

Abstract
Metal ions, usually bound by various amino-acid side chains in proteins, play multiple roles in protein folding, conformational change, cellular communication, and catalysis. Ca(II) and Mg(II), abundant among biologically relevant cations, execute their cellular functions associated with the conformational change of bound proteins. They bind with proteins where carboxylic acid residues are dominant ligands. To develop mass spectrometry for mapping protein-binding sites, we implemented a new carboxyl group footprinter, benzhydrazide, and refined it with isotope encoding. The method uses carbodiimide chemistry to footprint carboxylic residues, whereby 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide activates a carboxyl group followed by nucleophilic attack by benzhydrazide forming a stable labeled product. We tested the effectiveness of isotope-encoded benzhydrazide by studying Ca2+ and Mg2+ binding of calmodulin, an EF-hand protein. The footprinting results indicate that the four active sites for metal-ion binding (EF hands I, II, III, and IV) and the linker region (peptide 78–86) undergo conformational changes upon Ca(II) and Mg(II) binding, respectively. The outcome is consistent with previously reported results and 3-D structures, thereby validating a new reagent that is more reactive and discriminating for specific amino-acid protein footprinting. This reagent should be important for locating metal-binding sites of other metalloproteins.
Graphical Abstract
Metal ions are essential for many cellular functions.1 In the range of 25−50% of all proteins found within an organism contain metal ions.2,3 Metal ions bind via functional groups termed ligands that donate lone pairs to the metal ion. In the bound state, the metal ions in proteins can catalyze a variety of biochemical reactions or stabilize the protein structures.
Based on the properties of coordinated metals, we can classify two types of metals that interact with proteins: 1) borderline/soft ions (e.g., Zn(II), Fe(II) and Cu(II)) that coordinate with nitrogen- and sulfur-containing groups found in His and Cys, and 2) hard cations (e.g., Ca(II) and Mg(II)) that use oxygen ligands as the preferred coordination sites (e.g., Asp and Glu are preferred).4 Among all metalloproteins, those containing Mg(II), Ca(II), are relatively abundant.1–5 In many cases, Mg(II) and Ca(II) binding stabilizes the structure of folded proteins, and in some cases, locks in a physiologically active conformation. Specifically, Ca(II) plays a role in signal transduction, muscle contraction, nerve-impulse transmission, and calcium homeostasis. To maintain the correct Ca ion concentration in the intra- and extracellular space, the body uses Ca pumps. Most Ca-binding proteins possess a highly conserved Ca-binding motif, the EF hand, that selectively binds Ca in a background of up to 105-fold higher concentrations of Na(I), K(I), and Mg(II).6,7 Although divalent Mg has higher charge density and exists at higher concentration in the cytosol than Ca(II), it shares physical and chemical properties with Ca(II) to bind proteins and enable signaling, enzymatic activation and catalysis, and muscle relaxation.8,9 A clear knowledge of metal/protein interactions, including the location of the metal binding site and its binding characteristics, is essential to understand the functional mechanisms of metal-binding proteins.
Although nuclear resonance (NMR),10 X-ray crystallography,3 and Cryo-EM11 are commonly used to determine metal-binding sites, but they are limited by size, conformational flexibility, aggregation propensity, or limited sample amount, rendering them frequently inapplicable. Our goal is to develop mass spectrometry (MS)-based methods that complement these high-resolution techniques because MS is general, fast, sensitive, has high-throughout, and provides moderate resolution. One MS-based method, hydrogen deuterium exchange (HDX), has been widely applied in protein structure characterization.12-15 HDX can be extended to provide an approach termed protein-ligand interactions by MS, titration, and HD exchange (PLIMSTEX) to monitor the conformational changes of proteins upon Ca2+ binding and even measure the binding constants.16 HDX, however, cannot directly monitor the binding of side chains owing to their fast hydrogen deuterium exchange.
An alternative method for solving this challenge is irreversible covalent labeling whereby a variety of reactions can be invoked to modify amino-acid side chains. In this approach, reactive side chains are stably modified such that the label can be conveniently located by MS in peptides form from proteolysis. One approach is to use reactive species such as OH radicals (provided through electron beam or X-ray radiolysis, Fenton chemistry, or photolysis of peroxide),17 carbenes,18,19 iodide radicals,20 and trifluoromethyl radicals.21 Slower but still effective footprinting can use carbodiimide22, ICAT,23 or diethylpyrocarbonate (DEPC)24 chemistries. Because the reactions are slower than those of free radicals, there is always an issue that the labeling itself doesn’t perturb the native structures.
Carbodiimide chemistry provides the most versatile method for labeling or crosslinking carboxylic acids. Earlier, we employed 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), a crosslinking facilitator, to enable glycine ethyl ester (GEE) to footprint side chains of Asp and Glu. This approach was successfully applied to determine the orientation of the FMO antenna protein,25 to map the binding of Fab-1 and VEGF,26 to locate a membrane-associated tyrosine kinase binding site,27 to map the epitope of human interleukin-6 receptor,28 to determine the binding interface for the oligomeric apolipoprotein E,29 to characterize monoclonal antibody structure, 30 and ELISA antibody-antigen interactions.31 Nevertheless, GEE footprinting has limitations, including the requirement for a large excess of the GEE reagent (20,000 equivalents with respect to the protein), some hydrolysis of the product during work-up, and a limited opportunity to incorporate isotopes for encoding.
Herein, we explore application of carbodiimide compound (EDC), in the presence of new labeling reagent benzhydrazide (BHD), inspired by Leitner.32 To be specific, carboxylic acid residues are activated by EDC, then BHD, a nucleophilic modifying reagent, attacks the activated carboxyl group. Our goal is to avoid the hydrolysis of the modified product while still characterizing metal-binding sites. We focus on two hard metal ions Ca(II) and Mg(II) that chelate with carboxylic residues because they are ubiquitous in metalloproteins. We chose carbodiimide specifically because it reacts with carboxylic groups whose reactivity will be reduced when chelated with Ca(II) or Mg(II) ions. Furthermore, its size may allow high discrimination for metal-bound vs. unbound carboxylates. To improve the MS quantification, we incorporated five deuteriums in the structure to permit encoding.33 Results show that the method can accurately measure the binding site of Ca(II) and Mg(II) in a test protein, calmodulin (CaM). By assisting in the elucidation of metal-binding sites in proteins, this method should enrich the output of structural proteomics studies.
EXPERIMENTAL SECTION
Materials.
