

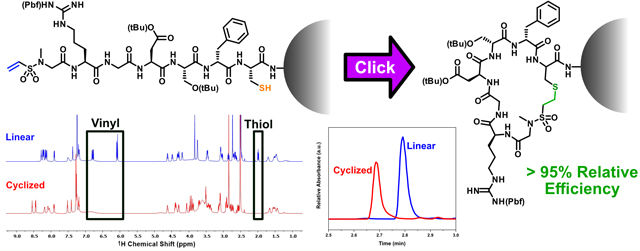
Abstract
Macrocyclization of linear peptides imparts improved stability to enzymatic degradation and increases potency of function. Many successful macrocyclization of peptides both in solution and on-resin have been achieved but are limited in scope as they lack selectivity, require long reaction times, or necessitate heat. To overcome these drawbacks a robust and facile strategy was developed employing thiol-Michael click chemistry via an N-methyl vinyl sulfonamide. We demonstrate its balance of reactivity and high stability through FTIR model kinetic studies, reaching 88% conversion over 30 min, and NMR stability studies, revealing no apparent degradation over an 8 day period in basic conditions. Using a commercially available reagent, 2-chloroethane sulfonyl chloride, the cell adhesion peptide, RGDS, was functionalized and macrocyclized on-resin with a relative efficiency of over 95%. The simplistic nature of this process demonstrates the effectiveness of vinyl sulfonamides as a thiol-Michael click acceptor and its applicability to many other bioconjugation applications.
Graphical Abstract
Peptide conjugation is an ever-expanding field that allows for the creation of complex structures1, molecular tagging2, and material functionalization.3 Intramolecular conjugation reactions within a peptide result in a cyclic structure, or macrocycle. Peptide macrocycles are of great interest owing to their enhanced resistance to protease degradation4 and improved potency.5 A well-known example highlighting cyclic peptide advantages over linear peptides is demonstrated in the simple cell adhesion peptide sequence, RGDS.6 Higher stability and efficacy of cyclic peptides have led to further exploration into the synthesis of macrocyclic motifs of various peptide sequences for biological relevance7 or self-assembling structures.8
Macrocyclic peptides are often synthesized using solid-phase peptide synthesis (SPPS) protocols and cyclizing the product either in solution or on a solid substrate (i.e., on-resin). The solution-phase macrocyclization route implements a conjugation reaction on the fully protected peptide in dilute solution. Examples of solution-phase strategies include native chemical ligation,9 imine formation,10 and palladium-catalyzed conjugation reactions.11–12 However, solution-phase strategies largely necessitate an additional deprotection step followed by purification to remove cleaved protecting groups and oligomerized byproducts. The solid-phase macrocyclization route implements a conjugation reaction on the peptides while attached to the resin, resulting in fewer side products and purification steps.13 The advantages of on-resin macrocyclization has made it the preferred strategy for synthesizing cyclic peptide structure.
Several on-resin conjugation reactions have been employed for the macrocyclization of peptides, including thiol-ene,14 azide-alkyne cycloaddition,15–16 amide coupling17–18, glacial coupling19, and ring-closing metathesis.20–21 In general, reactions demonstrating click-like characteristics are particularly attractive for the synthesis of cyclic peptides, as they are selective, proceed to full conversion, and produce little to no byproducts.22–23 One common click reaction used in peptide conjugation is the copper(I) catalyzed azide-alkyne cycloaddition (Cu-AAC).24 The CuAAC reaction is a bio-orthogonal reaction between an azide and alkyne in the presence of copper(I) to form a triazole product. For example, Ingale et al.15 demonstrated on-resin macrocyclization of peptides using copper bromide and excess sodium ascorbate; however, long reaction times of 18 h were required to reach sufficient conversions. Another common click reaction used in peptide conjugation is the thiolene reaction, which is the radical mediated addition reaction between a thiol and electron deficient alkene.25 For example, An-seth and coworkers14 demonstrated a simple thiol-ene conjugation strategy utilizing cysteine as a thiol and a commercially available allyoxycarbonyl (Alloc) protected lysine; however, this approach required multiple additions of photoinitiator and irradiation steps to perform the on-resin macrocyclization.
