

Abstract
An efficient, three-component strategy for Rh(III)-catalyzed annulation of readily available 3-aminopyrazoles, aldehydes, and sulfoxonium ylides to give diverse pyrazolo[1,5-a]pyrimidines is disclosed. The reactions were performed under straightforward benchtop conditions using microwave heating with short reaction times. Good yields were obtained for a number of substituted aminopyrazoles and a very large variety of aromatic and heteroaromatic aldehydes, including those incorporating electron-withdrawing, electron-donating, basic nitrogen, halide and acidic functionality. Ester and methoxy functionalities could also be directly installed on the pyrimidine ring by employing ethyl glyoxylate and trimethyl orthoformate in place of the aldehyde, respectively. Additionally, a range of sulfoxonium ylides provided products in good yields to establish that aryl, heteroaryl, and branched and unbranched alkyl substituents can be introduced with this reagent. Finally, the first use of a formyl sulfoxonium ylide in a chemical transformation enabled the preparation of products with only a single substituent on the pyrimidine ring as introduced by the aldehyde coupling partner. For the formyl ylide, a one-pot, stepwise reaction sequence was used to prevent competitive condensation of the formyl group with the aminopyrazole.
Graphical Abstract
INTRODUCTION
Fused [5,6]-bicyclic bridgehead nitrogen heterocycles are privileged pharmacophores in drug discovery. The azolopyrimidines are the most prevalent subclass, and are featured in many U.S. FDA approved drugs and even more clinical candidates.1,2 We have recently reported the first examples of imidoyl C–H activation via Rh(III)-catalyzed two-component annulations of N-azolo imines 1 with alkynes, diazoketones and sulfoxonium ylides to give azolopyrimidines 2 (Scheme 1A).3,4 Moreover, annulations with 1,4,2-dioxazol-5-one amidating reagents afforded a wide range of azolo[1,3,5]triazines 3.5,6 A particularly attractive feature of the aforementioned approach is the large number of diverse N-azolo imines 1 that can readily be prepared from commercially available aminoazoles and aldehydes.
Scheme 1.

Fused [5,6]-Bicyclic Bridgehead Nitrogen Heterocycle Synthesis
Multi-component reactions enable access to complex structures from simple precursors. In work relevant to N-fused [5,6]-bicyclic heterocycle synthesis, the Groebke–Blackburn–Bienaymé (GBBR) reaction is a popular and efficient approach (Scheme 1B).7 In this three-component reaction, imines formed in situ couple with isocyanides to afford a wide range of fused imidazole bridgehead nitrogen heterocycles 4.
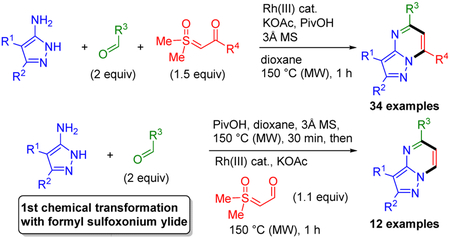
Herein, we report the multi-component synthesis of pyrazolopyrimidines 8 (Scheme 1C), which are found in more approved drugs and clinical candidates than any other subclass of [5,6]-bicyclic bridgehead nitrogen heterocycles.1 In this three-component coupling reaction, imines that are formed in situ from readily available aldehydes 5 and aminoazoles 6, undergo Rh-catalyzed annulation with sulfoxonium ylides 7, which are stable, crystalline carbene equivalents that are increasingly used in Rh(III)-catalyzed C–H functionalization.8,9 The rapid preparation of diverse pyrazolopyrimidines 8 is further facilitated by the use of a commercially available and air stable Rh(III) catalyst, convenient benchtop set up, and short reaction times with microwave heating.
High functional group compatibility is an important aspect of this three-component reaction, with unhindered basic heterocycle nitrogens, amides, esters, secondary and tertiary carbamates, halides, carboxylic acids and acidic secondary anilides all successfully incorporated. Ester and methoxy functionalities can be directly installed on the pyrimidine ring by employing ethyl glyoxylate and trimethyl orthoformate in place of the aldehyde, respectively.
We also report the efficient preparation of pyrazolopyrimidines 8 with only a single substituent on the pyrimidine ring (bottom of Scheme 1C). In a one-pot reaction, stepwise formation of N-azolo imines is followed by addition of the formyl ylide 7i and the Rh(III) catalyst. To our knowledge this is the first chemical transformation of a formyl sulfoxonium ylide.
RESULTS AND DISCUSSION
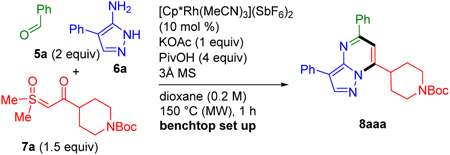
A large variety of reaction parameters were first examined for coupling benzaldehyde (5a), aminopyrazole 6a, and sulfoxonium ylide 7a to give pyrazolopyrimidine 8aaa (Table 1). This investigation established that good yields of 8aaa could be obtained by benchtop set up using the commercially available and air stable cationic catalyst [Cp*Rh(MeCN)3](SbF6)2 with KOAc, pivalic acid (PivOH) and 3Å sieves as additives in dioxane (0.2 M) under microwave (MW) conditions at 150 °C for 1 h (entry 1, Table 1). When the halide was not abstracted from [Cp*RhCl2]2, a slight reduction in yield of the product was observed (entry 2). As expected, when a Rh(III) catalyst was not added, no product was obtained (entry 3). NaOAc was found to be less effective than KOAc (entry 4), and completely removing the base led to a further reduction in the yield (entry 5). Removing the PivOH also resulted in a significantly lower yield (entry 6). Increasing the temperature to 160 °C did not improve the yield indicating that more forcing conditions are not necessary (entry 7). Chlorobenzene, a typical solvent for microwave reactions was tested and found to be less effective (entry 8). Reducing the amount of sulfoxonium ylide (1.1 equiv) led to only a slightly lower yield (entry 9). In comparison to Cp*Rh(III) catalysis, Cp*Ir(III) and Cp*Co(III) catalysts were much less effective (entries 10 and 11).
Table 1.
Reaction Parameters for Annulation to Pyrazolopyrimidine 8a
| entry | Variation | Yield %b |
|---|---|---|
| 1 | None | 75 |
| 2 | [Cp*RhCl2]2 (5%) | 61 |
| 3 | no Rh | 0 |
| 4 | NaOAc instead of KOAc | 60 |
| 5 | no KOAc | 39 |
| 6 | no PivOH | 27 |
| 7 | 160 °C | 73 |
| 8 | chlorobenzene as solvent | 57 |
| 9 | ylide 7a (1.1 equiv) | 67 |
| 10 | [Cp*IrCl2]2 (5%) and AgSbF6 (20%) | 28 |
| 11 | [Cp*Co(MeCN)3](SbF6)2 (10%) | 21 |
| 12 | 0.1 M | 62 |
| 13 | 0.4 M | 85 (82)c |
| 14 | 0.4 M, no sieves | 75 |
| 15 | 0.4 M, [Cp*Rh(MeCN)3](SbF6)2 (5%) | 77 |
| 16 | 0.4 M, [Cp*Rh(MeCN)3](SbF6)2 (2.5%) | 38 |
| 17 | 0.4 M, conventional heating, 100 °C, 16 h | 82 |
Conditions: 5a (0.20 mmol), 6a (0.10 mmol), 7a (0.15 mmol)
Yield determined by 1H-NMR relative to 1,3,5-trimethoxybenzene as external standard.
Isolated yield of a 0.30 mmol scale (see Scheme 2).
Concentration is important as might be expected for a multicomponent reaction (entries 12 and 13), with a higher 0.4 M concentration resulting in an appreciable increase in yield (entry 13). We did not explore further increases in the concentration due to the amount of 3Å sieves used (ca. 100 mg per 0.1 mmol of limiting reagent), although it should be noted that only a modest reduction in yield was observed when 3Å sieve were not added (entry 14).
Consistent with our goal to provide access to diverse pyrazolopyrimidines 8 at short reaction times, we chose 10 mol % catalyst loading for evaluating substrate scope. However, 5 and 2.5 mol % catalyst loading did afford significant amounts of the desired product (entries 15 and 16). Lower catalyst loadings are therefore likely to be applicable to many starting material combinations (vide infra) as well as by employing more forcing reaction conditions. Finally, an 82% yield for this three-component reaction was observed by employing conventional heating for 16 h at 100 °C, a temperature lower than the boiling point of the dioxane solvent (entry 17).
Using the optimal reaction conditions (i.e., entry 13, Table 1), we first explored aldehyde scope for three-component coupling with aminopyrazole 6a and sulfoxonium ylide 7a (Scheme 2). Benzaldehydes with electron withdrawing and donating groups at the para-position gave pyrazolopyrimidines 8baa and 8caa in 78% and 80% yields, respectively, establishing that the reaction is effective regardless of the electron properties of the aldehyde input. Benzaldehydes substituted at the meta- and ortho-positions were also effective coupling partners as exemplified by the halo substituted products 8daa and 8eaa, respectively. However, a moderate reduction in yield was observed for ortho substituted product 8eaa.
Scheme 2.

Aldehyde Scope for Three-component Synthesis of Pyrazolopyrimidines 8a
a Standard conditions with 5 (0.60 mmol), 6a (0.30 mmol), and 7a (0.45 mmol). b 0.2 M.
Benzaldehydes bearing a large variety of useful functional groups were effective inputs. For example, good yields were obtained for pyrazolopyrimidine products displaying ester (8faa), carboxylic acid (8gaa), and acidic secondary anilide (8haa) functionality. Pyridinecarboxaldehydes were also effective inputs. Notably, unhindered basic heterocyclic nitrogens do not interfere with this directed C-H functionalization reaction as demonstrated by the incorporation of 4-pyridinecarboxaldehyde to give 8iaa in 58% yield. The 4-acetamido and 4-chloro substituted 3-pyridinecarboxaldehydes also coupled effectively to give 8jaa and 8kaa in 77 and 66% yields, respectively. The chloropyridine substituent in 8kaa as well as the bromophenyl substituent in 8daa provide versatile sites for incorporating additional functionality. These examples highlight the compatibility of haloarene functionality with the three-component reaction conditions. This outcome contrasts with methods to elaborate pyrazolopyrimidine frameworks by using cross-coupling to introduce aromatic substituents, which inherently rely on reactions that proceed by aryl halide oxidative addition.10
Five-membered furan-, thiophene- and pyrazolecarboxaldehydes provided 8laa–naa in excellent yields (81–85%). Notably, the N-alkyl pyrazolyl motif in 8naa is incorporated into a number of approved kinase inhibitor drugs and drug candidates with [5,6]-bicyclic nitrogen heterocycle pharmacophores such as the approved janus kinase inhibitor drugs ruxolitinib and baricitinib.11 Lastly, enolizable aldehydes, provide the desired pyrazolopyrimidines 8 in poor yields as exemplified by 8oaa obtained in 13% yield from acetaldehyde.
We next investigated the scope for the sulfoxonium ylides 7 (Scheme 3). Significantly, these ylides can readily be prepared by straightforward one-step protocols from trimethylsulfoxonium iodide and acid chlorides or activated esters.8,9 In addition to employing the NBoc-piperidine ylide 7a input, another example of the incorporation of an α-branched substituent is provided by the isopropyl substituted pyrazolopyrimidine product 8aab, which was obtained in 79% yield. Methyl, β-branched and N-Boc-ethyl substituted products 8aac–aae were also obtained in excellent yields (80–94%). Electron-rich and -poor aryl-substituted ylides efficiently coupled to give 8aaf (89%) and 8aag (87%), respectively. These results when considered along with the successful incorporation of electron-poor and -rich benzaldehydes such as for 8baa and 8caa (Scheme 2), clearly establish that the regiospecific introduction of aromatic substituents regardless of electronic properties can be achieved. In contrast, while the efficient preparation of pyrazolopyrimidines can be accomplished by condensations of 3-aminopyrazole with symmetrical 1,3-dicarbonyl compounds, this approach generally provides mixtures of regioisomers when unsymmetrical diketones are employed.12 Finally, the effective incorporation of a heteroaryl-substituted ylide was demonstrated by the preparation of thiophenyl substituted 8aah in 85% yield.
Scheme 3.