Tris base, HEPES buffer (pH 7.5), urea, water, formic acid, glycine ethyl ester hydrochloride (GEE), calcium chloride, magnesium chloride, ammonium acetate, 1,1°-carbonyldiimidazole, benzoic acid, benzoic acid-2,3,4,5,6-d5, tetrahydrofuran (THF), hydrazine hydrate solution (78–82%), Z-Glu-OMe, and thin-layer chromatography supplies (TLC, silica gel 60 F254) were obtained from Sigma-Aldrich (St. Louis, MO). N-Benzyloxycarbonyl-L-aspartic acid 1-methyl ester was obtained from Alfa Aesar (Lancashire, UK) Sartorius™ Vivaspin™ 500 μL centrifugal concentrators were purchased from Sartorius Corporation (Goettingen, Germany). The protein bovine calmodulin (CaM) was from Ocean Biologics (Seattle, WA). The 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), trypsin and Zeba™ spin desalting columns (7K MWCO) were from Thermo Scientific (Rockford, IL). Cyclo(Arg-Gly-Asp-D-Phe-Glu) was from Peptides International, Inc. (Louisville, KY).
Synthesis and Characterization of Benzhydrazide and d5-Benzhydrazide.
Reagents (no known hazards) were synthesized based on protocol of Li,34 (Figure S1) except for the purification process. In 100 mL reaction flask, 1,1°-carbonyldiimidazole (1.73 g, 10.6 mmol) was added to a solution of benzoic acid (1.00 g, 8.19 mmol) in THF (15 mL). After stirring at 25 °C for 3 h, the mixture was added dropwise to the hydrazine hydrate solution (78–82%, 1.23 g, 24.6 mmol) in 6 mL THF over 45 min. The reaction was allowed to go to completion overnight at 25 °C, as monitored by TLC. Solvents were evaporated in vacuum to give a crude white product that was isolated and purified by silica gel column chromatography (hexane:ethyl acetate = 1:1) to give the benzhydrazide as a white solid (1.03 g, 93% yield). d5-Benzhydrazide was synthesized by using an identical procedure, giving the d5-benzhydrazide as a white solid (1.00 g, 87% yield). High-resolution mass spectra (HRMS) were recorded with a Bruker MaXis QToF (ESI). HRMS (ESI) m/z calcd. for protonated BHD (C7H8N2OH+): 137.0715. Found: 137.0714. HRMS (ESI) m/z calcd. for d5-BHD (C7H3D5N2OH+): 142.1029. Found: 142.1028. (Figure S2)
Preparation of Protected amino acids/Peptide/Protein Stock Solution.
All protected amino acids (Z-Glu-OMe and N-Benzyloxycarbonyl-L-aspartic acid 1-methyl ester) and cyclic peptide stock solutions of 100 uM concentration were prepared in 10 mM HEPES buffer. Ca2+-free CaM (50 μM) was prepared by centrifuging three times at 15,000 (×g) for 10 min using Vivaspin™ centrifugal concentrators, whereas Ca2+-bound CaM was prepared by incubating 50 μM CaM with 500 μM CaCl2 overnight at 25 °C. Similarly, Mg2+-bound CaM was prepared by incubating 50 μM CaM with 500 μM MgCl2 overnight at 25 °C. The concentration of CaM in 10 mM HEPES buffer (pH 7.5) was determined by using UV absorbance (molar absorptivity of 2980 Lmol−1 cm−1 at 280 nm) with a NanoDrop spectrophotometer (Thermo Scientific).
Carboxyl Group Modification.
The final concentrations of protected amino acids, peptide, or protein for all reactions were adjusted to 10 μM. For protected amino acids, the labeling was initiated by adding BHD and EDC to give final concentrations of 20 mM and 500 μM, respectively. The protected amino acid modification was quenched after 20 min by adding 20 μL ammonium acetate (1 M). For cyclic peptides, the labeling follows the same procedure as protected amino acids, except the labeling was quenched after 24 h. Experiments with CaM were performed in triplicate by using 20 mM BDH and 500 μM EDC, identical to the concentration used for cyclic peptide labeling. After 5 min of reaction at 37 °C, the solution was quenched by removing the reagents by using a Zeba spin desalting column according to the manufacturer’s protocol. (Figure S3)
Trypsin Proteolysis Protocol.
After desalting, the filtrate was evaporated to dryness in a vacuum centrifuge. The pellets were dissolved with 50 μL water to make an equal volume of protein. Aliquots of 10 μL filtrate of BHD and d5-BHD were combined at 1:1 ratio (v/v) and dried in vacuum (the dry sample can be stored in a freezer at −80 ℃ for months). Prior to analysis, the protein mixture pellet was denatured with urea (8 M, 5 μL) at 37 ℃ for 30 min, followed by adding 45 μL Tris buffer (pH 8.5). Protein samples were then digested with trypsin at a protease:protein ratio of 1:20 (w/w) at 37 ℃ overnight. Pure formic acid (1 μL) was added to stop the digestion.
Solvent Accessible Surface Area (SASA) calculation.
We used PDB files of Ca2+-free CaM (PDB ID: 1CFC) and Ca2+-bound CaM (PDB ID: 1CLL) to GETAREA (http://curie.utmb.edu/getarea.html) to calculate individual side-chain SASA.
Mass Spectrometry.
All mass spectra for intact proteins were acquired on a Bruker MaXis Q-TOF (Bremen, Germany) in the positive-ion mode, as was reported previously.18
For proteolytic peptides, digests were analyzed on a Q Exactive Plus hybrid quadrupole orbitrap mass spectrometer coupled with a Nanospray Flex ion source (Thermo Fisher, Santa Clara, CA). A C18 column was custom fabricated with an integrated emitter (75 μm × 150 mm, 1.8 μm, 100 Å). The gradient was from 2.0% solvent B (80% acetonitrile, 0.1% formic acid) to 20% solvent B over 55 min, then to 50% solvent B over 25 min, and finally to 90% solvent B for 10 min followed by a 15 min re-equilibration step. The 15 most abundant molecular ions were automatically chosen for fragmentation (DDA) by using the mass spectra scanned from m/z 380−2200 at a resolving power (RP) of 70 K (at m/z 200) throughout the chromatography. Precursor ions were isolated in the quadrupole with an isolation window of 2.0 m/z and fragmented with higher-energy collisional dissociation (HCD) with a normalized collision energy (NCE) of 32%. The automatic gain control (AGC) targets were 5 × 105 for MS and 5 × 104 for MS/MS acquisitions. Maximum injection times (maxIT) were 200 ms for MS and 100 ms for MS/MS.