An attractive alternative click reaction to the CuAAC and thiol-ene reactions is the thiol-Michael addition.26 In contrast to the radical-mediated thiol-ene reaction, thiol-Michael additions follow an anionic mechanism. Thiol-Michael reactions occur between a thiol and an electron deficient alkene to form a stable thioether bond in the presence of a base or nucleophile catalyst. Thiol-Michael reactions have been exploited in a wide range of biomolecular conjugation reaction, including peptides27, proteins28, and nucleic acids.29 Common thiol-Michael acceptors, such as maleimides and acrylates, exhibit fast kinetics and proceed to full conversion, but present complications when applied to peptide chemistry.30–31 Maleimides are known to undergo a ring opening reaction under aqueous conditions, both before and after conjugation,32–33 while acrylates are susceptible to hydrolytic cleavage, particularly after thiol addition.34 An alternative water stable thiol-Michael acceptor is the acrylamide functional group; however, they exhibit slow kinetics and incomplete conversions over long time periods.30 Here, we suggest the vinyl sulfonamide as an efficient, water stable thiol- Michael acceptor for peptide macrocyclization. Vinyl sulfonamides have been employed in both bioconjugations35 and polymer networks36 for its high reactivity and hydrolytic stability. Furthermore, simple installation of vinyl sulfonamides on primary or secondary amines is achieved using the commercially available and inexpensive 2-chloroethanesulfonyl chloride.
Solution-phase model reaction systems were performed to compare relative rates of vinyl sulfonamides with several commonly implemented Michael acceptors (Figure 1A), such as maleimide (E1) and acrylate (E2) as well as less reactive hydrolytically stable acrylamides (E3 and E4). The reaction kinetics of thiol Michael conjugations are highly dependent on alkene and thiol structure37. Model mono-vinyl sulfonamide compounds, (E5) and (E6), were synthesized and compared with maleimide, acrylate, and acrylamide functional groups. All thiol-Michael acceptors were conjugated with two commercially available thiols, n-butyl mercaptopropionate (T1) and a cysteine analog (T2). Mercaptopropionic acid is readily conjugated to the N-term inus of a peptide or amine side-group on an amino acid through amide coupling, whereas cysteine is a natural amino acid containing a thiol side-group.38
Figure 1.
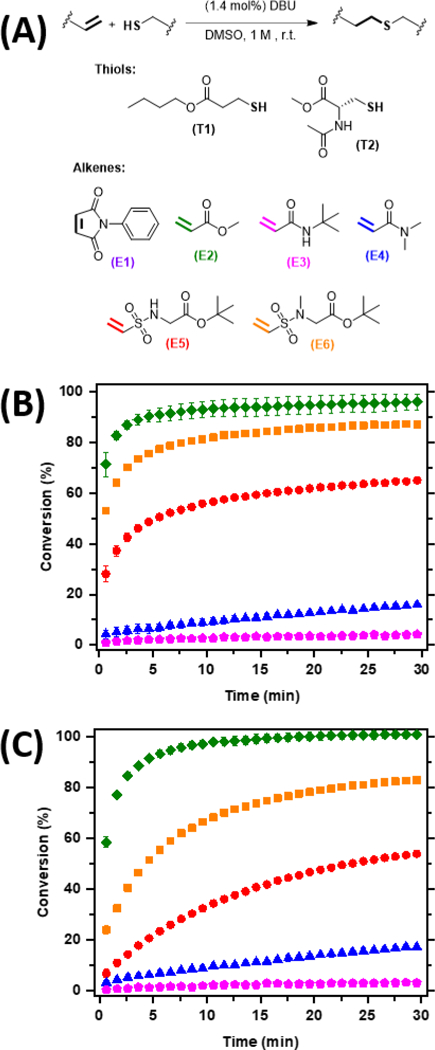
Reaction kinetics of various thiols and electron poor alkenes. A) The general thiol-Michael reaction scheme, and the model thiols and alkenes used in this study. B) Reaction kinetics were monitored at room temperature over 30 minutes using FTIR spectroscopy by tracking the disappearance of the thiol-peak between 2484 and 2545 cm−1. 1 eq of the thiol, T1, was reacted with 1 eq of each of the alkenes, E2 (green, filled diamond), E3 (magenta, filled pentagon), E4 (blue, filled triangle), E5 (red, filled circle), and E6 (orange, filled square) in the presence of DBU (1.4 mol%) in DMSO. (C) Similarly, 1 eq of the thiol, T2, was reacted with 1 eq of each alkene, E2 (green, filled diamond), E3 (magenta, filled pentagon), E4 (blue, filled triangle), E5 (red, filled circle), and E6 (orange, filled square). E1 is not shown as it reached full conversion with both T1 and T2 within the first 30 s of the experiment.