Sulfoxonium Ylide Scope for Three-Component Synthesis of Pyrazolopyrimidines 8a
a Standard conditions with 5a (0.60 mmol), 6a (0.30 mmol), and 7 (0.45 mmol)
As depicted in Scheme 4, we next evaluated the scope for the aminopyrazoles, many of which are commercially available or straightforward to prepare in high yields.13 Using standard conditions for 4-phenyl-substituted 3-aminopyrazole (6a), 4-methyl-substituted 3-aminopyrazole efficiently provided pyrazolopyrimidine 8aba in 71% yield. Disubstituted aminopyrazoles also coupled efficiently to give 8aca and 8ada. Unsubstituted and 5-tert-butyl-substituted aminopyrazoles were also effective inputs providing 8aea in 62% and 8afa in 73% yields, respectively. It should be noted that for these inputs, a modest improvement in the product yields were achieved by lowering the concentration to 0.2 M. Therefore, 0.2 M was chosen for the evaluation of other 5-substituted-3-aminopyrazoles, including those with methyl, isopropyl, cyclopropyl, and phenyl substituents, which gave 8aga–8aja in reasonable yields (49–65%). Additionally, electron deficient 5-trifluoromethyl-3-aminopyrazole (6k) efficiently underwent standard coupling reaction conditions at both concentrations (0.4 and 0.2 M) affording bicyclic product 8aka in good yield (76% and 71%, respectively). In contrast, 2-aminoimidazoles are not effective coupling partners, presumably because more forcing conditions are required for aldehyde condensation to give imines. However, we have previously reported that imines from 2-aminoimidazoles can be prepared using Ti(OEt)4 as an acid catalyst and water scavenger. After isolation, the imines can then be subjected to Rh(III)-catalyzed coupling with sulfoxonium ylides to give imidazopyrimidines in high yields.3
Scheme 4.

Aminopyrazole Scope for Three-Component Synthesis of Pyrazolopyrimidines 8a
a Standard conditions with 5a (0.60 mmol), 6 (0.30 mmol), and 7a (0.45 mmol). b 0.2 M
In some drug discovery efforts, pyrazolopyridimines with only a single substituent on the pyrimidine ring might be desired. We therefore explored formyl sulfoxonium ylide 7i as a potential input to give pyrazolopyrimidines 8 having only one pyrimidine ring substituent as introduced by the aldehyde (Scheme 5). To our knowledge, formyl sulfoxonium ylides have never previously been employed in a chemical transformation.
Scheme 5.

Annulations with Formyl Ylide to Give Pyrazolopyrimidines 8a
a Standard conditions 5 (0.60 mmol), 6 (0.30 mmol), 7i (0.33 mmol). b 0.4 M. c 0.1 M
Under the optimized one-step three-component reaction conditions, we found that the aldehyde functionality in formyl ylide 7i effectively competes with the aldehyde input for condensation with the aminopyrazoles. To circumvent this side reaction, a two-step one-pot sequence was instead implemented. The aldehyde 5 and aminopyrazole 6 are first condensed by microwave heating at 150 °C for 30 min in the presence of molecular sieves and PivOH. Formyl ylide 7i, KOAc, and [Cp*Rh(MeCN)3](SbF6)2 are then added followed by microwave heating at 150 °C for 1 h. It is worth noting that PivOH is added in the first step to catalyze imine formation. Additionally, use of only 1.1 equiv rather than 1.5 equiv of formyl ylide 7i resulted in moderately higher yields because it minimized competitive imine exchange during the annulation step.
The effect of reaction concentration on this two-step, one-pot protocol was evaluated for the preparation of pyrazolopyrimidine 8aai from benzaldehyde (5a), 4-phenyl-3-aminopyrazole (6a), and formyl ylide 7i. At 0.1 M and 0.2 M concentrations, comparable 80% and 78% yields were obtained, respectively. However, in contrast to the higher yield obtained at higher concentrations for the three-component reaction of β-keto sulfoxonium ylide 7a (see entries 12 and 13, Table 1), at 0.4 M a significantly lower 63% yield was observed. Therefore, we used a 0.2 M reaction concentration for evaluating the coupling of formyl ylide 7i with 4-phenyl-3-aminopyrazole 6a and different aldehydes 5. Good yields of pyrazolopyrimidines were obtained for benzaldehydes incorporating the electron-deficient trifluoromethyl (8bai), electron-rich methoxy (8cai), and bromo (8dai) functionality. Additionally, pyrazolopyrimidine 8kai was obtained from 6-chloronicotinaldehyde in 80% yield, and pyrazolopyrimidine 8nai was obtained from N-methyl-4-pyrazolecarboxaldehyde in 66% yield. Disubstituted aminopyrazoles also gave 8aci and 8adi in 82% and 72%, respectively.
Unsubstituted aminopyrazole and 5-substituted aminopyrazoles gave higher yields at lower concentrations. At 0.1 M concentration, pyrazolopyrimidines 8aei and 8afi were obtained in 30% and 61% yields, respectively.
To directly install an ester group onto the pyrimidine ring of the product we sought to employ ethyl glyoxylate (5p) as the aldehyde input (Scheme 6). The standard one-step three-component conditions provided the desired pyrazolopyrimidine 8paa, but in less than 15% yield. Due to the reactive nature of ethyl glyoxylate, we speculated that a two-step, one-pot procedure for imine formation followed by Rh(III)-catalyzed annulation might be more successful. Indeed, pyrazolopyrimidine 8paa was obtained in 82% yield by employing this stepwise approach using only slight excess of ethyl glyoxylate (i.e., 1.1 equiv). It is worth noting that the reaction was completely shut down at high glyoxylate loading (5 equiv). Additionally, this stepwise approach was successfully applied to formyl sulfoxonium ylide 7i to give pyrazolopyrimidine 8pai in 81%.
Scheme 6.

Ethyl Glyoxylate as the Aldehydea
a 5p (0.60 mmol), 6a (0.30 mmol), 7 (0.45 mmol). b 7i (1.1 equiv).
To directly install a methoxy group onto the pyrimidine ring, trimethyl orthoformate (5q) was used in place of the aldehyde input 5 (Scheme 7). For this transformation, a one-pot stepwise approach was also the most effective. Consistent with the use of trimethyl orthoformate as a low cost dehydrating agent, the highest yields were achieved by employing 10 equiv of trimethyl orthoformate without the addition of sieves. Under these conditions, pyrazolopyridimines 8qaa and 8qai were obtained in 78% and 36% yields, respectively.
Scheme 7.

Methoxy Group Introductiona
a 5q (3.0 mmol), 6a (0.30 mmol), 7 (0.45 mmol). b 7i (1.1 equiv).
Finally, pyrazolopyrimidine 8naa was prepared on three-fold larger 1 mmol scale with benchtop set up and a lower catalyst loading of 5 mol % (Scheme 8). It is noteworthy that this lower catalyst loading resulted in only a slight reduction in the yield to 77% at the standard reaction temperature of 150 °C and time of 1 h.
Scheme 8.

Benchtop Reaction Set Up at 1 mmol Scale
A plausible mechanism for this three-component coupling reaction is depicted in Scheme 9. In accordance to our publication on Rh(III)-catalyzed two-component annulations of N-azolo imines with alkynes, diazoketones and sulfoxonium ylides,3 we propose that imine A, formed in situ from aldehyde 5 and aminopyrazole 6, undergoes concerted metalation-deprotonation to give rhodacycle B. In this publication we reported the X-ray structural characterization of a catalytically competent neutral rhodacycle analogous to B.3 Carbene insertion of sulfoxonium ylide 7 with release of dimethyl sulfoxide (DMSO) then provides the six-membered rhodacycle C. Protodemetalation releases ketone D to regenerate the active Rh(III) catalyst. Finally, under the reaction conditions, ketone D undergoes cyclodehydration to afford the pyrazolopyrimidine 8.
Scheme 9.

Proposed Mechanism for Annulation
In conclusion, we have reported the Rh(III)-catalyzed three-component coupling of aldehydes 5, aminopyrazoles 6, and sulfoxonium ylides 7 to generate diverse pyrazolopyrimidines 8. This method is operationally convenient with benchtop set up and a short reaction time with microwave heating. A number of aromatic and heteroaromatic aldehydes 5 displaying diverse functionality are effective inputs as are a number of aminopyrazole derivatives 6. The reaction also proceeds in good yields for a range of aryl, heteroaryl, and alkyl ylides 7. In addition, the first examples of the application of a formyl sulfoxonium ylide in a metal-catalyzed transformation were implemented for the preparation of pyrazolopyrimidines 8 with only a single substituent on the pyrimidine ring. Ester and methoxy functionalities were also installed directly onto the pyrimidine ring of the bicyclic product by employing ethyl glyoxylate and trimethyl orthoformate in place of the aldehyde input, respectively.
EXPERIMENTAL SECTION
General Information.
Unless otherwise noted, all commercially available reagents were purchased and used as received. Solvents including 1,4-dioxane, tetrahydrofuran (THF), and dichloromethane (CH2Cl2) were deoxygenated by sparging with argon. Chlorobenzene was purified by passing through a plug of alumina before use. Microwave reactions were performed using a microwave reactor with an external IR sensor and in a closed reaction vessel. 1H, 13C, and 19F NMR spectra were recorded on 400 MHz or 600 MHz spectrometers. Chemical shifts [δ (ppm)], coupling constants [J (Hz)], multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, pent = pentet, m = multiplet, br = broad), and integration are reported. Chemical shifts for 1H and 13C NMR are reported relative to residual undeuterated solvent in CDCl3 (7.24 ppm for 1H NMR and 76.99 ppm for 13C NMR) and (CD3)2SO (2.47 ppm for 1H NMR and 39.94 ppm for 13C NMR). Flash chromatography was carried out with silica gel with 40–63 μm particle size and with 230–400 mesh. Partial data are provided for IR spectra. Melting points are reported uncorrected. High-resolution mass spectra (HRMS) were obtained using electrospray ionization (ESI) on a time of flight (TOF) mass spectrometer (Yale University) or electron ionization (EI) obtained by the University of Illinois SCS Mass Spectrometry Laboratory.
Preparation of catalysts and reactants.
All aldehydes 5 and aminopyrazoles 6 were purchased and used as received. [Cp*Rh(MeCN)3](SbF6)2 was synthesized according to literature procedures.14 Sulfoxonium ylides 7a–d and 7f–h were synthesized according to literature procedures.9f Sulfoxonium ylides 7e and 7i were prepared according to a literature procedure for a related compound with slight modifications.9f
tert-Butyl (4-(dimethyl(oxo)-λ6-sulfaneylidene)-3-oxobutyl)carbamate (7e):
Ylide 7e was prepared from a literature procedure for a related compound with slight modification.9f A mixture of trimethylsufoxonium iodide (3.52 g, 16.0 mmol, 3.2 equiv) and potassium tert-butoxide (1.68 g, 15.0 mmol, 3.0 equiv) in THF (125 mL) was heated to reflux (67 °C) for 2 h under nitrogen. After cooling to rt, to the above mixture was slowly added a solution of 4-nitrophenyl 3-((tert-butoxycarbonyl)amino)propanoate (1.55 g, 5.00 mmol, 1.00 equiv) in THF (25 mL). The resulting mixture was stirred at rt for 2 h, filtered through a pad of celite, washed thoroughly with CH2Cl2 and concentrated under reduced pressure. Purification by silica gel column chromatography (5% MeOH/CH2Cl2) afforded ylide 7e (591 mg, 45%) as a yellow solid. mp 114–116 °C. FTIR (neat) 3369, 3001, 1682, 1553, 1523, 1391, 1364, 1276, 1170, 1033, 990, 848, 449 cm−1. 1H NMR (600 MHz, CDCl3) δ 5.22 (s, 1H), 4.43 (s, 1H), 3.39 (s, 6H), 3.36 (q, J = 6.1 Hz, 2H), 2.37 (t, J = 6.1 Hz, 2H), 1.43 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 189.2, 156.1, 79.0, 70.2, 42.3, 40.0, 37.3, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C11H22NO4S+, 264.1264; found 264.1278.