LC-MS/MS Data Analysis
The LC-MS/MS data were searched for unmodified and modified CaM tryptic peptides by using Byonic™ Software (Protein Metrics, San Carlos, CA). All BHD and d5-BHD modifications on Asp and Glu were added as modifications to the database. The parameters were: 10 ppm precursor mass tolerance, 60 ppm fragment mass tolerance, and CID/HCD fragmentation. Modification-sites were validated by manually checking product-ion spectra and were double-checked with Thermo Xcalibur (a custom program from Thermo Fisher).
Results and Discussion
Evaluation of Isotope Effects.
A reliable approach in quantitative methods is to use stable isotope labeling for ratioing. The use of an isotope-encoded footprinter normalizes the MS intensity of analytes to their isotopic analogs and, therefore, effectively compensates for solvent effects, variable ion suppression from other coeluting analytes, and experimental variations accompanying sample preparation, injection, and instrument setup. To develop an isotope-encoded footprinter, certain criteria are required: 1) isotope effects should not significantly affect the kinetics of footprinting (i.e., the reaction rates of a footprinting reaction are nearly constant when one or more atoms of the footprinter are replaced by isotopes. 2) Analytes and their isotopic analogs (nearly) coelute during the LC; in this way, a pair of isotope-labeled analytes would experience the same ionization efficacy during MS analysis, which is important for accurate quantification. 3) A sufficient mass difference between two isotopically encoded adducts is needed to separate the peaks of the “light and heavy” analogs over a range of from m/z 400 to 2000. Unfortunately, only a limited number of isotopic footprinters are commercially available. Therefore, we prepared both the “light” and “heavy” reagents (see Experimental) that we used in this footprinting.
We first measured the relative labeling efficiency of D and E on protected-aspartic acid and glutamic acid. They show very similar modification extent (25.1% ± 0.4% for protected D v.s. 25.0% ± 0.1% for protected E). (Figure. S4)
To test whether the “intrinsic” chemical reactivity is the same for “light” vs. “heavy”, we chose a cyclic peptide cyclo(RGDFE), which has no secondary structure and no complicating N- or C-termini. The EICs (Figure 1a) for unmodified (black), BHD- (light blue), and d5-BHD (dark blue) labeled cyclic peptides were integrated, demonstrating that ratio of modified vs. unmodified was nearly the same, 43.2 ± 1.6% “light” and 44.6 ± 1.9% “heavy” labeling. Thus, the presence of isotopes in BHD did not affect the reactivity of the carboxylic residues in cyclo(RGDFE).
Figure 1.
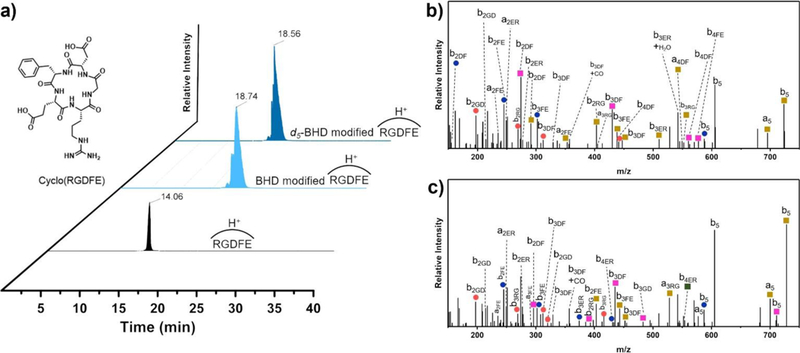
a) Extracted ion chromatograms (EIC) for unmodified (black), BHD-modified (light blue) and d5-BHD modified (dark blue) cyclo(RGDFE), product-ion (MS/MS) spectrum for BHD- b) and d5-BHD- c) modified cyclo(RGDFE). (Orange circle: loss of NH3, blue circle: loss of H2O, yellow square: addition 118.0524 or 123.0845 mass unit, green square: addition of 118.0524 or 123.0845 mass unit and loss of H2O, pink square: addition of 118.0524 or 123.0845 mass unit and loss of NH3.) (The nomenclature for cyclic peptides is that by Ngoka and Gross39, figure itself is original. Details are shown in Scheme S1.)
This cyclic peptide also helps determine whether isotopomer mixture of BHD can label both Asp and Glu if the sibling residues are adjoining. Metal ions (e.g., Ca, Mg) generally have multiple coordinated sites, and usually include several Asp and Glu in a single binding pocket. It is important that the footprinter can modify all carboxylic residues in a pocket-like site rather than react with one and be “silent” with the other. BHD labels both Asp and Glu even when they are adjoining in the cyclic peptide (cRGDFE). To validate the peptide assignments, we manually assigned peaks in the product-ion spectra (MS/MS) for the BHD- and d5-BHD modified cyclo(RGDFE), confirming that the modification occurs at both Asp and Glu (Figure 1b and 1c).35
We next determined whether there are any isotope effects in the separation of BHD- and d5-BHD modified peptides. If the light and heavy species coelute, matrix effects should not affect the relative efficiency of ionization of the analytes. Because there is a difference in the size of the atoms36,37, substitution of hydrogen with deuterium could lead to differences in the binding interactions (van der Waals interactions) with the stationary phase. Protiated compounds bind to hydrophobic stationary phase more strongly than their deuterated counterparts.36 When comparing the retention time of BHD- and d5-BHD modified peptide (Figure 1a), we found a small isotope effect that d5-BHD modified peptides eluted approximately 11 s after the BHD-modified counterpart.
Calmodulin Protein Footprinting.
Calmodulin (CaM) is a highly conserved, secondary messenger protein, which is ubiquitous in eukaryotic cells. More than thirty proteins and enzymes with various cellular functions are activated by CaM. CaM contains four highly conserved motifs, referred to as “EF-hands”, which show a typical helix–loop–helix motif. CaM binds various metals to execute its cellular functions, not only Ca2+, but also La3+, Tb3+, Pb2+, Sm3+, Sr2+, Hg2+, Cd2+, Zn2+, and Mn2+.38 Among them, Ca2+ binding is the most important because Ca2+ binding induces an allosteric communication of CaM to catalyze downstream enzymatic reactions.
