

Abstract
Immunotherapy has emerged as a key pillar of cancer treatment. To build upon the recent successes of immunotherapy, intense research efforts are aimed at a molecular understanding of anti-tumor immune responses, identification of biomarkers of immunotherapy response and resistance, and novel strategies to circumvent resistance. These studies are revealing new insight into the intricacies of tumor cell recognition by the immune system, in large part through Major Histocompatibility Complexes (MHCs). Though tumor cells widely express MHC-I, a subset of tumors originating from a variety of tissues also express MHC-II, an antigen presenting complex traditionally associated with professional antigen presenting cells (APCs). MHC-II is critical for antigen presentation to CD4+ T-lymphocytes, whose role in anti-tumor immunity is becoming increasingly appreciated. Accumulating evidence demonstrates that tumor-specific MHC-II associates with favorable outcomes in patients with cancer, including those treated with immunotherapies, and with tumor rejection in murine models. Herein, we will review current research regarding tumor-enriched MHC-II expression and regulation in a range of human tumors and murine models, and the possible therapeutic applications of tumor-specific MHC-II.
Keywords: Cancer Immunotherapy, Major Histocompatibility Complex Class-II, Immune Checkpoint Inhibition
Introduction
Recent advances in cancer therapy have shown clear benefits to targeting the immune system. Immune checkpoint inhibition (ICI) and other forms of immunotherapy have led to impressive gains in survival for many patients, including those with metastatic disease1,2. Despite these successes, most cancer patients treated with immunotherapy experience intrinsic or acquired resistance, and many have immune-related adverse events3,4. There are very few currently approved therapies to augment responsiveness to ICI and clinically useful biomarkers of response or resistance to target patients to appropriate treatment regimens. As novel immunotherapies and immunotherapy combinations are developed, understanding different facets of the anti-tumor immune response, including how tumors are recognized by T cells, may yield new insights and therapeutic applications.
ICI efficacy requires tumor antigens to be recognized by T cells. This critical step is mediated by Major Histocompatibility Complex (MHC): T Cell Receptor (TCR) interactions. MHC-class I (MHC-I) molecules are expressed by most nucleated cells and primarily present endogenously-derived peptide antigens to CD8+ T cells. MHC-class II (MHC-II) molecules are predominantly expressed by professional antigen presenting cells (pAPCs) such as dendritic cells (DCs), B cells and macrophages, and primarily present exogenously-derived peptide antigens to CD4+ T cells. Though cytotoxic CD8+ T cells are thought to be the primary effector cell type in the success of ICI, and thus, a major focus of immuno-oncology research, CD4+ T cells play critical roles in supporting CD8+ T cell activation, generation of memory T cells, and are now recognized as being necessary for an effective response to ICI5–13.
Despite the constitutive expression pattern of MHC-II on pAPCs, many other cell types, including some tumors, are capable of expressing MHC-II14. Tumor-specific MHC-II expression (tsMHC-II) may increase recognition of a tumor by the immune system, and therefore may play an important role in immunotherapy. TsMHC-II has been associated with superior prognosis, improved response to ICI in humans and increased tumor rejection in murine models15–22. Herein, we will review the association of tsMHC-II with outcomes in human tumors, the functional consequences of tsMHC-II upregulation in murine models, mechanisms of regulation of tsMHC-II, and the possible clinical applications of future research on tsMHC-II, including its development as a biomarker of response to immunotherapy, and therapeutic tsMHC-II upregulation as a possible treatment strategy in cancer.
The MHC-II pathway
MHC-II is a heterodimer consisting of an alpha and a beta chain. In humans, there are multiple MHC-II isotypes: HLA-DR, HLA-DP, and HLA-DQ23. MHC proteins are highly polymorphic, allowing for diversity of presented peptides within the population24. Interestingly, MHC-II may be able to bind a higher diversity of peptides than MHC-I, which could be therapeutically exploited to increase the likelihood tumor neoantigen recognition by T cells25,26. As compared to MHC-I, which binds peptides 8–10 amino acids in length, MHC-II binds peptides greater than 13 amino acids, and accommodates peptide side chains within its binding pocket25, two traits that increase MHC-II peptide diversity.
Though constitutively expressed primarily by mature pAPCs, MHC-II expression can be induced in a range of cell types, often due to inflammatory signaling processes. Expression of MHC-II and its related machinery is driven by the transcriptional master regulator Class II Transactivator (CIITA). Although CIITA does not directly bind DNA, its scaffolding activity recruits the necessary transcription factors at transcriptional start sites of MHC-II related genes. CIITA is necessary and sufficient for induction of a fully functional MHC-II pathway27,28 and, in some cases, for moderate MHC-I induction29,30. Importantly, CIITA is essential for MHC-II induction by interferon-gamma (IFN-γ)31. CIITA expression is governed by four distinct promoters, leading to transcripts with differing first exons and distinct CIITA isoforms. Promoters I (pI) and III (pIII) drive constitutive MHC-II expression in DCs and B cells, respectively32. Promoter IV (pIV) is inducible with IFN-γ stimulation in many cell types33. There is some evidence that pIII may be inducible with IFN-γ in endothelial cells and fibroblasts, but this inducibility appears to be weaker than that of pIV34,35. The function of promoter II is not well described and does not appear to be highly utilized in any human tissue36. Consistent with this observation, promoters I, III, and IV are more highly conserved between mouse and human32. Although CIITA isoforms derived from pI, pIII, and pIV all drive expression of MHC-II and related machinery, the isoform derived from pI is transcriptionally the most potent activator of the MHC-II gene, perhaps explaining the high density of MHC-II molecules on DCs37.
IFN-γ induction of MHC-II expression occurs by the following mechanism (Figure 1A): IFN-γ binds to its receptor at the cell surface, inducing activation of Janus Activated Kinases (JAK)-1 and JAK2, intracellular tyrosine kinases that phosphorylate on the transcription factor Signal Transducer and Activator of Transcription-1 (STAT1). Once phosphorylated, STAT1 dimerizes and translocates to the nucleus, where it binds to cis-acting GAS elements in the promoter regions of IFN responsive genes, including CIITA. Notably, CIITA upregulation does not occur following IFN-γ stimulation in JAK1- or STAT1-deficient cells, suggesting that JAK/STAT signaling is indispensable for IFNγ-mediated MHC-II induction38,39. Once bound to the GAS element, STAT1 is stabilized by interaction with the ubiquitous transcription factor USF-1 that binds to nearby E-box elements in the CIITA promoter32,33. The transcription factor interferon regulatory factor-1 (IRF-1) is also potently induced by IFN-γ, and like STAT1, loss of IRF-1 impairs IFNγ-mediated CIITA induction. Mutagenesis assays demonstrated that the GAS element, E box and the IRF-1 binding site of pIV are each essential for IFNγ-inducible CIITA expression33. Once CIITA is upregulated and transported to the nucleus, it acts as a scaffold, attracting RFX family transcription factors to the start sites of MHC-II related genes. Many RFX family proteins are ubiquitously expressed regardless of MHC-II status or cell type, indicating that MHC-II expression is generally regulated at the level of CIITA expression37.
Figure 1: Pathway of IFN-γ mediated upregulation of tsMHC-II.

A) IFN-γ binds to the IFN-γ receptor (IFNGR) leading to JAK1/2 phosphorylation. JAK1/2 phosphorylates STAT1 which translocates to the nucleus and cooperates with other transcription factors, like IRF-1 to activate promoter IV of CIITA. CIITA is then translated and returns to the nucleus (not shown) where it acts as a scaffold for RFX family members and drives transcription of MHC-II related genes such as HLA-DR, DP, DQ, invariant chain (Ii), and HLA-DM. MHC-II alpha and beta chains are assembled and complexed with Ii in the endoplasmic reticulum. MHC-II bound to Ii and associated with HLA-DM bud off in a vesicle. Ii targets this vesicle to the acidic endosomal compartment. In the acidic environment, Ii is degraded to CLIP. HLA-DM catalyzes the release of CLIP and binding of antigen. Stabilized peptide:MHC-II complexes translocate to the cell surface where MHC-II can present antigen to CD4 T cells. B) MHC-I is generally constitutively expressed. Cytosolic proteins are degraded by the proteasome into peptide antigens. These antigens are loaded into the ER by TAP1/TAP2 transporter proteins where they can be loaded onto the assembled MHC-I alpha chain and β2M complex. This complex is transported through the Golgi (not shown) to the cell surface where it can present peptide antigens to CD8+ T cells.
