

SUMMARY
The unfolded protein response (UPR) is induced by proteotoxic stress of the endoplasmic reticulum (ER). Here we report that ATF6, a major mammalian UPR sensor, is also activated by specific sphingolipids, dihydrosphingosine (DHS) and dihydroceramide (DHC). Single mutations in a previously undefined transmembrane domain motif that we identify in ATF6 incapacitate DHS/DHC activation while still allowing proteotoxic stress activation via the luminal domain. ATF6 thus possesses two activation mechanisms: DHS/DHC activation and proteotoxic stress activation. Reporters constructed to monitor each mechanism show that phenobarbital-induced ER membrane expansion depends on transmembrane domain-induced ATF6. DHS/DHC addition preferentially induces transcription of ATF6 target lipid biosynthetic and metabolic genes over target ER chaperone genes. Importantly, ATF6 containing a luminal achromatopsia eye disease mutation, unresponsive to proteotoxic stress, can be activated by fenretinide, a drug that upregulates DHC, suggesting a potential therapy for this and other ATF6-related diseases including heart disease and stroke.
Graphical Abstract
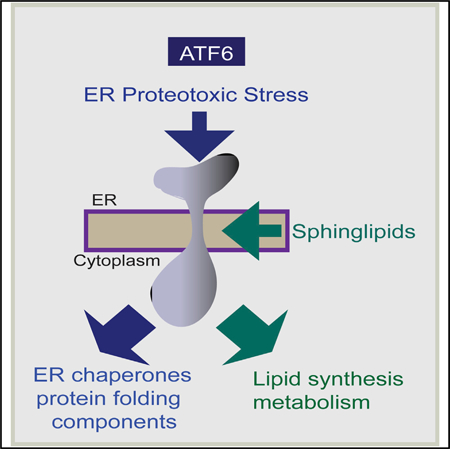
In Brief
The unfolded protein response (UPR) can respond to lipotoxic stress via unclear mechanisms. Tam et al. find that dihydrosphingosine and dihydroceramide, early sphingolipid biosynthetic pathway intermediates, directly activate the mammalian UPR sensor ATF6 via domains distinct from that targeted by ER proteotoxic stress for activation of ER lipid biosynthetic genes.
INTRODUCTION
In eukaryotic cells, the endoplasmic reticulum (ER) responds to changing cellular demands, environmental cues, and emergencies by constantly making adjustments to its constituents. The ER is the largest cellular organelle and performs a variety of critical functions, including synthesis of lipids, regulation of intracellular calcium, and synthesis and maturation of secreted and membrane-bound proteins (Ma and Hendershot, 2001; Voeltz et al., 2002). Such proteins enter the ER lumen as nascent polypeptides (Walter et al., 1984). Once the polypeptides enter the lumen, they associate with ER-resident chaperones and protein-folding enzymes to generate properly folded proteins. The need for ER protein-folding function often increases in response to changing cellular conditions and must be adjusted accordingly. An increased need for protein-folding components, signaled by the presence of high levels of nascent and unfolded secretory pathway proteins, is defined as ER proteotoxic stress. This stress triggers the unfolded protein response (UPR), which swings into action to increase ER protein-folding capacity (Ron and Walter, 2007; Mori, 2000; Rutkowski and Kaufman, 2004).
In mammalian cells, the UPR consists of three parallel signaling pathways, initiated respectively by the ER transmembrane sensors IRE1, PERK, and ATF6; in yeast IRE1 is the sole sensor for the UPR (Ron and Walter, 2007; Mori, 2000; Rutkowski and Kaufman, 2004). Activation of the sensors results in increased transcription of ER components, thereby increasing the protein-folding capacity of the ER. ATF6 is a cryptic transcription factor. Upon sensing proteotoxic stress via its ER luminal domain, the integral membrane protein ATF6 is transported via vesicular trafficking to the Golgi where it undergoes cleavage in its transmembrane domain to release the ATF6 cytoplasmic domain into the cytosol. This is transported to the nucleus, where it acts as a major UPR-specific transcription factor to induce increased expression of genes encoding ER chaperones and other protein-folding components.
In addition to its response to the accumulation of unfolded proteins, the UPR is thought to respond to a parallel need for more lipids, which is termed ER lipotoxic stress (Fu et al., 2011, 2012; Volmer and Ron, 2015; Lee et al., 2008; Rutkowski et al., 2008; Promlek et al., 2011; Miller et al., 2017; Thibault et al., 2012; Yamamoto et al., 2010). The synthesis of most major cellular lipids, including phospholipids, sterols, and sphingolipids, is known to start in the ER (Jacquemyn et al., 2017; Ron and Hampton, 2004). A series of observations indicate that the UPR components IRE1 and PERK can be activated by a lipotoxic stress that is caused by adding free fatty acids; in those instances activation has been proposed to occur by the fatty acids increasing membrane fluidity, with the increased fluidity being the signal for UPR activation (Volmer et al., 2013; Halbleib et al., 2017). While membrane synthesis has long been described as an integral part of the UPR pathway, the molecular mechanism by which such coordination is achieved has remained largely elusive. In an example of coordination, when antigen stimulation induces differentiation of resting B cells into plasma cells that now secrete vast quantities of antibodies, this process is accompanied by massive ER membrane expansion (Schuck et al., 2009; van Anken et al., 2003).
Here, we show that UPR induction in vivo is accompanied by an increase in specific sphingolipids, dihydrosphingosine (DHS) and dihydroceramide (DHC). We further find that exogenous addition of these specific sphingolipids to unstressed cells preferentially activates the ATF6 arm of the UPR pathway and does so independently of proteotoxic stress. We identify a required peptide sequence within the ATF6 transmembrane domain that we show is needed for its activation by these sphingolipids. Our results thus reveal an unexpected dual mechanism for activating ATF6, and provide mechanistic insight into the possibility of coordinating proteotoxic and lipotoxic stress through the ATF6 arm of the UPR pathway.
RESULTS
Sphingolipid Pathway Intermediates Dihydrosphingosine and Dihydroceramide Are Increased in Response to ER Stress
Sphingolipid signaling has been observed to play important roles in turning on cellular pathways (Olson et al., 2015; Hannun and Obeid, 2018). However, it has only recently been possible to achieve the sensitivity of mass spectrometry to measure the levels of early sphingolipid intermediates. To investigate whether sphingolipid levels are elevated in mammalian cells by ER stress, we performed liquid chromatography-mass spectrometry (LC-MS) analysis (Benjamin et al., 2015) on HEK293 cells treated with the canonical UPR inducer, thapsigargin (Tg), which induces ER stress by depleting Ca2+ from the ER. The levels of DHS, an early intermediate in sphingolipid synthesis (Figure 1A), increased ~2.5-fold within 90 min of Tg addition (Figure 1B); many other sphingolipid intermediates showed little change (figure 1B and S1A). ER stress induction by another UPR inducer, DTT, which causes breakage of disulfide bonds, resulted in an increase in DHC, the intermediate immediately downstream of DHS (Figures S1C and S1D). Biosynthesis of both DHS and DHC is known to occur at the ER. The differences in increase in DHS by Tg and DHC by DTT may reflect subtle differences in the way ER stress was induced. Although the MS analysis used here was capable of detecting all the sphingolipids in Figure 1A from dihydrosphingosine onward, we found that only DHS and DHC were uniquely increased during ER stress; the levels of ceramide, sphingosine, and sphingomyelin did not change upon UPR induction (figure 1A, 1B, and S1A–S1D).
Figure 1. ER Stress Specifically Induces a DHS Increase in Mammals.

(A) Schematic of the human sphingolipid/ceramide biosynthetic pathway. De novo sphingolipid synthesis up to ceramide occurs in the ER, while generation of sphingosine and sphingomyelin occurs in the Golgi. Key biosynthetic enzymes and inhibitors are shown. (B) DHS increases after Tg treatment. Representative lipid profiles of HEK293 cells 0, 30, and 90 min after Tg treatment, as determined by LC-MS, are shown for DHS, DHC (C18:0, C20:0, C22:0, and C24:0), and ceramide (C16:0, C18:0, and C20:4). See Figure S1A for sphingosine and sphingomyelin levels. In all cases, the relative lipid levels (fold induction) with respect to the values at 0 min are shown. Note that the distance between 30- and 90-min time points is foreshortened to save space; in all cases, the fold induction over 0 min is the key comparison. Error bars represent values from five independent samples. (C) Sphingolipid production is increased in vitro. Microsomes isolated from untreated (Untr) or Tg-treated HEK293 cells were incubated with increasing amounts of [3H]serine and palmitoyl-CoA. The sum of the 3H-labeled 3-ketodihydrosphingosine, DHS, DHC, and ceramide lipids produced were separated from free [3H]serine by extraction and measured. Control reactions were also performed with the inhibitor myriocin (Myr). Unpaired two-tailed t tests comparing untreated and treated samples: **p < 0.01; *p < 0.05; ns, not significant.
The increase in DHS and DHC suggested that ER stress induces the early steps in ER sphingolipid biosynthesis. To test this in vitro, we prepared microsomes from HEK293 cells that had been incubated for 30 min with [3H]L-serine, unlabeled palmitoyl-coenzyme A (CoA), plus or minus Tg (Rutti et al., 2009). We found the levels of 3H-labeled lipids produced were consistently higher in ER stressed microsomes than in those from unstressed cells (Figure 1C), arguing that lipid synthesis by early sphingolipid biosynthetic enzymes such as SPT and FVT-1 (Figure 1A) was increased by ER stress. Importantly, the addition of myriocin, a well-established inhibitor of SPT, blocked the increase of [3H] lipids in both stressed and unstressed cells to identical low levels (Figure 1C). Taken together, the MS analysis and enzymatic analysis revealed that ER stress increases the synthesis of DHS and DHC.
ATF6 Is Activated by the Early Biosynthetic Intermediates Dihydrosphingosine and Dihydroceramide
Given the increased levels of DHS and DHC in response to ER stress, we tested whether addition of exogenous DHS or DHC activates any UPR components in HEK293 cells. Using a previously validated ATF6-GFP reporter (Shen et al., 2002; Gallagher and Walter, 2016; Ye et al., 2000), we found that either DHS or DHC addition induced ATF6-GFP activation and movement to the nucleus (figure 2B–2G and S2A–S2C). Moreover, activation of ATF6 by DHS or DHC occurred in a concentration-dependent manner (Figures S2D and S2E). Strikingly, the kinetics of ATF6 activation and movement closely mirrored those seen after Tg or DTT addition (figure 2A–2G and S2A–S2C).
Figure 2. DHS and DHC Activate the UPR Sensor ATF6.

(A–C) Representative images of HEK293 cells with ATF6-GFP upon treatment with (A) Tg (200 nM), (B) DHS (50 µM), or (C) DHC (50 µM). ATF6-GFP localization is shown at the ER (green), Golgi (red; anti-GM130), and nucleus (blue; DAPI). (D–G) Kinetic changes in ATF6-GFP localization in HEK293 cells treated with DHC (D) or DHS (E) versus Tg, or with DHC (F) or DHS (G) versus DTT. As incubation time with DHC or DHS increased, ER-localized ATF6-GFP decreased and, ultimately, ATF6-GFP appeared in the nucleus, following transit through the Golgi. Data represent the mean ± SD of at least three independent experiments, each performed with ≥50 cells per time point. Unpaired two-tailed t tests comparing untreated and treated samples: **p < 0.01; *p < 0.05. (H) DHC or DHS treatment of HEK293 cells did not induce IRE1 as measured by XBP1 mRNA splicing, while Tg treatment induced efficient splicing of XBP1 mRNA, as expected. Spliced (XBP1s) and unspliced (XBP1u) forms of XBP1 mRNA are marked. (I) eIF2α is phosphorylated following Tg, but not DHC or DHS treatment of HEK293 cells. The fold increase in levels of phosphorylated eIF2α, visualized by western blotting using an anti-phospho-eIF2α antibody, was normalized to those of total eIF2α. Data represent the mean ± SD of three independent experiments.