2-(Dimethyl(oxo)-λ6-sulfaneylidene)acetaldehyde (7i):
Ylide 7i was prepared from a literature procedure for a related compound with slight modification.9f A mixture of trimethylsufoxonium iodide (10.6 g, 48.0 mmol, 3.2 equiv) and potassium tert-butoxide (5.05 g, 45.0 mmol, 3.0 equiv) in THF (100 mL) was heated at reflux (67 °C) for 2 h under nitrogen. After cooling to rt, to the above mixture was slowly added a solution of ethyl formate (1.11 g, 15.0 mmol, 1.0 equiv) in THF (10 mL). The resulting mixture was stirred at rt for 1 h, filtered through a pad of celite, washed thoroughly with CH2Cl2 and concentrated under reduced pressure. Purification by silica gel column chromatography (10% MeOH/CH2Cl2) afforded formyl ylide 7i as a 97:3 cis/trans ratio of stereoisomers (1.12 g, 62%) as a white solid. mp 71–73 °C. FTIR (neat) 3394, 3082, 3005, 2914, 2788, 1565, 1309, 1296, 1162, 1028, 949, 833, 466 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.09 (trans, d, J = 9.6 Hz, 0.03H), 8.61 (cis, d, J = 2.6 Hz, 1H), 4.61 (trans, d, J = 9.6 Hz, 0.03H), 4.35 (cis, d, J = 2.6 Hz, 1H), 3.41 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 178.1, 71.9, 42.5. 1H NMR in (CD3)2SO was also obtained to provide a different ratio of sulfoxonium ylide cis/trans isomers (0.74:0.26), which also equilibrated slowly on the NMR time scale as has previously been noted by Kondo and Tunemoto:15 1H NMR (400 MHz, (CD3)2SO) δ 8.84 (trans, d, J = 9.9 Hz, 0.24H), 8.46 (cis, d, J = 2.6 Hz, 0.64H), 4.70 (cis, d, J = 2.3 Hz, 0.74H), 4.53 (trans, d, J = 9.9 Hz, 0.26H), 3.47 (s, 6H). HRMS (EI): m/z [M]+ calcd for C4H8O2S, 120.0245; found 120.0244.
General Procedure for Three-Component Reaction of Aminopyrazoles, Aldehydes and β-Keto Sulfoxonium Ylides (0.3 mmol scale).
To a flame-dried 2–5 mL Biotage microwave vial (#351521) charged with a stir bar on the benchtop were added aminopyrazole (0.300 mmol, 1.00 equiv), aldehyde (0.600 mmol, 2.00 equiv), [Cp*Rh(MeCN)3](SbF6)2 (10 mol %, 0.030 mmol, 25 mg), PivOH (1.20 mmol, 4.00 equiv, 123 mg), KOAc (0.30 mmol, 1.0 equiv, 29 mg), 3Å molecular sieves (approximately 300 mg), sulfoxonium ylide (0.450 mmol, 1.50 equiv) and dioxane (0.400 M, 0.750 mL). The vial was capped with a Teflon-lined cap, flushed with nitrogen for about 2 min, and then heated with a Biotage Initiator+ (#356007), which employs an external IR sensor and a closed reaction vessel. The resultant mixture was stirred in the microwave reactor for 1 h at 150 °C using the following settings (absorption level, low; vial type, 2−5 mL; prestirring, 0; initial power, 0; dynamic deflector optimization, ON; pressure: OFF; power, OFF; fixed hold time, ON; stir rate, 600). After cooling to rt, the crude mixture was transferred to a separatory funnel with CH2Cl2 (50 mL). PivOH was removed by extracting with satd. NaHCO3 (100 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were washed with satd. NaHSO3 (10 % wt, 100 mL) to remove remaining aldehyde.16 The organic layer was dried (anhyd. Na2SO4) and concentrated under reduced pressure. The product was purified by silica gel column chromatography.
tert-Butyl 4-(3,5-diphenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aaa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–30% Et2O/hexanes) afforded 8aaa (112 mg, 82%) as a yellow solid. 1H and 13C NMR spectra matched with previously reported literature.3 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.22–8.09 (m, 4H), 7.59–7.39 (m, 5H), 7.26 (t, J = 7.3 Hz, 1H), 7.12 (s, 1H), 4.51–4.14 (m, 2H), 3.78 (tt, J = 12.2, 3.4 Hz, 1H), 2.98 (m, 2H), 2.32–2.11 (m, 2H), 1.86–1.65 (m, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 155.9, 154.6, 152.1, 145.1, 142.3, 137.5, 132.3, 130.4, 128.9, 128.7, 127.3, 126.2, 126.1, 110.7, 101.4, 79.8, 43.7, 36.6, 29.4, 28.5.
tert-Butyl 4-(3-phenyl-5-(4-(trifluoromethyl)phenyl)pyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8baa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 82.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5b and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–30% Et2O/hexanes) afforded 8baa (123 mg, 78%) as a yellow foam. FTIR (neat) 1681, 1422, 1314, 1237, 1157, 1113, 1063, 768, 725, 692 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.26 (d, J = 8.2 Hz, 2H), 8.18–8.09 (m, 2H), 7.77 (d, J = 8.2 Hz, 2H), 7.52–7.43 (m, 2H), 7.32–7.26 (m, 1H), 7.12 (s, 1H), 4.51–4.17 (m, 2H), 3.79 (tt, J = 12.1, 3.3 Hz, 1H), 3.10–2.87 (m, 2H), 2.28–2.15 (m, 2H), 1.85–1.69 (m, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.6, 154.2, 152.6, 144.9, 142.6, 140.8, 132.0, 131.9 (q, J = 32.6 Hz), 128.8, 127.6, 126.34, 126.30, 125.8 (q, J = 3.8 Hz), 124.0 (q, J = 272.2 Hz), 111.3, 101.3, 79.9, 43.7, 36.7, 29.3, 28.4. 19F NMR (376 MHz, CDCl3) δ −62.8. HRMS (ESI): m/z [M + H]+ calcd for C29H30F3N4O2+, 523.2315; found 523.2291.
tert-Butyl 4-(5-(4-methoxyphenyl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8caa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 73.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5c and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–30% Et2O/hexanes) afforded 8caa (116 mg, 80%) as a yellow solid. mp 191–193 °C. FTIR (neat) 1693, 1602, 1426, 1388, 1250, 1173, 1128, 1032, 945, 823, 699, 563, 509 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 8.23–8.05 (m, 4H), 7.45 (t, J = 7.8 Hz, 2H), 7.25 (t, J = 7.3 Hz, 1H), 7.11–6.90 (m, 3H), 4.49–4.17 (m, 2H), 3.88 (s, 3H), 3.75 (tt, J = 12.2, 3.4 Hz, 1H), 2.98 (broad t, J = 10.6 Hz, 2H), 2.28–2.15 (m, 2H), 1.86–1.64 (m, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 161.6, 155.5, 154.7, 151.9, 145.1, 142.2, 132.5, 130.0, 128.8, 128.7, 126.2, 125.9, 114.3, 110.3, 100.9, 79.8, 55.4, 43.8, 36.6, 29.4, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C29H33N4O3+, 485.2547; found 485.2547.
tert-Butyl 4-(5-(3-bromophenyl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8daa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 70.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5d and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–40% Et2O/hexanes) afforded 8daa (114 mg, 71%) as a yellow solid. mp 168–171 °C. FTIR (neat) 1682, 1563, 1429, 1234, 1166, 1126, 1078, 946, 795, 760, 623, 533 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.26 (t, J = 1.8 Hz, 1H), 8.16–8.11 (m, 2H), 8.09 (ddd, J = 7.9, 1.8, 1.0 Hz, 1H), 7.61 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.47 (t, J = 7.8 Hz, 2H), 7.39 (t, J = 7.9 Hz, 1H), 7.31–7.25 (m, 1H), 7.06 (s, 1H), 4.55–4.20 (m, 2H), 3.78 (tt, J = 12.1, 3.4 Hz, 1H), 3.15–2.77 (m, 2H), 2.28–2.16 (m, 2H), 1.84–1.66 (m, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.6, 154.2, 152.5, 144.9, 142.6, 139.5, 133.2, 132.1, 130.4, 130.3, 128.8, 126.3, 126.3, 125.9, 123.1, 111.1, 101.2, 79.8, 43.9, 36.7, 29.3, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C28H30BrN4O2+, 533.1547; found 533.1573.
tert-Butyl 4-(5-(2-chlorophenyl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8eaa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 67.6 μL (0.600 mmol, 2.00 equiv) of aldehyde 5e and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (5–20% EtOAc/hexanes) afforded 8eaa (67.5 mg, 46%) as a yellow solid. mp 194–196 °C. FTIR (neat) 1688, 1609, 1564, 1425, 1231, 1163, 1126, 946, 758, 696, 516 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.14–8.05 (m, 2H), 7.81–7.74 (m, 1H), 7.55–7.47 (m, 1H), 7.46–7.37 (m, 4H), 7.26–7.20 (m, 1H), 7.10 (s, 1H), 4.49–4.20 (m, 2H), 3.79 (tt, J = 12.1, 3.3 Hz, 1H), 3.08–2.89 (m, 2H), 2.30–2.17 (m, 2H), 1.80–1.65 (m, 2H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 156.1, 154.7, 151.1, 144.9, 142.2, 137.8, 132.3, 132.1, 131.8, 130.6, 130.4, 128.7, 127.2, 126.3, 126.2, 111.3, 106.0, 79.8, 43.8, 36.5, 29.2, 28.4. HRMS (ESI): m/z [M + H]+ calcd for C28H30ClN4O2+, 489.2052; found 489.2042.
tert-Butyl 4-(5-(4-(methoxycarbonyl)phenyl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8faa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 98.5 mg (0.600 mmol, 2.00 equiv) of aldehyde 5f and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (2–10% EtOAc/CH2Cl2) afforded 8faa (108 mg, 70%) as a yellow solid. mp > 240 °C. FTIR (neat) 1716, 1692, 1406, 1274, 1233, 1166, 1119, 1109, 947, 767, 699 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.26–8.12 (m, 6H), 7.47 (t, J = 7.7 Hz, 2H), 7.28 (t, J = 7.4 Hz, 1H), 7.14 (s, 1H), 4.49–4.22 (m, 2H), 3.95 (s, 3H), 3.78 (tt, J = 12.1, 3.4 Hz, 1H), 3.09–2.86 (m, 2H), 2.29–2.16 (m, 2H), 1.86–1.68 (m, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 166.6, 154.6, 154.5, 152.5, 144.9, 142.6, 141.4, 132.1, 131.5, 130.1, 128.8, 127.2, 126.3, 111.3, 101.4, 79.8, 52.3, 43.8, 36.7, 29.3, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C30H33N4O4+, 513.2496; found 513.2520.
4-(7-(1-(tert-Butoxycarbonyl)piperidin-4-yl)-3-phenylpyrazolo[1,5-a]pyrimidin-5-yl)benzoic acid (8gaa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 90.1 mg (0.600 mmol, 2.00 equiv) of aldehyde 5g and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane) and work-up procedure; the reaction mixture after diluting with CH2Cl2 was washed with citric acid monohydrate (1 % wt, 100 mL) instead of satd. NaHCO3. Purification by silica gel column chromatography (10–50% EtOAc/CH2Cl2) afforded 8gaa (92.7 mg, 62%) as a yellow solid. mp > 240 °C. FTIR (neat) 1716, 1615, 1439, 1366, 1243, 1163, 1131, 1113, 787, 692, 663, 515, 526 cm−1. 1H NMR (600 MHz, (CD3)2SO) δ 13.12 (br s, 1H), 8.76 (s, 1H), 8.43 (d, J = 8.5 Hz, 2H), 8.23–8.16 (m, 2H), 8.08 (d, J = 8.5 Hz, 2H), 7.62 (s, 1H), 7.45 (d, J = 7.8 Hz, 2H), 7.26–7.21 (m, 1H), 4.27–4.05 (m, 2H), 3.72 (tt, J = 12.1, 3.4 Hz, 1H), 3.08–2.73 (m, 2H), 2.13–1.99 (m, 2H), 1.89–1.72 (m, 2H), 1.41 (s, 9H). 13C NMR (151 MHz, (CD3)2SO) δ 167.4, 154.8, 154.1, 153.2, 144.6, 143.1, 141.0, 132.7, 132.5, 130.2, 129.2, 128.0, 126.4, 126.2, 110.2, 102.8, 79.2, 44.2, 43.2, 36.7, 29.0, 28.6. HRMS (ESI): m/z [M + H]+ calcd for C29H31N4O4+, 499.2340; found 499.2358.