Following CIITA-mediated expression of MHC-II related machinery, MHC-II alpha and beta chains assemble in the endoplasmic reticulum (ER) in complex with the MHC class II-associated invariant chain (Ii, CD74). Ii occupies the peptide-binding groove of MHC-II to prevent peptide loading within the ER. In the absence of Ii, MHC-II may be loaded with endogenously-derived ER peptides, or may be retained in the ER as a misfolded protein40–43. Like other membrane bound proteins, MHC-II is transported via vesicles which bud from the ER. Ii targets MHC-II containing vesicles to acidic endosomes, where MHC-II is loaded with exogenously-derived antigens acquired through endocytosis, or with antigens produced within vesicles (e.g., from endosomally-localized microbes). MHC-II-containing vesicles may also fuse with autophagosomes, allowing for cross presentation of endogenously-derived peptides44. In acidic vesicles, Ii is degraded into a short fragment called the class II-associated invariant chain peptide (CLIP). If MHC-II fails to bind peptide in the endosome, it is degraded in the acidic environment. Release of CLIP and binding of peptide antigen is catalyzed by the co-chaperone HLA-DM. HLA-DM also catalyzes the release of weakly bound peptides, ensuring that only strongly bound peptide:MHC-II (pMHC-II) complexes reach the cell surface. In some cells types such as splenic B cells, thymic epithelial cells and certain DC lineages, this process is aided by another co-chaperone, HLA-DO, which antagonizes the binding of peptides to MHC-II, further ensuring that only strong pMHC-II bonds can form37,45. MHC-II complexes are stable only when antigen-loaded, and importantly, only stable MHC-II complexes remain at the cell surface46. Once on the cell surface, pMHC-II complexes can be recognized T-lymphocytes, primarily CD4+ T cells. There is some evidence that CD8+ T cells may recognize MHC-II in the absence of CD4+ T-cells (e.g., due to CD4 knockout, or advanced HIV infection)47, although the implications of this are unclear.
Comparison between MHC-II and MHC-I
Though both MHC-I and MHC-II present peptide antigen to T cells, there are many differences in their structure and function (Figure 1B). Unlike the heterodimeric MHC-II, MHC-I consists of a single polymorphic alpha chain which associates with the non-polymorphic beta-2-microglobulin (β2M). MHC-I canonically presents endogenously produced antigens, which are generated by the proteasome and shuttled by TAP1/TAP2 transporter proteins into the ER, where MHC-I is loaded with peptide. MHC-I is typically constitutively expressed by all nucleated cells, including cancer cells. In most tissues, MHC-I expression is not controlled by a master regulator, in contrast to MHC-II regulation by CIITA. However, in certain immune cells, particularly T cells, NOD-like receptor family CARD domain containing 5 (NLRC5) acts as a master transcriptional regulator of MHC-I and related machinery48,49. Though generally constitutively expressed, MHC-I expression is inducible by IFN-γ and NF-κB pathway activation48,50.
Despite the near ubiquitous expression pattern of MHC-I, some cancer cells may lose MHC-I expression as a mechanism of immune escape. Although loss of MHC-I expression is a trigger for NK-mediated cell killing, many tumors are able to evade immunosurveillance without expression of MHC-I. Loss of MHC-I expression, often mediated by loss of β2M, has been reported as a mechanism of resistance to anti-PD-1 therapy51,52. Interestingly, some tumors which lose expression of MHC-I may retain expression of MHC-II, though the functional significance of this is unclear22. Likewise, several melanoma cell lines which do not express MHC-II in response to IFN-γ still express high levels of MHC-I15. These observations suggest that MHC-I and MHC-II are independently regulated in cancer and that expression of MHC-I and MHC-II may have independent implications for cancer immunotherapy.
CD4+ T cells in cancer immunotherapy
TsMHC-II may play a role in stimulation of CD4+ T cell subsets, which have diverse functions in immunity. T helper 1 (Th1) CD4+ cells secrete IFN-γ and other activating cytokines, while regulatory T cells (Tregs) suppress immunity and inflammation, playing a central role in tolerance53. Other CD4+ T cell subsets such as Th2 and Th17 have less clear roles in anti-cancer immunity, with both pro- and anti- tumor effects reported53, and are reviewed more comprehensively elsewhere53–56. Importantly, CD4+ T cells have been shown to recognize cancer-associated antigens and, in some instances, recognize a wider array of antigens than CD8+ T cells57–59. This may be due to the greater repertoire of antigens which can be presented on MHC-II and may be functionally significant for tumors which have fewer candidate neoantigens25. Intriguingly, a recent report showed that mutations predisposed for MHC-II presentation are negatively selected during tumorigenesis. Notably, selective pressure against MHC-II-restricted neoantigens was stronger than that against MHC-I restricted neoantigens, indicating that CD4+ T cell immunosurveillance is an important suppressor of cancer development and progression60. Additionally, CD8+ T cells cannot generate an effective, long-lasting memory response without CD4+ help5–7,61. As evidence of this, response to anti-PD-1 therapy, which is thought to reinvigorate CD8+ cytotoxic T cell responses, requires CD4+ T cells in several murine models13. Additionally, therapeutic adoptive cell transfer of CD4+ T cells is an emerging concept with promising results62–64. Complete melanoma regressions have been documented following infusion of CD4+ T cells specific for a melanoma-associated antigen (BRAF-V600E or NY-ESO-1)62,63. Recent work in CAR-T based therapies also demonstrated improved responses when CD8+:CD4+ T cell ratios are controlled for optimal T cell help65–67. In addition to their role as helper T cells, CD4+ T cells can be directly cytotoxic and tumoricidal in some instances11,68–70. These data, generated in a variety of immunotherapy modalities, demonstrate that CD4+ T cells are active and critical players in anti-tumor immunity. Thus, novel strategies to enhance CD4+ T cell activation may produce tangible benefits.
An unresolved consideration is what effect, if any, tsMHC-II has on CD4+ Tregs. Tregs are abundant in many tumors and can be potently suppressive71,72. Tregs also require T cell receptor (TCR) stimulation by MHC-II to maintain their suppressive activity73. However, the favorable association of tsMHC-II with improved immune-mediated outcomes would suggest that tsMHC-II may fail to activate Tregs, though robust evidence for this is lacking. In murine tumors transduced with CIITA, no increase in the Treg marker FoxP3 was seen74. Treg infiltration in MHC-II+ human tumors has not been robustly assessed.
An intriguing open question related to tsMHC-II is whether or not CD4+ T cells require classical co-stimulation (i.e. via CD80 and CD86) in all cases for initial priming of naïve cells and/or subsequent reactivation, or if non-classical co-stimulatory molecules may substitute. Canonically, T cell activation requires signal 1 (MHC:T cell receptor binding) and signal 2 (co-stimulation with CD80/86 binding to CD28). Signal 1 in the absence of signal 2 may lead to anergy or T cell tolerance75. Tumor cells typically do not express CD80/8676; however, many co-stimulatory receptors beyond CD80/86 have been identified (reviewed in Reference75). Some tumor cells express non-classical co-stimulatory molecules which may be sufficient to activate certain subsets of T cells14,76. An example is CD70, a member of the tumor necrosis factor super family 7 expressed by renal cell carcinoma and other tumor types. CD70 binds to CD27 on T cells and can induce lymphocyte proliferation or apoptosis77. OX40-ligand, another co-stimulatory receptor, is expressed by some glioblastomas and other tumors78. T cells in the tumor microenvironment (TME) may also receive co-stimulation from adjacent, juxtaposed immune cells56 or may have been previously primed and activated in the tumor draining lymph node prior to trafficking to the tumor site. Requirements for additional co-stimulation in previously educated T cells are unclear. The role of various co-stimulatory factors in anti-tumor immunity merits further study.
MHC-II in human cancers
Expression of MHC-II and related pathway components by cancer cells has been seen in a variety of human tumors including: melanoma15,22,79, breast cancer17,80–88, colorectal cancer89,90, ovarian cancer91,92, prostate cancer93, classic Hodgkin lymphoma21, glioma94, and non-small cell lung cancer95 (Figure 2). Notably, this includes many solid tumors where the tissue of origin does not ordinarily express MHC-II molecules. Several studies, in multiple cancer types, have found an association between tsMHC-II and favorable prognosis. TsMHC-II expression has been associated with improved progression-free (PFS) and overall survival (OS) in melanoma and classic Hodgkin lymphoma patients treated with anti-PD-1/anti-PD-L1, but not anti-CTLA-415,21,22,79. Importantly, these studies used immunohistochemistry (IHC) and co-staining with a tumor specific marker (e.g. SOX-10 for melanoma) to delineate tsMHC-II versus MHC-II expressed by infiltrating immune cells or stroma. TsMHC-II did not predict survival in an unselected cohort of melanoma patients, suggesting a specificity of tsMHC-II toward immune-mediated tumor outcomes in melanoma15. In a study of 681 triple negative breast cancer (TNBC) patients, approximately 30% had some degree of tsMHC-II positivity by IHC on treatment-naïve resection specimens, and tsMHC-II was correlated with better disease-free survival (DFS) in patients with lymph node metastases following adjuvant radiotherapy and/or chemotherapy17. In another study using RNA-sequencing of 47 TNBC tumors, MHC-II pathway genes were the most strongly correlated with improved PFS. High expression of a 13 gene composite of the pathway (including CIITA, CD74, HLA-DPA1, HLA-DPB1, HLA-DPB2, HLA-DQA1, HLA-DRB1, HLA-DRB5, and HLA-DRB6) as well as CIITA or CD74 alone was significantly correlated with improved PFS. This finding was validated in an independent publicly-available Affymetrix microarray dataset85. A limitation of this study is the use of RNA-sequencing, which does not inform on which cell types express the MHC-II-related genes of interest. IHC analysis of 112 unselected primary breast cancers showed that tumors positive for HLA-DR, Ii, and HLA-DM had significantly better PFS and OS than tumors negative for HLA-DR or expressing HLA-DR without Ii and HLA-DM. A significant limitation of this study is the lack of stratification by subtype and small sample size in individual groups: only 9 tumors expressed all three molecules82. A separate group found that HLA-DMB RNA expression was correlated with improved survival in 38 cases of advanced-stage serous ovarian cancer. Immunofluorescence performed on a subset of tumors from this cohort showed that HLA-DR staining was present on both the epithelial cancer cells and infiltrating CD8+ T cells91.