The model maleimide and acrylate compounds proceeded rapidly, while both acrylamides compounds showed minimal conversion after 30 min under the same conditions. A summary of the thiol-Michael kinetics is presented in Figure 1B and Figure 1C. The reaction of phenyl maleimide, E1, with T1 or T2 reacted to full conversion within the short time period of adding and mixing them with 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) and transferring to the FTIR (30 seconds). Maleimide functional groups showed enhanced reactivity for two distinct reasons: alleviation of ring strain and the electron deficiency of the olefin caused by the two adjacent activating carbonyl groups.39 The kinetics of the methyl acrylate, E2, reacted with either the alkyl or cysteine-based thiols proceeded rapidly, reaching conversions of 92 ± 3% or 91 ± 1%, respectively, at 5 min. In comparison, reactions of either acrylamide functional groups with either type of thiol exhibited sluggish kinetics. The reaction of the secondary acrylamide, E3, with T1 or T2 reached conversions of only 2 ± 1% or 1.5 ± 0.3% within 5 min, respectively. Surprisingly, the reaction of the N-methyl tertiary acrylamide (denoted henceforth as a tertiary acrylamide), E4, with either T1 or T2 had improved reactivity, reaching 7 ± 1% within 5 min. We attribute the poor reactivity of both acrylamides to the decrease in electronegativity of the amides as compared with esters.40 High electronegative moieties adjacent to olefins result in a more reactive center for thiol-Michael conjugations. Furthermore, the enolate that forms in amides is known to be less stable as compared with ester-based groups.41–42 It is believed that an induction effect reduces the stability of the eno-late species thereby resulting in slower kinetics of the secondary acrylamide in comparison with the tertiary acrylamides.
Both secondary and tertiary vinyl sulfonamides exhibited comparable kinetics to the acrylate, and much faster kinetics than the secondary and tertiary acrylamides. The reaction of secondary vinyl sulfonamide, E5, with either T1 or T2 reached a conversion of 49 ± 1% or 21 ± 1%, respectively, over 5 minutes. As observed with secondary and tertiary vinyl acrylamides, the tertiary vinyl sulfonamide demonstrated faster kinetics than the secondary vinyl sulfonamides. The reaction of tertiary vinyl sulfonamide, (E6), reacted more rapidly than E5 within the same timeframe with conversions reaching 76.4±0.6% and 53.4± 0.7% with T1 and T2, respectively. The increased reactivity of vinyl sulfonamides over vinyl acrylamides and comparable reactivity to acrylates is attributed to two main factors. First, sulfur atoms possess high polarizability leading to a stronger inductive effect for stabilizing the α-carbanion that forms during the thiol-Michael reaction mechanism.43 Second, antibonding orbitals of sulfur atoms create σ-effects that further stabilize the anionic center.44–46 The added stability of the α-carbanion results in an increase in the kinetic performance for vinyl sulfonamides in thiol-Michael conjugations. Additionally, it is noteworthy that slower kinetics were experienced across all the alkenes when reacting with the cysteine-based thiol T2. The slower kinetics were an unexpected result based on the apparent thiol pKa (between 6–9). However, the slower kinetics could be attributed to neighboring groups providing steric hinderance or affecting thiol pKa.
Water stability of the thioether product from the thiol reaction with either the secondary and tertiary vinyl sulfonamides was evaluated and compared with the thioether product of an acrylate. Sinha et al.36 showed that tertiary vinyl sulfonamide demonstrated superior stability to acidic and basic environments in polymer networks as compared with networks formed using secondary sulfonamides and acrylates. Using NMR spectroscopy, we confirmed the stability of both secondary- and tertiary-vinyl sulfonamides in both acidic and basic media (Figures S16 to S21). Under basic conditions (in pH 10 borate buffer), the model acrylate underwent rapid hydrolytic cleavage, while both vinyl sulfonamide compounds remained stable over an 8 day period.
Owing to its rapid reaction kinetics and hydrolytic stability, tertiary vinyl sulfonamide was selected to cyclize a ubiquitous cell-adhesion peptide, RGDS; a process which has been shown to both increase its stability to enzymatic degradation and efficacy to promote cell spreading.6 The procedure for macrocy- clizing the RGDS peptide is summarized in Figure 2. The peptide sequence was preceded by a cysteine residue, bearing a monomethoxy trityl (Mmt) protected thiol. The eventual removal of the Mmt group through well-established protocols provides a thiol chemical handle for the thiol-Michael addition.47 A D-phenylalanine residue was coupled after the cysteine and prior to the rest of the sequence, which has been shown to help promote macrocyclization of peptides.48–49 To install the tertiary vinyl sulfonamide, a low-cost commercially available Fmoc-protected sarcosine was added at the end of the peptide. The Fmoc protecting group was removed revealing a secondary amine to which the vinyl sulfonamide was added. To generate the vinyl sulfonamide, a commercially available reagent, 2-chlorosulfonyl chloride, was applied to the resin mixture in the presence of triethylamine (TEA), yielding the Michael-acceptor product. Macrocyclization of the peptide was catalyzed using DBU, a common reagent used in Fmoc SPPS.50 Upon macrocy-clization, a non-natural sulfonamide linkage is introduced into the backbone of the macrocyclic peptide.