We evaluated the BHD reactivity with CaM by fixing the concentration of EDC at 500 uM, 50 fold excess relative to CaM (10 uM). Because each CaM molecule contains 37 Asp and Glu, all Asp and Glu on the protein surface have a chance to react. With increasing amounts of BHD from 5–200 mM, the modification extent of CaM increased from 43% to 65% (Figure S5). When the concentration of BHD was increased further, the modification extent did not significantly change, probably because EDC is no longer the limiting reagent. Thus, we chose 200 mM as the optimum concentration of BHD. We also tested the relative reactivity of “light” and “heavy” BHD to CaM. Similar to cyclo(RGDFE), we found that “light” and “heavy” BHD show nearly identical modification extents for the intact CaM protein (Figure 2).
Figure 2.
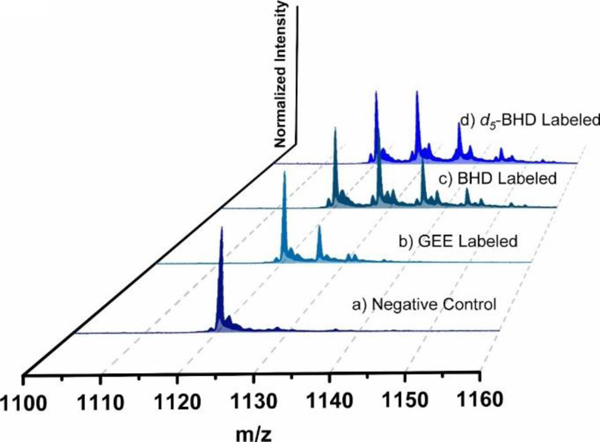
Mass spectra of modified CaM at +15 charge state: a) control: Ca2+-free CaM, b) Ca2+-free CaM labeled by GEE, c) Ca2+-free CaM labeled by BHD, d) Ca2+-free CaM labeled by d5-BHD (25 °C, 5 min).
To evaluate whether BHD improves the labeling efficiency with respect to GEE, we compared the reactivity of both reagents. BHD is more reactive when compared to GEE (Figure 2, compare b to c), 67 ± 3% BHD and 25 ± 3% GEE labeling.
BHD as a footprinting reagent.
Our next goal is to test BHD as a footprinter to establish whether its reactions report the metal-binding site for CaM. Isotope-encoding provided a “light” and “heavy” labeling of the two different states of CaM. In this protocol (Figure S6), Ca-free CaM was labeled by d5-BHD in one Ependorf tube and Ca-bound CaM was labeled by do-BHD in another tube. Following the footprinting, the tube contents were combined in a 1:1 ratio prior to trypsin digestion and LC/MS/MS analysis. In this way, we could maintain all experimental conditions (e.g., protein digestion, MS analysis) nearly identical for both light and heavy samples following protein modification.
Data processing showed 100% sequencing coverage of the tryptic peptides. We took advantage of accurate-mass measurements to obtain reliable EICs for integrating the signal intensities representing (Figure 3a) modifications by d5-BHD and d0-BHD (seen as two distinct masses, +123.0845 (C7HD5N2) and +118.0531 (C7H6N2) on peptide 78–86, respectively (Figure 3b, 3c)). When comparing their retention time, however, we did observe, as before, a small isotope effect (Figure 3a) that d5-BHD modified peptide 78–86 eluted 15.6 s earlier than its d0-BHD-modified counterpart. This deviation is consistent with the previously reported effect of deuteration.36,37 In addition, we found that the labeled peptides are more hydrophobic and eluted approximately 14 min later than their unmodified counterparts (Figure S7). This chromatographic pattern assists the identification of the modified peptides, sometimes facilitating separation of isomeric peptides with modifications on different residues.
Figure 3.
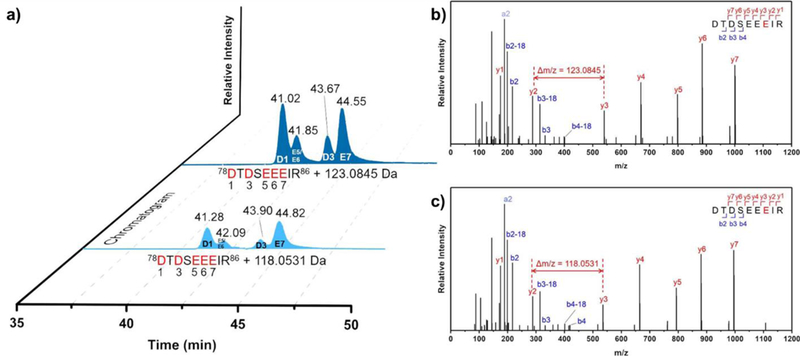
a) EIC for BHD- (light blue) and d5-BHD- (dark blue) modified peptide 78–86, b) product-ion (MS/MS) spectrum for peptide 78–86: unique mass shift of b) 123.0845 Da, and c) 118.0531 Da is consistent with d5-BHD modified and d0-BHD modification on E84.
Overall, we found that eight regions of the protein undergo large conformational changes with modification extent difference over 50% and p-values less than 0.01.40 These regions are represented by peptides 14–21, 22–30, 31–37, 38–74, 76–86, 78–86, 95–106, and 127–148 (Figure 4), consistent with Ca2+-induced regional structural transitions that dramatically alter the molecular surface of CaM. For example, there is only trace amount of BHD-labeled peptides 22–30, and 38–74 for the holo state, whereas modification extents of these two peptides for the apo-state is > 8% (Figure 4, Table S1).
Figure 4.
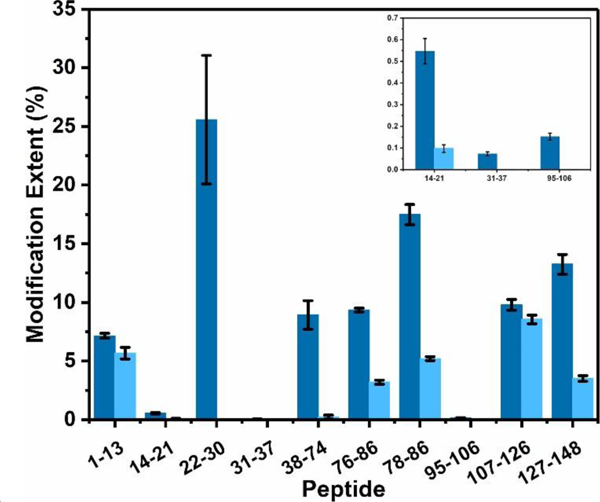
Comparison of the modification extents of constituent peptides upon footprinting Ca2+-free CaM (dark blue) and Ca2+-bound CaM (light blue).