Figure 2: Cancer types which have been shown to express MHC-II.

A diverse subset of human tumors has been shown to express MHC-II. Those tumor types, and the outcomes associated with tsMHC-II are shown here.
In addition to survival, tsMHC-II has been associated with higher number of both CD4+ and CD8+ tumor infiltrating lymphocytes (TILs), absence of lymphovascular invasion, increased formation of tertiary lymphoid structures, upregulation of genes associated with IFN-γ pathway activation (including CD274 which encodes PD-L1)17,91, and higher levels of IFNG, IL2, and IL12 mRNA (Th1 cytokines)82. IL4, IL10, and TGFB1 mRNA (Th2 cytokines) did not differ by tsMHC-II, suggesting a skewing toward Th1 polarization82. Taken together, these data suggest that increased expression of MHC-II or related pathway components by tumor cells (or in the bulk tumor population) is associated with better prognosis and enhanced anti-tumor immunity. This leads to the hypothesis that strategies to increase tsMHC-II may be therapeutic, particularly in combination with immunotherapies.
Another intriguing study in breast cancer raises the possibility of such a therapeutic application. Targeted next-generation sequencing was performed on 74 clinically defined TNBC tumors who all had residual disease burden in the breast following neoadjuvant chemotherapy, which is the group at highest risk for disease recurrence following surgery. Ras/MAPK pathway alterations and a high transcriptional MEK signature were significantly assoicated with low TIL burden. A high MEK signature was further associated with low tumor specific MHC-I, MHC-II and PD-L1. Inhibition of the Ras/MAPK pathway increased anti-PD-1 sensitivity in mouse models of breast cancer and increased MHC-I and MHC-II expression in mouse and human breast cancer cell lines. These data suggest that MEK inhibition may be a means of sentizing breast tumors to anti-PD-1/anti-PD-L1 therapies via upregulation of tumor specific antigen presentation, but stop short of establishing a causal link between the two80. Clinical trials are underway testing combinations of MEK inhibitors with anti-PD-L1 therapy in metastatic breast cancer.
TsMHC-II may be a clinically useful biomarker of a T cell-inflamed tumor, as MHC-II upregulation is downstream of IFN-γ, and tsMHC-II can be measured by IHC, a technique which is routinely used clinically and is more efficient than RNA-sequencing for an IFN-γ signature96. Though PD-L1 can also be measured by IHC, its use as a biomarker is complicated by multiple available assays, imperfect concordance between assays and mixed results in clinical trials, with robust responses to anti-PD-1/anti-PD-L1 seen in some patients with no or low PD-L1 expression97,98. Notably, tsMHC-II may also be regulated independently of IFN-γ. Not all tsMHC-II expression can be explained solely as a byproduct of IFN-γ and immune activation, as many melanoma cell lines grown in vitro in the absence of any immune stimuli express high levels of MHC-II. Melanoma cell line-specific MHC-II expression can be categorized by three phenotypes: 1) constitutive expression, 2) no/low baseline expression with IFN-γ inducible population, and 3) no baseline expression with no induction with IFN-γ15,84,85. The inducible IFN-γ phenotype has also been replicated in a patient derived xenograft (PDX) model of melanoma, suggesting that inducible MHC-II expression is likely a phenotype of human tumors in vivo, not only cultured cells15. Importantly, whether tsMHC-II serves as a biomarker of IFN-γ response is distinct from a possible functional role of tsMHC-II itself in augmenting an immune response. Many studies in murine model systems have investigated the functional role of tsMHC-II through genetic manipulation, summarized below.
Consequences of tsMHC-II upregulation in murine models
Correlative associations in human tumors do not establish causality, and thus, the use of animal models is required to experimentally delineate the role of tsMHC-II. TsMHC-II in mouse models has been studied by multiple groups, with most finding that transduction of tumor cells with ectopic MHC-II or CIITA increases immune-mediated tumor rejection18,99–102. However, some contrasting reports have shown that MHC-II or CIITA has no effect or, in some cases, accelerates tumor growth103–105. These studies and possible hypotheses for their conflicting results are summarized below and in Table 1.
Table 1:
Effect of tsMHC-II in murine models
| Tumor Site(s) of Origin | MHC-II induced by transduction of | tsMHC-II associated with | Ref(s) |
|---|---|---|---|
| Breast, sarcoma, lung, colon | IFNG, MHC-II (Ak), Ciita | Tumor rejection and/or inhibited growth | 18,100,101,106–108 |
| Breast, lung, colon | Ciita | Resistance to challenge with parental cells | 18,107 |
| Lung | Ciita | Increased tumor growth (only in high MHC-II expressing clones) | 103 |
| Sarcoma, Lung | MHC-II and Ii or Ciita | No change in tumor growth | 28,103,104 |
| Breast, Lung | IFNG, Ciita | Vaccination with killed tumor cells leads to resistance to re-challenge with parental | 100,103,107,134,135 |
| Breast | Ciita | IFN-γ production by splenocytes | 18 |
In the majority of studies, transduction of tumor cells with Ciita increased tumor rejection and led to resistance to challenge with parental cells in mouse models of breast cancer, sarcoma, lung cancer, and colon cancer18,100,101,106,107. In a mouse model of breast cancer, Ciita+ clones with the highest surface expression of MHC-II were rejected at the highest rate18, while depletion studies revealed that both CD4+ and CD8+ T cells, but not B-cells or natural killer (NK) cells, were necessary for tumor rejection. Furthermore, splenocytes harvested from animals that rejected Ciita+ tumor cells produced significantly more IFN-γ when stimulated ex vivo than control tumor bearing animals, suggesting systemic immune activation18. Surprisingly, mice rejecting Ciita+ tumors were immunized against re-challenge with parental (Ciita-) tumor cells, which could be conferred to tumor-naïve mice through co-injection of parental tumor cells with CD4+ or CD8+ splenocytes harvested from mice rejecting Ciita+ tumor cells107. Anti-CD80 and anti-CD86 blocking antibodies were used to test the importance of co-stimulation in MHC-II induced tumor rejection. Mice treated with the co-stimulation blocking antibodies failed to reject an MHC-II-transduced sarcoma line, which was rejected by control mice. Transduction of the sarcoma line with CD86 abrogated tumor growth and provided resistance to re-challenge with parental tumors similarly to transduction of MHC-II108. These results suggest that, as expected, co-stimulation can augment anti-tumor immunity, but that at least in some cases, tumor cells themselves do not need to express CD86 in order to elicit an immune response. Notably, depletion of DCs or macrophages had no impact on the ability of mice to reject Ciita+ tumor cells, raising the possibility that MHC-II+ tumor cells act as APCs to prime anti-tumor immunity107. This is a surprising result, given the widely accepted crucial nature of lymphatic pAPCs in initiating an adaptive immune response. As macrophages and DCs were not simultaneously depleted, it is possible that these cell types have redundancy in their functions. Furthermore, the role of B cells was not assessed. It is also possible that only the small quantity of residual DCs or macrophages which survived depletion are necessary to carry out their function.
Not all studies of tsMHC-II in murine models have shown increased immunogenicity. In a mouse model of lung cancer, single cell clones from a Ciita transduced population grew more aggressively in mice when cell surface MHC-II was highest. The polyclonal cell line and clones with low MHC-II expression grew more slowly than the parental line. A confounding variable in this model was that transduction with Ciita also increased MHC-I expression which was hypothesized to decrease NK cell-mediated surveillance, although characterization of the tumor immune infiltrate was not reported103. Another study reported that, while transduction of mouse sarcoma cells with MHC-II alone increased tumor rejection, combined transduction with MHC-II and invariant chain, or transduction with Ciita abrogated MHC-II-mediated rejection104. Although not directly reported, it is possible that high expression of Ii or H2-M (the mouse analog of human HLA-DM) and Ii (via CIITA) led to insufficient presentation of endogenous antigens, producing a blunted immunologic response20.