Examination of fluorescently labeled DHS and DHC (NBD-DHS and NBD-DHC) confirmed that DHS and DHC have the capacity to rapidly enter cells and initially localize to the ER (Figures S2F, S2G, and S2I). The NBD-labeled sphingolipids were subsequently found to move to the Golgi, co-localizing with Golph3 (Figures S2H and S2J), a well-established Golgi marker (Dippold et al., 2009).
We also found ATF6-GFP was activated by DHS or DHC in other mammalian cells, including the human tumor cell lines MCF7 and HeLa (Figures S2K and S2L), as well as in mouse embryonic fibroblasts (MEFs) (Figures S2M–S2O). Additionally, the magnitude and kinetics of ATF6-GFP reporter activation was unaffected by the presence of endogenous ATF6, since DHS, DHC, and Tg all had essentially the same effect on ATF6-GFP activation whether the reporter was transfected into wild-type MEFs or Atf6—/— MEFs (Figures S2M–S2O). We confirmed that the activation events observed with transfected ATF6-GFP recapitulate activation of endogenous ATF6, as we observed the appearance of cleaved ATF6 N terminus for both endogenous ATF6 and the transfected ATF6-GFP (Figure S2P), indicating that ATF6-GFP is indeed a faithful reporter of the activation status of ATF6, as shown previously (Shen et al., 2002; Gallagher and Walter, 2016). These experiments demonstrate that DHS and DHC, increased in response to ER stress, can activate ATF6.
Notably, the abilities of DHS and DHC to activate ATF6 were specific to these early intermediates, since addition of the downstream sphingolipids and ceramides in the biosynthetic pathway did not effectively activate ATF6 (Figures S2Q and S2R). Furthermore, we found that the carbon chain lengths of DHC did not affect ATF6 activation as both C2:0- and C16:0-DHC were equally active (Figures S2Q and S2R). The fact that mammalian ATF6 activation was limited to DHS and DHC and was not effectively activated by the structurally similar ceramides or sphingosine (Figure 1A) suggests again that ATF6 activation by either DHS or DHC is unlikely to be caused by pleiotropic disturbances of the ER membrane. Furthermore, DHS and DHC had little effect on ATF6β (Figure S3A) or on the unrelated ER transmembrane transcription factor, SREBP (Figures S3B and S3C). Taken together, the data indicate that the activation of ATF6 is induced by DHS and DHC.
DHS and DHC Do Not Activate IRE1 or PERK
Upon testing, we found that DHS and DHC did not activate IRE1, the initiator of another arm of the mammalian UPR response. Normally when activated by Tg, IRE1 becomes an active riboendonuclease that specifically cleaves out the intron of XBP1 mRNA; this spliced mRNA is then translated into the UPR transcription factor XBP1. After DHS or DHC addition, no spliced XBP1 RNA was observed (Figure 2H). Similarly, we found that DHS and DHC do not appear to activate the third arm of the mammalian UPR, PERK, which upon activation by Tg becomes a functional kinase for eIF2α. Specifically, no eIF2α phosphorylation above background (normalized to total eIF2α) was observed following DHS or DHC addition to HEK293 cells (Figure 2I). We conclude that DHS and DHC activate ATF6, but not IRE1 or PERK.
Differences Observed between DHS/DHC Activation of ATF6 and Proteotoxic Stress Activation
ATF6 activation by canonical proteotoxic stress inducers involves a well-choreographed set of biochemical changes and movements from the ER to the Golgi to the nucleus, culminating in UPR-specific gene expression. We compared the early steps seen with proteotoxic stressors with the steps seen with DHS or DHC. Tg and DTT induce ATF6 by causing the accumulation of unfolded proteins in the ER. To first determine whether DHC might activate ATF6 by increasing unfolded proteins within the ER, we utilized a VSVG-GFP reporter previously described as a ‘‘folding status’’ reporter (Nehls et al., 2000; Hirschberg et al., 1998; Cole et al., 1998). VSVG, the vesicular stomatitis virus glycoprotein G, is a secretory pathway protein, ultimately targeted to the cell surface. Upon synthesis, VSVG enters the ER for folding and maturation before being transported to the Golgi and cell surface. The folding status of VSVG can be determined using an antibody that recognizes only its folded form (Cole et al., 1998; Nehls et al., 2000). As was done in those studies to facilitate detection of the folded status of VSVG, we blocked all the VSVG-GFP in the ER using FLI-06, an inhibitor of COP II vesicle formation (Figure 3 panel 2, and S3D). Following addition of DTT for 1 hr, folded VSVG-GFP (red) quickly disappeared while the overall amount of VSVG-GFP (green) remained unchanged throughout the 3-hr time course (Figure 3A, panels 3–5). In contrast, incubation with DHC appeared not to decrease the amount of folded VSVG-GFP during the first hour of DHC treatment (red, Figure 3A, panel 6). The folded form (red) gradually disappeared as VSVG-GFP unfolded (Figure 3A, panels 7 and 8). Since ATF6 activation occurs within 30 min after DHC activation (figure 2A–2G and S2A–S2C), which precedes the appearance of unfolded VSVG seen here, we concluded that proteotoxic stress is unlikely to be the major cause of ATF6 activation by DHS or DHC.
Figure 3. Distinct Differences Are Observed Early in ATF6 Activation by DHC versus Proteotoxic Stress.

(A) DHC does not cause VSVG-GFP unfolding until 2–3 hr after its addition (red, panels 7–8), compared with DTT treatment, which causes rapid unfolding of VSVG-GFP (red; 1 hr, panel 3). Folding was detected with an antibody that binds only to folded VSVG-GFP. In unstressed cells, VSVG-GFP was localized throughout the secretory pathway (red, panel 1). For this experiment, both the DTT- and DHC-treated cells were incubated with FLI-06, an inhibitor of transport to the Golgi, to ensure retention in the ER for better detection of both folded (red) and total (green) VSVG-GFP. Scale bar, 10 µm. (B) Treatment with the chemical chaperone 4-PBA is known to reduce ATF6 activation as it helps to alleviate ER proteotoxic stress in Tg-treated HEK293 cells (lane 4). 4-PBA had a significantly lesser effect on ATF6 activation by DHC (lane 6). Data represent the mean ± SD of at least three independent experiments, each performed with ≥50 cells. (C) DHC does not induce significant BiP dissociation from WT-ATF6 immunoprecipitates from HeLa cells, while Tg, as expected, does. BiP does not dissociate from ATF6 mutated in the luminal domain (Y567N-ATF6), after either Tg or DHC treatment. Quantitation of BiP-bound ATF6 levels in ATF6 immunoprecipitates is shown (graph). The mean ± SD of three independent experiments is shown. (D) ATF6 is glycosylated to a similar extent after activation by either Tg, DTT, DHC, or DHS. HEK293 cells were treated with Tg, DHS, DHC, or DTT for 2 hr, before ATF6 was immunoprecipitated, treated with or without Endo H, and subjected to SDS-PAGE. (E) Oligomerization/monomer status of ATF6 in HEK293 cells treated with either Tg or DHC was similar, as visualized by immunoblotting with anti-ATF6 antibody under non-reducing (NR) or reducing (R) conditions, as described in Nadanaka et al. (2007). In (B) and (C), **p < 0.01; n.s. indicates statistically insignificant differences using unpaired two-tailed t tests comparing untreated and treated samples.
These results are also consistent with experiments we performed using the chemical chaperone 4-phenylbutyric acid (4-PBA), a chemical known to reduce the level of unfolded proteins in the ER (Cao et al., 2013). 4-PBA strongly suppressed ATF6 activation in response to Tg (Figure 3B: 4PBA + Tg, compare lanes 3 and 4; Figure S3E). Addition of 4-PBA showed much less effect on the DHC-induced activation of ATF6 (Figure 3B, compare lanes 5 and 6; Figure S3E), measured as the rate of appearance of ATF6 in the nucleus.
We next asked whether DHC causes BiP to dissociate from ATF6. Upon UPR activation by the proteotoxic stressor Tg, reduced levels of BiP co-immunoprecipitate with ATF6, as BiP dissociates from the ATF6 luminal domain and instead binds to the unfolded proteins accumulating in the ER lumen (Shen et al., 2002; Bertolotti et al., 2000; Schindler and Schekman, 2009), as also seen here in Figure 3C (wild-type ATF6 [WT-ATF6], compare lanes 1 and 2). In contrast, DHC treatment caused little or no reduction in BiP co-immunoprecipitating with ATF6 (Figure 3C; WT-ATF6, compare lanes 1 and 3). This indicates that an initial step in activation by DHC differs from that induced by the proteotoxic ER stressor Tg.
We also found that while pretreatment of cells with the Ca2+ chelator BAPTA inhibits Tg-mediated activation of ATF6 (Senkal et al., 2011), BAPTA did not inhibit ATF6 activation by DHC (Figures S3F and S3G). Thus, we conclude that DHS and DHC activation of ATF6 is not caused by accumulation of ER unfolded proteins or depletion of ER Ca2+.
Dihydroceramide-Activated ATF6 Is Still Packaged into COPII Vesicles and Cleaved by S1P and S2P
In contrast, we found that the known downstream steps of proteotoxic activation of ATF6 (Schindler and Schekman, 2009; Higa et al., 2014; Nadanaka et al., 2007), including glycosylation state (Figure 3D, compare lanes 2, 8 versus 4, 6), oligomerization status (Figure 3E, lanes 1–3 versus 7–9), COPII vesicle association (co-localization with Sec 16), and localization at the ER exit sites (co-localization with Sec 31A), all appeared indistinguishable whether activation occurred by Tg, DTT, DHS, or DHC (figure 4A, 4B, and S4A). Additionally, FLI-06 hindered DHC-induced ATF6 from appearing in the nucleus, just as it had for Tg-induced ATF6 (Figure 4F). In a further similarity, treatment of cells with Ceapin A7, an inhibitor of Tg activation of ATF6 (Gallagher et al., 2016; Gallagher and Walter, 2016), also blocked DHC activation of ATF6 (figure 4C–4E, S4B, and S4C). Lastly, we asked whether DHC activation must induce cleavage of the transmembrane domain of ATF6 for effective activation (freeing of the N-terminal transcription factor domain). Indeed, pretreatment of cells with AEBSF, a chemical inhibitor of the S1P/S2P protease (Okada et al., 2003), markedly decreased the percentage of DHC-treated cells containing nuclear ATF6-GFP (figure 4G and S4D). These data argue that DHC/DHS differ by not inducing or requiring unfolded proteins or the loss of BiP for activation (figure 3A–3C), but after that point many of the intermediate steps involved in activation of ATF6 occur similarly in response to Tg and DHC.
Figure 4. DHS/DHC Activation of ATF6 Requires Transport to the Golgi via COP II Vesicles and S1P/S2P Cleavage of the ATF6 Transmembrane Domain.