Figure 2.
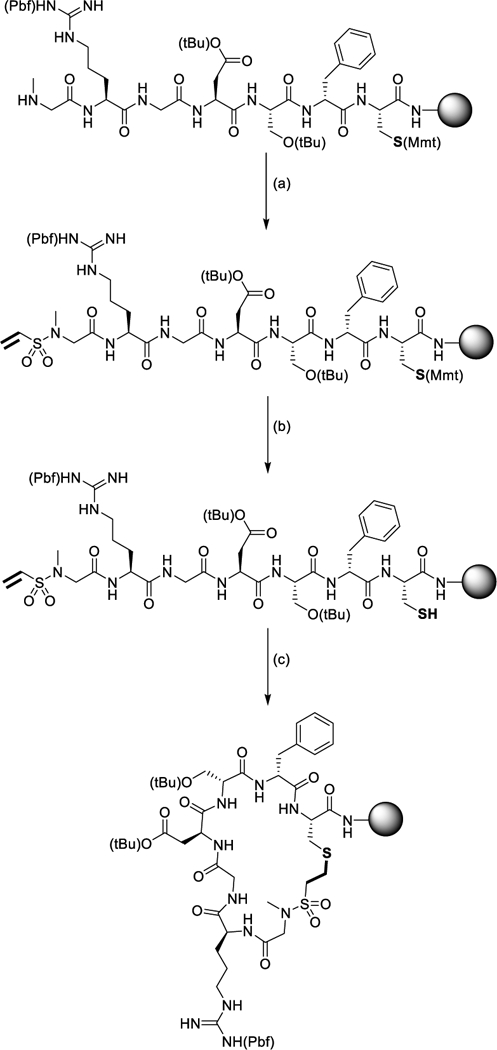
On-resin macrocyclization of an RGDS peptide. The following procedure was used to functionalize and macrocyclize the peptide: (a) (4 eq) 2-chloroethanesulfonyl chloride, (8 eq) TEA, r.t., for 2 h, 2 times, (b) 3% TFA/5% TES in DCM, r.t., 30 s, 15 times, and (c) 10 mol% DBU in DMF, r.t., 1 h.
The macrocyclization of the peptide is achieved on-resin by reacting the thiol of the cysteine with the tertiary vinyl sulfonamide end group. First, the resin was washed followed by the selective deprotection of the Mmt protected cysteine, using a dilute acid solution of 3% trifluoroacetic acid (TFA)/5% triethylsilane (TES) in dichloromethane (DCM). The peptide resin was then neutralized using a dilute TEA solution and was collected. One third of the resin was subjected to cleavage from the resin using a 95% TFA cleavage cocktail. This peptide was collected, precipitated, and purified by HPLC to produce the linear peptide with an overall yield of 10.0 mg or 14.2% based on the initial resin’s molarity. The other two-thirds of the resin was subjected to the macrocyclization procedure. A catalytic amount of DBU in dimethylformamide (DMF) was added and mixed for one hour. The resin was then washed multiple times with DMF and DCM and was subjected to the resin cleavage cocktail, precipitated, and purified by reversed-phase high-performance liquid chromatography (RP-HPLC). The final product was lyophilized resulting in 19.4 mg or 13.8% yield of fully macrocyclized peptide. The structure of the fully cleaved macrocyclized peptide is presented in Figure 3A. Based on the final yield of the peptide the overall relative efficiency of the macro-cyclization was >95%. Such a high efficiency of macrocyclization demonstrates the effectiveness of using vinyl sulfonamides as a thiol-Michael acceptor for peptide macrocyclizations. All attempts at cyclizing the peptide using a tertiary acrylamide were unsuccessful at macrocyclizing on-resin further demonstrating the superior reactivity of vinyl sulfonamides over acrylamides. As expected, attempts at performing solution-phase macrocyclizations under highly dilute conditions utilizing the same vinyl sulfonamide chemistry resulted in minimal macrocycle product.13
Figure 3.