There are a number of factors that may account for the discrepancies noted above in terms of whether ectopic MHC-II or Ciita expression by tumor cells enhances anti-tumor immunity. A likely confounding variable is the uncertainty about which tumor associated antigens are capable of being presented on MHC-II in each model system. Elution and characterization of antigens bound to MHC-II remains difficult. Mouse tumor cell lines are also variable in terms of number of mutations and therefore number of candidate neoantigens. Because MHC-II canonically presents exogenously derived peptides, the mechanism and efficiency of loading of endogenously-derived peptides is unclear. This may depend on the degree of autophagy carried out in a given cell line under particular conditions44. Published studies also differ in the number of tumor cells injected, the injection site, and the mouse strains used – all of which could have immunologic consequences. Tumor model and mouse strain may also affect the expression of non-classical co-stimulatory molecules75, many of which have not been extensively studied in cancer. It is also difficult to compare levels of expression of MHC-II and pathway components induced by ectopic expression across studies. In a small cohort of unselected melanoma patients, tsMHC-II was not associated with survival, but has been associated with improved response to anti-PD-1/anti-PD-L115, leading to the possibility that some MHC-II+ murine models generate inflammation, leading to rapid T cell suppression via PD-1/PD-L1. Thus, ICI therapy may be required to unmask the pro-immunogenic effect of tsMHC-II. An intriguing but underexplored hypothesis is that induction of tsMHC-II may lead to selective pressure to upregulate immunoinhibitory molecules on TILs, such as Lymphocyte Activation Gene 3 (LAG-3; an immunoinhibitory receptor which binds MHC-II) and others.
Finally, there are a variety of novel proposed mechanisms by which tsMHC-II exerts immunogenic effects (Figure 3). Ciita+ tumor cells which either express a model antigen or are pulsed with model antigen peptide or protein can activate antigen specific-transgenic CD4+ T cells, showing that tsMHC-II can be directly recognized by CD4+ T cells in vitro18,109. One report described MHC-II-containing exosomes derived from MHC-II+ tumor cells which were immunostimulatory110; a finding consistent with the contrasting role of PD-L1-loaded exosomes that has been recently reported111. However, independent validation of these mechanisms is lacking at his time.
Figure 3: Mechanisms of tsMHC-II-mediated immunostimulation.
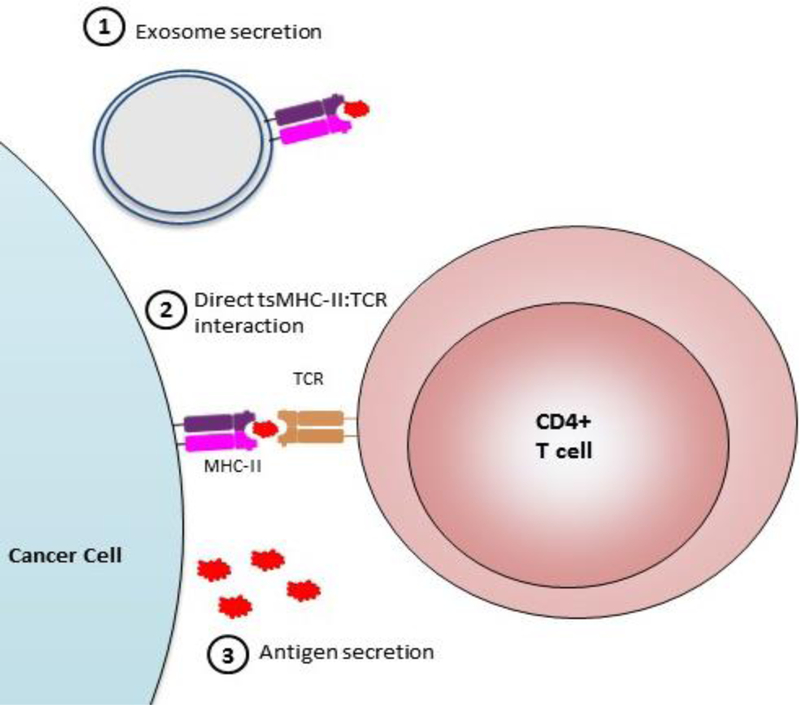
Mechanisms by which tsMHC-II has been proposed to affect immunity. 1) Cancer cells expressing MHC-II may secrete exosomes which can be immunostimulatory. 2) tsMHC-II may directly interact with CD4+ T cells to affect polarization and activation. 3) Cancer cells may secrete antigens which can be endocytosed and presented by pAPCs.
Regulation of MHC-II expression in cancer cells
As reviewed above, MHC-II expression can be regulated at the level of IFN-γ-driven Ciita expression, but some tumor cells are deficient in their IFN-γ response15. There are multiple pathways which may interact with and modulate MHC-II expression in cancer cells. JAK/STAT signaling is indispensable for MHC-II upregulation38,39, and JAK mutations are known to contribute to blunted interferon signaling and immunotherapy resistance112,113. Another intriguing example is MHC-II suppression by RAS/MAPK activation in breast cancer, which can be therapeutically reversed by MEK inhibition80. In contrast, MAPK activation in HLA-DR+ melanoma lines activated CIITA promoter pIII, a promoter activated primarily in B cells114. The opposite effects of MAPK on HLA-DR expression in these two studies80,114 may be explained by different tumor models (breast vs melanoma) or by differential mechanisms of regulation for constitutive, inducible, or stably null expression of MHC-II. Some melanoma cell lines which constitutively express HLA-DR have aberrant CIITA expression driven from pIII115 or aberrant constitutive activation of pIV, which has not been shown to be constitutively active in any other context116. Other tumor cells regulate inducible HLA-DR expression at pIV of CIITA, like other cell types117. In addition to IFN-γ, type I interferons, GM-CSF, IL-4 and TNF-α are known to upregulate MHC-II on dendritic cells118–120. Conversely, IL-10 downregulates MHC-II expression on pAPCs121. However, the effect of these cytokines on tsMHC-II is not known.
Inducible HLA-DR expression may also be modulated by retinoblastoma (Rb) protein122–124. Some cells can induce CIITA expression with IFN-γ stimulation, but not produce functional MHC-II at the cell surface. In some instances where Rb function is lost through genomic deletion or mutation, the defect from CIITA to cell-surface MHC-II can be rescued by reconstitution of functional Rb protein. However, in other cell types in which CIITA induction itself is defective in response to IFN-γ, Rb reconstitution does not result in full MHC-II expression122–125. These results suggest that expression of MHC-II at the cell surface may be regulated by CIITA and also at the post-CIITA level (e.g., by Rb).
Tumor cells may also select for mechanisms to downregulate MHC-II expression. For example, genomic alterations in CIITA are common in Hodgkin Lymphoma and primary mediastinal large B-cell lymphoma and are associated with reduced tsMHC-II expression126–128. Other non-genomic mechanism of silencing CIITA have also been found. Small cell lung cancer and neuroblastoma overexpress L-myc, N-myc, and human achaete-scute complex homologue-1 (HASH-1) which can bind to promoter IV of CIITA and repress the transcriptional response to IFN-γ 95,129. Epigenetic silencing may also be a mechanism of repression of MHC-II expression in human tumors130. Repression of MHC-II expression by tumor cells may be a mechanism of immune evasion, through avoiding recognition by anti-tumor CD4+ T cell subsets. Given the favorable association of tsMHC-II expression with response to immunotherapy, downregulation of tsMHC-II may contribute to resistance to ICI.
Therapeutic implications of tsMHC-II and future directions
TsMHC-II associates with improved prognosis, including in response to ICI, increased TILs, and pro-inflammatory interferon signaling in human tumors. Murine models suggest that there may be a causative relationship between tsMHC-II expression and increased immunogenicity. TsMHC-II may be a clinically actionable biomarker of response to ICI and novel strategies to upregulate tsMHC-II may improve response to immunotherapies.
Currently, there are very few clinically used biomarkers to predict response to immunotherapies and to target patients to single versus double agent regimens131. TsMHC-II expression correlates with response to anti-PD-1/anti-PD-L1 in melanoma and classic Hodgkin lymphoma15,21,22,79. Therefore, expression of tsMHC-II may portend a high likelihood of response to single agent anti-PD-1/anti-PD-L1, rendering the addition of anti-CTLA-4, and the consequent increased likelihood of immune-related adverse events, unnecessary. Conversely, absence of tsMHC-II suggests a lower probability of response to single agent anti-PD-1/anti-PD-L1 and a higher likelihood of additional benefit with combination anti-PD-1/anti-PD-L1 and anti-CTLA-4. Furthermore, novel immune checkpoint inhibitors, such as those which target LAG-3, are currently in development. As LAG-3 is an inhibitory receptor which binds to MHC-II132, it is reasonable to posit that tsMHC-II may exert a selective pressure to upregulate LAG-3 in the TME and therefore may be an appropriate biomarker for anti-LAG-3 therapies133. However, this hypothesis requires further study.
