Dewey WL; Zhang Y 6β-N-Heterocyclic substituted naltrexamine derivative as potential lead to develop peripheral mu opioid receptor selective antagonists. Bioorg. Med. Chem. Lett 2012, 22, 4731–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).(a) Yuan Y; Elbegdorj O; Chen J; Akubathini SK; Zhang F; Stevens DL; Beletskaya IO; Scoggins KL; Zhang Z; Gerk PM; Selley DE; Akbarali HI; Dewey WL; Zhang Y Design, synthesis, and biological evaluation of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4’-pyridyl)carboxamido]morphinan deriv atives as peripheral selective μ opioid receptor Agents. J. Med. Chem 2012, 55, 10118–10129. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yakhontov LY; Lapan EI; Rubtsov MV The synthesis of isomeric 4- and 6-ethoxy-3-ethylpyridines. Chem. Heterocycl. Compd 1967, 3, 829–831. [Google Scholar]; (c) Xin BW Phosphine-free cross-coupling reaction of halopyridines with arylbor onic acids in an ionic liquid: water mixture. J. Chem. Res 2008, 2008, 412–415. [Google Scholar]; (d) Okawa T; Aramaki Y; Yamamoto M; Kobayashi T; Fukumoto S; Toyoda Y; Henta T; Hata A; Ikeda S; Kaneko M; Hoffman ID; Sang BC; Zou H; Kawamoto T Design, synthesis, and evaluation of the highly selective and potent G-protein-coupled receptor kinase 2 (GRK2) inhibitor for the potential treatment of heart failure. J. Med. Chem 2017, 60, 6942–6990. [DOI] [PubMed] [Google Scholar]
- (58).Wang H; Zaidi SA; Zhang Y Binding mode analyses of NAP derivatives as mu opioid receptor selective ligands through docking studies and molecular dynamics simulation. Bioorg. Med. Chem 2017, 25, 2463–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hagmann WK The many roles for fluorine in medicinal chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]
- (60).Mahar Doan KM; Humphreys JE; Webster LO; Wring SA; Shampine LJ; Serabjit-Singh CJ; Adkison KK; Polli JW Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther 2002, 303, 1029–1037. [DOI] [PubMed] [Google Scholar]
- (61).Christie MJ Cellar neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br. J. Pharmacol 2008, 154, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Fleming FF; Yao L; Ravikumar PC; Funk L; Shook BC Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J. Med. Chem 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Brack A; Bottiger BW; Schafer M New insights in postoperative pain therapy. Anasthesiol. Intensivmed. Schmerzther 2004, 39, 157–164. [DOI] [PubMed] [Google Scholar]
- (64).Manglik A; Kruse AC; Kobilka TS; Thian FS; Mathiesen JM; Sunahara RK; Pardo L; Weis WI; Kobilka BK; Granier S Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wu H; Wacker D; Mileni M; Katritch V; Han GW; Vardy E; Liu W; Thompson AA; Huang XP; Carroll FI; Mascarella SW; Westkaemper RB; Mosier PD; Roth BL; Cherezov V; Stevens RC Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Granier S; Manglik A; Kruse AC; Kobilka TS; Thian FS; Weis WI; Kobilka BK Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- (68).Jones G; Willett P; Glen RC Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol 1995, 245, 43–53. [DOI] [PubMed] [Google Scholar]
- (69).Ballesteros JA; Weinstein H Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- (70).Clark M; Cramer RD; Van Opdenbosch N Validation of the general purpose tripos 5.2 force field. J. Comput. Chem 1989, 10, 982–1012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.