To establish that these MS results are structurally meaningful, we analyzed them with respect to the high-resolution NMR and X-ray structures. CaM binds Ca2+ at four, nonidentical sites that contain the EF-hand structural motif, two in the N-domain (EF-1 and EF-2) and two in the C-domain (EF-3 and EF-4). Each hand contains an acidic, Ca2+-coordinating loop, or “EF-loop” where Ca2+ is chelated with seven oxygens from six residues in a pentagonal bipyramidal fashion. 6,41 EF-1 chelates Ca2+ with ligands D20, D22, D24, and E31 from peptide 14–21, 22–30, 31–37. (Figure 5a and 5f). For this binding site, the reagent is extraordinarily discriminating such that the carboxylates of this region undergo little modification when in the holo state (peptide 14–21, 22–30, 31–37).
Figure 5.
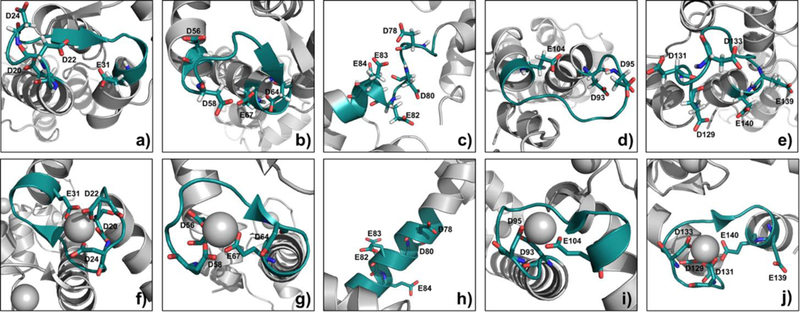
3D structure of Ca2+-free CaM from the average NMR structure (PDB ID, 1CFD) and Ca2+-bound CaM from the X-ray crystal structure (PDB ID, 1CLL), Asp and Glu shown as sticks. a) and f) region 20–31 in Ca2+-free CaM and Ca2+-bound CaM, b) and g) region 56–67 in Ca2+-free CaM and Ca2+-bound CaM, c) and h) region 78–86 in Ca2+-free CaM and Ca2+-bound CaM, d) and i) region 93–104 in Ca2+-free CaM and Ca2+-bound CaM, e) and j) region 129–140 in Ca2+-free CaM and Ca2+-bound CaM. (Gray sphere is the metal ion.)
Likewise, EF-2 region, which is represented by peptide 38–74 and chelates Ca2+ via D56, D58, D64 and E67 (Figure 5b and 5g), also becomes unreactive in the holo state. Because Asp and Glu lose nearly completely their chemical reactivity, the modification extent difference for EF hand II reaches 97%.
As pointed out earlier, the binding of Ca2+ to the EF hand induces a dramatic change of the linker region (peptide 76–86 and 78–86), changing from a flexible loop (Figure 5c) to a rigid α-helix (Figure 5h). Although the conformation of this linker region was under debate that the formation of the α-helical conformation is a crystallization artifact, 42 many studies 43 report that the Ca2+ binding does induce domain opening in CaM, indicating the linker region is extended and becomes more rigid than in the apo-CaM. The plasticity of the linker region in Ca2+-bound CaM allows EF-hand domains to take up various orientations with respect to each other.44 Our footprinting results also reveal a significant modification change, evidenced by two overlapping peptides that show approximately 70% modification difference (see Table S1). The D78, D80, E82, E83, and E84 on a flexible loop in holo-CaM can easily react with BHD, whereas when this region becomes a rigid α-helix region in the apo form, those residues are less reactive.
EF-3, as represented by peptide 95–106 (Figure 5d), uses D95 and E104 to chelate Ca2+ (Figure 5i). Although the modification ratio for this region in apo CaM is only 0.1%, there is negligible modification for the Ca2+-bound CaM, indicating a large conformational change of EF-3 hand. Finally, for EF-4 peptide 127–148 (Figure 5e) shows a 74% modification decrease upon binding. All the Asp and Glu in this region, except a distant E139, are candidates for binding with Ca2+ (Figure 5j).
To investigate whether changes in SASA agree with our result, we compared the residue-level quantification and the calculated SASA of Ca2+-free and Ca2+-bound CaM (Figure S8). The modification extent of all D and E residues at the EF-hands significantly decreases upon Ca2+ binding. The results correlate well with the similar trend that most D and E residues of Ca2+-bound CaM at the binding sites dramatically lose their SASAs. Nevertheless, the decreased modification extent of E82/E83/E84 does not correlate well with the their theoretical increasing SASAs. This may be due to the reaction in this region not only depends on solvent accessibility but also on stereochemistry. Compared with previous GEE study, 45 BHD with 10% amount of GEE labels 27 D and E in CaM, showing higher reactivity. The significant differences of labeling efficiency for GEE and BHD are that the alpha effect in BHD increases the nucleophilicity of the nitrogen owing to the presence of adjacent nitrogen atoms with nonbonding electron pairs, leading BHD to be more reactive. 46 In addition, BHD is more discriminating given that it successfully reports four binding-sites and that the linker region undergoes large conformational changes.
Mg2+ Binding.
In addition to Ca2+-binding, we extended the approach to study Mg2+ binding with CaM. Like Ca2+, divalent Mg is also a “hard” ion and prefers “hard” ligands with oxygen being most preferred. In contrast to the well-characterized Ca2+-bound structure, the binding of Mg2+ to CaM is still unresolved. Conclusions from microcalorimetric studies indicate four identical Mg2+-binding sites that do not completely overlap with the Ca2+-binding sites in CaM. On the other hand, conclusions from flow dialysis, NMR, and fluorescence indicate that Mg2+ shares same binding sites with Ca2+.47 Only recently, the structure of Mg2+-bound CaM 6 was partially resolved by X-ray crystallography to show the N-terminal Mg2+ binding sites are similar to those of Ca2+. Nevertheless, the full high-resolution structure of Mg2+-bound CaM remains unknown.