(A) DHC treatment resulted in co-localization of ATF6-GFP with Sec31A, a COPII vesicle component, and Sec16, an ER exit site (ERES) component, to an extent similar to ATF6-GFP in Tg-treated cells (arrowheads indicate co-localization of all three molecules). Following treatment of HEK293 cells with Tg or DHC for 60 min, immunofluorescence was done with anti-Sec31A and anti-Sec16 antibodies. Representative images of cells treated with Tg, DHC, or untreated are shown, with high magnification views of the white-boxed areas at the right. ATF6-GFP in green, Sec31A in red, and Sec16 in blue. Scale bar, 10 µm. (B) Percent co-localization of ATF6-GFP with Sec31A (magenta) and Sec16 (dark purple) is shown. See STAR Methods for a detailed description of the quantification. Each co-localization experiment was repeated at least three independent times. (C) Ceapin A7, which blocks Tg activation of ATF6 as defined by nuclear localization, inhibits DHC and DHS activation of ATF6 to a similar extent. (D and E) Ceapin A7 inhibits co-localization of DHC-, DHS-, and Tg-induced ATF6-GFP with Sec31A (D) and with both Sec31A and Sec16 (E) upon treatment with ATF6. (F) DHC activation of ATF6 requires transport to the Golgi. Pretreatment of HEK293 cells with FLI-06, an inhibitor of COPII transport, blocked ATF6-GFP activation by Tg and DHC. (G) ATF6 activation by either Tg or DHC is similarly blocked by AEBSF, a known chemical inhibitor of the membrane-bound proteases S1P and S2P. In (C)–(G), **p < 0.01 and *p < 0.05 using unpaired two-tailed t tests comparing untreated and treated samples.
DHS- and DHC-Mediated Activation of ATF6 Is Uniquely Dependent on the ATF6 Transmembrane Domain
Given that DHS and DHC are sphingolipids synthesized with their fatty acid tail(s) tethered in the outer leaflet of the ER bilayer, this suggested the possibility that transmembrane residues of ATF6 might act to participate in recognizing DHS and DHC. In search of a published example of a protein demonstrated to recognize a specific sphingolipid via its transmembrane domain, we found that the protein p24, an integral membrane protein of COP I vesicles, does indeed bind the downstream lipid sphingomyelin via its transmembrane domain (Contreras et al., 2012). Upon visual inspection of the transmembrane sequence of human ATF6, we found a conserved sequence VXXFIXXNY, which showed both spacing homology and some sequence homology to the essential residues for p24 binding to sphingomyelin (VXXTLXXIY) (Figure 5A). Use of the secondary structure program Phyre2 predicted that the three key p24 transmembrane domain residues (L17, I20, and Y21) for sphingomyelin binding and the equivalent residues of the ATF6 transmembrane domain (I388, N391, and Y392) were predicted to map in the same structural relationship (figure 5C and 5D).
Figure 5. A Conserved Dihydroceramide- and Dihydrosphingosine-Recognition Motif Is Found within the ATF6 Transmembrane Domain.

(A) Alignment of residues within the transmembrane domain of ATF6 (yellow). The previously reported consensus binding site (VXXTLXXIY) for sphingomyelin (SM) in the COP I component p24 (Contreras et al., 2012) is shown for comparison with a putative DHS-binding site in ATF6 (VXXFIXXNY, red), proposed here. Residues within the DHS recognition motif that map in equivalent locations to the essential residues of p24 are highlighted in blue. (B) Sequence alignment of the transmembrane domains of ATF6α, ATF6β, and other transmembrane transcription factors, including OASIS, BBF2H7, LUMAN, CREBH, and ALBZIP, is shown. For comparison, the transmembrane domains of IRE1 and PERK are also shown. The VXXFIXXNY motif was not seen in IRE1 or PERK. (C and D) Predicted structural models of the p24 sphingomyelin-binding motif within its transmembrane domain (C) and the putative DHS-binding motif within the ATF6 transmembrane domain (D) were derived from the computational program Phyre2. The N termini are at the bottom of the image and the C termini at the top. The sphingomyelin-binding motif of p24 is shown (C), with the key residues L17, I20, and Y21 indicated (Contreras et al., 2012). The spatial relationships of three conserved residues, I388, N391, and Y392 in the DHS-recognition motif are highlighted in red (D). (E) Each residue within the putative DHS or DHC recognition motif of ATF6 was mutated to the amino acid residues shown in dark blue. Within the motif, critical amino acid residues for activation by DHS and DHC are shown in red. The activation rate of the individual ATF6 mutants was measured upon Tg, DHC, or DHS treatment of HEK293 cells carrying the mutants. In particular, I388F-ATF6 and Y392C-ATF6 were diminished for activation by DHC and DHS, but retained the ability to be activated by Tg. Data represent the mean ± SD of three independent experiments, each with at least 50 cells per sample. **p < 0.01, *p < 0.05, n.s. indicates statistically insignificant differences, using unpaired two-tailed t tests comparing activation rate of WT versus mutants samples. Note that an amino acid change at the valine (V384) caused constitutive activation of ATF6 even in the absence of Tg or DHS/DHC, and thus was omitted from mutational analysis (Figure S5A). (F) WT-ATF6 binds to [3H]DHS in a concentration-dependent manner and reaches a plateau at 100 pmol. The I388F-ATF6 mutant was severely diminished in its ability to bind to [3H]DHS. **p < 0.01, *p < 0.05 using unpaired two-tailed t tests comparing WT-ATF6 and I388F-ATF6 at each [3H]DHS concentration added. Each experiment was repeated at least three independent times. (G) Association of WT-ATF6 with [3H]DHS was effectively competed by addition of excess cold DHS, but unchanged by addition of the excess unrelated lipid cholesterol. **p < 0.01 using unpaired two-tailed t tests comparing the 3H-bound values of no competitor versus addition of cold DHS, while n.s. (not significant) compares the values between no competitor and added cold cholesterol. Each experiment was repeated at least three independent times.
To test the importance of this putative ATF6 transmembrane motif, we generated mutants of human ATF6-GFP carrying single substitutions within the human sequence VLAFIILNY (Figure 5E). Strikingly, I388F-ATF6 and Y392C-ATF6 mutants were dramatically reduced in their ability to be activated by DHS or DHC when compared with WT-ATF6 (Figure 5E, see dotted squares; Figures S5A–S5I). Our changes of other residues either did not significantly alter ATF6 activation by DHS or DHC (L385I, A386V, I389L, L390I) or reduced activation by one but not the other sphingolipid (F387A) (figure 5E and S5A–S5I). Importantly, the most severe mutations, I388F-ATF6 and Y392C-ATF6, which were greatly decreased in their activation by DHS or DHC, were still activated by Tg treatment (Figure 5E, blue, S5E and S5I), demonstrating that these mutations did not alter overall ATF6 protein structure. We conclude that the I388F- and Y392C-ATF6 mutants are greatly reduced in their response to the sphingolipids DHS and DHC, but continue to respond to proteotoxic stress.
The above mutational analysis suggested that the ATF6 transmembrane motif could be directly binding to DHS or DHC. To test this, we performed DHS binding assays in vitro. WT-ATF6 was isolated by immunoprecipitation from HEK293 microsomes derived from cells transfected with WT-ATF6; immunoprecipitated WT-ATF6 was then incubated with increasing levels of [3H]DHS. We found that the level of bound [3H]DHS increased in a concentration-dependent manner (Figure 5F, WT-ATF6). The binding of [3H]DHS reached saturation, as it was seen to plateau with added [3H]DHS beyond 100 pmol. In contrast, when immunoprecipitated I388F-ATF6 was used in the assay, the level of [3H]DHS binding was greatly diminished (Figure 5F, I388F-ATF6, and S5K). Finally, the binding of [3H]DHS to ATF6 was effectively competed by unlabeled DHS, but not by the unrelated lipid, cholesterol (Figure 5G). This strongly indicates that the reason why ATF6 proteins with the most severe mutations in their predicted DHS/DHC-binding domain are unable to be activated is that these mutants cannot bind these specific lipids.
The Existence of Two ATF6 Activation Pathways Is Confirmed by Use of a Human Achromatopsia Disease Mutation
Point mutations in the luminal domain of ATF6 have been reported in the hereditary eye disease achromatopsia (Kohl et al., 2015). We found that introducing the achromatopsia Y567N mutation into the ER luminal domain (Figure 6A) disrupts Tg activation of ATF6-GFP (Figure 6B, lane 4; Figure S5J). We further found that the luminal domain of Y567N-ATF6 does not dissociate from BiP upon Tg treatment (Figure 3C, lanes 4–6), demonstrating that Y567N-ATF6 is unable to either sense or respond to ER proteotoxic stress induced by Tg (Figure 6B). However, Y567N-ATF6 is still activatable by DHC (Figure 6B, lane 8 and S5J). Thus, ATF6 activation by DHC via the ER transmembrane domain can occur effectively even when the ATF6 ER luminal domain is impaired in its response to the ER proteotoxic stressor Tg.
Figure 6. Mutants Allow Differential Dissection of the Two Modes of ATF6 Activation.

(A) Schematics of WT-ATF6, the transmembrane domain mutants I388F-ATF6 and Y392C-ATF6, and an achromatopsia-related ATF6 ER luminal mutation, Y567N-ATF6. (B) Cells expressing I388F- and Y392C-ATF6 are unable to be activated by treatment with DHC or with the drug fenretinide (FEN; 10 µM). However, the same mutants are activatable by Tg. FEN blocks the activity of dihydroceramide desaturase 1, resulting in the accumulation of DHC (Figure 1A). Since Tg treatment also induces an increase in DHS and DHC (figure 1B and S1), myriocin (25 µM) was included with Tg incubation to block this increase in DHS/DHC levels. The Y567N-ATF6 mutant was activated by DHC or FEN, but not by Tg. **p < 0.01 using unpaired two-tailed t tests comparing WT, I388F, Y392C, and Y567N for DHC-, Tg-, and FEN-treated samples. Each experiment was repeated at least three independent times. (C and D) Electron microscopy pictures of atf6—/— MEFs transfected with WT or I388F-ATF6. Cells with WT-ATF6, if treated with phenobarbital (PB) for 16 hr, showed the appearance of distended ER (arrowheads) (C). However, phenobarbital-treated cells carrying I388F-ATF6 did not show distended ER. Quantitation of distended ER found in WT versus I388F-ATF6 cells with or without PB treatment (D). *p < 0.05 using unpaired two-tailed t tests comparing WT and I388F. Additional electron microscopy images and quantitation are shown in Figure S6I. (E and F) Overexpression of cytochrome b(5)tail activates ATF6 via the DHS/DHC domain. The activation of either an I388F-ATF6-tdTomato reporter or a Y567N-ATF6-tdTomato reporter was monitored in HeLa-tetOFF cells induced to express the cytochrome b(5) C-terminal tail (GFP-b(5)tail). Upon induction of GFP-b(5)tail expression by removal of doxycycline, Y567N-ATF6 tdTomato started to appear in the nucleus within 24 hr and increased with time, in a manner similar to the WT-ATF6tdTomato reporter. In contrast, I388F-ATF6 tdTomato, which lacks ability to be induced through its transmembrane domain, appeared in the nucleus at a much reduced level until 48 hr. Quantitation of the nuclear-localized ATF6 for each condition is shown in (F). SD of all the experiments is calculated from at least three different experiments.
Conversely, I388F-ATF6 and Y392C-ATF6, which cannot be activated by DHS or DHC (Figure 6B, lanes 6 and 7), can still be activated by ER proteotoxic stress via the ER luminal domain (Figure 6B, lanes 2 and 3 Tg, and S5E and S5I).