TsMHC-II has been studied in murine models as a possible tool in anti-tumor vaccines. Inoculation with non-viable tumor cells that express MHC-II protected mice from challenge with parental tumor cells103,107,134,135. An intriguing area of future research is therapeutic upregulation of MHC-II. Given the murine studies which find that upregulation of tsMHC-II often leads to tumor rejection, it is reasonable to hypothesize that upregulation of tsMHC-II in human tumors will enhance anti-tumor immunity and increase sensitivity to immune checkpoint blockade, but this remains to be rigorously tested. Likewise, endogenous antigens presented by tsMHC-II and the direct effect of tsMHC-II on antigen-specific CD4+ T cells are unknown. One hurdle in this space is a lack of sufficient understanding of what regulates MHC-II expression in the diverse landscape of human tumors and what prevents tsMHC-II expression. Studies in divergent tumor types and phenotypes have alternately shown that MEK inhibition upregulates80 or downregulates114 MHC-II expression. An intriguing possibility for therapeutic upregulation of MHC-II lies in epigenetic modifiers which have been shown to upregulate MHC-II expression in human and murine ovarian cancer cell lines, as well as ovarian cancer PDX models92. Other strategies to upregulate tsMHC-II are actively being investigated.
Summary & Conclusions
MHC-II is necessary for the activation of CD4+ T cells, which play diverse and crucial roles in anti-tumor immunity. Although MHC-II is canonically expressed by pAPCs, its expression is also noted on wide range of tumor cells. TsMHC-II has been associated with better patient outcomes, either overall or in response to immunotherapy with anti-PD-1/anti-PD-L1 agents. Upregulation of tsMHC-II and related pathway components often, but not always, leads to enhanced tumor rejection in murine models. TsMHC-II may be constitutively expressed, inducible with IFN-γ, or stably null. Each of these phenotypes may be the consequence of distinct molecular events. TsMHC-II holds promise as a biomarker of inflamed tumors and a higher likelihood of response to anti-PD-1/anti-PD-L1 agents and may be more easily translated as a predictive biomarker into clinical practice than other parameters. Finally, upregulation of tsMHC-II may be a novel means of enhancing anti-tumor immune responses. Further study on the regulation of MHC-II expression in tumors and the functional effects of tsMHC-II may yield new insights into cancer immunotherapy.
Acknowledgments:
This article is supported by the U.S. Department of Defense (BC170037; J. M. Balko) and the National Institute of General Medical Sciences (T32GM007347; M. L. Axelrod).
D. B. Johnson reports receiving commercial research grants from Bristol-Myers Squibb and Incyte, is a consultant/advisory board member for Array, Bristol-Myers Squibb, Genoptix, Merck, Incyte, and Novartis, and is inventor on a provisional patent on the use of MHC-II as a biomarker of immunotherapy response. J. M. Balko is a consultant/advisory board member for Novartis, reports receiving commercial research grants from Incyte, Bristol-Myers Squibb, and Genentech, and is inventor on a provisional patent on the use of MHC-II as a biomarker of immunotherapy response and a patent on HLADR as a biomarker for ICI response.
Footnotes
Conflicts and Disclosures: No potential conflicts of interest were disclosed by the other authors.
References Cited
- 1.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(1):23–34. 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015;372(26):2521–2532. 10.1056/NEJMoa1503093 [DOI] [PubMed] [Google Scholar]
- 3.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (80- ) 2018;359(6382):1350–1355. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. Longo DL, ed. N Engl J Med 2018;378(2):158–168. 10.1056/NEJMra1703481 [DOI] [PubMed] [Google Scholar]
- 5.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003;421(6925):852–856. 10.1038/nature01441 [DOI] [PubMed] [Google Scholar]
- 6.Sun JC, Bevan MJ. Defective CD8 T Cell Memory Following Acute Infection Without CD4 T Cell Help. Science (80- ) 2003;300(5617):339–342. 10.1126/science.1083317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat Rev Immunol 2016;16(2):102–111. 10.1038/nri.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ossendorp F, Mengedé E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med 1998;187(5):693–702. http://www.ncbi.nlm.nih.gov/pubmed/9480979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 2017;168(3):487–502. 10.1016/j.cell.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahrends T, Baba a N, Xiao Y, Yagita H, van Eenennaam H, Borst J. CD27 Agonism Plus PD-1 Blockade Recapitulates CD4+ T-cell Help in Therapeutic Anticancer Vaccination. Cancer Res 2016;76(10):2921–2931. 10.1158/0008-5472.CAN-15-3130 [DOI] [PubMed] [Google Scholar]
- 11.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4 + T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010;207(3):637–650. 10.1084/jem.20091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bos R, Sherman LA. CD4+ T-Cell Help in the Tumor Milieu Is Required for Recruitment and Cytolytic Function of CD8+ T Lymphocytes. Cancer Res 2010;70(21):8368–8377. 10.1158/0008-5472.CAN-10-1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer Immunol Res 2016;4(10):845–857. 10.1158/2326-6066.CIR-16-0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol 2014;14(11):719–730. 10.1038/nri3754 [DOI] [PubMed] [Google Scholar]
- 15.Johnson DB, Estrada MV., Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 2016;7:10582 10.1038/ncomms10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forero A, Li Y, Chen D, Grizzle WE, Updike KL, Merz ND, et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol Res 2016;4(5):390–399. 10.1158/2326-6066.CIR-15-0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park IA, Hwang S-H, Song IH, Heo S-H, Kim Y-A, Bang WS, et al. Expression of the MHC class II in triple-negative breast cancer is associated with tumor-infiltrating lymphocytes and interferon signaling. Ahmad A, ed. PLoS One 2017;12(8):e0182786 10.1371/journal.pone.0182786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbaro A, Procopio FA, et al. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res 2006;12(11 Pt 1):3435–3443. 10.1158/1078-0432.CCR-06-0165 [DOI] [PubMed] [Google Scholar]
- 19.Baskar S, Clements VK, Glimcher LH, Nabavi N, Ostrand-Rosenberg S. Rejection of MHC class II-transfected tumor cells requires induction of tumor-encoded B7–1 and/or B7–2 costimulatory molecules. J Immunol 1996;156(10):3821–3827. [PubMed] [Google Scholar]
- 20.Armstrong TD, Clements VK, Martin BK, Ting JP, Ostrand-Rosenberg S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc Natl Acad Sci U S A 1997;94(13):6886–6891. 10.1073/PNAS.94.13.6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, Ouyang J, et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J Clin Oncol 2018;36(10):942–950. 10.1200/JCO.2017.77.3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med 2018;10(450):eaar3342 10.1126/scitranslmed.aar3342 [DOI] [PubMed] [Google Scholar]
- 23.Trowsdale J Genomic structure and function in the MHC. Trends Genet 1993;9(4):117–122. http://www.ncbi.nlm.nih.gov/pubmed/8516845. Accessed September 24, 2018. [DOI] [PubMed] [Google Scholar]
- 24.Unanue ER, Turk V, Neefjes J. Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annu Rev Immunol 2016;34(1):265–297. 10.1146/annurev-immunol-041015-055420 [DOI] [PubMed] [Google Scholar]
- 25.Arnold PY, La Gruta NL, Miller T, Vignali KM, Adams PS, Woodland DL, et al. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J Immunol 2002;169(2):739–749. http://www.ncbi.nlm.nih.gov/pubmed/12097376. [DOI] [PubMed] [Google Scholar]
- 26.Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, et al. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol 1998;160(7):3363–3373. http://www.ncbi.nlm.nih.gov/pubmed/9531296. [PubMed] [Google Scholar]
- 27.Sartoris S, Valle MT, Barbaro AL, Tosi G, Cestari T, D’Agostino A, et al. HLA class II expression in uninducible hepatocarcinoma cells after transfection of AIR-1 gene product CIITA: acquisition of antigen processing and presentation capacity. J Immunol 1998;161(2):814–820. http://www.ncbi.nlm.nih.gov/pubmed/9670958. [PubMed] [Google Scholar]