Comparing the footprinting results for EF-1 and the X-ray structure, we found that although D20, D22, and D24 chelate Mg2+ (Figure 7a), E31 is too far from Mg2+ to contribute to the coordination. In contrast to pentagonal bipyramid coordination for Ca2+, octahedrally coordinated Mg2+ does not directly bind to Glu12 of the EF-hand. Footprinting results shows that E31 undergoes 0.008% BHD modification in Mg2+-bound CaM, but little modification in Ca2+-bound CaM, correlating well with the coordination geometry. Furthermore, the modification-extent differences for Mg2+ are significant, but not as significant as for Ca2+-binding (89% for Mg2+- vs. ~100% for Ca2+- bound ones), suggesting weaker binding for Mg2+. This difference is consistent with the enthalpy changes upon Ca2+ binding being exothermic, whereas endothermic, and thus unfavorable, for Mg2+ binding to CaM. Consequently, Mg2+ does not bind to CaM as tightly as Ca2+. 48
Figure 7.
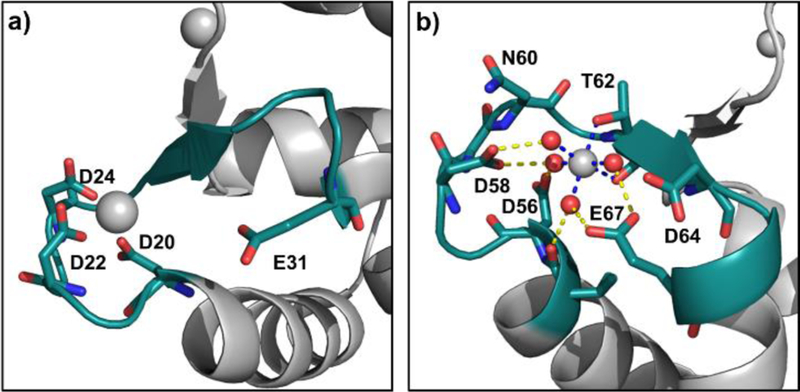
3D structure of Mg2+-bound CaM (PDB ID, 3UCW). a) Region 20–31, b) region 55–67. (Gray sphere is the metal ion; red sphere is water molecule.)
Although previous reports disagree on details, they do indicate that Mg2+ indeed binds to CaM, likely at the calcium sites. When we compared our footprinting results for Mg2+-bound with those of Ca2+-bound, we found that eight peptides regions have modification extent difference more than 0.5 and p-value less than 0.01 (Figure 6 and Table S2). Those eight peptides regions belong to EF-1 to 4 plus to the linker region. Our results support the conclusion that the binding site of Mg2+ resembles that of Ca2+.5,41,47
Figure 6.
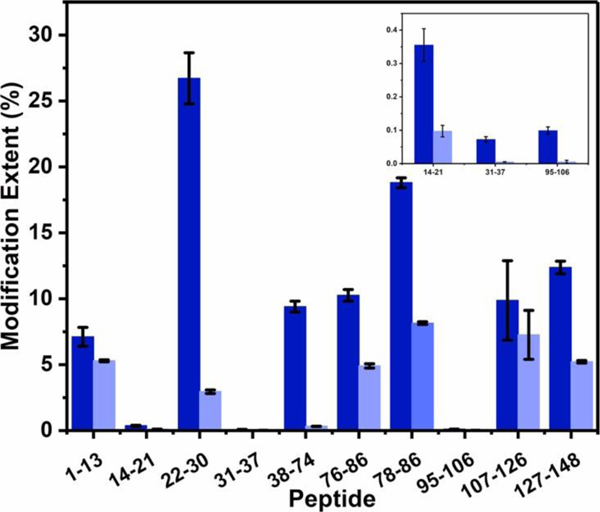
Comparison of the modification extent of constituent peptides from Mg2+-free CaM (dark blue) and Mg2+-bound CaM (light blue).
For EF-2 regions, however, Mg2+ shows a very different pattern than Ca2+ for binding with CaM. From the X-ray structure, Mg2+ coordinates with two oxygens from T62 and a backbone carbonyl group. (Figure 7b) The remaining four coordinating positions are occupied instead, by four water molecules that form strong hydrogen bonds with D58 and E67, allowing Asp and Glu residues to gain some protection. In other words, Mg2+ directly binds to first-shell water molecules, allowing it to bind in an indirect way to D58 and E67 (the binding is called a second-shell ligand interaction). Our MS results show no modification extent difference in peptide 38–74 with that in Ca2+-CaM even though there is an indirect interaction between Mg2+ and the nearby carboxylic residues. This identical footprinting, however, also reminds us that a more sensitive reagent is needed if we want to resolve the pattern between first-shell and second-shell ligand binding.
Furthermore, the linker region of Mg2+-bound CaM does show significant change, but the changes are not as large as those of Ca2+-bound CaM. (Figure 6, peptide 76–86 and 78–86). The modification extent differences of Mg2+-bound CaM are on the borderline (57%) whereas the Ca2+-bound shows differences at 70%. One expects that Ca2+-bound CaM should undergo larger conformation changes in this region on the basis of previous studies.5,43,49
Finally, we can evaluate EF-3 and EF-4 regions even though there is no crystal structure for this part in Mg2+-bound CaM. Like for Ca2+-bound CaM, we detected trace amount of modification for peptide 95–106 (EF-3). Peptide 127–148, containing EF-4, undergoes relatively less conformational change with Mg2+ compared to Ca2+-binding, with modification extent differences 58% vs. 74% for Ca2+-binding. The ionic radius of Mg2+ 50 is too small to allow some carboxylic acid residues to coordinate with Mg2+, eliminating any large differences in the reaction of BHD with Mg2+ binding.
In summary, peptide 14–21, 22–30, 31–37,76–86, 78–86, and 127–148 of Mg2+-bound CaM experience conformational changes, but they are less significant than those of Ca2+-bound CaM. This indicates that Mg2+ with smaller ionic radii binds to CaM without chelating with the 12th residues of the EF-hand, consequently causing smaller structural changes.
Conclusion
With this new footprinting reagent, we can readily identify the four Ca binding sites in CaM and correlate them with 3-D structures. In contrast to other irreversible labeling strategies that either monitor the backbone or side chains around the metal-binding site, this reaction directly footprints carboxyl groups at the binding site. For Mg2+ binding, which is weaker and more difficult to discriminate, the footprinting still shows that Mg2+ binds with four EF hands similar to Ca2+.