Our initial studies of sphingolipid induction began when we found that treatment of HEK293 cells with Tg, the well-established proteotoxic stress inducer, also increased levels of DHS (Figure 1B). Here we find that addition of myriocin, an inhibitor of SPT, to cells with WT-ATF6-GFP decreased the extent of ATF6 activation (Figure 6B, compare lane 1 with lane 9). A decrease does not occur with cells containing the ATF6 transmembrane domain mutations I388F-ATF6 or Y392C-ATF6 (Figure 6B, compare lanes 2 and 3 with lanes 10 and 11). These results suggest that WT-ATF6 is activated via the sum of the ER luminal domain sensing ER proteotoxic stress and the transmembrane motif sensing the DHS/DHC induction.
The conclusion that ATF6 can be independently activated by two activating signals (or inputs) suggests that drugs could be developed that bypass the effects of a debilitating mutation within either domain. One relevant drug that could theoretically be useful in this arena is fenretinide (FEN), which increases intracellular DHC concentrations by blocking the conversion of DHC to ceramide, resulting in an increase in intracellular DHC concentration (Figure 1A) (Bikman et al., 2012). Interestingly, we found that WT-ATF6 GFP was activated to the same extent in FEN-treated cells as it was after DHC addition (Figure 6B, compare lanes 5 and 13). FEN could not, however, activate the transmembrane mutants I388F-ATF6 or Y392C-ATF6 (Figure 6B, lanes 14 and 15; Figures S6E–S6H). It did still effectively activate the luminal Y567N-ATF6 mutant (Figure 6B, lane 16; S6E-H). Thus FEN, already being tested in humans as a potential anti-cancer drug for reasons unrelated to ATF6 (Cooper et al., 2017), could be tested for efficacy in ATF6 mutant-related diseases.
ATF6 Activation via Its Transmembrane Domain Is Involved in Physiological Settings that Show ER Membrane Expansion
One of the most compelling changes in the ER occurs when cells encounter events that challenge the ER to expand massively. Notably, treatment of hepatocytes with phenobarbital (PB), an anti-seizure drug, has been shown to result in expansion of the smooth ER, as the cells work to detoxify the phenobarbital (Amatsu et al., 1995). Based on our results above, we asked whether there is role for transmembrane domain-dependent activation of ATF6 in phenobarbital-induced ER membrane change. We used atf6—/— MEFs transfected with either WT-ATF6 or I388F-ATF6, the latter of which cannot be activated by DHS/DHC. Following transfection, phenobarbital was added for 24 hr, after which electron microscopy was performed. Phenobarbital addition caused detectable dilation of the ER in cells with WT ATF6, but not in cells carrying I388F-ATF6, the form that cannot respond to DHS/DHC (figure 6C, 6D, and S6I). Thus, these results argue that this change in ER structure depends on the DHS/DHC-binding motif-dependent activation of ATF6.
During detoxification by the liver, the genes encoding detoxifying ER membrane proteins such as cytochrome P450 and cytochrome b(5) become highly upregulated. This leads to the large expansion in the amount of smooth ER that others have observed (Sprocati et al., 2006). In fact, simple overexpression of the C-terminal tail of cytochrome b(5), termed b(5)tail, has been shown to be sufficient to expand the amount of ER membranes in cells and also to activate ATF6 (Maiuolo et al., 2011). Here we used a fluorescently tagged proteotoxic stress-activat- able ATF6 reporter (I388F-ATF6-tdTomato) or a DHS/DHC-activatable ATF6 reporter (Y567N-ATF6-tdTomato) to ask whether either became activated by cytochrome b(5)tail overexpression. For this, the reporters were transfected separately into a HeLa-TetOff cell line that carries a GFP-tagged cytochrome b(5)tail anchored to the ER membrane (Sprocati et al., 2006). Upon removal of doxycycline, b(5)tail-GFP expression was observed to be gradually induced in the cells over time (Figure 6E). Cells containing WT-ATF6-tdTomato showed robust activation of ATF6 (i.e., nuclear localization) as the b(5)tail amount increased, consistent with the b(5)tail activation of ATF6 reported previously (Maiuolo et al., 2011). By 24 hr, b(5)tail-GFP expression also induced a significant increase in nuclear Y567N-ATF6-tdTomato, the DHS/DHC-pathway reporter (figure 6E and 6F, right panels and S6J). In contrast, little activation was observed for I388F-ATF6-Tomato, the proteotoxic stress-pathway reporter, at 24 hr, as shown by its lack of nuclear localization. Ultimately, at time points ≥48 hr, we observed that both pathways became activated (i.e., showed nuclear-localized ATF6) to similar levels. However, the kinetics show that the increased expression of cytochrome b(5)tail, an ER transmembrane protein, starts with the preferential activation of ATF6 via its transmembrane domain at early times.
Dihydroceramide-Activated ATF6 Induces Distinct Transcriptional Responses
A clear question of interest is whether differential ATF6 activation causes differential transcription. First, we demonstrated that DHS or DHC treatment of atf6—/— MEFs, which lack ATF6, does not induce transcription of the chaperones BiP and HERP (blue), which are ATF6 target genes (Figure 7B, lanes 1–3) (Shoulders et al., 2013). Neither did DHS or DHC treatment of atf6—/— MEFs induce significant transcription of PPARα and LRP (purple), previously known ATF6 target genes involved in lipid synthesis and regulation (Figure 7A, lanes 1–3). As a positive control, we showed that all these ATF6 target genes were induced by expression of the active transcription factor form of ATF6 (the soluble N-terminal domain) in atf6—/— MEFs (figure 7A and 7B, lane 5). We concluded that in the absence of ATF6 expression, DHS and DHC do not induce ATF6 target genes.
Figure 7. Differential Transcription Output Induced by Tg-Activated ATF6 and by DHC/DHS-Activated ATF6.

(A and B) Transcription activation by DHC, DHS, and Tg depends on the presence of ATF6, as activation is severely reduced in atf6—/— MEFs (lanes 2–4 in A and B). Transcription levels of the ATF6 target lipid metabolism genes PPARα and LRP (A) and the ER chaperone genes BiP and HERP (B) were analyzed by qRT-PCR of RNA isolated after each treatment. The atf6—/— MEFs contained a transfected N-terminal domain of ATF6 fused to a labile mutant DHFR. This ATF6 N-terminal-mutant DHFR fusion protein was inactive, unless stabilized by addition of trimethoprim (TMP) where it then accumulated, and activated the ATF6 target genes, which served as a positive control (lane 5). Data represent the mean ± SD of three independent experiments, each with at least 50 cells per sample per point. (C and D) Expression of the ATF6 targets, LRP1 and ACOX1, involved in lipid biogenesis and metabolism (C) are effectively activated by DHS/DHC, but the ATF6 target ER chaperone genes, BiP and GERPUD (D), are significantly decreased. Data were obtained by qRT-PCR analyses of transcripts induced during the time-course treatment of HEK203 cells with Tg, DHC, or DHS for up to 8 hr. Each experiment was performed at least three independent times. **p < 0.01 and *p < 0.05 using unpaired two-tailed t tests comparing indicated values; ns indicates no significant difference. (E and F) DHS preferentially induced ATF6 target lipid biosynthetic and function genes (E) more than ATF6 target ER chaperone genes (F). Heatmap of transcription profiles of either DHS- or Tg-induced ATF6. The same RNA used for qRT-PCR experiments was used for microarray experiments. Transcription levels of either DHS- or Tg-treated HEK293 cells are compared with those of untreated cells. Lists of ATF6 target lipid biosynthetic and function genes and ER chaperone genes are taken from previous studies (Shoulders et al., 2013). (G) ATF6 can be activated independently by ER proteotoxic stress via its ER luminal domain and by DHS/DHC-induced lipotoxic stress through the unique motif within the ATF6 transmembrane domain, leading to transcription activation of lipid genes.
However, in HEK293 cells which are wild-type for ATF6, both DHS and DHC upregulated a number of previously identified ATF6 target genes, as measured by qRT-PCR. Strikingly, DHS and DHC did not induce ER chaperone genes, such as BiP and HerpUD, anywhere near as efficiently as did the proteotoxic stressor Tg (figure 7D and S7B). However, DHS and DHC induced the ATF6 target genes involved in lipid biogenesis and metabolism, such as LRP1 and ACOX1 (Figure 7C), and six other genes in the same category (Figure S7A), in large part as efficiently as Tg induced them. This unique DHS/DHC pattern of transcriptional induction was confirmed for the 8-hr DHS time point at the genome scale by microarray analysis: for Tg activation, transcription of both lipid-related genes and ER chaperone genes was high, while for DHS activation, induced transcription of lipid-related genes was high but that of ER chaperones was significantly lower than seen with Tg (figure 7E, 7F, and S7C).
DHC caused no increase in the transcription of ERdj4 and Asn-S, which are IRE1/XBP1- and PERK-specific target genes, respectively (Figure S7D), and no increase of HMG-CoA and INSIG, which are SREBP target genes (Figure S7E) (Gjymishka et al., 2009; Shoulders et al., 2013). Taken together, our results indicate that DHS/DHC activation of ATF6, which is initiated in the ER by a distinct mechanism, i.e., binding of DHS/DHC to the ATF6 transmembrane domain, culminates in the nucleus by promoting a distinct transcriptional program.
DISCUSSION
The UPR, in addition to maintaining protein homeostasis, is known to regulate cellular lipids (Fu et al., 2011; Lee et al., 2008; Volmer and Ron, 2015). Mechanistically, how UPR components mediate this lipid regulation remains largely unexplored. Here our discoveries indicate that ATF6 responds not only to protetoxic stressors but also to the sphingolipids DHS and DHC. Mutations in the ATF6 transmembrane domain that block this activation have been identified and used for functional analyses. Moreover, DHS shows direct binding to this conserved transmembrane domain.
The mechanism of DHS/DHC-dependent activation of ATF6, while retaining some similarities, differs significantly from proteotoxic stress activation of ATF6. One of the key differences involves the ATF6 transcription targets. ER proteotoxic-induced ATF6 results in the effective induction of ATF6 target lipid synthetic and metabolic genes, as well as ER chaperone and protein-folding genes. However, DHS and DHC, while inducing strong transcription of the lipid-related ATF6 target genes, cause proportionately less transcription of the ATF6 target ER chaperones and protein-folding genes. These different preferences in ATF6 transcription target genes suggest the interesting possibility that ATF6 somehow retains a molecular ‘‘memory’’ of how it was initially activated, i.e., activation either by ER proteotoxic stress sensed via the luminal domain or by DHS/DHC-induced activation sensed via the transmembrane domain. At the molecular level, such ‘‘memory’’ might result from different post-translational modification of ATF6 or from conformational changes induced by DHS/DHC binding to ATF6 that are maintained after cleavage of the transmembrane domain. Understanding how activation by DHS/DHC or ER proteotoxic stress leads to different transcriptional responses will require a deeper analysis of the time course of differential gene expression, as well as of the ATF6 transcription factor generated by the two different upstream signals.
Recent studies have reported that IRE1 or PERK respond to more global structural changes in the ER membrane, such as an increase in ER membrane fluidity or lipid packing, following free fatty acid addition. Such global change is proposed to be the driving force for the activation of IRE1 and PERK (Kono et al., 2017; Halbleib et al., 2017). In those cases, instead of specific amino acid sequences being required in the transmembrane domains, replacement of the entire ER transmembrane domain of IRE1 or PERK with a stretch of leucines still retained their ability to become activated by an increase in free fatty acids. Any cell would presumably need to integrate different types of lipid stresses to tailor the correct UPR response. We conclude that ATF6 is activated by specific sphingolipid inducers, DHS and DHC, while IRE1 and PERK are tasked with sensing more general membrane characteristics.