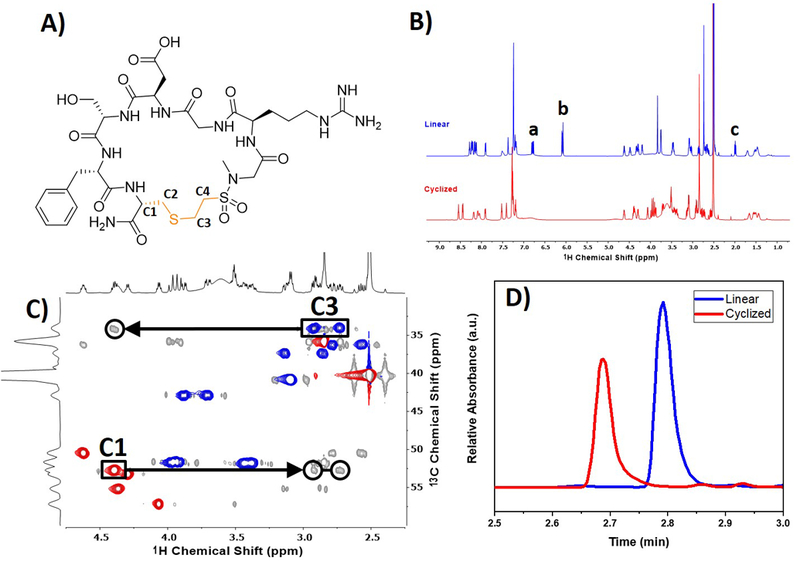
Characterization of macrocyclic RGDS peptide. (A) Structure of the macrocyclized peptide cleaved from the resin with important carbons for confirming macrocyclization numbered C1 through C4. The proton NMR (B) shows the linear peptide in blue and the cyclic peptide in red. Alkenyl protons, denoted by ‘a’ and ‘b,’ disappear after macrocyclization. Additionally, the thiol peak ‘c’ disappears after performing the macrocyclization. An overlay of 1H-13C HSQC (red/blue) and HMBC (grey) is shown in (C) with black arrows highlighting the correlation between the newly formed thioether (C1) and cysteine residue (C3) for the macrocyclized peptide. For the edited 1H-13C HSQC, the blue and red peaks correspond to CH2 or CH/CH3 carbons, respectively. The ESI-UPLC trace shown in (D) of the microcleaved linear and macrocyclized peptides shows a shift in the elution time of the two peptide structures.
lH NMR was performed to verify the disappearance of the vinyl sulfonamide protons, denoted by ‘a’ and ‘b‘ in Figure 3B. As expected after undergoing cyclization on-resin, the vinyl proton shifts (δ = 6.07, 6.78 ppm) disappeared from the cyclized peptide spectrum. The thiol peak (δ = 2.00), denoted as ‘c,’ also disappeared post cyclization. Furthermore, the shifts in the 1H spectra post-cyclization further signifying a structural change to the peptide has occurred. For example, lH chemical shifts in the amide region (δ = 7.25 to 9.00 ppm) are broader prior to macrocyclization. As linear peptides adopt a random coil conformation, amide peaks form a broad distribution of conformations demonstrated by the clustering of the protons between 8.00 and 8.25 ppm. Once a peptide forms a macrocycle, the discrete conformations are observed from the spreading of the amide protons as they adopt distinct orientations.
Correlation between protons using 2D NMR techniques further confirmed the structure of the cyclic peptide. 1H-13C HSQC and HMBC of the cyclic peptide were overlaid to compare the single and multiple bond correlations between 1H and 13C peaks, shown in Figure 3C. With these correlations, protons were assigned to the resulting thioether product and cysteine residue. Upon macrocyclization, the thioether carbon (C3; δ = 34.08; 2.73/2.92 ppm) was used to determine the correlation with the cysteine α-proton (C1; δ = 52.69; 4.39 ppm), as denoted by the black arrows. These correlations confirm that the thiol-Michael reaction has occurred. Furthermore, COSY, NOSEY, and TOCSY NMR data were collected on both the linear and macrocyclized peptides to confirm these findings (Figure S28 to S43).
On-resin microcleavages were performed on the linear and macrocyclic peptides for further confirmation of macrocycliza- tion. A shift was observed when analyzing the macrocyclized peptide which eluted at 2.69 min, while the linear peptide eluted at 2.79 min. Upon macrocyclization, the structural changes in the peptide results in a faster elution time on the UPLC column. The R groups of the amino acid residues are forced outward allowing for more direct interactions of the polar R groups with the hydrophobic C18 column packing resulting in a faster elution time. A shift in retention time was again observed during purification of the linear and macrocyclized peptide using RPHPLC (Figures S2 and S3).