This approach has several advantages and some disadvantages: 1) BHD coupled with EDC enables improved chemical reactivity and a stable modified product. 2) The isotope-encoded benzylic acid used for d5-BHD synthesis is commercially available and inexpensive; high-yield d5-BHD can be obtained conveniently with a two-step synthesis. 3) The d5-BHD, incorporating five deuterium atoms, improves the resolution and accuracy of quantification, especially for large proteolytic peptides. On the other hand, there is a small isotope effect on LC elution when a long chromatographic gradient was used for our custom-made C18 column. Further, the reagent is unable to differentiate first- and second-shell ligand interactions, and a more discriminating labeling protocol or reagent are needed. For the future, we wish to extend this new footprinting approach to other hard metal-ion binding and, via a titration protocol, to test its ability to determine metal-binding affinity.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge research supported by grants NIH P41RR103422, as well as Dr. Gerstenecker from Washington University in St. Louis for his advice for this project and Protein Metrics for software.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Synthetic route and MS spectra for BHD and d5-BHD; modification reaction by BHD coupling reaction; condition screening for BHD; workflow of isotope-encoded covalent labeling; EIC for unmodified peptide 78–86; modification extent and calculated SASA of D and E residues at the EF-hand; summary of carboxylic acid modification percentage for Ca2+-free/Ca2+bound CaM and Mg2+-free/Mg2+-bound CaM.
Notes
Any additional relevant notes should be placed here.
REFERENCES
- (1).Dudev T, Lim C, Competition among metal ions for protein binding sites: Determinants of metal ion selectivity in proteins. Chem. Rev. 2014, 114, 538–556. [DOI] [PubMed] [Google Scholar]
- (2).Tainer JA; Roberts VA; Getzoff ED, Protein metal-binding sites. Curr. Opin. Chem. Biol. 1992, 3, 378–387. [DOI] [PubMed] [Google Scholar]
- (3).Bowman SJ; Bridwell-Rabb J Drennan CL Metalloprotein crystallography: More than a structure., Acc. Chem. Res. 2016, 49, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gregory DS; Martin AC; Cheetham JC; Rees AR, The prediction and characterization of metal binding sites in proteins. Protein Eng. 1993, 6, 29–35. [DOI] [PubMed] [Google Scholar]
- (5).Katz AK; Glusker JP; Beebe SA; Bock CW, Calcium ion coordination: A comparison with that of beryllium, magnesium, and zinc. J. Am. Chem. Soc. 1996, 118, 5752–5763. [Google Scholar]
- (6).Senguen FT; Grabarek Z, X‑ray structures of magnesium and manganese complexes with the N‑terminal domain of calmodulin: Insights into the mechanism and specificity of metal ion binding to an EF-hand. Biochemistry, 2012, 51, 6182–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Denessiouk K; Permyakov S; Denesyuk A; Permyakov E; Johnson MS, Two structural motifs within canonical EF-hand calcium-binding domains identify five different classes of calcium buffers and sensors. PLoS One 2014, 9, e109287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dudev T; Chang L; Lim C, Factors governing the substitution of La3+ for Ca2+ and Mg2+ in metalloproteins: A DFT/CDM study. J. Am. Chem. Soc. 2005, 127, 4091–4103. [DOI] [PubMed] [Google Scholar]
- (9).Payandeh J; Pfoh R; Pai EF, The structure and regulation of magnesium selective ion channels. Biochim. Biophys. Acta 2013, 1828, 2778–2792. [DOI] [PubMed] [Google Scholar]
- (10).Jesnsen MR, Petersen G; Lauritzen C; Pedersen J; Led JJ, Metal binding sites in proteins: Identification and characterization by paramagnetic NMR relaxation. Biochemistry 2005, 44, 11014–11023. [DOI] [PubMed] [Google Scholar]
- (11).Zhang Q; Dai X; Cong Y; Zhang J; Chen D; Dougherty MT; Wang J; Ludtke SJ; Schmid MF; Chiu W, Cryo-EM structure of a molluscan hemocyanin suggests its allosteric mechanism. Structure, 2013, 21, 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kojetin DJ; Matta-Camacho E; Hughes TS; Srinivasan S; Nwachukwu JC; Cavvet V; Nowak J; Chalmers MJ; Marciano DP; Kamenecka TM; Shulman AI; Rance M; Griffin PR; Bruning JB; Nettles KW, Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nat. Commun. 2015, 6, 8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fast CS; Vahidi S; Konermann L, Changes in enzyme structural dynamics studied by hydrogen exchange-mass spectrometry: Ligand binding effects or catalytically relevant motions? Anal. Chem. 2017, 89, 13326–13333. [DOI] [PubMed] [Google Scholar]
- (14).Jensen PF; Comamala G; Trelle MB; Madsen JB; JØrgensen TJD; Rand KD, Removal of N‑linked glycosylations at acidic pH by PNGase A facilitates hydrogen/deuterium exchange mass spectrometry analysis of N‑linked glycoproteins. Anal. Chem. 2016, 88, 12479–12488. [DOI] [PubMed] [Google Scholar]
- (15).Wang L; Chance MR, Protein footprinting comes of age: Mass spectrometry for biophysical structure assessment. Mol. Cell. Proteomics 2017, 16, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhu M; Rampel DL; Du Z; Gross ML, Quantification of protein-ligand interactions by mass spectrometry, titration, and H/D exchange: PLIMSTEX. J. Am. Chem. Soc. 2003, 125, 5252–5253. [DOI] [PubMed] [Google Scholar]
- (17).Zhang B; Cheng M; Rampel D; Gross ML, Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods 2018, 144, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang B; Rempel DL; Gross ML, Protein footprinting by carbenes on a fast photochemical oxidation of proteins (FPOP) platform. J. Am. Soc. Mass Spectrom. 2016, 27, 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jumper CC; Schriemer DC, Mass spectrometry of laser-initiated carbene reactions for protein topographic analysis. Anal. Chem. 2011, 83, 2913–2920. [DOI] [PubMed] [Google Scholar]
- (20).Chen J; Cui W; Gross ML, New protein footprinting: Fast photochemical iodination combined with top-down and bottom-up mass spectrometry. J Am Soc Mass Spectrom. 2012, 23, 1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cheng M; Zhang B; Cui W; Gross ML, Laser-initiated radical trifluoromethylation of peptides and proteins: Application to mass-spectrometry-based protein footprinting. Angew. Chem. Int. Ed. 2017, 56, 14007–14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hoare DG; Koshland DE Jr., A method for the quantitative modification and estimation of carboxylic acid groups in proteins. J. Biol. Chem. 1967, 242, 2447–2453. [PubMed] [Google Scholar]