Future questions include what are the physiological condition(s) whereby ATF6 is activated through its transmembrane domain by DHS/DHC? Furthermore, what are the functional consequences of activating ATF6 by DHS/DHC? For example, does such activation contribute ultimately to ER expansion? Liver cells challenged by hydrophobic toxic chemicals, such as phenobarbital, undergo increased expression of the detoxifying membrane protein cytochrome P450, which requires membrane expansion to house the increased enzymes (Staubli et al., 1969; Kemp et al., 2005). Elevated expression of the cytochrome b(5)tail has also been shown previously to expand the amount of ER membrane and is a more tractable experimental system for the study of ER expansion (Maiuolo et al., 2011). We found that within the first 24 hr of b(5)tail overexpression, DHS/DHC-inducible ATF6 (Y567N-ATF6) was activated while the ER proteotoxic-inducible ATF6 (I388F-ATF6) was not. This strongly suggests that activation of ATF6 by DHS/DHC could play an important role in ER membrane expansion.
Massive ER membrane expansion also occurs during the terminal differentiation of B lymphocytes when they are stimulated by antigen to become antibody-secreting plasma cells (van Anken et al., 2003; Brewer and Hendershot, 2005). UPR components, including ATF6, have indeed been shown to be activated during B cell differentiation (Iwakoshi et al., 2003; Brunsing et al., 2008), but which mode of ATF6 activation is used remains unknown. It will be interesting to test whether DHS/DHC-dependent ATF6 activation occurs during B cell differentiation and ER membrane expansion. The genome-wide transcription results described here suggest that DHS/DHC-induced ATF6 activation could ultimately contribute to the increase in phospholipids needed for expanding the ER membrane during B cell differentiation. For future progress, this described experimental system, which allows us to dissect two different types of ATF6 activation, may hold the key to understanding how UPR activation in some physiological settings increases both ER functional capacity and the amount of ER membrane, as in B cells, while in other cases leads solely to increased ER functional capacity. Additionally, the described ATF6 reporters that can differentiate between the two modes of ATF6 activation should contribute to allowing us to evaluate the extent of ER proteotoxic-induced ATF6 and DHS/DHC-induced ATF6 in normal and diseased states.
A key finding of our study of ATF6 was the discovery of the VXXFIXXNY sequence motif within its transmembrane domain, which is conserved from zebrafish to humans, through resemblance to a motif VXXTLXXIY found in the COP I vesicle component p24 (Contreras et al., 2012). The p24 motif is known to bind to sphingomyelin (SM), a downstream lipid in the sphingolipid biosynthetic pathway (Figure 1). A computer-simulated structure of the human ATF6 transmembrane domain sequence predicts a sphingolipid binding surface similar to that of p24 (figure 5B and 5C). The aforementioned DHS/DHC-binding motif of ATF6 is not found in IRE1 or PERK (Figure 5B). In addition, other ER membrane transcription factors, such as SREBP and the β-isoform of ATF6, do not have this motif and are not activated by DHS or DHC (Figure 5B). Mutation of an isoleucine in the fifth position of the motif VXXF(L/I)XXNY, when changed to phenylalanine (I388F), substantially diminishes the ability of ATF6 to respond to DHS or DHC. Similarly, a change in the ninth position from tyrosine to cystine also diminishes the ability of ATF6 to be activated by DHS or DHC (Figure 5E). Thus we conclude that, uniquely, ATF6 has a transmembrane domain that can respond to the specific lipids DHS and DHC to initiate this key arm of the mammalian UPR response. Mutations in this ATF6 motif promise to be valuable for probing for other physiological roles for DHS/DHC activation.
ATF6 in Human Disease
Changes in ATF6 expression and activity have been described in multiple human disorders, including the eye disease achromatopsia, myocardial infarction, stroke, amyloidosis, and a number of cancers (Ariyasu et al., 2017; Chiang et al., 2012; Manie et al., 2014; Plate et al., 2016; Urra et al., 2016; Zhou and Tabas, 2013). The finding that atf6—/— mice have a higher incidence of neuronal cell death upon brain ischemia compared with wild-type mice argues for a protective effect of ATF6 (Yoshikawa et al., 2015). Indeed, ectopic overexpression of ATF6 in wild-type mice has been shown to protect heart tissue from ischemic stress (Doroudgar et al., 2009). Our finding that ATF6 is activated by DHS and DHC may provide a basis for the development of new therapeutic drugs that combat human diseases by activating ATF6 via its transmembrane domain. Broadly speaking, patients carrying mutations that render ATF6 insensitive or less sensitive to canonical proteotoxic UPR triggers, such as in achromatopsia or heart disease, might benefit from drugs such as FEN that induce ATF6 activation via its transmembrane domain. Our observations suggest that, in general, drugs that induce DHS or DHC may be a viable alternative for these and related diseases.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-GFP antibody 11–814-460–001 | Roche | 11–814-460–001 |
| Mouse anti-GM130 antibody | BD Biosciences | 610823 |
| Mouse Anti-phospho eIF2α antibody | Cell Signaling Technology | 9721S |
| Anti-total eIF2α antibody | Cell Signaling Technology | 9722S |
| Rabbit Anti-GRP78 antibody | Santa Cruz Biotech | sc-13968 |
| Mouse Anti-VSVG antibody | Kerafast | Clone IE9F9 |
| Rabbit Anti-Sec31A antibody | Fisher Scientific | BDB612350 |
| Rabbit Anti-Sec16 antibody | BETHYL | A300648A |
| Goat-anti-RABBIT IgG H +L | Life Technologies | A11036 |
| Goat-anti-MOUSE IgG H +L | Life Technologies | A11031 |
| Anti-ATF6 antibody | Cosmo Bio | 73–505EX |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| Dihydrosphingosine (DHS) | Sigma Aldrich | D3314 |
| Dihydroceramide (DHC) | Sigma Aldrich | C7980 |
| Thapsigargin | Calbiochem | 586005 |
| DTT | Fisher Scientific | 3483–12-3 |
| Tunicamycin (Tm) | Calbiochem | 654330 |
| ceramides | Enzo Life Sciences | 89164–592 |
| sphingosine | Sigma Aldrich | S7049 |
| sphingomyelin | Avanti Polar Lipids | 860062P |
| palmitoyl-CoA | Sigma Aldrich | P9716 |
| 4-phenylbutyric acid (4-PBA) | Fisher Scientific | P21005 |
| Aprotinin | Fisher Scientific | 9078–70-1 |
| Fenretinide [FEN] | Torics | 65646–68-6 |
| 1,2-bis(2-aminophenoxy)ethane-N,N,N0,N0- tetraacetic acid (BAPTA) | Sigma Aldrich | A1076 |
| phenobarbital (PB) | Sigma Aldrich | P1578 |
| PMSF | AMRESCO | M145 |
| MG132 | Fisher Scientific | 508338 |
| Myriocin | Sigma Aldrich | M1177 |
| Pepstatin | Bachem | N1125.0025BA |
| FLI-06 | Sigma | SML0975–5MG |
| CeapinA7 | (Gallagher and Walter, 2016) | N/A |
| 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) | Sigma | 101500 |
| ER tracker (DiOC6(3)) | Enzo Life Sciences | 52303 |
| para-formaldehyde (PFA) | Sigma | P6148 |
| NBD-DHC | Cayman | 10007957 |
| NBD-DHS | Avanti Polar Lipids | 810206 |
| DAPI | Pierce | D1306 |
| DMEM | Cellgro | 4500–304 |
| FBS | Cellgro | 10437028 |
| Penicillin/ streptomycin | Mediatech | 45000–650 |
| Lipofectamine 2000 | Invtrogen | 11668027 |
| Effectene | Qiagen | 301425 |
| C2-DHC (≥98% purity) | Sigma Aldrich | C7980 |
| C16-DHC | Santa Cruz Biotechnology | 5966–29-0 |
| Sphingomyelin | Avanti Polar Lipids | 8600062P |
| NP-40 | Nonidet | P-872 |
| BSA | Sigma Aldrich | A2153 |
| Mounting media (Aqua-Mount) | Lerner Laboratories | 13800 |
| HEPES | Sigma Aldrich | H0527 |
| TritonX-100 | Sigma Aldrich | T8787 |
| sodium deoxycholate | Fisher Scientific | H0527 |
| Maxima Reverse Transcriptase | Thermo | FEREP0752 |
| TRIzol | Invtrogen | 15596026 |
| SYBR Green PCR Master Mix | Applied Biosystems | 4309155 |
| Fos-Choline 16 | Fisher Scientific | 5010 |
| GFP-nAb beads | Allele | ABP-NABGFPA050 |
| 3H DHS, 1mCi/ml | American Radiolabeled Chemicals | ART0460 |
| 3H L-serine, 30.0 Ci/mmol | Perkin Elmer | NET248001 |
| GeneChip Human Transcriptome Array 2.0 | Affymetrix | 902914 |
| PMSF | AMRESCO | M145 |
| Trimethoprim | Sigma Aldrich | 92131 |
|
Critical Commercial Assays | ||
| BCA Assays | Thermo | 23225 |
|
Deposited Data | ||
| Microarray data | N/A | N/A |
|
Experimental Models: Cell Lines | ||
| MCF-7 | ATCC | HTB22 |
| HeLa | Niwa et al., 1999 | N/A |
| HEK293 | Lin et al., 2007 | N/A |
| ATF6+/+ and atf6−/− | Wu et al., 2007 | N/A |
| HeLa TetOff cytochrome (b)5 | Maiuolo et al., 2011 | N/A |
|
Oligonucleotides | ||
| Oligo (dT)12–18 for Reverse transcription | Thermo Fisher | 18418012 |
| Oligonucleotides for qPCR | Table S1 | N/A |
|
Recombinant DNA | ||
| VSVG-GFP | Dippold et al., 2009 | N/A |
| WT ATF6-GFP | Shen et al., 2002 | N/A |
| Y567N ATF6-GFP | this study | N/A |
| L385I-ATF6-GFP | this study | N/A |
| A3896V-ATF6-GFP | this study | N/A |
| F387A-ATF6-GFP | this study | N/A |
| F387Y-ATF6-GFP | this study | N/A |
| I388F ATF6-GFP | this study | N/A |
| I389L-ATF6-GFP | this study | N/A |
| L390I-ATF6-GFP | this study | N/A |
| N391F-ATF6-GFP | this study | N/A |
| Y392C-ATF6-GFP | this study | N/A |
| GOLPH3-mcherry | Dippold et al., 2009 | N/A |
|
Software and Algorithms | ||
| Zen software | Carl Zeiss | N/A |
|
Other | ||
| Observer Z1 microscope | Carl Zeiss | N/A |
| 100X or 63X 1.4 NA objectives | Carl Zeiss | N/A |
| Typhoon 9400 | GE Healthcare | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maho Niwa (niwa@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T, HeLa, ATF6+/+ MEFs, atf6−/−MEFs and MCF-7 cells used in this study were not authenticated. Cells were cultured in DMEM medium (Cellgro) supplemented with 10% fetal calf serum (Gibco), 100 U/ml penicillin, and 100 µg/ml streptomycin (both Mediatech) at 37○C in 5% CO2.