tert-Butyl 4-(5-(4-acetamidophenyl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8haa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 97.9 mg (0.600 mmol, 2.00 equiv) of aldehyde 5h and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–40% EtOAc/CH2Cl2) afforded 8haa (128 mg, 83%) as a yellow solid. mp 130–133 °C. FTIR (neat) 1667, 1596, 1522, 1387, 1366, 1235, 1161, 1124, 945, 768, 693, 507 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1H), 8.16–8.06 (m, 4H), 7.73 (br s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.44 (t, J = 7.8 Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 7.02 (s, 1H), 4.45–4.19 (m, 2H), 3.71 (tt, J = 12.2, 3.4 Hz, 1H), 3.07–2.81 (m, 2H), 2.21 (s, 3H), 2.25–2.14 (m, 2H), 1.82–1.62 (m, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 168.6, 155.0, 154.7, 152.0, 145.0, 142.2, 140.0, 132.9, 132.3, 128.7, 128.1, 126.1, 126.0, 119.7, 110.5, 101.0, 79.9, 43.9, 36.6, 29.3, 28.5, 24.7. HRMS (ESI): m/z [M + H]+ calcd for C30H34N5O3+, 512.2656; found 512.2661.
tert-Butyl 4-(3-phenyl-5-(pyridin-4-yl)pyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8iaa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 56.5 μL (0.600 mmol, 2.00 equiv) of aldehyde 5i and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–40% EtOAc/CH2Cl2) afforded 8iaa (78.8 mg, 58%) as a yellow solid. mp 169–171 °C. FTIR (neat) 1682, 1604, 1566, 1428, 1387, 1295, 1234, 1166, 1129, 942, 817, 768, 692 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.79 (d, J = 5.6 Hz, 2H), 8.47 (s, 1H), 8.16–8.08 (m, 2H), 8.06–7.96 (m, 2H), 7.47 (t, J = 7.8 Hz, 2H), 7.33–7.24 (m, 1H), 7.12 (s, 1H), 4.51–4.22 (m, 2H), 3.80 (tt, J = 12.1, 3.3 Hz, 1H), 3.10–2.89 (m, 2H), 2.30–2.19 (m, 2H), 1.86–1.68 (m, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.6, 153.0, 152.9, 150.7, 144.8, 144.4, 142.8, 131.8, 128.8, 126.5, 126.4, 121.1, 111.7, 101.0, 79.9, 43.8, 36.7, 29.3, 28.4. HRMS (ESI): m/z [M + H]+ calcd for C27H30N5O2+, 456.2394; found 456.2401.
tert-Butyl 4-(5-(6-acetamidopyridin-3-yl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8jaa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 98.5 mg (0.600 mmol, 2.00 equiv) of aldehyde 5j and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–40% EtOAc/CH2Cl2) afforded 8jaa (118 mg, 77%) as a yellow solid. mp > 240 °C. FTIR (neat) 1696, 1598, 1520, 1384, 1364, 1305, 1232, 1167, 1122, 9945, 767, 691, 509 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.07 (d, J = 1.8 Hz, 1H), 8.47 (s, 1H), 8.46–8.40 (m, 2H), 8.39–8.31 (m, 1H), 8.15–8.07 (m, 2H), 7.45 (t, J = 7.8 Hz, 2H), 7.29–7.22 (m, 1H), 7.04 (s, 1H), 4.47–4.18 (m, 2H), 3.77 (tt, J = 12.1, 3.3 Hz, 1H), 3.06–2.88 (m, 2H), 2.26 (s, 3H), 2.29–2.17 (m, 2H), 1.82–1.66 (m, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 168.8, 154.6, 152.9, 152.6, 152.6, 147.1, 144.9, 142.5, 137.1, 132.1, 129.1, 128.7, 126.3, 126.2, 113.5, 110.9, 100.5, 79.8, 43.7, 36.7, 29.3, 28.5, 24.8. HRMS (ESI): m/z [M + H]+ calcd for C29H33N6O3+, 513.2609; found 513.2619.
tert-Butyl 4-(5-(6-chloropyridin-3-yl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8kaa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 84.9 mg (0.600 mmol, 2.00 equiv) of aldehyde 5k and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (2–10% EtOAc/CH2Cl2) afforded 8kaa (96.3 mg, 66%) as a yellow solid. mp 199–202 °C. FTIR (neat) 1692, 1567, 1426, 1235, 1164, 1129, 1105, 945, 826, 771, 692, 497, 474 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.11 (d, J = 2.1 Hz, 1H), 8.45 (s, 1H), 8.43 (dd, J = 8.3, 2.5 Hz, 1H), 8.14–8.05 (m, 2H), 7.52–7.40 (m, 3H), 7.32–7.25 (m, 1H), 7.05 (s, 1H), 4.49–4.20 (m, 2H), 3.78 (tt, J = 12.1, 3.3 Hz, 1H), 3.09–2.83 (m, 2H), 2.31–2.13 (m, 2H), 1.84–1.68 (m, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.6, 153.1, 153.0, 152.0, 148.5, 144.8, 142.7, 137.3, 132.0, 131.8, 128.77, 126.5, 126.3, 124.5, 111.4, 100.6, 79.9, 43.7, 36.8, 29.2, 28.4. HRMS (ESI): m/z [M + H]+ calcd for C27H29ClN5O2+, 490.2004; found 490.2022.
tert-Butyl 4-(5-(furan-2-yl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8laa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 49.7 μL (0.600 mmol, 2.00 equiv) of aldehyde 5l and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (10–40% Et2O/hexanes) afforded 8laa (108 mg, 81%) as a yellow solid. mp 201–203 °C. FTIR (neat) 1681, 1610, 1426, 1292, 1229, 1167, 1005, 937, 765, 743, 515 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 8.15–8.07 (m, 2H), 7.59 (dd, J = 1.8, 0.8 Hz, 1H), 7.44 (t, J = 7.8 Hz, 2H), 7.31 (dd, J = 3.5, 0.8 Hz, 1H), 7.29–7.20 (m, 1H), 7.10 (s, 1H), 6.59 (dd, J = 3.5, 1.8 Hz, 1H), 4.48–4.21 (m, 2H), 3.74 (tt, J = 12.1, 3.4 Hz, 1H), 3.06–2.86 (m, 2H), 2.27–2.13 (m, 2H), 1.84–1.64 (m, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.6, 152.5, 152.3, 147.7, 144.9, 144.5, 142.3, 132.3, 128.7, 126.2, 126.1, 112.7, 111.5, 110.4, 100.1, 79.7, 43.8, 36.5, 29.3, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C26H29N4O3+, 445.2240; found 445.2263.
tert-Butyl 4-(3-phenyl-5-(thiophen-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8maa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 52.6 μL (0.600 mmol, 2.00 equiv) of aldehyde 5m and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (5–25% EtOAc/hexanes) afforded 8maa (117 mg, 85%) as a yellow solid. mp 182–185 °C. FTIR (neat) 1691, 1672, 1608, 1418, 1366, 1291, 1237, 1163, 1134, 1068, 762, 691, 515 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 8.17–8.09 (m, 2H), 8.03 (dd, J = 3.0, 1.3 Hz, 1H), 7.84 (dd, J = 5.1, 1.3 Hz, 1H), 7.50–7.40 (m, 3H), 7.30–7.22 (m, 1H), 6.96 (s, 1H), 4.50–4.17 (m, 2H), 3.75 (tt, J = 12.1, 3.4 Hz, 1H), 3.08–2.84 (m, 2H), 2.27–2.17 (m, 2H), 1.83–1.64 (m, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.7, 152.1, 151.9, 144.9, 142.3, 140.9, 132.4, 128.7, 126.7, 126.7, 126.2, 126.0, 125.9, 110.5, 101.7, 79.8, 43.8, 36.5, 29.4, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C26H29N4O2S+, 461.2006; found 461.2000.
tert-Butyl 4-(5-(1-methyl-1H-pyrazol-4-yl)-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8naa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 66.1 mg (0.600 mmol, 2.00 equiv) of aldehyde 5n and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (5–30% EtOAc/CH2Cl2) afforded 8naa (114 mg, 83%) as a light yellow solid. mp > 240 °C. FTIR (neat) 1684, 1616, 1551, 1427, 1235, 1168, 1126, 999, 863, 767, 751, 695, 516 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 8.12–8.09 (m, 2H), 8.07 (s, 1H), 8.05 (s, 1H), 7.47–7.43 (m, 2H), 7.30–7.21 (m, 1H), 6.78 (s, 1H), 4.51–4.14 (m, 2H), 3.99 (s, 3H), 3.73 (tt, J = 12.2, 3.4 Hz, 1H), 3.12–2.86 (m, 2H), 2.27–2.15 (m, 2H), 1.79–1.65 (m, 2H), 1.49 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 154.6, 152.0, 150.9, 145.1, 142.2, 138.4, 132.5, 130.1, 128.7, 126.1, 125.9, 122.5, 109.7, 101.4, 79.8, 44.1, 43.4, 39.4, 36.4, 29.4, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C26H31N6O2+, 459.2503; found 459.2507.
tert-Butyl 4-(5-methyl-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8oaa):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 26.5 mg (0.600 mmol, 2.00 equiv) of aldehyde 5o and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane) and in order of addition; after nitrogen flush, acetaldehyde was added in one portion as a solution in dioxane (1.50 mL) via syringe. Purification by preparative TLC plate (UV254, 20×20 cm, 1000 micron; 20:80:1 EtOAc/hexanes/Et3N) afforded 8oaa (15.2 mg, 13%) as a white solid. mp 194–196 °C. FTIR (neat) 1679, 1605, 1566, 1441, 1427, 1365, 1289, 1234, 1156, 1125, 864, 771, 699 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 8.10–7.95 (m, 2H), 7.41 (t, J = 7.8 Hz, 2H), 7.27–7.18 (m, 1H), 6.51 (s, 1H), 4.45–4.15 (m, 2H), 3.69 (tt, J = 12.1, 3.4 Hz, 1H), 2.94 (t, J = 13.2 Hz, 2H), 2.61 (s, 3H), 2.21–2.10 (m, 2H), 1.69–1.58 (m, 2H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 159.1, 154.6, 151.3, 144.9, 141.9, 132.4, 128.7, 126.2, 126.0, 109.6, 105.1, 79.8, 43.7, 36.2, 29.3, 28.4, 25.2. HRMS (ESI): m/z [M + H]+ calcd for C23H29N4O2+, 393.2285; found 393.2289.
7-Isopropyl-3,5-diphenylpyrazolo[1,5-a]pyrimidine (8aab):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a, and 78.8 mg (0.486 mmol, 1.5 equiv) of ylide 7b. Purification by silica gel column chromatography (25–50% CH2Cl2/hexanes) afforded 8aab (74.1 mg, 79%) as a yellow solid. mp 105–107 °C. FTIR (neat) 1606, 1564, 1523, 1492, 1386, 1253, 1201, 1176, 828, 763, 692, 657, 603 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.48 (s, 1H), 8.23–8.20 (m, 4H), 7.58–7.47 (m, 5H), 7.29 (tt, J = 7.3, 1.2 Hz, 1H), 7.20 (s, 1H), 3.95 (hept, J = 6.9 Hz, 1H), 1.54 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 155.9, 155.2, 145.2, 142.3, 137.7, 132.6, 130.2, 128.9, 128.7, 127.4, 126.2, 126.0, 110.5, 100.9, 28.7, 20.1. HRMS (ESI): m/z [M + H]+ calcd for C21H20N3+, 314.1652; found 314.1654.
7-Isobutyl-3,5-diphenylpyrazolo[1,5-a]pyrimidine (8aac):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a, and 80.1 mg (0.454 mmol, 1.5 equiv) of ylide 7c. Purification by silica gel column chromatography (25–50% CH2Cl2/hexanes) afforded 8aac (85.0 mg, 87%) as a yellow solid. mp 114–116 °C. FTIR (neat) 1607, 1564, 1523, 1389, 1197, 763, 685, 643, 595, 516 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.47 (s, 1H), 8.24–8.18 (m, 4H), 7.59–7.46 (m, 5H), 7.31–7.26 (m, 1H), 7.16 (s, 1H), 3.12 (d, J = 7.3 Hz, 2H), 2.54–2.43 (m, 1H), 1.09 (d, J = 6.6 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 155.6, 149.1, 145.4, 142.6, 137.7, 132.7, 130.4, 129.0, 128.8, 127.5, 126.3, 126.1, 110.6, 104.9, 40.2, 26.0, 22.8. HRMS (ESI): m/z [M + H]+ calcd for C22H22N3+, 328.1808; found 328.1803.