- 28.Armstrong TD, Clements VK, Martin BK, Ting JP, Ostrand-Rosenberg S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc Natl Acad Sci U S A 1997;94(13):6886–6891. 10.1073/PNAS.94.13.6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin BK, Chin KC, Olsen JC, Skinner CA, Dey A, Ozato K, et al. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity 1997;6(5):591–600. http://www.ncbi.nlm.nih.gov/pubmed/9175837. [DOI] [PubMed] [Google Scholar]
- 30.Gobin SJ, Peijnenburg A, Keijsers V, van den Elsen PJ. Site alpha is crucial for two routes of IFN gamma-induced MHC class I transactivation: the ISRE-mediated route and a novel pathway involving CIITA. Immunity 1997;6(5):601–611. http://www.ncbi.nlm.nih.gov/pubmed/9175838. [DOI] [PubMed] [Google Scholar]
- 31.Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 1994;265(5168):106–109. http://www.ncbi.nlm.nih.gov/pubmed/8016643. [DOI] [PubMed] [Google Scholar]
- 32.Muhlethaler-Mottet A, Otten LA, Steimle V, Mach B. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J 1997;16(10):2851–2860. 10.1093/emboj/16.10.2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muhlethaler-Mottet A, Di Berardino W, Otten LA, Mach B. Activation of the MHC class II transactivator CIITA by interferon-gamma requires cooperative interaction between Stat1 and USF-1. Immunity 1998;8(2):157–166. 10.1016/S1074-7613(00)80468-9 [DOI] [PubMed] [Google Scholar]
- 34.van der Stoep N, Quinten E, van den Elsen PJ. Transcriptional regulation of the MHC class II trans-activator (CIITA) promoter III: identification of a novel regulatory region in the 5’-untranslated region and an important role for cAMP-responsive element binding protein 1 and activating transcription factor-1 in CIITA-promoter III transcriptional activation in B lymphocytes. J Immunol 2002;169(9):5061–5071. 10.4049/JIMMUNOL.169.9.5061 [DOI] [PubMed] [Google Scholar]
- 35.Piskurich JF, Linhoff MW, Wang Y, Ting JP. Two distinct gamma interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor beta. Mol Cell Biol 1999;19(1):431–440. 10.1128/MCB.19.1.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harton JA, Ting JP. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol 2000;20(17):6185–6194. 10.1128/MCB.20.17.6185-6194.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ting JP-Y, Trowsdale J. Genetic Control of MHC Class II Expression. Cell 2002;109(2):S21–S33. 10.1016/S0092-8674(02)00696-7 [DOI] [PubMed] [Google Scholar]
- 38.Chang CH, Fontes JD, Peterlin M, Flavell RA. Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. J Exp Med 1994;180(4):1367–1374. http://www.ncbi.nlm.nih.gov/pubmed/7931070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996;84(3):431–442. http://www.ncbi.nlm.nih.gov/pubmed/8608597. [DOI] [PubMed] [Google Scholar]
- 40.Busch R, Cloutier I, Sékaly RP, Hämmerling GJ. Invariant chain protects class II histocompatibility antigens from binding intact polypeptides in the endoplasmic reticulum. EMBO J 1996;15(2):418–428. http://www.ncbi.nlm.nih.gov/pubmed/8617217. [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, Arase N, Kohyama M, Hirayasu K, Suenaga T, Jin H, et al. Transport of misfolded endoplasmic reticulum proteins to the cell surface by MHC class II molecules. Int Immunol 2013;25(4):235–246. 10.1093/intimm/dxs155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiltbold EM, Roche PA. Trafficking of MHC class II molecules in the late secretory pathway. Curr Opin Immunol 2002;14(1):30–35. http://www.ncbi.nlm.nih.gov/pubmed/11790530. [DOI] [PubMed] [Google Scholar]
- 43.Bikoff EK, Huang LY, Episkopou V, van Meerwijk J, Germain RN, Robertson EJ. Defective major histocompatibility complex class II assembly, transport, peptide acquisition, and CD4+ T cell selection in mice lacking invariant chain expression. J Exp Med 1993;177(6):1699–1712. http://www.ncbi.nlm.nih.gov/pubmed/8098731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valečka J, Almeida CR, Su B, Pierre P, Gatti E. Autophagy and MHC-restricted antigen presentation. Mol Immunol 2018;99:163–170. 10.1016/j.molimm.2018.05.009 [DOI] [PubMed] [Google Scholar]
- 45.Poluektov YO, Kim A, Sadegh-Nasseri S. HLA-DO and Its Role in MHC Class II Antigen Presentation. Front Immunol 2013;4:260 10.3389/fimmu.2013.00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 2015;15(4):203–216. 10.1038/nri3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ranasinghe S, Lamothe PA, Soghoian DZ, Kazer SW, Cole MB, Shalek AK, et al. Antiviral CD8 + T Cells Restricted by Human Leukocyte Antigen Class II Exist during Natural HIV Infection and Exhibit Clonal Expansion. Immunity 2016;45(4):917–930. 10.1016/j.immuni.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jongsma MLM, Guarda G, Spaapen RM. The regulatory network behind MHC class I expression. Mol Immunol December 2017. 10.1016/J.MOLIMM.2017.12.005 [DOI] [PubMed]
- 49.Meissner TB, Liu Y-J, Lee K-H, Li A, Biswas A, van Eggermond MCJA, et al. NLRC5 Cooperates with the RFX Transcription Factor Complex To Induce MHC Class I Gene Expression. J Immunol 2012;188(10):4951–4958. 10.4049/jimmunol.1103160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van den Elsen PJ. Expression Regulation of Major Histocompatibility Complex Class I and Class II Encoding Genes. Front Immunol 2011;2:48 10.3389/fimmu.2011.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 2016;375(9):819–829. 10.1056/NEJMoa1604958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 2017;8(1):1136 10.1038/s41467-017-01062-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim H-J, Cantor H. CD4 T-cell Subsets and Tumor Immunity: The Helpful and the Not-so-Helpful. Cancer Immunol Res 2014;2(2):91–98. 10.1158/2326-6066.CIR-13-0216 [DOI] [PubMed] [Google Scholar]
- 54.Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol July 2018:1 10.1038/s41577-018-0044-0 [DOI] [PubMed]
- 55.Zanetti M Tapping CD4 T Cells for Cancer Immunotherapy: The Choice of Personalized Genomics. J Immunol 2015;194(5):2049–2056. 10.4049/jimmunol.1402669 [DOI] [PubMed] [Google Scholar]
- 56.Haabeth OAW, Tveita AA, Fauskanger M, Schjesvold F, Lorvik KB, Hofgaard PO, et al. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front Immunol 2014;5:174 10.3389/fimmu.2014.00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 2015;21(1):81–85. 10.1038/nm.3773 [DOI] [PubMed] [Google Scholar]
- 58.Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520(7549):692–696. 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dadmarz R, Sgagias MK, Rosenberg SA, Schwartzentruber DJ. CD4+ T lymphocytes infiltrating human breast cancer recognise autologous tumor in an MHC-class-II restricted fashion. Cancer Immunol Immunother 1995;40(1):1–9. http://www.ncbi.nlm.nih.gov/pubmed/7828162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marty R, Thompson WK, Salem RM, Zanetti M, Carter H. Evolutionary Pressure against MHC Class II Binding Cancer Mutations. Cell 2018;0(0). 10.1016/j.cell.2018.08.048 [DOI] [PubMed] [Google Scholar]
- 61.Cassell D, Forman J. Linked recognition of helper and cytotoxic antigenic determinants for the generation of cytotoxic T lymphocytes. Ann N Y Acad Sci 1988;532:51–60. http://www.ncbi.nlm.nih.gov/pubmed/2460011. [DOI] [PubMed] [Google Scholar]