A facile protocol for macrocyclization of peptides using tertiary vinyl sulfonamides was established and shown to be compatible with standard Fmoc-based SPPS protecting groups and methods. This functional group is easily generated using any secondary amine handle utilizing a low-cost commercially available reagent, 2-chlorosulfonyl chloride. Additionally, it was shown to possess comparable reactivity to acrylates and was considerably more reactive than acrylamides. This functional handle is stable in harsh basic environments unlike acrylates. The higher stability of tertiary vinyl sulfonamides suggests that it can be applied in applications where the conjugation bond stability is desired. Due to the wide availability, low-cost, high selectivity, and increased stability of the tertiary vinyl sulfonamide, we expect the scope of this thiol-Michael click reaction to expand beyond peptide macrocyclization and broaden the toolbox of reaction handles for thiol-Michael conjugations.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by DOE grant DE-SC0019355. This project was partially supported by the Delaware COBRE program, with a grant from the NIH NIGMS (5 P30 GM110758–02).
Supporting Information
Supporting information is available free of charge via the Internet at http://pubs.acs.org.
Synthetic procedures for peptide synthesis and functionalization, NMR characterization for peptides and small molecules, and model degradation study
Notes
The Authors declare no competing financial interest.
REFERENCES
- 1.Wuo MG; Mahon AB; Arora PS, An Effective Strategy for Stabilizing Minimal Coiled Coil Mimetics. Journal of the American Chemical Society 2015, 137 (36), 11618–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ni JH; Singh S; Wang LX, Synthesis of maleimide-activated carbohydrates as chemoselective tags for site-specific glycosylation of peptides and proteins. Bioconjugate Chemistry 2003, 14 (1), 232–238. [DOI] [PubMed] [Google Scholar]
- 3.Kloxin AM; Kasko AM; Salinas CN; Anseth KS, Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science 2009, 324 (5923), 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark RJ; Fischer H; Dempster L; Daly NL; Rosengren KJ; Nevin ST; Meunier FA; Adams DJ; Craik DJ, Engineering stable peptide toxins by means of backbone cyclization: Stabilization of the alpha-conotoxin MII. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (39), 13767–13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierschbacher MD; Ruoslahti E, Influence of stereochemistry of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. Journal ofBiological Chemistry 1987, 262 (36), 17294–17298. [PubMed] [Google Scholar]
- 6.Kumagai H; Tajima M; Ueno Y; Yuhko GH; Ohba M, Effect of Cyclic RGD Peptide on Cell-Adhesion and Tumor-Metastasis. Biochemical and Biophysical Research Communications 1991, 177 (1), 74–82. [DOI] [PubMed] [Google Scholar]
- 7.Bokesch HR; Pannell LK; Cochran PK; Sowder RC; McKee TC; Boyd MR, A novel anti-HIV macrocyclic peptide from Palicourea condensata. Journal of Natural Products 2001, 64 (2), 249–250. [DOI] [PubMed] [Google Scholar]
- 8.Ranganathan D; Lakshmi C; Karle IL, Hydrogen-bonded self-assembled peptide nanotubes from cystine-based macrocyclic bisureas. Journal of the American Chemical Society 1999, 121 (26), 6103–6107. [Google Scholar]
- 9.Ollivier N; Toupy T; Hartkoorn RC; Desmet R; Monbaliu JCM; Melnyk O, Accelerated microfluidic native chemical ligation at difficult amino acids toward cyclic peptides. Nature Communications 2018, 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malins LR; deGruyter JN; Robbins KJ; Scola PM; Eastgate MD; Ghadiri MR; Baran PS, Peptide Macrocyclization Inspired by Non-Ribosomal Imine Natural Products. Journal of the American Chemical Society 2017, 139 (14), 5233–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai ZB; Cai CX; Yu ZL; Wang H, Backbone-Enabled Directional Peptide Macrocyclization through Late-Stage Palladium-Catalyzed delta-C(sp(2))-H Olefination. Angewandte Chemie-InternationalEdition 2018, 57 (42), 13912–13916. [DOI] [PubMed] [Google Scholar]