- (23).Underbakke ES; Zhu Y; Kiessling LL, Isotope-coded affinity tags with tunable reactivities for protein footprinting. Angew. Chem. Int. Ed. 2008, 47, 9677–9680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Borotto NB; Zhou YP; Hollingsworth SR; Hale JE; Graban EM; Vaughan RC; Vachet RW, Investigating therapeutic protein structure with diethylpyrocarbonate labeling and mass spectrometry. Anal. Chem. 2015, 87, 10627–10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wen J; Zhang H; Gross ML; Blankenship RE, Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc. Natl. Acad. Sci. USA 2009, 106, 6134–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wecksler AT; Kalo MS; Deperalta G, Mapping of Fab-1:VEGF interface using carboxyl group footprinting mass spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 2077–2080. [DOI] [PubMed] [Google Scholar]
- (27).Zhang H; Shen W; Rempel D; Monsey J; Vidasky I; Gross ML; Bose R, Carboxyl-group footprinting maps the dimerization interface and phosphorylationinduced conformational changes of a membrane-associated tyrosine kinase. Mol. Cell. Proteomics. 2011, 10, M110.005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li K; Chen G; Mo J; Huang R; Deyanova EG; Beno BR; O’Neil SR; Tymiak AA; Gross ML, Orthogonal mass spectrometry-based footprinting for epitope mapping and structural characterization: The IL-6 receptor upon binding of protein therapeutics. Anal. Chem. 2017, 89, 7742–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gau B; Garai K; Frieden C; Gross ML, Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry 2011, 50, 8117–8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kaur P; Tomechko SE; Kiselar J; Shi W; Deperalta G; Wecksler AT; Gokulrangan G; Ling V; Chance MR, Characterizing monoclonal antibody structure by carboxyl group footprinting. mAbs 2015, 7, 540–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lin M; Krawitz D; Callahan MD; Deperalta G; Wecksler AT, Characterization of ELISA antibody-antigen interaction using footprinting-mass spectrometry and negative staining transmission electron microscopy. J. Am. Soc. Mass Spectrom. 2018, 29, 961–971. [DOI] [PubMed] [Google Scholar]
- (32).Leitner A; Joachimiak LA; Unverdorben P; Walzthoeni T; Frydmam J; Förster F; Aebersold R, Chemical cross-linking/mass spectrometry targeting acidic residues in proteins and protein complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9455–9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kahsai AW; Rajagopal S; Sun J; Xiao K, Monitoring protein conformational changes and dynamics using stable-isotope labeling and mass spectrometry. Nat. Protoc. 2014, 9, 1301–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li Z; Bai X; Deng Q; Zhang G; Zhou L; Liu Y; Wang J, Wang Y, Preliminary SAR and biological evaluation of antitubercular triazolothiadiazine derivatives against drug-susceptible and drug-resistant Mtb strains. Bioorganic Med. Chem. 2017, 25, 213–220. [DOI] [PubMed] [Google Scholar]
- (35).Liu W; Ng J; Meluzzi D; Banderia N; Gutierrez M; Simmons TL; Schultz AW; Linington RG; Moore BG; Gerwick WH; Pevzner PA; Dorrestein PC, Interpretation of tandem mass spectra obtained from cyclic nonribosomal peptides. Anal. Chem. 2009, 81, 4200–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Turowski M; Yamakawa N; Meller J Kimata K; Ikegami T; Hosoya K; Tanaka N; Thornton ER, Deuterium isotope effects on hydrophobic interactions: The importance of dispersion interactions in the hydrophobic phase. J. Am. Chem. Soc, 2003, 125, 13836–13849. [DOI] [PubMed] [Google Scholar]
- (37).Guo K; Ji C; Li L, Stable-isotope dimethylation labeling combined with LC-ESI MS for quantification of amine-containing metabolites in biological samples. Anal. Chem, 2007, 79, 8631–8638. [DOI] [PubMed] [Google Scholar]
- (38).Ouyang H; Vogel HJ, Metal ion binding to calmodulin: NMR and fluorescence studies. Bio. Metals 1998, 11, 213–222. [DOI] [PubMed] [Google Scholar]
- (39).Ngoka LC; Gross ML, A nomenclature system for labeling cyclic peptide fragments. J. Am. Soc. Mass Spectrom. 1999, 10, 360–363. [DOI] [PubMed] [Google Scholar]
- (40).McCarthy DJ, Smyth GK, Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ohashi W; Hirota H; Yamazaki T, Solution structure and fluctuation of the Mg2+-bound form of calmodulin C-terminal domain. Protein Sci. 2011, 20, 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Vetter SW, Leclerc E, Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem. 2003, 270, 404–414. [DOI] [PubMed] [Google Scholar]
- (43).Grabarek Z, Insights into modulation of calcium signaling by magnesium in calmodulin, troponin C and related EF-hand proteins. Biochim. Biophys. Acta. 2011, 1813, 913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gifford JL; Walsh MP; Vogel HJ, Structures and metal-ion-binding properties of the Ca2+-binding helix–loop–helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. [DOI] [PubMed] [Google Scholar]
- (45).Zhang H; Wen J; Huang R; Blankenship RE; Gross ML, Mass spectrometry-based carboxyl footprinting of proteins: Method evaluation. Int. J. Mass Spectrom.2012, 312, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Singh N; Karpichev Y; Sharma R; Gupta B; Sahu AK; Satnami M; Ghosh KK, From α-nucleophiles to functionalized aggregates: exploring the reactivity of hydroxamate ion towards esterolytic reactions in micelles. Org. Biomol. Chem. 2015, 13, 2827–2848. [DOI] [PubMed] [Google Scholar]
- (47).Ohki S; Ikura M; Zhang M, Identification of Mg2+-binding sites and the role of Mg2+ on target recognition by calmodulin. Biochem. 1997, 36, 4309–4316. [DOI] [PubMed] [Google Scholar]
- (48).Gilli R; Lafitte D; Lopez C; Kihoffer M; Makarov A; Briand C; Haiech J, Thermodynamic analysis of calcium and magnesium binding to calmodulin. Biochemistry, 1998, 37, 5450–5456. [DOI] [PubMed] [Google Scholar]
- (49).Cates MS, Teodoro ML, Phillips GN, Molecular mechanisms of calcium and magnesium binding to parvalbumin. Biophys. J. 2002, 82, 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shannon RD, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.