7-Methyl-3,5-diphenylpyrazolo[1,5-a]pyrimidine (8aad):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 65.2 mg (0.486 mmol, 1.5 equiv) of ylide 7d. Purification by silica gel column chromatography (50% CH2Cl2/hexanes) afforded 8aad (68.5 mg, 80%) as a yellow solid. mp 142–144 °C. FTIR (neat) 1605, 1563, 1369, 1200, 765, 692, 640, 596, 515 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.23–8.10 (m, 4H), 7.56–7.41 (m, 5H), 7.31–7.22 (m, 1H), 7.17 (s, 1H), 2.84 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 155.5, 146.0, 145.0, 142.5, 137.4, 132.5, 130.3, 128.9, 128.7, 127.3, 126.2, 126.0, 110.6, 105.0, 17.5. HRMS (ESI): m/z [M + H]+ calcd for C19H16N3+, 286.1339; found 286.1338.
tert-Butyl (2-(3,5-diphenylpyrazolo[1,5-a]pyrimidin-7-yl)ethyl)carbamate (8aae):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 129 mg (0.488, 1.5 equiv mmol) of ylide 7e. Purification by silica gel column chromatography (25% EtOAc/hexanes) afforded 8aae (118 mg, 94%) as a yellow solid. mp 153–155 °C. FTIR (neat) 1698, 1565, 1530, 1271, 1160, 1137, 765, 685, 596, 495 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.44 (s, 1H), 8.25–8.13 (m, 4H), 7.56–7.45 (m, 5H), 7.31–7.26 (m, 2H), 4.93 (br s, NH, 1H), 3.76 (q, J = 6.6 Hz, 2H), 3.46 (t, J = 6.8 Hz, 2H), 1.42 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 155.9, 155.7, 146.6, 145.1, 142.5, 137.2, 132.3, 130.4, 128.9, 128.7, 127.4, 126.2, 126.1, 110.8, 104.9, 79.6, 37.5, 31.6, 28.3. HRMS (ESI): m/z [M + H]+ calcd for C25H27N4O2+, 415.2129; found 415.2138.
7-(4-Methoxyphenyl)-3,5-diphenylpyrazolo[1,5-a]pyrimidine (8aaf):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 107 mg (0.473 mmol, 1.5 equiv) of ylide 7f. Purification by silica gel column chromatography (25–50% CH2Cl2/hexanes) afforded 8aaf (101 mg, 89%) as a yellow solid. 1H and 13C NMR spectra matched with previously reported literature.3 1H NMR (600 MHz, CDCl3) δ 8.49 (s, 1H), 8.28–8.20 (m, 4H), 8.13–8.07 (m, 2H), 7.58–7.47 (m, 5H), 7.38 (s, 1H), 7.32–7.27 (m, 1H), 7.15–7.09 (m, 2H), 3.92 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 161.9, 156.0, 146.9, 146.2, 143.0, 137.7, 132.7, 131.1, 130.5, 129.1, 128.9, 127.5, 126.5, 126.2, 123.8, 114.3, 110.6, 104.5, 55.7.
3,5-Diphenyl-7-(4-(trifluoromethyl)phenyl)pyrazolo[1,5-a]pyrimidine (8aag):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 124 mg (0.469 mmol, 1.5 equiv) of ylide 7g. Purification by silica gel column chromatography (25–50% CH2Cl2/hexanes) afforded 8aag (109 mg, 87%) as a yellow solid. 1H, 13C and 19F NMR spectra matched with previously reported literature.3 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.27–8.16 (m, 6H), 7.87 (d, J = 8.2 Hz, 2H), 7.60–7.47 (m, 5H), 7.39 (s, 1H), 7.32 (t, J = 7.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 156.0, 146.0, 145.5, 143.2, 137.2, 135.0, 132.9 (q, J = 32.9 Hz), 132.2, 130.8, 129.9, 129.2, 128.9, 127.5, 126.6, 126.5, 125.9 (q, J = 3.7 Hz), 123.9 (q, J = 272.5 Hz), 111.3, 105.5. 19F NMR (376 MHz, CDCl3) δ – 63.0.
3,5-Diphenyl-7-(thiophen-2-yl)pyrazolo[1,5-a]pyrimidine (8aah):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 91.1 mg (0.450, 1.50 equiv mmol) of ylide 7h. Purification by silica gel column chromatography (25–50% CH2Cl2/hexanes) afforded 8aah (89.8 mg, 85%) as a red solid. mp 162–164 °C. FTIR (neat) 1603, 1554, 1529, 1494, 1380, 820, 763, 715, 690, 668, 491 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.56 (s, 1H), 8.39–8.37 (m, 1H), 8.26–8.19 (m, 4H), 7.74–7.71 (m, 1H), 7.69 (s, 1H), 7.59–7.47 (m, 5H), 7.33–7.27 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 155.4, 146.1, 142.6, 140.2, 137.6, 132.5, 132.1, 131.9, 131.3, 130.4, 129.0, 128.8, 127.7, 127.4, 126.5, 126.2, 110.7, 101.7. HRMS (ESI): m/z [M + H]+ calcd for C22H16N3S+, 354.1059; found 354.1063.
tert-Butyl 4-(3-methyl-5-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aba):
The reaction was performed according to the general procedure employing 29.2 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6b, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (2–5% EtOAc/CH2Cl2) afforded 8aba (83.6 mg, 71%) as a light yellow solid. mp 150–152 °C. FTIR (neat) 1673, 1616, 1430, 1381, 1238, 1162, 1125, 968, 773, 690, 639, 538 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.10–8.04 (m, 2H), 7.95 (s, 1H), 7.53–7.41 (m, 3H), 7.01 (s, 1H), 4.47–4.15 (m, 2H), 3.73 (tt, J = 12.1, 3.3 Hz, 1H), 3.07–2.83 (m, 2H), 2.42 (s, 3H), 2.27–2.14 (m, 2H), 1.80–1.61 (m, 2H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.6, 154.6, 151.5, 146.5, 144.5, 137.9, 130.0, 128.8, 127.2, 106.2, 100.8, 79.7, 43.7, 36.4, 29.3, 28.5, 7.7. HRMS (ESI): m/z [M + H]+ calcd for C23H29N4O2+, 393.2285; found 393.2298.
tert-Butyl 4-(2-methyl-3,5-diphenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aca):
The reaction was performed according to the general procedure employing 52.0 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6c, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (5–20% acetone/hexanes) afforded 8aca (84.4 mg, 60%) as a light yellow solid. mp 156–158 °C. FTIR (neat) 1680, 1606, 1420, 1364, 1233, 1165, 1125, 745, 768, 696, 621 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.11 (d, J = 6.7 Hz, 2H), 7.85 (d, J = 6.8 Hz, 2H), 7.53–7.40 (m, 5H), 7.31 (t, J = 7.4 Hz, 1H), 7.06 (s, 1H), 4.50–4.20 (s, 2H), 3.80 (tt, J = 12.2, 3.4 Hz, 1H), 3.12–2.89 (bs, 2H), 2.68 (s, 3H), 2.35–2.16 (m, 2H), 1.82–1.70 (m, 2H), 1.50 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 155.6, 154.7, 152.2, 151.4, 146.3, 137.6, 132.8, 130.1, 128.8, 128.8, 128.4, 127.2, 126.0, 109.4, 100.9, 79.8, 44.2, 43.4, 36.3, 29.4, 28.5, 14.7. HRMS (ESI): m/z [M + H]+ calcd for C29H33N4O2+, 469.2598; found 469.2601.
tert-Butyl 4-(2-ethyl-3-methyl-5-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8ada):
The reaction was performed according to the general procedure employing 37.6 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6d, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (5–20% EtOAc/hexanes) afforded 8ada (82.0 mg, 65%) as a light yellow solid. mp 120–122 °C. FTIR (neat) 1689, 1617, 1425, 1364, 1292, 1232, 1165, 1119, 770, 688 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.13–8.01 (m, 2H), 7.53–7.38 (m, 3H), 6.92 (s, 1H), 4.50–4.14 (m, 2H), 3.74 (tt, J = 12.1, 3.4 Hz, 1H), 3.08–2.90 (m, 2H), 2.85 (q, J = 7.6 Hz, 2H), 2.36 (s, 3H), 2.26–2.13 (m, 2H), 1.80–1.63 (m, 2H), 1.48 (s, 9H), 1.33 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 158.4, 154.7, 154.3, 151.2, 147.0, 138.2, 129.8, 128.8, 127.1, 102.9, 99.9, 79.7, 44.1, 43.4, 36.2, 29.3, 28.5, 21.0, 13.7, 7.0. HRMS (ESI): m/z [M + H]+ calcd for C25H33N4O2+, 421.2598; found 421.2604.
tert-Butyl 4-(5-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aea):
The reaction was performed according to the general procedure employing 25.0 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6e, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane). Purification by silica gel column chromatography (5–20% EtOAc/CH2Cl2) afforded 8aea (69.9 mg, 62%) as an off-white foam. FTIR (neat) 1670, 1613, 1432, 1240, 1163, 1129, 1002, 903, 771, 693, 640, 540 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 2.4 Hz, 1H), 8.08–7.97 (m, 2H), 7.54–7.40 (m, 3H), 7.06 (s, 1H), 6.71 (d, J = 2.3 Hz, 1H), 4.47–4.21 (m, 2H), 3.77 (tt, J = 12.2, 3.2 Hz, 1H), 3.06–2.87 (m, 2H), 2.27–2.16 (m, 2H), 1.81–1.66 (m, 2H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 156.2, 154.6, 152.0, 148.9, 144.6, 137.6, 130.2, 128.9, 127.2, 101.5, 97.1, 79.7, 43.7, 36.6, 29.3, 28.4. HRMS (ESI): m/z [M + H]+ calcd for C22H27N4O2+, 379.2129; found 379.2131.
tert-Butyl 4-(2-(tert-butyl)-5-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8afa):
The reaction was performed according to the general procedure employing 41.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6f, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane). Purification by silica gel column chromatography (2–5% EtOAc/CH2Cl2) afforded 8afa (95.0 mg, 73%) as a white foam. FTIR (neat) 2961, 1690, 1610, 1533, 1449, 1364, 1233, 1164, 1120, 771, 692, 591 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.10–7.92 (m, 2H), 7.54–7.38 (m, 3H), 6.96 (s, 1H), 6.54 (s, 1H), 4.46–4.19 (m, 2H), 3.73 (tt, J = 12.1, 3.4 Hz, 1H), 3.06–2.86 (m, 2H), 2.28–2.16 (m, 2H), 1.82–1.65 (m, 2H), 1.48 (s, 9H), 1.41 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 167.7, 155.5, 154.7, 151.5, 149.2, 138.0, 129.9, 128.8, 127.1, 100.5, 92.9, 79.7, 43.9, 36.7, 32.9, 30.4, 29.0, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C26H35N4O2+, 435.2755; found 435.2760.
tert-Butyl 4-(2-methyl-5-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aga):
The reaction was performed according to the general procedure employing 29.2 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6g, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane). Purification by silica gel column chromatography (2–10% EtOAc/CH2Cl2) afforded 8aga (57.6 mg, 49%) as an off-white solid. mp 165–167 °C. FTIR (neat) 1685, 1610, 1533, 1470, 1415, 1364, 1235, 1156, 999, 867, 772, 695, 586 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.05–7.99 (m, 2H), 7.52–7.41 (m, 3H), 6.97 (s, 1H), 6.48 (s, 1H), 4.48–4.13 (m, 2H), 3.76 (tt, J = 12.2, 3.4 Hz, 1H), 3.12–2.84 (m, 2H), 2.52 (s, 3H), 2.25–2.15 (m, 2H), 1.80–1.62 (m, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 155.9, 154.9, 154.7, 151.5, 149.7, 137.9, 130.0, 128.8, 127.2, 100.6, 96.4, 79.7, 43.8, 36.4, 29.4, 28.5, 14.8. HRMS (ESI): m/z [M + H]+ calcd for C23H29N4O2+, 393.2285; found 393.2286.