- 62.Veatch JR, Lee SM, Fitzgibbon M, Chow I-T, Jesernig B, Schmitt T, et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J Clin Invest 2018;128(4). 10.1172/JCI98689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of Metastatic Melanoma with Autologous CD4+ T Cells against NY-ESO-1. N Engl J Med 2008;358(25):2698–2703. 10.1056/NEJMoa0800251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li K, Donaldson B, Young V, Ward V, Jackson C, Baird M, et al. Adoptive cell therapy with CD4+ T helper 1 cells and CD8+ cytotoxic T cells enhances complete rejection of an established tumour, leading to generation of endogenous memory responses to non-targeted tumour epitopes. Clin Transl Immunol 2017;6(10):e160 10.1038/cti.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016;126(6):2123–2138. 10.1172/JCI85309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016;30(2):492–500. 10.1038/leu.2015.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 2016;8(355):355ra116 10.1126/scitranslmed.aaf8621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kitano S, Tsuji T, Liu C, Hirschhorn-Cymerman D, Kyi C, Mu Z, et al. Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol Res 2013;1(4):235–244. 10.1158/2326-6066.CIR-13-0068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, et al. Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 2012;209(11):2113–2126. 10.1084/jem.20120532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon E-KM, et al. Naive tumor-specific CD4 + T cells differentiated in vivo eradicate established melanoma. J Exp Med 2010;207(3):651–667. 10.1084/jem.20091921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016;45(5):1122–1134. 10.1016/j.immuni.2016.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jang J-E, Hajdu CH, Liot C, Miller G, Dustin ML, Bar D, et al. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep 2017;20:558–571. 10.1016/j.celrep.2017.06.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 2014;15(11):1070–1078. 10.1038/ni.3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCaw TR, Li M, Starenki D, Cooper SJ, Liu M, Meza-Perez S, et al. The expression of MHC class II molecules on murine breast tumors delays T-cell exhaustion, expands the T-cell repertoire, and slows tumor growth. Cancer Immunol Immunother October 2018. 10.1007/s00262-018-2262-5 [DOI] [PMC free article] [PubMed]
- 75.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013;13(4):227–242. 10.1038/nri3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev 2009;229(1):126–144. 10.1111/j.1600-065X.2009.00771.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jilaveanu LB, Sznol J, Aziz SA, Duchen D, Kluger HM, Camp RL. CD70 expression patterns in renal cell carcinoma. Hum Pathol 2012;43(9):1394–1399. 10.1016/j.humpath.2011.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shibahara I, Saito R, Zhang R, Chonan M, Shoji T, Kanamori M, et al. OX40 ligand expressed in glioblastoma modulates adaptive immunity depending on the microenvironment: a clue for successful immunotherapy. Mol Cancer 2015;14:41 10.1186/s12943-015-0307-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson DB, Bordeaux JM, Kim J-Y, Vaupel CA, Rimm DL, Ho TH, et al. Quantitative Spatial Profiling of PD-1/PD-L1 Interaction and HLA-DR/IDO-1 Predicts Improved Outcomes of anti-PD-1 Therapies in Metastatic Melanoma. Clin Cancer Res July 2018: 10.1158/1078-0432.CCR-18-0309 [DOI] [PMC free article] [PubMed]
- 80.Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res 2016;22(6):1499–1509. 10.1158/1078-0432.CCR-15-1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Concha A, Ruiz-Cabello F, Cabrera T, Nogales F, Collado A, Garrido F. Different patterns of HLA-DR antigen expression in normal epithelium, hyperplastic and neoplastic malignant lesions of the breast. Eur J Immunogenet 1995;22(4):299–310. http://www.ncbi.nlm.nih.gov/pubmed/7495782. [DOI] [PubMed] [Google Scholar]
- 82.Oldford SA, Robb JD, Codner D, Gadag V, Watson PH, Drover S. Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients. Int Immunol 2006;18(11):1591–1602. 10.1093/intimm/dxl092 [DOI] [PubMed] [Google Scholar]
- 83.Oldford SA, Robb JD, Watson PH, Drover S. HLA-DRB alleles are differentially expressed by tumor cells in breast carcinoma. Int J Cancer 2004;112(3):399–406. 10.1002/ijc.20441 [DOI] [PubMed] [Google Scholar]
- 84.Jabrane-Ferrat N, Faille A, Loiseau P, Poirier O, Charron D, Calvo F. Effect of gamma interferon on HLA class-I and -II transcription and protein expression in human breast adenocarcinoma cell lines. Int J cancer 1990;45(6):1169–1176. http://www.ncbi.nlm.nih.gov/pubmed/2112515. [DOI] [PubMed] [Google Scholar]
- 85.Forero A, Li Y, Chen D, Grizzle WE, Updike KL, Merz ND, et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol Res 2016;4(5):390–399. 10.1158/2326-6066.CIR-15-0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.da Silva GBRF, Silva TGA, Duarte RA, Neto NL, Carrara HHA, Donadi EA, et al. Expression of the Classical and Nonclassical HLA Molecules in Breast Cancer. Int J Breast Cancer 2013;2013:1–9. 10.1155/2013/250435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feinmesser M, Sulkes A, Morgenstern S, Sulkes J, Stern S, Okon E. HLA-DR and beta 2 microglobulin expression in medullary and atypical medullary carcinoma of the breast: histopathologically similar but biologically distinct entities. J Clin Pathol 2000;53(4):286–291. http://www.ncbi.nlm.nih.gov/pubmed/10823125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bártek J, Petrek M, Vojtĕsek B, Bártková J, Kovarík J, Rejthar A. HLA-DR antigens on differentiating human mammary gland epithelium and breast tumours. Br J Cancer 1987;56(6):727–733. http://www.ncbi.nlm.nih.gov/pubmed/3435699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Michel S, Linnebacher M, Alcaniz J, Voss M, Wagner R, Dippold W, et al. Lack of HLA class II antigen expression in microsatellite unstable colorectal carcinomas is caused by mutations in HLA class II regulatory genes. Int J cancer 2010;127(4):889–898. 10.1002/ijc.25106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bustin SA, Li S-R, Phillips S, Dorudi S. Expression of HLA Class II in Colorectal Cancer: Evidence for Enhanced Immunogenicity of Microsatellite-Instability-Positive Tumours. Tumor Biol 2001;22(5):294–298. 10.1159/000050630 [DOI] [PubMed] [Google Scholar]
- 91.Callahan MJ, Nagymanyoki Z, Bonome T, Johnson ME, Litkouhi B, Sullivan EH, et al. Increased HLA-DMB Expression in the Tumor Epithelium Is Associated with Increased CTL Infiltration and Improved Prognosis in Advanced-Stage Serous Ovarian Cancer. Clin Cancer Res 2008;14(23):7667–7673. 10.1158/1078-0432.CCR-08-0479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Turner TB, Meza-Perez S, Londoño A, Katre A, Peabody JE, Smith HJ, et al. Epigenetic modifiers upregulate MHC II and impede ovarian cancer tumor growth. Oncotarget 2017;8(27):44159–44170. 10.18632/oncotarget.17395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Younger AR, Amria S, Jeffrey WA, Mahdy AEM, Goldstein OG, Norris JS, et al. HLA class II antigen presentation by prostate cancer cells. Prostate Cancer Prostatic Dis 2008;11(4):334–341. 10.1038/sj.pcan.4501021 [DOI] [PubMed] [Google Scholar]
- 94.Soos JM, Krieger JI, Stüve O, King CL, Patarroyo JC, Aldape K, et al. Malignant glioma cells use MHC class II transactivator (CIITA) promoters III and IV to direct IFN-gamma-inducible CIITA expression and can function as nonprofessional antigen presenting cells in endocytic processing and CD4(+) T-cell activation. Glia 2001;36(3):391–405. http://www.ncbi.nlm.nih.gov/pubmed/11746775. [DOI] [PubMed] [Google Scholar]
- 95.Yazawa T, Kamma H, Fujiwara M, Matsui M, Horiguchi H, Satoh H, et al. Lack of class II transactivator causes severe deficiency of HLA‐DR expression in small cell lung cancer. J Pathol 187(2):191–199. [DOI] [PubMed] [Google Scholar]
- 96.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016;165(1):35–44. 10.1016/J.CELL.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rimm DL, Han G, Taube JM, Yi ES, Bridge JA, Flieder DB, et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non–Small Cell Lung Cancer. JAMA Oncol 2017;3(8):1051 10.1001/jamaoncol.2017.0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther 2015;14(4):847–856. 10.1158/1535-7163.MCT-14-0983 [DOI] [PubMed] [Google Scholar]
- 99.Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol 2003;33(5):1183–1192. 10.1002/eji.200323712 [DOI] [PubMed] [Google Scholar]
- 100.Panelli MC, Wang E, Shen S, Schluter SF, Bernstein RM, Hersh EM, et al. Interferon γ (IFNγ) gene transfer of an EMT6 tumor that is poorly responsive to IFNγ stimulation: increase in tumor immunogenicity is accompanied by induction of a mouse class II transactivator and class II MHC. Cancer Immunol Immunother 1996;42(2):99–107. 10.1007/s002620050258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ostrand-Rosenberg S, Thakur A, Clements V. Rejection of mouse sarcoma cells after transfection of MHC class II genes. J Immunol 1990;144(10):4068–4071. http://www.ncbi.nlm.nih.gov/pubmed/2332639. [PubMed] [Google Scholar]
- 102.Ostrand-Rosenberg S, Clements VK, Thakur A, Cole GA. TRANSFECTION OF MAJOR HISTOCOMPATIBLITY COMPLEX CLASS I AND CLASS II GENES CAUSES TUMOUR REJECTION. Eur J Immunogenet 1989;16(4–5):343–349. 10.1111/j.1744-313X.1989.tb00481.x [DOI] [PubMed] [Google Scholar]
- 103.Martin BK, Frelinger JG, Ting JP. Combination gene therapy with CD86 and the MHC class II transactivator in the control of lung tumor growth. J Immunol 1999;162(11):6663–6670. http://www.ncbi.nlm.nih.gov/pubmed/10352284. [PubMed] [Google Scholar]