METHOD DETAILS
Mass Spectrometry (MS) Analysis
Analysis of sphingolipid changes during ER stress. HEK293 cells (2 × 106 cells per point) were untreated (0 min) or treated with 200 nM thapsigargin (Tg) for 30 or 90 min. XBP1 splicing assays showed that ER stress was successfully induced (Figures S1A and S1B). At each time point, cells were harvested and a sample was counted to ensure uniform cell numbers in each sample. Lipid analyses were conducted using previously described methods (Benjamin et al., 2013; 2015 (PMID 23980144; 25871544)). Briefly, 2 million cells were plated overnight, serum starved for 2 hours prior to harvesting, after which cells were washed twice with PBS, harvested by scraping, and flash frozen. For lipidomic analyses, flash frozen cell pellets were extracted in 4 mL of 2:1:1 chloroform/methanol/PBS with internal standards dodecylglycerol (10 nmoles) and pentadecanoic acid (10 nmoles), after which organic and aqueous layers were separated by centrifugation, and organic layer was extracted. Aqueous layer was acidified with 0.1% formic acid followed by re-extraction with 2 mL chloroform. The second organic layer was combined with the first extract and dried under nitrogen, after which lipids were resuspended in chloroform, and an aliquot was then analyzed by single-reaction monitoring (SRM)-based LC-MS/MS. Relative levels of lipids were quantified by integrating the area under the curve for each lipid, normalizing to internal standard values, and then normalizing to the average values of the control groups.
Cell Treatments
HEK293, HeLa, ATF6+/+ MEFs, atf6−/−MEFs, and MCF-7 cells were placed in 6-well plates containing poly-L-lysine–treated cover slips and grown to 70% confluency. Cells were transfected with 800 ng of plasmid using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. For drug treatments, Tg (Calbiochem), DTT (Fisher), DHC (Sigma, ≥98% purity), or DHS (Sigma, ≥98% purity) at final concentrations of 200 nM, 1 mM, 50 µM, or 50 µM, respectively (unless otherwise indicated) was added to the media. C2- DHC (Sigma, ≥98% purity), C16- DHC (Santa Cruz Biotechnology), C2-ceramide (Enzo, ≥98% purity), C16-ceramide (Enzo, ≥98% purity), sphingosine (Sigma, ≥98% purity), palmitic acid (Sigma, ≥99% purity), and cholesterol (Sigma, ≥99% purity) were added at 50 µM, unless otherwise noted.
For inhibitor treatments, cells were pre-treated with 10 µM dihydropyridine (FLI-06; Sigma), an inhibitor of ER to Golgi transport (Kramer et al., 2013), 300 µM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF; Sigma), 10 mM 4-phenyl butyric acid (4-PBA; Sigma), 10 or 20 µM 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA; Sigma), 10 mM Ceapin A7 (Gallagher et al., 2016), or 10 µM Fenretinide (Torics) for 60 min, unless otherwise indicated, followed by addition of Tg, DHC, or DHS. For Myriocin experiments, cells were pre-incubated with 25 µM Myriocin for 60 min, followed by treatment with 200 nM Tg; samples were collected during the time course for up to 90 min.
Immunofluorescence Microscopy
For immunofluorescence microscopy, cells were treated and fixed with 4% para-formaldehyde (PFA; Sigma P6148) for 5 min, permeablized with 0.2% NP-40 in PBS for 5 min, and blocked in 5% BSA in PBS for at least 1 h. The primary anti-GM130 antibody (BD Biosciences), followed by secondary antibody (Alexa Fluor 568, Life Technologies), was used for visualization of the Golgi. For the experiments shown in Figures S2G and S2H, we transfected a GOLPH3-mcherry plasmid to visualize the Golgi (Dippold et al., 2009). For visualization of the nucleus, 1µg/ml DAPI (Pierce) was added to the mounting media (Aqua-Mount; Lerner Laboratories). Coverslips were mounted onto slides and the cells were imaged with an Observer Z1 microscope (Carl Zeiss MicroImaging) with either a 100X or a 633 1.4 NA objective. Images were acquired with a monochrome digital camera (Axiocam; Carl Zeiss MicroImaging) and analyzed using Zen software (Carl Zeiss Microimaging).
ATF6-GFP Co-Localization with COPII Vesicles
To visualize co-localization of ATF6-GFP with Sec31A, a COPII component, and/or Sec16, an ER exit site (ERES)-localized protein, HEK293 cells transfected with ATF6-GFP were treated with DMSO, Tg or DHC for 60 min. After treatment, cells were fixed with icecold methanol and incubated at −20○C for 5 min. Following incubation with 2% goat serum for 1h, antibody against either anti-Sec31A (1:1000) or anti-Sec 16 (1:500) was incubated overnight at 4○C, before secondary antibody (Alexa Fluor 568, Life Technologies) was added for 1 h. Cover slips were mounted onto slides and the cells were imaged with an Olympus IX70 deconvolution scope with a 100X objective, and images were deconvolved using softWoRx 6.5.2. ATF6-GFP co-localized with Sec31A foci, representing COPII vesicles, was quantitated. Similarly, COPII vesicles containing ATF6-GFP co-localized at ER exit sites was determined by counting the discrete foci containing all three signals of ATF6-GFP, Sec31A and Sec16. At least, 30 cells were analyzed per experiment and the experiment was repeated three independent times. Similarly, the effect of Ceapin A7 on ATF6-GFP co-localized with COPII vesicles or with COPII vesicles at ERES was quantitated.
Immunoprecipitation of BiP-Associated ATF6
Immunoprecipitation (IP) and western blotting of BiP and ATF6 was performed as described in (Shen et al., 2002). Briefly, cells were treated as indicated and lysed in IP Buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, and protease inhibitor cocktail [PMSF, aprotinin, leupeptin, and pepstatin]). ATF6-GFP was immunoprecipitated using Anti-GFP antibody (Sigma G1544), subjected to 8% SDS-PAGE and transferred to nitrocellulose membrane. The levels of BiP associated with ATF6 were analyzed by blotting with anti-GRP78 antibody (Santa Cruz Biotech sc-13968), as described in (Nadanaka et al., 2007). Blots were imaged on a Typhoon 9400 (GE Healthcare) fluorescence imager and BiP-associated ATF6 was quantitated using ImageQuant Software (GE Healthcare).
Examination of ATF6 Oligomerization Status
For westerns with anti-ATF6 antibody (Cosmo Bio 73–505EX), a similar protocol was used except cells were lysed in ATF6 extraction buffer (50 mM Tris-HCl, pH 6.8, containing 2% SDS, 10% glycerol, 10 µM proteasome inhibitor (MG-132), 1 mM PMSF, 100mM DTT, 100 U/ml aprotinin, 1.4 µg/ml pepstatin, and 1 µg/ml leupeptin), boiled at 95○C for 5 min and vortexed for 1 min (Nadanaka et al., 2007). For analyses of ATF6 oligomerization states, cell lysates were prepared in a similar protocol except cell lysis was carried out in a non-reducing extraction buffer (50 mM Tris-HCl, pH 6.8, containing 2% SDS, 10% glycerol, 10 µM proteasome inhibitor (MG-132), 1 mM PMSF, 100 U/ml aprotinin, 1.4 µg/ml pepstatin, and 1 µg/ml leupeptin) as described in (Nadanaka et al., 2007). To maintain non-reducing conditions for SDS-PAGE, samples were prepared in SDS Loading Buffer without reducing agent (50mM Tris pH 6.8, 2% SDS, 10% glycerol, 0.1% Bromophenol Blue). Both reduced and non-reduced samples were visualized using a 7% SDS-PAGE gel.
Examination of ATF6 Glycosylation States
For EndoH treatment, cells were lysed in RIPA buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate, 0.1% SDS, 10 mM NaF, 1 mM sodium orthovanadate, 1 mM PMSF, 100 U/ml aprotinin, 1.4 µg/ml pepstatin, and 1 µg/ml leupeptin). Some lysates were treated with 10,000 unit EndoH (New England Biolabs) for 1hr according to manufacturer’s instructions. Samples were run on a 7% SDS PAGE gel and western blots were performed as described above.
Detection of Phosphorylated eIF2α
The activation status of PERK was monitored by the appearance of phosphorylated eIF2α. For phospho-eIF2α blotting, cells were lysed in RIPA buffer as described above for testing ATF6 glycosylation states. Western membranes were probed with antibodies against phospho-eIF2α or total eIF2α (Cell Signaling Technology), followed by incubation with an HRP-conjugated anti-rabbit secondary antibody (GE Healthcare). Blots were imaged on a Typhoon 9400 (GE Healthcare) fluorescence imager using ImageQuant Software (GE Healthcare).
XBP1 mRNA Splicing Assay
Activation of IRE1 was tested by the presence of the spliced form of XBP1 mRNA. Determination of spliced XBP1 mRNA was performed as described in detail (Lin et al., 2007). Briefly, cDNA was amplified by PCR using XBP1-specific primers: (5’-TTACGGGAGAA AACTCACGGC-3’ and 5’-GGGTCCAACTTGTCCAGAATGC-3’), which yielded a 289 bp unspliced XBP1 fragment and a 263 bp spliced XBP1 fragment. Fragments were resolved on a 2.5% agarose gel and signals were quantified using a Typhoon fluorescence scanner (GE Healthcare).
RNA Isolation and qRT/PCR
RNA was prepared using TRIzol (Invitrogen) according to the manufacturer’s instructions. Samples of 1 µg total RNA were reverse transcribed using Maxima RT (Thermo), according to the manufacturer’s instructions. qPCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) and 400 nM of each primer in a total reaction volume of 25 µl. Melting curves were generated after each run to confirm amplification of a single product. RT/qPCR primers of ER chaperone genes were as described in (Shoulders et al., 2013; Wu et al., 2013) Primer sequences used here for PCR are included in Table S1.
Alignment and Modeling
Protein sequence alignment was performed using Clustal Omega. For modeling of p24 and ATF6 transmembrane domains, we used Phyre2 (Kelley et al., 2015). Phyre2 uses known structural motifs to generate predicted structures. At this point, X-ray crystal structure data are not available for either p24 or ATF6 transmembrane domains. Thus, we modeled both transmembrane domains using Phyre2 in reference to a single-pass transmembrane domain of VAMP2 (vesicle associated membrane protein 2), whose crystal structure has been solved (Kummel et al., 2011). Specifically, the transmembrane portion of the VAMP2 sequence (residues 91–116) was replaced with VVCVMIVLAFIILNYGPMSMLEQ for ATF6 or VVLWSFFEALVLVAMTLGQIYY for p24 for Phyre2 analyses. The generated p24 structure is in agreement with the simulated p24 structure previously generated by the BALL program (Contreras et al., 2012). The images shown in figure 5C and 5D were created with Chimera (UCSF) (Pettersen et al., 2004).
Lipid Binding Assay
Lipid binding assays were modified from those previously established (Radhakrishnan et al., 2004). HEK293 cells were transfected with either ATF6-GFP or cytosolic GFP for 24h. To minimize the amount of endogenous DHS, we treated cells with 25µM Myriocin for 1hr to block de novo synthesis of DHS. Furthermore, the buffer used to generate microsomes contained 25µM Myriocin. Cells were lysed in FC Buffer (50mM Tris pH 7.4, 150mM NaCl, 1mM EDTA, 5% Fos-Choline 16, protease inhibitors, 25µM Myriocin). BCA (Thermo) assays were performed to determine protein concentration. 1000 µg of protein lysate was used to immunoprecipitate ATF6-GFP using 10µl GFP-nAb beads (Allele). The purity of the protein was checked by silver staining of SDS-PAGE. Before immunoprecipitation, non-specific binding of GFP-nAb beads was blocked by incubation with 50µM cold DHS for 30 min at 4○C and washed 3 times with FC Buffer containing 0.1% Fos-Choline as described (Radhakrishnan et al., 2004). Immunoprecipitated protein was washed 3 times with FC Buffer with 0.1% Fos-Choline and incubated with 0, 10, 50, 75, 100, or 150 pmol of 3H-DHS (American Radiolabeled Chemicals, 1mCi/ml), as indicated, in 100ml FC Buffer containing 0.1% Fos-Choline. 3H-DHS was allowed to bind to ATF6–GFP bound to anti-GFP-nAB beads for 4 hrs at room temperature to reach equilibrium conditions. After 4 hrs, samples were then washed 4 times in 1ml FC Buffer with 0.1% Fos-Choline. The ATF6-Beads were eluted with 100 µl SDS Loading Buffer (50 mM Tris-HCl, pH 6.8, containing 2% SDS, 10% glycerol) to release ATF6 and lipids from the beads. The levels of incorporated 3H were measured using a scintillation counter to measure radioactive lipid. Using standard curves of 3H, our scintillation counter was performing at 10% efficiency. For competition assays, 10nmol of cold DHS or cold cholesterol was added to the beads for 15 min before addition of 3H-DHS. To generate the lipid binding graphs (figure 5F and 5G), the amount of 3H-DHS bound in pmol to WT-ATF6 or I388F-ATF6 protein (y-axis) was graphed versus added 3H-DHS in pmol (x-axis), similarly to previous lipid binding studies (Radhakrishnan et al., 2004).