tert-Butyl 4-(2-isopropyl-5-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aha):
The reaction was performed according to the general procedure employing 37.6 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6h, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane). Purification by silica gel column chromatography (10–40% Et2O/hexanes) afforded 8aha (81.7 mg, 65%) as a white solid. mp 100–102 °C. FTIR (neat) 2957, 1704, 1613, 1554, 1417, 1365, 1232, 1170, 1124, 766, 683, 439 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.06–7.96 (m, 2H), 7.51–7.41 (m, 3H), 6.97 (s, 1H), 6.51 (s, 1H), 4.43–4.17 (m, 2H), 3.75 (tt, J = 12.1, 3.4 Hz, 1H), 3.17 (hept, J = 6.9 Hz, 1H), 3.02–2.92 (m, 2H), 2.27–2.16 (m, 2H), 1.81–1.64 (m, 2H), 1.48 (s, 9H), 1.37 (d, J = 7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 165.2, 155.7, 154.7, 151.6, 149.4, 137.9, 129.9, 128.8, 127.2, 100.6, 93.3, 79.7, 43.9, 36.4, 29.2, 28.8, 28.5, 22.9. HRMS (ESI): m/z [M + H]+ calcd for C25H33N4O2+, 421.2598; found 421.2596.
tert-Butyl 4-(2-cyclopropyl-5-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aia):
The reaction was performed according to the general procedure employing 37.0 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6i, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane). Purification by silica gel column chromatography (2–10% EtOAc/CH2Cl2) afforded 8aia (70.4 mg, 56%) as a white solid. mp 108–110 °C. FTIR (neat) 1964, 1611, 1557, 1419, 1366, 1272, 1226, 1159, 1120, 981, 769, 685, 661 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.07–7.95 (m, 2H), 7.54–7.37 (m, 3H), 6.95 (s, 1H), 6.30 (s, 1H), 4.46–4.16 (m, 2H), 3.74 (tt, J = 12.1, 3.2 Hz, 1H), 2.96 (m, 2H), 2.27–2.08 (m, 3H), 1.77–1.64 (m, 2H), 1.48 (s, 9H), 1.11–1.01 (m, 2H), 0.95–0.85 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 161.5, 155.8, 154.7, 151.3, 149.6, 137.9, 130.0, 128.8, 127.1, 100.5, 92.4, 79.7, 43.8, 36.4, 29.3, 28.5, 10.2, 9.4. HRMS (ESI): m/z [M + H]+ calcd for C25H31N4O2+, 419.2442; found 419.2445.
tert-Butyl 4-(2,5-diphenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aja):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6j, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a with slight modification in reaction concentration (0.200 M, 1.50 mL of dioxane). Purification by silica gel column chromatography (2–10% EtOAc/CH2Cl2) afforded 8aja (70.6 mg, 52%) as an off-white solid. mp 171–173 °C. FTIR (neat) 1662, 1612, 1464, 1420, 1236, 1165, 1134, 764, 744, 691 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 6.7 Hz, 2H), 8.03 (d, J = 6.9 Hz, 2H), 7.55–7.44 (m, 5H), 7.40 (t, J = 7.4 Hz, 1H), 7.06 (s, 1H), 7.01 (s, 1H), 4.51–4.23 (m, 2H), 3.86 (tt, J = 12.1, 3.4 Hz, 1H), 3.09–2.90 (m, 2H), 2.33–2.24 (m, 2H), 1.86–1.71 (m, 2H), 1.50 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 156.1, 155.9, 154.7, 151.8, 150.1, 137.7, 133.1, 130.2, 128.9, 128.7, 127.2, 126.6, 101.5, 93.8, 79.8, 44.3, 43.4, 36.7, 29.3, 28.5. HRMS (ESI): m/z [M + H]+ calcd for C28H31N4O2+, 455.2442; found 455.2434.
tert-Butyl 4-(5-phenyl-2-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8aka):
The reaction was performed according to the general procedure employing 45.6 mg (0.300 mmol) of aminopyrazole 6k, 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a and 137 mg (0.450 mmol, 1.50 equiv) of ylide 7a. Purification by silica gel column chromatography (100% CH2Cl2, then 10–25% EtOAc/hexanes) afforded 8aka (102 mg, 76%) as an off-white solid. mp 164–167 °C. FTIR (neat) 1684, 1612, 1423, 1232, 1161, 1128, 1103, 955, 774, 692 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.10–8.05 (m, 2H), 7.56–7.50 (m, 3H), 7.23 (s, 1H), 6.98 (s, 1H), 4.52–4.19 (m, 2H), 3.80 (tt, J = 12.1, 3.4 Hz, 1H), 3.10–2.91 (m, 2H), 2.28–2.19 (m, 2H), 1.83–1.68 (m, 2H), 1.50 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 157.8, 154.8, 152.9, 149.2, 146.8 (q, J = 38.2 Hz), 137.1, 131.0, 129.2, 127.5, 121.4 (q, J = 269.7 Hz), 103.8, 95.9, 80.0, 43.8, 36.6, 29.4, 28.6. 19F NMR (376 MHz, CDCl3) δ –62.4. HRMS (ESI): m/z [M + H]+ calcd for C23H26F3N4O2+, 447.2002; found 447.1996.
General Procedure for Two-Step, One-Pot Annulations with Aminopyrazoles, Aldehydes and Formyl Sulfoxonium Ylide 7i (0.3 mmol scale).
To a flame-dried 2–5 mL Biotage microwave vial (#351521) charged with a stir bar on the benchtop were added aminopyrazole (0.300 mmol, 1.00 equiv), aldehyde (0.600 mmol, 2.00 equiv), PivOH (1.20 mmol, 4.00 equiv, 123 mg), 3Å molecular sieves (approximately 300 mg) and dioxane (0.200 M, 1.50 mL). The vial was capped with a Teflon-lined cap, flushed with nitrogen for about 2 min, and then heated with a Biotage Initiator+ (#356007), which employs an external IR sensor and a closed reaction vessel. The resultant mixture was stirred in the microwave reactor for 30 min at 150 °C using the following settings (absorption level, low; vial type, 2−5 mL; prestirring, 0; initial power, 0; dynamic deflector optimization, ON; pressure: OFF; power, OFF; fixed hold time, ON; stir rate, 600). After cooling to rt, the cap was opened, and [Cp*Rh(MeCN)3](SbF6)2 (10 mol %, 0.030 mmol, 25 mg), KOAc (0.30 mmol, 1.0 equiv, 29 mg) and formyl sulfoxonium ylide 7i (0.330 mmol, 1.10 equiv, 39.7 mg) were added. The vial was capped with a Teflon-lined cap, flushed with nitrogen for about 2 min and then heated in the microwave reactor for 1 h at 150 °C using the above settings. After cooling to rt, the crude mixture was transferred to a separatory funnel with CH2Cl2 (50 mL). PivOH was removed by extracting with satd. NaHCO3 (100 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were washed with satd. NaHSO3 (10 % wt, 100 mL) to remove remaining aldehyde.16 The organic layer was dried (anhyd. Na2SO4) and concentrated under reduced pressure. The product was purified by silica gel column chromatography.
3,5-Diphenylpyrazolo[1,5-a]pyrimidine (8aai):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a and 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a. Purification by silica gel column chromatography (2–5% EtOAc/CH2Cl2) afforded 8aai (63.5 mg, 78%) as a yellow solid. mp 135–137 °C. FTIR (neat) 1612, 1559, 1521, 1493, 1413, 1267, 1192, 972, 815, 758, 682, 635, 549 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J = 7.4 Hz, 1H), 8.44 (s, 1H), 8.22–8.12 (m, 4H), 7.57–7.42 (m, 5H), 7.32–7.25 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 155.9, 144.7, 143.2, 137.0, 135.3, 132.2, 130.6, 128.9, 128.7, 127.3, 126.2, 126.2, 110.7, 105.3. HRMS (ESI): m/z [M + H]+ calcd for C18H14N3+, 272.1182; found 272.1183.
3-Phenyl-5-(4-(trifluoromethyl)phenyl)pyrazolo[1,5-a]pyrimidine (8bai):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a and 82.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5b. Purification by silica gel column chromatography (2–5% EtOAc/CH2Cl2) afforded 8bai (77.9 mg, 77%) as a yellow solid. mp 130–132 °C. FTIR (neat) 1619, 1414, 1329,1318, 1122, 1104, 1071, 1015, 810, 766, 752, 691 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.69 (d, J = 7.3 Hz, 1H), 8.46 (s, 1H), 8.24 (d, J = 8.2 Hz, 2H), 8.16–8.08 (m, 2H), 7.77 (d, J = 8.2 Hz, 2H), 7.47 (t, J = 7.8 Hz, 2H), 7.32–7.23 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 154.1, 144.5, 143.5, 140.2, 135.6, 132.1 (q, J = 32.7 Hz), 131.8, 128.8, 127.6, 126.5, 126.3, 125.9 (q, J = 3.8 Hz), 123.9 (q, J = 272.3 Hz), 111.3, 105.1. 19F NMR (376 MHz, CDCl3) δ –62.8. HRMS (ESI): m/z [M + H]+ calcd for C19H13F3N3+, 340.1056; found 340.1053.
5-(4-Methoxyphenyl)-3-phenylpyrazolo[1,5-a]pyrimidine (8cai):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a and 73.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5c. Purification by silica gel column chromatography (2–5% EtOAc/CH2Cl2) afforded 8cai (66.4 mg, 73%) as a yellow solid. mp 136–138 °C. FTIR (neat) 1604, 1563, 1412, 1251, 1180, 1023, 845, 789, 767, 694, 547 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 7.4 Hz, 1H), 8.41 (s, 1H), 8.21–8.06 (m, 4H), 7.46 (t, J = 7.8 Hz, 2H), 7.31–7.17 (m, 2H), 7.02 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.7, 155.5, 144.7, 143.0, 135.1, 132.3, 129.5, 128.9, 128.7, 126.1, 126.0, 114.3, 110.2, 104.8, 55.4. HRMS (ESI): m/z [M + H]+ calcd for C19H16N3O+, 302.1288; found 302.1300.
5-(3-Bromophenyl)-3-phenylpyrazolo[1,5-a]pyrimidine (8dai):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a and 70.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5d. Purification by silica gel column chromatography (2–5% EtOAc/CH2Cl2) afforded 8dai (91.4 mg, 87%) as a yellow solid. mp 115–117 °C. FTIR (neat) 1610, 1558, 1492, 1406, 1260, 1194, 780, 762, 686, 548, 535 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.68 (d, J = 7.4 Hz, 1H), 8.45 (s, 1H), 8.28 (t, J = 1.8 Hz, 1H), 8.17–8.10 (m, 2H), 8.08 (dt, J = 7.6, 1.2 Hz, 1H), 7.61 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.48 (t, J = 7.8 Hz, 2H), 7.39 (t, J = 7.9 Hz, 1H), 7.32–7.21 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 154.2, 144.5, 143.4, 139.0, 135.5, 133.4, 131.9, 130.4, 130.3, 128.8, 126.4, 126.3, 125.9, 123.2, 111.1, 105.1. HRMS (ESI): m/z [M + H]+ calcd for C18H13BrN3+, 350.0287; found 350.0294.
5-(6-Chloropyridin-3-yl)-3-phenylpyrazolo[1,5-a]pyrimidine (8kai):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a and 84.9 mg (0.600 mmol, 2.00 equiv) of aldehyde 5k. Purification by silica gel column chromatography (2–10% EtOAc/CH2Cl2) afforded 8kai (73.9 mg, 80%) as a yellow solid. mp > 240 °C. FTIR (neat) 1612, 1560, 1413, 1314, 1129, 1104, 1022, 971, 817, 768, 500, 472 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.10 (d, J = 2.5 Hz, 1H), 8.71 (d, J = 7.4 Hz, 1H), 8.46 (s, 1H), 8.43 (dd, J = 8.4, 2.6 Hz, 1H), 8.16–8.02 (m, 2H), 7.52–7.40 (m, 3H), 7.31–7.25 (m, 1H), 7.23 (d, J = 7.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 153.3, 152.0, 148.4, 144.4, 143.6, 137.2, 135.9, 131.62, 131.58, 128.8, 126.6, 126.2, 124.6, 111.3, 104.5. HRMS (ESI): m/z [M + H]+ calcd for C17H12ClN4+, 307.0745; found 307.0748.