- 104.Clements VK, Baskar S, Armstrong TD, Ostrand-Rosenberg S. Invariant chain alters the malignant phenotype of MHC class II+ tumor cells. J Immunol 1992;149(7):2391–2396. http://www.ncbi.nlm.nih.gov/pubmed/1527384. [PubMed] [Google Scholar]
- 105.Armstrong TD, Clements VK, Ostrand-Rosenberg S. Class II-transfected tumor cells directly present endogenous antigen to CD4+ T cells in vitro and are APCs for tumor-encoded antigens in vivo. J Immunother 1998;21(3):218–224. http://www.ncbi.nlm.nih.gov/pubmed/9610914. [DOI] [PubMed] [Google Scholar]
- 106.Ostrand-Rosenberg S, Clements VK, Thakur A, Cole GA. TRANSFECTION OF MAJOR HISTOCOMPATIBLITY COMPLEX CLASS I AND CLASS II GENES CAUSES TUMOUR REJECTION. Eur J Immunogenet 1989;16(4–5):343–349. 10.1111/j.1744-313X.1989.tb00481.x [DOI] [PubMed] [Google Scholar]
- 107.Bou Nasser Eddine F, Forlani G, Lombardo L, Tedeschi A, Tosi G, Accolla RS. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4 + TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cells. Oncoimmunology 2017;6(1):e1261777 10.1080/2162402X.2016.1261777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baskar S, Clements VK, Glimcher LH, Nabavi N, Ostrand-Rosenberg S. Rejection of MHC class II-transfected tumor cells requires induction of tumor-encoded B7–1 and/or B7–2 costimulatory molecules. J Immunol 1996;156(10):3821–3827. http://www.ncbi.nlm.nih.gov/pubmed/8621919. [PubMed] [Google Scholar]
- 109.Haabeth OAW, Fauskanger M, Manzke M, Lundin KU, Corthay A, Bogen B, et al. CD4+ T cell-mediated rejection of MHC class II-positive tumor cells is dependent on antigen secretion and indirect presentation on host APCs. Cancer Res May 2018: 10.1158/0008-5472.CAN-17-2426 [DOI] [PubMed]
- 110.Lee YS, Kim SH, Cho JA, Kim CW. Introduction of the CIITA gene into tumor cells produces exosomes with enhanced anti-tumor effects. Exp Mol Med 2011;43(5):281 10.3858/emm.2011.43.5.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018;560(7718):382–386. 10.1038/s41586-018-0392-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 2017;7(2):188–201. 10.1158/2159-8290.CD-16-1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 2016;375(9):819–829. 10.1056/NEJMoa1604958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Martins I, Deshayes F, Baton F, Forget A, Ciechomska I, Sylla K, et al. Pathologic expression of MHC class II is driven by mitogen-activated protein kinases. Eur J Immunol 2007;37(3):788–797. 10.1002/eji.200636620 [DOI] [PubMed] [Google Scholar]
- 115.Deffrennes V, Vedrenne J, Stolzenberg MC, Piskurich J, Barbieri G, Ting JP, et al. Constitutive expression of MHC class II genes in melanoma cell lines results from the transcription of class II transactivator abnormally initiated from its B cell-specific promoter. J Immunol 2001;167(1):98–106. http://www.ncbi.nlm.nih.gov/pubmed/11418637. Accessed August 4, 2018. [DOI] [PubMed] [Google Scholar]
- 116.Goodwin BL, Xi H, Tejiram R, Eason DD, Ghosh N, Wright KL, et al. Varying functions of specific major histocompatibility class II transactivator promoter III and IV elements in melanoma cell lines. Cell Growth Differ 2001;12(6):327–335. http://www.ncbi.nlm.nih.gov/pubmed/11432807. Accessed August 4, 2018. [PubMed] [Google Scholar]
- 117.Mostafa AA, Codner D, Hirasawa K, Komatsu Y, Young MN, Steimle V, et al. Activation of ERα Signaling Differentially Modulates IFN-γ Induced HLA-Class II Expression in Breast Cancer Cells. Kovats S, ed. PLoS One 2014;9(1):e87377 10.1371/journal.pone.0087377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kittur DS, Wilasrusmee C, Han W-F, Xu R, Burdick JF, Adler W. Locally derived cytokines and upregulation of MHC class II genes in allografts. J Heart Lung Transplant 2002;21(8):882–889. http://www.ncbi.nlm.nih.gov/pubmed/12163088. [DOI] [PubMed] [Google Scholar]
- 119.Lee J, Tam H, Adler L, Ilstad-Minnihan A, Macaubas C, Mellins ED. The MHC class II antigen presentation pathway in human monocytes differs by subset and is regulated by cytokines. Boissonnas A, ed. PLoS One 2017;12(8):e0183594 10.1371/journal.pone.0183594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct Effects of Type I Interferons on Cells of the Immune System. Clin Cancer Res 2011;17(9):2619–2627. 10.1158/1078-0432.CCR-10-1114 [DOI] [PubMed] [Google Scholar]
- 121.Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol 2015;34:22–27. 10.1016/J.COI.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lu Y, Ussery GD, Muncaster MM, Gallie BL, Blanck G. Evidence for retinoblastoma protein (RB) dependent and independent IFN-gamma responses: RB coordinately rescues IFN-gamma induction of MHC class II gene transcription in noninducible breast carcinoma cells. Oncogene 1994;9(4):1015–1019. [PubMed] [Google Scholar]
- 123.Tschickardt ME, Lu Y, Jacim M, Ussery G de W, Steimle V, Mach B, et al. RB and A novel E2F‐1 binding protein in MHC class II deficient B‐cell lines and normal IFN‐γ induction of the class II transactivator ciita in class II non‐inducible RB‐defective tumor lines. Int J Cancer 62(4):461–465. 10.1002/IJC.2910620417 [DOI] [PubMed] [Google Scholar]
- 124.Lu Y, Boss JM, Hu SX, Xu HJ, Blanck G. Apoptosis-independent retinoblastoma protein rescue of HLA class II messenger RNA IFN-gamma inducibility in non-small cell lung carcinoma cells. Lack of surface class II expression associated with a specific defect in HLA-DRA induction. J Immunol 1996;156(7):2495–2502. http://www.ncbi.nlm.nih.gov/pubmed/8786310. [PubMed] [Google Scholar]
- 125.Lu Y, Tschickardt ME, Schmidt BJ, Blanck G. IFN-γ inducibility of class II transactivator is specifically lacking in human tumour lines: Relevance to retinoblastoma protein rescue of IFN-γ inducibility of the HLA class II genes. Immunol Cell Biol 1997;75(4):325–332. 10.1038/icb.1997.50 [DOI] [PubMed] [Google Scholar]
- 126.Mottok A, Woolcock B, Chan FC, Tong KM, Chong L, Farinha P, et al. Genomic Alterations in CIITA Are Frequent in Primary Mediastinal Large B Cell Lymphoma and Are Associated with Diminished MHC Class II Expression. Cell Rep 2015;13(7):1418–1431. 10.1016/j.celrep.2015.10.008 [DOI] [PubMed] [Google Scholar]
- 127.Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011;471(7338):377–381. 10.1038/nature09754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Steidl C, Woolcock B, Rogic S, Ben-Neriah S, Telenius A, Drake M, et al. Inactivating Gene Alterations of MHC Class II Transactivator CIITA Are Recurrent in Primary Mediastinal B Cell Lymphoma and Hodgkin Lymphoma. Blood 2011;118(21). http://www.bloodjournal.org/content/118/21/437?sso-checked=true. [Google Scholar]
- 129.Yazawa T, Ito T, Kamma H, Suzuki T, Okudela K, Hayashi H, et al. Complicated mechanisms of class II transactivator transcription deficiency in small cell lung cancer and neuroblastoma. Am J Pathol 2002;161(1):291–300. 10.1016/S0002-9440(10)64181-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Holling TM, Bergevoet MWT, Wilson L, Van Eggermond MCJA, Schooten E, Steenbergen RDM, et al. A role for EZH2 in silencing of IFN-gamma inducible MHC2TA transcription in uveal melanoma. J Immunol 2007;179(8):5317–5325. http://www.ncbi.nlm.nih.gov/pubmed/17911618. [DOI] [PubMed] [Google Scholar]
- 131.Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol 2016;9(1):47 10.1186/s13045-016-0277-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev 2017;276(1):80–96. 10.1111/imr.12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Balko JM, Johnson DB, Wang DY, Ericsson-Gonzalez P, Nixon M, Salgado R, et al. MHC-II expression to drive a unique pattern of adaptive resistance to antitumor immunity through receptor checkpoint engagement. J Clin Oncol 2018;36(5_suppl):180–180. 10.1200/JCO.2018.36.5_suppl.18029220290 [DOI] [Google Scholar]
- 134.Frangione V, Mortara L, Castellani P, De Lerma Barbaro A, Accolla RS. CIITA-driven MHC-II positive tumor cells: Preventive vaccines and superior generators of antitumor CD4+ T lymphocytes for immunotherapy. Int J Cancer 2010;127(7):1614–1624. 10.1002/ijc.25183 [DOI] [PubMed] [Google Scholar]
- 135.Pulaski BA, Ostrand-Rosenberg S, Bosch JJ, Clements VK, Chen PW, Ksander BR, et al. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res 1998;58(7):1486–1493. 10.1158/0008-5472.can-03-2634 [DOI] [PubMed] [Google Scholar]