- 12.Noisier AFM; Garcia J; Ionut IA; Albericio F, Stapled Peptides by Late-Stage C(sp(3))-H Activation. Angewandte Chemie-InternationalEdition 2017, 56 (1), 314–318. [DOI] [PubMed] [Google Scholar]
- 13.Mazur S; Jayalekshmy P, Chemistry of polymer-bound ortho-benzyne - frequency of encounter between substituents on crosslinked polystyrenes. Journal of the American Chemical Society 1979, 101 (3), 677–683. [Google Scholar]
- 14.Aimetti AA; Shoemaker RK; Lin CC; Anseth KS, On-resin peptide macrocyclization using thiol-ene click chemistry. Chemical Communications 2010, 46 (23), 4061–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingale S; Dawson PE, On Resin Side-Chain Cyclization of Complex Peptides Using CuAAC. Organic Letters 2011, 13 (11), 2822–2825. [DOI] [PubMed] [Google Scholar]
- 16.Jagasia R; Holub JM; Bollinger M; Kirshenbaum K; Finn MG, Peptide Cyclization and Cyclodimerization by Cu-I-Mediated Azide-Alkyne Cycloaddition. Journal of Organic Chemistry 2009, 74 (8), 2964–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundquist JT; Pelletier JC, A new tri-orthogonal strategy for peptide cyclization. Organic Letters 2002, 4 (19), 32193221. [DOI] [PubMed] [Google Scholar]
- 18.Asfaw H; Wetzlar T; Martinez-Martinez MS; Imming P, An efficient synthetic route for preparation of antimycobacterial wollamides and evaluation of their in vitro and in vivo efficacy. Bioorganic & Medicinal Chemistry Letters 2018, 28 (17), 2899–2905. [DOI] [PubMed] [Google Scholar]
- 19.Cistrone PA; Silvestri AP; Hintzen JCJ; Dawson PE, Rigid Peptide Macrocycles from On-Resin Glaser Stapling. Chembiochem 2018, 19 (10), 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangold SL; Grubbs RH, Stereoselective synthesis of macrocyclic peptides via a dual olefin metathesis and ethenolysis approach. Chemical Science 2015, 6 (8), 4561–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cromm PM; Wallraven K; Glas A; Bier D; Furstner A; Ottmann C; Grossmann TN, Constraining an Irregular Peptide Secondary Structure through Ring-Closing Alkyne Metathesis. Chembiochem 2016, 17 (20), 1915–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolb HC; Finn MG; Sharpless KB, Click chemistry: Diverse chemical function from a few good reactions. Angewandte Chemie-International Edition 2001, 40 (11), 2004–2021. [DOI] [PubMed] [Google Scholar]
- 23.Tang W; Becker ML, “Click” reactions: a versatile toolbox for the synthesis of peptide-conjugates. Chemical Society Reviews 2014, 43 (20), 7013–7039. [DOI] [PubMed] [Google Scholar]
- 24.Liang LY; Astruc D, The copper(I)-catalyzed alkyneazide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coordination Chemistry Reviews 2011, 255 (23–24), 29332945. [Google Scholar]
- 25.Hoyle CE; Bowman CN, Thiol-Ene Click Chemistry. Angewandte Chemie-International Edition 2010, 49 (9), 1540–1573. [DOI] [PubMed] [Google Scholar]
- 26.Nair DP; Podgorski M; Chatani S; Gong T; Xi WX; Fenoli CR; Bowman CN, The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chemistry of Materials 2014, 26 (1), 724–744. [Google Scholar]
- 27.Tsurkan MV; Chwalek K; Prokoph S; Zieris A; Levental KR; Freudenberg U; Werner C, Defined Polymer-Peptide Conjugates to Form Cell-Instructive starPEG-Heparin Matrices In Situ. Advanced Materials 2013, 25 (18), 2606–2610. [DOI] [PubMed] [Google Scholar]
- 28.Hao ZY; Song YQ; Lin SX; Yang MY; Liang YJ; Wang J; Chen PR, A readily synthesized cyclic pyrrolysine analogue for site-specific protein “click” labeling. Chemical Communications 2011, 47 (15), 4502–4504. [DOI] [PubMed] [Google Scholar]
- 29.El-Sagheer AH; Brown T, Click Nucleic Acid Ligation: Applications in Biology and Nanotechnology. Accounts of Chemical Research 2012, 45 (8), 1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen L-TT; Gokmen MT; Du Prez FE, Kinetic comparison of 13 homogeneous thiol-X reactions. Polymer Chemistry 2013, 4 (22), 5527–5536. [Google Scholar]
- 31.Renault K; Fredy JW; Renard PY; Sabot C, Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjugate Chemistry 2018, 29 (8), 2497–2513. [DOI] [PubMed] [Google Scholar]
- 32.Alley SC; Benjamin DR; Jeffrey SC; Okeley NM; Meyer DL; Sanderson RJ; Senter PD, Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjugate Chemistry 2008, 19 (3), 759–765. [DOI] [PubMed] [Google Scholar]