5-(1-Methyl-1H-pyrazol-4-yl)-3-phenylpyrazolo[1,5-a]pyrimidine (8nai):
The reaction was performed according to the general procedure employing 47.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6a and 66.1 mg (0.600 mmol, 2.00 equiv) of aldehyde 5n. Purification by silica gel column chromatography (10–35% EtOAc/CH2Cl2) afforded 8nai (54.3 mg, 66%) as a yellow solid. mp 172–174 °C. FTIR (neat) 1616, 1583, 1551, 1514, 1236, 1187, 992, 887, 863, 808, 768, 690, 555, 516 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.54 (d, J = 7.3 Hz, 1H), 8.37 (s, 1H), 8.12–8.06 (m, 2H), 8.04 (d, J = 1.9 Hz, 2H), 7.44 (t, J = 7.8 Hz, 2H), 7.30–7.20 (m, 1H), 6.93 (d, J = 7.3 Hz, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 151.0, 144.7, 143.0, 138.5, 135.1, 132.3, 130.1, 128.7, 126.1, 126.0, 122.2, 109.6, 105.4, 39.4. HRMS (ESI): m/z [M + H]+ calcd for C16H14N5+, 276.1244; found 276.1247.
2-Methyl-3,5-diphenylpyrazolo[1,5-a]pyrimidine (8aci):
The reaction was performed according to the general procedure employing 52.0 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6c and 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a. Purification by silica gel column chromatography (5–25% EtOAc/hexanes) afforded 8aci (70.2 mg, 82%) as a yellow solid. mp 138–140 °C. FTIR (neat) 1609, 1559, 1493, 1422, 816, 767, 700, 689, 594 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.59 (d, J = 7.3 Hz, 1H), 8.16–8.06 (m, 2H), 7.88–7.81 (m, 2H), 7.54–7.41 (m, 5H), 7.35–7.28 (m, 1H), 7.23 (d, J = 7.4 Hz, 1H), 2.67 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 155.7, 153.0, 145.9, 137.1, 134.4, 132.5, 130.3, 128.8, 128.7, 128.4, 127.2, 126.1, 109.3, 104.7, 14.5. HRMS (ESI): m/z [M + H]+ calcd for C19H16N3+, 286.1339; found 286.1343.
2-Ethyl-3-methyl-5-phenylpyrazolo[1,5-a]pyrimidine (8adi):
The reaction was performed according to the general procedure employing 37.6 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6d and 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a. Purification by silica gel column chromatography (5–20% EtOAc/hexanes) afforded 8adi (51.1 mg, 72%) as a yellow solid. mp 77–79 °C. FTIR (neat) 1611, 1515, 1494, 1260, 1086, 1020, 795, 764, 742, 682, 645, 534 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.52 (d, J = 7.3 Hz, 1H), 8.14–8.03 (m, 2H), 7.54–7.40 (m, 3H), 7.09 (d, J = 7.3 Hz, 1H), 2.84 (q, J = 7.6 Hz, 2H), 2.34 (s, 3H), 1.34 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.2, 154.3, 146.6, 137.6, 134.3, 130.0, 128.8, 127.0, 103.8, 103.0, 20.8, 13.3, 6.8. HRMS (ESI): m/z [M + H]+ calcd for C15H16N3+, 238.1339; found 238.1338.
5-Phenylpyrazolo[1,5-a]pyrimidine (8aei):
The reaction was performed according to the general procedure employing 25.0 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6e and 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a with slight modification in reaction concentration (0.100 M, 3.00 mL of dioxane). Purification by silica gel column chromatography (5–20% EtOAc/hexanes) afforded 8aei (17.5 mg, 30%) as a yellow solid. 1H and 13C NMR spectra matched with previously reported literature.17 1H NMR (600 MHz, CDCl3) δ 8.69 (d, J = 7.3 Hz, 1H), 8.12 (d, J = 2.3 Hz, 1H), 8.09–8.05 (m, 2H), 7.55–7.45 (m, 3H), 7.26 (d, J = 7.4 Hz, 1H), 6.71 (d, J = 2.2 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 156.2, 148.5, 145.6, 137.1, 135.1, 130.5, 129.0, 127.2, 105.5, 97.0.
2-(tert-Butyl)-5-phenylpyrazolo[1,5-a]pyrimidine (8afi):
The reaction was performed according to the general procedure employing 41.8 mg (0.300 mmol, 1.00 equiv) of aminopyrazole 6f and 61.0 μL (0.600 mmol, 2.00 equiv) of aldehyde 5a with slight modification in reaction concentration (0.100 M, 3.00 mL of dioxane). Purification by silica gel column chromatography (10–30% Et2O/hexanes) afforded 8afi (45.7 mg, 61%) as a tan solid. mp 88–90 °C. FTIR (neat) 1609, 1544, 1415, 1358, 1242, 793, 765, 739, 689 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.61 (d, J = 7.3 Hz, 1H), 8.09–7.97 (m, 2H), 7.54–7.42 (m, 3H), 7.15 (d, J = 7.3 Hz, 1H), 6.54 (s, 1H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 168.8, 155.7, 148.9, 137.4, 134.7, 130.2, 128.9, 127.1, 104.5, 93.2, 32.8, 30.4. HRMS (ESI): m/z [M + H]+ calcd for C16H18N3+, 252.1495; found 252.1485.
General Procedure for Two-Step, One-Pot Annulations Using Ethyl Glyoxylate 5p as the Aldehyde (0.3 mmol scale).
To a flame-dried 2–5 mL Biotage microwave vial (#351521) charged with a stir bar on the benchtop were added aminopyrazole 6a (0.300 mmol, 1.00 equiv, 47.8 mg), PivOH (1.20 mmol, 4.00 equiv, 123 mg) and 3Å molecular sieves (approximately 300 mg). The vial was capped with a Teflon-lined cap and flushed with nitrogen for about 2 min. Under positive nitrogen pressure, the solution of ethyl glyoxylate 5p (65.4 μL of a 50% v/v in toluene, 0.330 mmol, 1.10 equiv) in dioxane (0.100 M, 3.00 mL) was quickly added. The reaction mixture was then heated with a Biotage Initiator+ (#356007), which employs an external IR sensor and a closed reaction vessel. The resultant mixture was stirred in the microwave reactor for 30 min at 150 °C using the following settings (absorption level, low; vial type, 2−5 mL; prestirring, 0; initial power, 0; dynamic deflector optimization, ON; pressure: OFF; power, OFF; fixed hold time, ON; stir rate, 600). After cooling to rt, the cap was opened, and [Cp*Rh(MeCN)3](SbF6)2 (10 mol %, 0.030 mmol, 25 mg), KOAc (0.30 mmol, 1.0 equiv, 29 mg) and sulfoxonium ylide 7a (0.450 mmol, 1.50 equiv, 137 mg) or formyl sulfoxonium ylide 7i (0.330 mmol, 1.10 equiv, 39.7 mg) were added. The vial was capped with a Teflon-lined cap, flushed with nitrogen for about 2 min and then heated in the microwave reactor for 1 h at 150 °C using the above settings. After cooling to rt, the crude mixture was transferred to a separatory funnel with CH2Cl2 (50 mL). PivOH was removed by extracting with satd. NaHCO3 (100 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were dried (anhyd. Na2SO4) and concentrated under reduced pressure. The product was purified by silica gel column chromatography.
Ethyl 7-(1-(tert-butoxycarbonyl)piperidin-4-yl)-3-phenylpyrazolo[1,5-a]pyrimidine-5-carboxylate (8paa):
The reaction was performed according to the general procedure employing 137 mg (0.450 mmol, 1.50 equiv) of sulfoxonium ylide 7a. Purification by silica gel column chromatography (10–30% EtOAc/hexanes) afforded 8paa (111 mg, 82%) as a yellow solid. mp 140–142 °C. FTIR (neat) 1717, 1691, 1668, 1427, 1239, 1171, 866, 765, 728, 693 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 8.12–8.06 (m, 2H), 7.45 (t, J = 7.7 Hz, 2H), 7.39 (s, 1H), 7.31–7.25 (m, 1H), 4.49 (q, J = 7.1 Hz, 2H), 4.43–4.21 (m, 2H), 3.76 (tt, J = 12.1, 3.4 Hz, 1H), 3.06–2.88 (m, 2H), 2.23–2.15 (m, 2H), 1.80–1.66 (m, 2H), 1.48 (s, 9H), 1.46 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 164.2, 154.6, 152.8, 146.5, 144.2, 142.9, 131.4, 128.8, 126.8, 126.6, 113.2, 103.7, 79.8, 62.4, 43.9, 36.7, 29.0, 28.4, 14.2. HRMS (ESI): m/z [M + H]+ calcd for C25H31N4O4+, 451.2340; found 451.2339.
Ethyl 3-phenylpyrazolo[1,5-a]pyrimidine-5-carboxylate (8pai):
The reaction was performed according to the general procedure employing 39.7 mg (0.330 mmol, 1.10 equiv) of formyl sulfoxonium ylide 7i. Purification by silica gel column chromatography (10–30% EtOAc/hexanes) afforded 8pai (64.6 mg, 81%) as a yellow solid. mp 120–122 °C. FTIR (neat) 1724, 1297, 1138, 1015, 761, 685, 602, 556 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.74 (d, J = 7.3 Hz, 1H), 8.52 (s, 1H), 8.13–8.01 (m, 2H), 7.53 (d, J = 7.3 Hz, 1H), 7.45 (t, J = 7.8 Hz, 2H), 7.33–7.25 (m, 1H), 4.49 (q, J = 7.1 Hz, 2H), 1.46 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 163.7, 146.5, 143.8, 143.7, 135.5, 131.1, 128.9, 126.9, 126.5, 113.3, 107.5, 62.5, 14.2. HRMS (ESI): m/z [M + H]+ calcd for C15H14N3O2+, 268.1081; found 268.1078.
General Procedure for Two-Step, One-Pot Annulations Using Trimethyl Orthoformate 5q as the Aldehyde (0.3 mmol scale).
To a flame-dried 2–5 mL Biotage microwave vial (#351521) charged with a stir bar on the benchtop were added aminopyrazole 6a (0.300 mmol, 1.00 equiv, 47.8 mg) and PivOH (1.20 mmol, 4.00 equiv, 123 mg). The vial was capped with a Teflon-lined cap and flushed with nitrogen for about 2 min. Under positive nitrogen pressure, the solution of trimethyl orthoformate 5q (0.33 mL, 3.0 mmol, 10 equiv) in dioxane (0.100 M, 3.00 mL) was quickly added. The reaction mixture was then heated with a Biotage Initiator+ (#356007), which employs an external IR sensor and a closed reaction vessel. The resultant mixture was stirred in the microwave reactor for 30 min at 150 °C using the following settings (absorption level, low; vial type, 2−5 mL; prestirring, 0; initial power, 0; dynamic deflector optimization, ON; pressure: OFF; power, OFF; fixed hold time, ON; stir rate, 600). After cooling to rt, the cap was opened, and [Cp*Rh(MeCN)3](SbF6)2 (10 mol %, 0.030 mmol, 25 mg), KOAc (0.30 mmol, 1.0 equiv, 29 mg) and sulfoxonium ylide 7a (0.450 mmol, 1.50 equiv, 137 mg) or formyl sulfoxonium ylide 7i (0.330 mmol, 1.10 equiv, 39.7 mg) were added. The vial was capped with a Teflon-lined cap, flushed with nitrogen for about 2 min and then heated in the microwave reactor for 1 h at 150 °C using the above settings. After cooling to rt, the crude mixture was transferred to a separatory funnel with CH2Cl2 (50 mL). PivOH was removed by extracting with satd. NaHCO3 (100 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were dried (anhyd. Na2SO4) and concentrated under reduced pressure. The product was purified by silica gel column chromatography.
tert-Butyl 4-(5-methoxy-3-phenylpyrazolo[1,5-a]pyrimidin-7-yl)piperidine-1-carboxylate (8qaa):
The reaction was performed according to the general procedure employing 137 mg (0.450 mmol, 1.50 equiv) of sulfoxonium ylide 7a. Purification by silica gel column chromatography (10–20% EtOAc/hexanes) afforded 8qaa (95.4 mg, 78%) as a white solid. mp 135–137 °C. FTIR (neat) 1684, 1629, 1545, 1426, 1392, 1229, 1167, 1120, 945, 865, 762, 692 cm−1. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.06–7.97 (m, 2H), 7.41 (t, J = 7.7 Hz, 2H), 7.24–7.16 (m, 1H), 6.17 (s, 1H), 4.41–4.17 (m, 2H), 4.06 (s, 3H), 3.62 (tt, J = 12.1, 3.4 Hz, 1H), 3.05–2.83 (m, 2H), 2.19–2.08 (m, 2H), 1.69–1.54 (m, 2H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 162.2, 154.7, 153.9, 143.7, 141.8, 132.5, 128.6, 125.6, 108.4, 95.9, 79.7, 53.9, 43.7, 36.4, 29.4, 28.4. HRMS (ESI): m/z [M + H]+ calcd for C23H29N4O3+, 409.2234; found 409.2236.