VSVG Folding Experiment
HEK293 cells expressing VSVG-GFP (Dippold et al., 2009; Hirschberg et al., 1998) were grown on poly-L-lysine cover slips. For this VSVG-GFP reporter, a vesicular stomatitis virus glycoprotein (VSVG) fragment was fused with GFP, and shown to be a ‘folding status’ reporter. The folded VSVG can be visualized by staining with a specific anti-VSVG monoclonal antibody (Kerafast, clone IE9F9) (Nehls et al., 2000). In contrast, the folding state of the GFP portion of the reporter is not affected by the folding status of the VSVG fragment and thus, remains detectable regardless of folding status of the VSVG domain. Using this reporter, we tested whether DHC addition negatively impacts the folding of VSVG.
VSVG-GFP is normally localized throughout the secretory pathway compartment including cell surface. To effectively assess the folding status of all the VSVG-GFP, previous studies have used Brefeldin A to keep all the cellular VSVG-GFP in the ER. In this study, we pre-treated cells with 10 µM FLI-06 for 2 hrs to block the protein traffic from the ER to the Golgi, which facilitated visualization of all VSVG-GFP regardless of its folding status. Upon pre-treatment of HEK293 cells with 10 µM FLI-06 (2 hrs), the folding states of VSVG-GFP were monitored during a time course up to 3 hrs following treatment with either 1 mM DTT or 50 µM DHC. Cells were then fixed with 4% PFA for 5 min, permeablized with 0.2% NP-40 in PBS for 5 min, and blocked in 5% BSA in PBS for at least 1 h. After staining with the anti-VSVG monoclonal antibody that only binds to folded VSVG, cells were imaged with an Observer Z1 microscope (Carl Zeiss MicroImaging) with either a 100X or a 633 1.4 NA objective. Images were acquired with a monochrome digital camera (Axiocam; Carl Zeiss MicroImaging) and analyzed using Zen software (Carl Zeiss Microimaging).
In Vitro Serine Palmitoyl-Transferase Assay
In order to corroborate our mass spec data showing increased levels of DHC or DHS during DTT or Tg treatment of HEK293 cells, we performed a modified in vitro SPT assay to measure activities of sphingolipid biosynthetic pathway enzymes that are localized in the ER in response to ER stress. The SPT assay was slightly modified from the well-established assay (Rutti et al., 2009). Briefly, HEK293 cells were left untreated or pre-treated with or without 25mM Myriocin for 60 min, then either left untreated or treated with 200nM Tg for 90 min. Microsomes were prepared from homogenization of these cells re-suspended in 500 µl Microsome buffer (Hepes pH8.0, 1mM EDTA, and protease inhibitors). Homogenization of cells was performed by passage through a 27 gauge needle 15 times and centrifugation at 500xg for 3 min at 4○C. Microsomes were collected by centrifugation at 100,000 g for 30 min with resuspension of pellet in Microsome buffer. BCA (Thermo) assays were performed to determine protein concentration. The enzyme activity assays were performed using 400 µg of microsome protein in 200 µl volume of 1X Assay Buffer (20X Assay Buffer: 10 mM L-serine, 400 µM pyroxidal 5’ phosphate, 1 mM palmitoyl Co-A) and incubated with 3H-Serine, a starting component of the sphingolipid biosynthetic pathway (Figure 1A). To generate activity curves, 0, 1, 2, 5, and 10 mM 3H-Serine (Perkin Elmer, 30.0 Ci/mmol) was added to the reactions. At each time point, lipids were extracted and radiolabeled lipid production was determined by scintillation counting. Lipids were extracted using the chloroform/methanol procedure by adding 0.5 ml methanol/KOH:CHCl3, 0.5 ml CHCl3, and 0.5 ml alkaline dH2O, and 100 µl 2N NH4OH. The lower phase was washed in 900 ml alkaline dH2O, and the lipid phase was measured by scintillation counting. For each condition and concentration of 3H-Serine, a curve was plotted of pmol radiolabeled lipid generated versus time in minutes. The initial rate of production was then determined by measuring the linear part of the graph using linear regression. To calculate ‘Activity’, this slope was divided by the amount of protein (0.4 µg) to get units of pmol/min/mg. Activity was graphed with [3H-Serine] in mM to generate the SPT activity assay curves (figure 1C and S1E).
Cytochrome b(5) Tail HeLa TetOff Cells
To demonstrate the effect of ER expansion on ATF6 activation, we used HeLa TetOff cells expressing the C-terminal tail of cytochrome b(5) as described in (Maiuolo et al., 2011). Removal of Dox from these cells results in expansion of the ER (Maiuolo et al., 2011). b(5)tail expressing HeLa cells were grown on poly-L-Lysine treated slides transfected for 24 hrs with either WT-ATF6-Tomato, I388F-ATF6-Tomato, or Y567N-ATF6-Tomato using Effectene (Qiagen). Dox was then removed to induce ER expansion. At 0, 24h, 38h, 42h, 48h, after removal of Dox, samples were taken and fixed with 4% PFA for 5 min. For visualization of the nucleus, 1µg/ml DAPI (Pierce) was added to the mounting media (Aqua-Mount; Lerner Laboratories). Cover slips were mounted onto slides and imaged as described above for immunofluorescence. For each cell, the total fluorescence signal of ATF6 (red channel for Tomato) is measured. In each cell, if 35% or more of the fluorescence co-localized with the DAPI channel (marking the nucleus), that cell was considered to have nuclear localization of ATF6. The ATF6 activation status of at least 50 cells for each time point/condition was counted and ‘% of cells’ with nuclear localized ATF6 was calculated and graphed over time.
Microarray Analysis
HEK293 cells were treated with either 200 nM Tg or 50 µM DHS for 8 hrs. RNA was extracted using Trizol (Invitrogen) according to manufacturer’s instructions and quantified using NanoDrop (Invitrogen). Samples were analyzed using GeneChip Human Transcriptome Array 2.0 (Affymetrix).
Electron Microscopy and Image Analysis
Atf6−/− MEFs were transfected with either WT-ATF6 or I388F-ATF6 for 24h and either left untreated or treated with 1 mM phenobarbital (Sigma P1578) for 16 hrs. Cells were processed for Transmission EM using embedding for routine morphology EM by the UCSD Cellular and Molecular Medicine Electron Microscope Facility. Images were taken with a FEI Tecnai Spirit G2 BioTWIN Transmission Electron Microscope equipped with an Eagle 4k (16 megapixel) camera. Images were analyzed using ImageJ, and total ER in the cell was measured by tracing the ER using the Freehand Line tool and measuring the length of the ER in microns with the ‘Measure’ function using the scale bar as a reference. The typical width of ER was measured to be 0.1 micron (data not shown), so we defined ‘distended ER’ to be areas of ER that were greater than 0.3 micron in width. For each cell, the length of ‘distended ER’ was measured using ImageJ. To calculate ‘% Distended ER’, we determined the microns of ‘distended ER’ / total ER in microns for each cell. We graphed averages and standard deviations of 10 cells for each condition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Imaging Quantification Parameters
ATF6-GFP was visualized in the green channel (525nm), Golgi (anti-GM130) in the red channel (605nm), and nuclei (DAPI) in the blue channel (455nm). Images were taken in 3D sections using 0.12 micron stacks for each channel, followed by deconvolution using Zen Software (Zeiss) of the Adjustable Constrained Iterative function with an adjusted value of 7. The areas of the Golgi, and the nucleus were defined using GM130 and DAPI, respectively. The ATF6-GFP fluorescence signal in total and each area (ER, Golgi, Nucleus) was quantitated using the Zen software Measure function (Carl Zeiss Microimaging). ATF6-GFP localized to the nucleus, the Golgi, and the ER for each cell, was determined as a percentage value over total signal and based on the following criteria, a cell was regarded to have specific localization of ATF6. If a specific cell had 35% or more nuclear ATF6-GFP, we defined such cell as a cell with the nuclear ATF6. Upon defining the specific ATF6-GFP localization for each cell, ATF6 activation status of at least 50 cells for each time point/condition were counted and ‘% ER’ was calculated by # cells with ER localization divided by the total # of cells; ‘% Golgi’ was calculated by # cells with Golgi localization divided by the total # of cells; and ‘% Nucleus’ was calculated by # cells with nuclear localization divided by the total # of cells. All quantifications were performed on minimum of 50 cells and three independent experimental replicates unless otherwise stated in specific figure legends.
To assess the activation rate of ATF6, the ‘% Nucleus’ was determined as described above and graphed on the y-axis of a linear graph. Time in min was graphed on the x-axis of the same graph and Linear regression was used to calculate the slope (in %/min), which was the calculated ‘ATF6 activation rate’.
Mass Spec Data Analyses
Lipid profiles were analyzed and quantified by LC/MS as described in (Benjamin et al., 2013, 2015). As noted above in the Method Details section on Mass Spec Analysis, relative levels of lipids were quantified by integrating the area under the curve for each lipid, normalizing to internal standard values, and then normalizing to the average values of the control groups.
Microarray Data Analysis
Data was analyzed using Affymetrix Transcriptome Analysis Console (TAC) and GeneChip Expression Console Software (Affymetrix). Two-fold up regulated and down regulated genes were selected for further analysis. Heat maps were generated using Multiple Experiment Viewer (MeV) and pathway annotation of genes was performed with DAVID software from NIH (Miyazaki et al., 2015). The same RNA samples were used for qPCR of selected genes to confirm the outcomes of the microarray data. Primers used for qRT/PCR are listed in Table 1. GO analyses and visualization files were generated by HOMER (http://biowhat.ucsd.edu/homer) as described previously (Miyazaki et al., 2015).
Supplementary Material
Highlights.