- 33.Baldwin AD; Kiick KL, Tunable Degradation of Maleimide-Thiol Adducts in Reducing Environments. Bioconjugate Chemistry 2011, 22 (10), 1946–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rydholm AE; Anseth KS; Bowman CN, Effects of neighboring sulfides and pH on ester hydrolysis in thiol-acrylate photopolymers. Acta Biomaterialia 2007, 3 (4), 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dadova J; Orsag P; Pohl R; Brazdova M; Fojta M; Hocek M, Vinylsulfonamide and Acrylamide Modification of DNA for Cross-linking with Proteins. Angewandte Chemie-International Edition 2013, 52 (40), 10515–10518. [DOI] [PubMed] [Google Scholar]
- 36.Sinha J; Podgorski M; Huang SJ; Bowman CN, Multifunctional monomers based on vinyl sulfonates and vinyl sulfonamides for crosslinking thiol-Michael polymerizations: monomer reactivity and mechanical behavior. Chemical Communications 2018, 54 (24), 3034–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang SJ; Sinha J; Podgorski M; Zhang XP; Claudino M; Bowman CN, Mechanistic Modeling of the Thiol-Michael Addition Polymerization Kinetics: Structural Effects of the Thiol and Vinyl Monomers. Macromolecules 2018, 51 (15), 59795988. [Google Scholar]
- 38.Cemazar M; Craik DJ, Microwave-assisted Boc-solid phase peptide synthesis of cyclic cysteine-rich peptides. Journal of Peptide Science 2008, 14 (6), 683–689. [DOI] [PubMed] [Google Scholar]
- 39.Northrop BH; Frayne SH; Choudhary U, Thiolmaleimide “click” chemistry: evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity. Polymer Chemistry 2015, 6 (18), 3415–3430. [Google Scholar]
- 40.Reddick JJ; Cheng JM; Roush WR, Relative rates of Michael reactions of 2 ‘-(phenethyl)thiol with vinyl sulfones, vinyl sulfonate esters, and vinyl sulfonamides relevant to vinyl sulfonyl cysteine protease inhibitors. Organic Letters 2003, 5 (11), 1967–1970. [DOI] [PubMed] [Google Scholar]
- 41.Bernardi F; Bottoni A; Rossi I; Robb MA, Theoretical-study of substituent-effect on the stability of enolate ions. Journal of Molecular Structure 1993, 300, 157–169. [Google Scholar]
- 42.Rosenberg RE, Enols and enolates of carboxylic acid derivatives. A G2(MP2) ab initio study. Journal of Organic Chemistry 1998, 63 (16), 5562–5567. [Google Scholar]
- 43.Bernardi F; Csizmadia IG; Mangini A; Schlegel HB; Whangbo MH; Wolfe S, Irrelevance of d-orbital. I. alpha-thiocarbanion - comparative quantum chemical study of static and dynamic properties and proton affinities of carbanions adjacent to oxygen and to sulfur. Journal of the American Chemical Society 1975, 97 (8), 2209–2218. [Google Scholar]
- 44.Bernardi F; Bottoni A; Venturini A; Mangini A, The stabilization of alpha-substituted oxycarbanions and thiocarbanions. Journal of the American Chemical Society 1986, 108 (26), 8171–8175. [Google Scholar]
- 45.Borden WT; Davidson ER; Andersen NH; Denniston AD; Epiotis ND, Regarding the mechanism of C-H bond acidification by sulfur. Journal of the American Chemical Society 1978, 100 (5), 1604–1605. [Google Scholar]
- 46.Lehn JM; Wipff G, Stereoelectronic effects. 5. Stereoelectronic properties, stereospecificity, and stabilization of alpha-oxa and alpha-thia carbanions. Journal of the American Chemical Society 1976, 98 (24), 7498–7505. [Google Scholar]
- 47.Barlos K; Gatos D; Hatzi O; Koch N; Koutsogianni S, Synthesis of the very acid-sensitive Fmoc-Cys(Mmt)-OH and its application in solid-phase peptide synthesis. International Journal of Peptide and Protein Research 1996, 47 (3), 148–153. [DOI] [PubMed] [Google Scholar]
- 48.White CJ; Yudin AK, Contemporary strategies for peptide macrocyclization. Nature Chemistry 2011, 3 (7), 509–524. [DOI] [PubMed] [Google Scholar]
- 49.Brady SF; Varga SL; Freidinger RM; Schwenk DA; Mendlowski M; Holly FW; Veber DF, Practical synthesis of cyclic-peptides, with an example of dependence of cyclization yield upon linear sequence. Journal of Organic Chemistry 1979, 44 (18), 3101–3105. [Google Scholar]
- 50.Ralhan K; KrishnaKumar VG; Gupta S, Piperazine and DBU: a safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. Rsc Advances 2015, 5 (126), 104417104425. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.