5-Methoxy-3-phenylpyrazolo[1,5-a]pyrimidine (8qai):
The reaction was performed according to the general procedure employing 39.7 mg (0.330 mmol, 1.10 equiv) of formyl sulfoxonium ylide 7i. Purification by silica gel column chromatography (10–20% EtOAc/hexanes) afforded 8qai (24.3 mg, 36%) as a yellow solid. mp 84–86 °C. FTIR (neat) 1633, 1535, 1434, 1405, 1285, 1184, 1019, 946, 793, 757, 686, 512 cm−1. 1H NMR (600 MHz, CDCl3) δ 8.40 (d, J = 7.4 Hz, 1H), 8.30 (s, 1H), 8.06–7.97 (m, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.26–7.18 (m, 1H), 6.35 (d, J = 7.4 Hz, 1H), 4.08 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 161.8, 143.3, 142.8, 137.0, 132.3, 128.6, 125.7, 125.6, 108.3, 100.2, 54.1. HRMS (ESI): m/z [M + H]+ calcd for C13H12N3O+, 226.0975; found 226.0973.
Procedure for Three-Component Reaction of Aminopyrazole 6a, Aldehyde 5n and β-Keto Sulfoxonium Ylide 7a (1 mmol scale).
To a flame-dried 2–5 mL Biotage microwave vial (#351521) charged with a stir bar on the benchtop were added aminopyrazole 6a (1.00 mmol, 1.00 equiv, 159 mg), aldehyde 5n (2.00 mmol, 2.00 equiv, 220 mg), [Cp*Rh(MeCN)3](SbF6)2 (5 mol %, 0.0500 mmol, 41.7 mg), PivOH (4.00 mmol, 4.00 equiv, 409 mg), KOAc (1.00 mmol, 1.00 equiv, 98.1 mg), 3Å molecular sieves (approximately 1 g), sulfoxonium ylide 7a (1.50 mmol, 1.50 equiv, 455 mg) and dioxane (0.400 M, 2.50 mL). The vial was capped with a Teflon-lined cap, flushed with nitrogen for about 5 min, and then heated with a Biotage Initiator+ (#356007), which employs an external IR sensor and a closed reaction vessel. The resultant mixture was stirred in the microwave reactor for 1 h at 150 °C using the following settings (absorption level, low; vial type, 2−5 mL; prestirring, 0; initial power, 0; dynamic deflector optimization, ON; pressure: OFF; power, OFF; fixed hold time, ON; stir rate, 600). After cooling to rt, the crude mixture was transferred to a separatory funnel with CH2Cl2 (150 mL). PivOH was removed by extraction with satd. NaHCO3 (300 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 150 mL). The combined organic extracts were washed with satd. NaHSO3 (10 % wt, 300 mL) to remove the remaining aldehyde.16 The organic layer was dried (anhyd. Na2SO4) and concentrated under reduced pressure. Purification by silica gel column chromatography (5–30% EtOAc/CH2Cl2) afforded 8naa (351 mg, 77%) as a light yellow solid. 1H and 13C NMR spectra matched with 8naa obtained from small scale (0.3 mmol).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (R35GM122473 to J.A.E).
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
NMR spectra (PDF)
Notes
The authors declare no competing financial interest
REFERENCES
- (1).For representative approved drugs and clinical candidates of the pyrazolopyrimidine subclass, see: zaleplon, anagliptin, larotrectinib sulfate, indiplon, remeglurant, verucerfont, lorediplon, ropotrectinib, decoglurant, presatovir, ocinaplon, dinaciclib, TAK-43, IPI-549, OT-7100, AVN-322, AVN-211, MK-8776. The compound structure, bioactivity, list of literature, and access to ongoing clinical trials, applications, and usage can be obtained by searching the compound name in PubChem.
- (2).For representative approved drugs and clinical candidates within this heterocycle framework outside of the pyrazolopyrimidine subclass, see: zolpidem, alpidem, olprinone, zolimidine, miroprofen, necopidem, saripidem, minodronic acid, GSK812397, soraprazan, divaplon, fasiplon. The compound structure, bioactivity, list of literature, and access to ongoing clinical trials, applications, and usage can be obtained by searching the compound name in PubChem.
- (3).Halskov KS; Witten MR; Hoang GL; Mercado BQ; Ellman JA Rhodium(III)-Catalyzed Imidoyl C–H Activation for Annulations to Azolopyrimidines. Org. Lett 2018, 20, 2464–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For reviews on the synthesis of nitrogen heterocycles by C–H functionalization, see; For select reviews, see:Satoh T; Miura M Oxidative Coupling of Aromatic Substrates with Alkynes and Alkenes under Rhodium Catalysis. Chem. Eur. J 2010, 16, 11212–11222.Yamaguchi J; Yamaguchi AD; Itami K C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed 2012, 51, 8960–9009.Song G; Wang F; Li X C–C, C–O and C–N Bond Formation via Rhodium(III)-Catalyzed Oxidative C-H Activation. Chem. Soc. Rev 2012, 41, 3651–3678.Gulías M; Mascareñas JL Metal-Catalyzed Annulations through Activation and Cleavage of C–H Bonds. Angew. Chem. Int. Ed 2016, 55, 11000–11019.Yoshino T; Matsunaga S (Pentamethylcyclopentadienyl)cobalt(III)-Catalyzed C–H Bond Functionalization: From Discovery to Unique Reactivity and Selectivity. Adv. Synth. Catal 2017, 359, 1245–1262.Hummel JR; Boerth JA; Ellman JA Transition-Metal-Catalyzed C–H Bond Addition to Carbonyls, Imines, and Related Polarized π Bonds. Chem. Rev 2017, 117, 9163–9227.
- (5).Hoang GL; Halskov KS; Ellman JA Synthesis of Azolo[1,3,5]triazines via Rhodium(III)-Catalyzed Annulation of N-Azolo Imines and Dioxazolones. J. Org. Chem 2018, 83, 9522–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Park Y; Kim Y; Chang S Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301; and references cited therein. [DOI] [PubMed] [Google Scholar]
- (7).Shaanban S; Abdel-Wahab BF Groebke–Blackburn–Bienaymé Multicomponent Reaction: Emerging Chemistry for Drug Discovery. Mol. Divers 2016, 20, 233–254; and references cited therein. [DOI] [PubMed] [Google Scholar]
- (8).For leading references on sulfoxonium ylides as carbene precursors, see:Baldwin JE; Adlington RM, Godfrey CRA; Gollins DW; Vaughan JG J. A Novel Entry to Carbenoid Species via β-Ketosulfoxonium Ylides. Chem. Soc., Chem. Commun 1993, 1434–1435.Mangion IK; Nwamba IK; Shevlin M; Huffman MA Iridium-Catalyzed X−H Insertions of Sulfoxonium Ylides. Org. Lett 2009, 11, 3566–3569.Burtoloso ACB; Dias RMP; Leonarczyk IA Sulfoxonium and Sulfonium Ylides as Diazocarbonyl Equivalents in Metal-Catalyzed Insertion Reactions. Eur. J. Org. Chem 2013, 5005–5016.Neuhaus JD; Oost R; Merad J; Maulide N Sulfur-Based Ylides in Transition-Metal-Catalysed Processes. Top. Curr. Chem 2018, 317, 1–47.
- (9).For leading references on the application of sulfoxonium ylides in Rh(III)-catalyzed C–H functionalization, see:
- (a).Xu Y; Zhou X; Zheng G; Li X Sulfoxonium Ylides as a Carbene Precursor in Rh(III)-Catalyzed C–H Acylmethylation of Arenes. Org. Lett 2017, 19, 5256–5259. [DOI] [PubMed] [Google Scholar]; (b) Barday M; Janot C; Halcovitch NR; Muir J; Aïssa C Cross-Coupling of α-Carbonyl Sulfoxonium Ylides with C–H Bonds. Angew. Chem. Int. Ed 2017, 56, 13117–13121. [DOI] [PubMed] [Google Scholar]; (c) Wu X; Xiong H; Sun S; Cheng J Rhodium-Catalyzed Relay Carbenoid Functionalization of Aromatic C–H Bonds toward Fused Heteroarenes. Org. Lett 2018, 20, 1396–1399. [DOI] [PubMed] [Google Scholar]; (d) Zheng G; Tian M; Xu Y; Chen X; Li X Rhodium(III)-Catalyzed Annulative Coupling Between Arenes and Sulfoxonium Ylides via C–H Activation. Org. Chem. Front 2018, 5, 998–1002. [Google Scholar]; (e) Xu Y; Zheng G; Yang X; Li X Rhodium(III)-Catalyzed Chemodivergent Annulations Between N-Methoxybenzamides and Sulfoxonium Ylides via C–H Activation. Chem. Commun 2018, 54, 670–673. [DOI] [PubMed] [Google Scholar]; (f) Hoang GL; Ellman JA Rhodium(III)-Catalyzed C–H Functionalization of C-alkenyl Azoles with Sulfoxonium Ylides for the Synthesis of Bridgehead N-fused [5,6]-Bicyclic Heterocycles. Tetrahedron 2018, 74, 3318–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Oh H; Han S; Pandey AK; Han SH; Mishra NK; Kim S; Chun R; Kim HS; Park J; Kim IS Synthesis of (2H)-Indazoles through Rh(III)-Catalyzed Annulation Reaction of Azobenzenes with Sulfoxonium Ylides. J. Org. Chem 2018, 83, 4070–4077. [DOI] [PubMed] [Google Scholar]
- (10).Manach CL; Paquet T; Brunschwig C; Njoroge M; Han Z; Cabrera DG; Bashyam S; Dhinakaran R; Taylor D; Reader J; Botha M; Churchyard A; Lauterbach S; Coetzer TL; Birkholtz L-M; Meister S; Winzeler EA; Waterson D; Witty MJ; Wittlin S; Jiménez-Díaz M-B; Martínez MS; Ferrer S; Angulo-Barturen I; Street LJ; Chibale K A Novel Pyrazolopyridine with in Vivo Activity in Plasmodium berghei- and Plasmodium falciparum-Infected Mouse Models from Structure−Activity Relationship Studies around the Core of Recently Identified Antimalarial Imidazopyridazines. J. Med. Chem 2015, 58, 8713–8722. [DOI] [PubMed] [Google Scholar]
- (11).The compound structure, bioactivity, list of literature, and access to ongoing clinical trials, applications, and usage can be obtained by searching the compound name in PubChem.
- (12).Maquestiau A; Taghret H; Vanden Eynde J-J Preparation and characterization of pyrazolo[1,5-a]pyrimidines. Bull. Soc. Chim. Belg 1992, 101, 131–136. [Google Scholar]
- (13).Kelada M; Walsh JMD; Devine RW; McArdle P; Stephens JC Synthesis of Pyrazolopyrimidinones using a “One-pot” Approach under Microwave Irradiation. Beilstein J. Org. Chem 2018, 14, 1222–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Park Y; Park KT; Kim JG; Chang S Mechanistic Studies on the Rh(III)-Mediated Amido Transfer Process Leading to Robust C−H Amination with a New Type of Amidating Reagent. J. Am. Chem. Soc 2015, 137, 4534–4542. [DOI] [PubMed] [Google Scholar]
- (15).Kondo K; Tunemoto D Nuclear Magnetic Resonance Evidence for Restricted Rotation in Stable Oxosulphonium Ylides. Chem. Commun 1970, 1361–1362. [Google Scholar]
- (16).Boucher MM; Furigay MH; Quach PK; Brindle CS Liquid−Liquid Extraction Protocol for the Removal of Aldehydes and Highly Reactive Ketones from Mixtures. Org. Process Res. Dev 2017, 21, 1394–1403. [Google Scholar]
- (17).Jismy B; Guillaumet G; Allouchi H; Akssira M; Abarbri. M. Concise and Efficient Access to 5,7-Disubstituted Pyrazolo[1,5-a]pyrimidines by Pd-Catalyzed Sequential Arylation, Alkynylation and SNAr Reaction. Eur. J. Org. Chem 2017, 6168–6178. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.