ATF6 is activated not only by proteotoxic stress but also by sphingolipids
Proteotoxic stress and sphingolipids activate ATF6 via separate domains
Mutations in a transmembrane motif in ATF6 block lipid but not proteotoxic activation
Sphingolipids DHS and DHC upregulate ER lipid biosynthetic genes over chaperone genes
ACKNOWLEDGMENTS
We thank Dr. Marilyn Farquhar for help with EM analysis, Dr. Nica Borgese for cytochrome (b)5 HeLa cells, Dr. Seth Field for VSVG-GFP and GOLPH3-mCherry plasmids, Dr. Peter Walter for CeapinA7, and Dr. Luke Wiseman for TMP-inducible DHFR-ATF6 cells. We also thank Drs. Peter E. Geiduschek and Randy Hampton for critical reading of the manuscript. This work was funded by NIH RO1 grants GM087415 (M.N.), GM033279 (D.J.F.), NIH/NCI R01CA172667 (D.N.), and NIHT32, UCSD/LIAI allergy postdoctoral training grant (A.B.T.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Amatsu T, Watanabe J, Asaka Y, and Kanamura S (1995). Endoplasmic reticulum proliferates without an increase in cytochrome P-450 in hepatocytes of mice treated with phenobarbital and cobalt chloride. Eur. J. Cell Biol 68, 256–262. [PubMed] [Google Scholar]
- Ariyasu D, Yoshida H, and Hasegawa Y (2017). Endoplasmic reticulum (ER) stress and endocrine disorders. Int. J. Mol. Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DI, Cozzo A, Ji X, Roberts LS, Louie SM, Mulvihill MM, Luo K, and Nomura DK (2013). Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc. Natl. Acad. Sci. USA 110, 14912–14917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DI, Li DS, Lowe W, Heuer T, Kemble G, and Nomura DK (2015). Diacylglycerol metabolism and signaling is a driving force underlying FASN inhibitor sensitivity in cancer cells. ACS Chem. Biol 10, 1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Zhang YH, Hendershot LM, Harding HP, and Ron D (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol 2, 326–332. [DOI] [PubMed] [Google Scholar]
- Bikman BT, Guan Y, Shui G, Siddique MM, Holland WL, Kim JY, Fabrias G, Wenk MR, and Summers SA (2012). Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J. Biol. Chem 287, 17426–17437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JW, and Hendershot LM (2005). Building an antibody factory: a job for the unfolded protein response. Nat. Immunol 6, 23–29. [DOI] [PubMed] [Google Scholar]
- Brunsing R, Omori SA, Weber F, Bicknell A, Friend L, Rickert R, and Niwa M (2008). B- and T-cell development both involve activity of the unfolded protein response pathway. J. Biol. Chem 283, 17954–17961. [DOI] [PubMed] [Google Scholar]
- Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE, Eaton KA, and Kaufman RJ (2013). The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 144, 989–1000.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Hiramatsu N, Messah C, Kroeger H, and Lin JH (2012). Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest. Ophthalmol. Vis. Sci 53, 7159–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Ellenberg J, Song J, Dieuliis D, and Lippincott-Schwartz J (1998). Retrograde transport of Golgi-localized proteins to the ER. J. Cell Biol 140, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras FX, Ernst AM, Haberkant P, Bjorkholm P, Lindahl E, Gonen B, Tischer C, Elofsson A, Von Heijne G, Thiele C, et al. (2012). Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 481, 525–529. [DOI] [PubMed] [Google Scholar]
- Cooper JP, Reynolds CP, Cho H, and Kang MH (2017). Clinical development of fenretinide as an antineoplastic drug: pharmacology perspectives. Exp. Biol. Med. (Maywood) 242, 1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippold HC, Ng MM, Farber-Katz SE, Lee SK, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith KA, Madhavarapu S, et al. (2009). GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, and Glembotski CC (2009). Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem 284, 29735–29745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Watkins SM, and Hotamisligil GS (2012). The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab 15, 623–634. [DOI] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, and Hotamisligil GS (2011). Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher CM, Garri C, Cain EL, Ang KK, Wilson CG, Chen S, Hearn BR, Jaishankar P, Aranda-Diaz A, Arkin MR, et al. (2016). Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. Elife 5, 10.7554/eLife.11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher CM, and Walter P (2016). Ceapins inhibit ATF6alpha signaling by selectively preventing transport of ATF6alpha to the Golgi apparatus during ER stress. Elife 5, 10.7554/eLife.11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjymishka A, Su N, and Kilberg MS (2009). Transcriptional induction of the human asparagine synthetase gene during the unfolded protein response does not require the ATF6 and IRE1/XBP1 arms of the pathway. Biochem. J 417, 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hanelt I, Hummer G, and Ernst R (2017). Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 67, 673–684.e8. [DOI] [PubMed] [Google Scholar]
- Hannun YA, and Obeid LM (2018). Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol 19, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa A, Taouji S, Lhomond S, Jensen D, Fernandez-Zapico ME, Simpson JC, Pasquet JM, Schekman R, and Chevet E (2014). Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol 34, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, and Lippincott-Schwartz J (1998). Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol 143, 1485–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, and Glimcher LH (2003). Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol 4, 321–329. [DOI] [PubMed] [Google Scholar]
- Jacquemyn J, Cascalho A, and Goodchild RE (2017). The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep 18, 1905–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CA, Marechal JD, and Sutcliffe MJ (2005). Progress in cytochrome P450 active site modeling. Arch. Biochem. Biophys 433, 361–368. [DOI] [PubMed] [Google Scholar]
- Kohl S, Zobor D, Chiang WC, Weisschuh N, Staller J, Gonzalez Menendez I, Chang S, Beck SC, Garcia Garrido M, Sothilingam V, et al. (2015). Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet 47, 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono N, Amin-Wetzel N, and Ron D (2017). Generic membrane-spanning features endow IRE1alpha with responsiveness to membrane aberrancy. Mol. Biol. Cell 28, 2318–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Mentrup T, Kleizen B, Rivera-Milla E, Reichenbach D, Enzensperger C, Nohl R, Tauscher E, Gorls H, Ploubidou A, et al. (2013). Small molecules intercept Notch signaling and the early secretory pathway. Nat. Chem. Biol 9, 731–738. [DOI] [PubMed] [Google Scholar]
- Kummel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, Pincet F, Rothman JE, and Reinisch KM (2011). Complexin cross-links prefusion SNAREs into a zigzag array. Nat. Struct. Mol. Biol 18, 927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, and Glimcher LH (2008). Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, and Walter P (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Hendershot LM (2001). The unfolding tale of the unfolded protein response. Cell 107, 827–830. [DOI] [PubMed] [Google Scholar]
- Maiuolo J, Bulotta S, Verderio C, Benfante R, and Borgese N (2011). Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. USA 108, 7832–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manie SN, Lebeau J, and Chevet E (2014). Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. Orchestrating the unfolded protein response in oncogenesis: an update. Am. J. Physiol. Cell Physiol 307, C901–C907. [DOI] [PubMed] [Google Scholar]
- Miller M, Tam AB, Mueller JL, Rosenthal P, Beppu A, Gordillo R, Mcgeough MD, Vuong C, Doherty TA, Hoffman HM, et al. (2017). Cutting edge: targeting epithelial ORMDL3 increases, rather than reduces, airway responsiveness and is associated with increased sphingosine-1-phosphate. J. Immunol 198, 3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Chen S, Chandra V, Wagatsuma K, Agata Y, Rodewald HR, Saito R, Chang AN, Varki N, et al. (2015). The E-Id protein axis modulates the activities of the PI3K-AKT-mTORC1-Hif1a and c-myc/p19Arf pathways to suppress innate variant TFH cell development, thymocyte expansion, and lymphomagenesis. Genes Dev 29, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K (2000). Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101, 451–454. [DOI] [PubMed] [Google Scholar]
- Nadanaka S, Okada T, Yoshida H, and Mori K (2007). Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol 27, 1027–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls S, Snapp EL, Cole NB, Zaal KJ, Kenworthy AK, Roberts TH, Ellenberg J, Presley JF, Siggia E, and Lippincott-Schwartz J (2000). Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol 2, 288–295. [DOI] [PubMed] [Google Scholar]
- Niwa M, Sidrauski C, Kaufman RJ, and Walter P (1999). A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell 99, 691–702. [DOI] [PubMed] [Google Scholar]
- Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, and Mori K (2003). A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem 278, 31024–31032. [DOI] [PubMed] [Google Scholar]
- Olson DK, Frohlich F, Farese RV Jr., and Walther TC (2015). Taming the sphinx: mechanisms of cellular sphingolipid homeostasis. Biochim. Biophys. Acta 1861 (8 Pt B), 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Plate L, Cooley CB, Chen JJ, Paxman RJ, Gallagher CM, Madoux F, Genereux JC, Dobbs W, Garza D, Spicer TP, et al. (2016). Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 5, 10.7554/eLife.15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, and Kimata Y (2011). Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 22, 3520–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, and Goldstein JL (2004). Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell 15, 259–268. [DOI] [PubMed] [Google Scholar]
- Ron D, and Hampton RY (2004). Membrane biogenesis and the unfolded protein response. J. Cell Biol 167, 23–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, and Kaufman RJ (2004). A trip to the ER: coping with stress. Trends Cell Biol 14, 20–28. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, et al. (2008). UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppres- sion of transcriptional master regulators. Dev. Cell 15, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutti MF, Richard S, Penno A, Von Eckardstein A, and Hornemann T (2009). An improved method to determine serine palmitoyltransferase activity. J. Lipid Res 50, 1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler AJ, and Schekman R (2009). In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 106, 17775–17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S, Prinz WA, Thorn KS, Voss C, and Walter P (2009). Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol 187, 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkal CE, Ponnusamy S, Manevich Y, Meyers-Needham M, Saddoughi SA, Mukhopadyay A, Dent P, Bielawski J, and Ogretmen B (2011). Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J. Biol. Chem 286, 42446–42458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, and Prywes R (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111. [DOI] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR 3rd, Su AI, Kelly JW, and Wiseman RL (2013). Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3, 1279–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprocati T, Ronchi P, Raimondi A, Francolini M, and Borgese N (2006). Dynamic and reversible restructuring of the ER induced by PDMP in cultured cells. J. Cell Sci 119, 3249–3260. [DOI] [PubMed] [Google Scholar]
- Staubli W, Hess R, and Weibel ER (1969). Correlated morphometric and biochemical studies on the liver cell. II. Effects of phenobarbital on rat hepatocytes. J. Cell Biol 42, 92–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault G, Shui G, Kim W, Mcalister GC, Ismail N, Gygi SP, Wenk MR, and Ng DT (2012). The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol. Cell 48, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra H, Dufey E, Avril T, Chevet E, and Hetz C (2016). Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2, 252–262. [DOI] [PubMed] [Google Scholar]
- van Anken E, Romijn EP, Maggioni C, Mezghrani A, Sitia R, Braakman I, and Heck AJR (2003). Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity 18, 243–253. [DOI] [PubMed] [Google Scholar]
- Voeltz GK, Rolls MM, and Rapoport TA (2002). Structural organization of the endoplasmic reticulum. EMBO Rep 3, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R, and Ron D (2015). Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol 33, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R, Van der Ploeg K, and Ron D (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 110, 4628–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Gilmore R, and Blobel G (1984). Protein translocation across the endoplasmic-reticulum. Cell 38, 5–8. [DOI] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GDY, and Kaufman RJ (2007). ATF6alpha optimizes longterm endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364. [DOI] [PubMed] [Google Scholar]
- Wu N, Sarna LK, Hwang SY, Zhu Q, Wang P, Siow YL, and O K (2013). Activation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase during high fat diet feeding. Biochim. Biophys. Acta 1832, 1560–1568. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, and Mori K (2010). Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 21, 2975–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, and Goldstein JL (2000). ER stress induces cleavage of membrane- bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364. [DOI] [PubMed] [Google Scholar]
- Yoshikawa A, Kamide T, Hashida K, Ta HM, Inahata Y, Takarada- Iemata M, Hattori T, Mori K, Takahashi R, Matsuyama T, et al. (2015). Deletion of Atf6alpha impairs astroglial activation and enhances neuronal death following brain ischemia in mice. J. Neurochem 132, 342–353. [DOI] [PubMed] [Google Scholar]
- Zhou AX, and Tabas I (2013). The UPR in atherosclerosis. Semin. Immunopathol 35, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.