

Abstract
Background
Phenotypic characterization of immune cells in the bone marrow (BM) of patients with acute myeloid leukemia (AML) is lacking.
Methods
T‐cell infiltration was quantified on BM biopsies from 13 patients with AML, and flow cytometry was performed on BM aspirates (BMAs) from 107 patients with AML who received treatment at The University of Texas MD Anderson Cancer Center. The authors evaluated the expression of inhibitory receptors (programmed cell death protein 1 [PD1], cytotoxic T‐lymphocyte antigen 4 [CTLA4], lymphocyte‐activation gene 3 [LAG3], T‐cell immunoglobulin and mucin‐domain containing‐3 [TIM3]) and stimulatory receptors (glucocorticoid‐induced tumor necrosis factor receptor‐related protein [GITR], OX40, 41BB [a type 2 transmembrane glycoprotein receptor], inducible T‐cell costimulatory [ICOS]) on T‐cell subsets and the expression of their ligands (41BBL, B7‐1, B7‐2, ICOSL, PD‐L1, PD‐L2, and OX40L) on AML blasts. Expression of these markers was correlated with patient age, karyotype, baseline next‐generation sequencing for 28 myeloid‐associated genes (including P53), and DNA methylation proteins (DNA methyltransferase 3α, isocitrate dehydrogenase 1[IDH1], IDH2, Tet methylcytosine dioxygenase 2 [TET2], and Fms‐related tyrosine kinase 3 [FLT3]).
Results
On histochemistry evaluation, the T‐cell population in BM appeared to be preserved in patients who had AML compared with healthy donors. The proportion of T‐regulatory cells (Tregs) in BMAs was higher in patients with AML than in healthy donors. PD1‐positive/OX40‐positive T cells were more frequent in AML BMAs, and a higher frequency of PD1‐positive/cluster of differentiation 8 (CD8)‐positive T cells coexpressed TIM3 or LAG3. PD1‐positive/CD8‐positive T cells were more frequent in BMAs from patients who had multiply relapsed AML than in BMAs from those who had first relapsed or newly diagnosed AML. Blasts in BMAs from patients who had TP53‐mutated AML were more frequently positive for PD‐L1.
Conclusions
The preserved T‐cell population, the increased frequency of regulatory T cells, and the expression of targetable immune receptors in AML BMAs suggest a role for T‐cell–harnessing therapies in AML.
Keywords: acute myeloid leukemia, flow cytometry, immune checkpoint, immunotherapy, T cell
Short abstract
T‐cell subsets are preserved in the bone marrow of patients with acute myeloid leukemia. The expression of targetable immune checkpoints by T cells suggests that therapies harnessing T cells may benefit these patients.
Introduction
Immune checkpoint therapy has revolutionized the treatment of patients with cancer.1, 2 Checkpoint receptors and their ligands play an important role in T‐cell stimulation and exhaustion and are currently the focus of significant efforts in understanding and modulating antitumor immune responses.3 In hematologic malignancies, clinical benefits have been observed with single‐agent or combination checkpoint‐based therapies in patients with Hodgkin lymphoma4, 5, 6 and non‐Hodgkin lymphoma7, 8 and after allogeneic stem cell transplantation (ASCT) patients with acute myeloid leukemia (AML).9, 10 Recent data suggest that programmed cell death protein 1 (PD1) and cytotoxic T‐lymphocyte antigen 4 (CTLA4) inhibitors, in combination with hypomethylating agents (HMAs), may be effective in patients who have relapsed AML and high‐risk myelodysplastic syndrome (MDS).11, 12 A randomized phase 3 study to evaluate the efficacy of combining HMAs with PD1 blockade in the frontline setting for elderly patients with AML has been initiated (clinicaltrials.gov identifier NCT03092674).
Patients with AML have an increased frequency of regulatory T cells (Tregs) in their peripheral blood (PB) that persists after achieving a complete remission and has been associated with an increased risk of relapse.13, 14 Increased expression of (PD1) ligand (PD‐L1) was identified as an independent, negative prognostic factor for survival among patients with French‐American‐British classification M5 AML.15 PD‐L1 and PD‐L2 upregulation in patients with MDS and AML who received 5‐azacitidine was correlated negatively with response and survival in combination with HMAs.16 Patients who relapsed after ASCT for AML had a higher frequency of PD1hi/T‐cell immunoglobulin and mucin‐domain containing‐3 (TIM3)‐positive T cells in their PB, and these T cells exhibited evidence of exhaustion in response to cluster of differentiation 3 (CD3)/CD28 stimulation.17 In murine models, AML progression was associated with increased PD1 expression by CD8‐positive T cells and increased Treg infiltration into organs and bone marrow (BM).18 The PD1‐positive/CD8‐positive T‐cell exhaustion was reversible with PD1 and PD‐L1 blockade.19
Limited data have been published that comprehensively describe the composition of T cells in the BM niche, the expression of clinically actionable checkpoint receptors on different T‐cell subsets, and the expression of checkpoint ligands by blasts in patients with AML. Better defining the T‐cell and immune checkpoint landscape of human AML may be important in guiding the selection, timing, and combinatorial partners for T‐cell–harnessing therapies in ongoing and future clinical trials. In the current report, we attempt to characterize the composition of different T‐cell subsets, the expression of several clinically actionable checkpoint receptors on T cells, and the expression of ligands on blasts isolated from BM aspirates (BMAs) and to determine how these correlate with each other and with clinical and molecular characteristics in 107 patients with AML.
Materials and Methods
Immunohistochemistry Staining and Quantification of CD3‐Positive Cells
BM core biopsies were obtained with informed consent and were fixed in buffered formalin, decalcified, paraffin‐embedded, and cut into 4‐μm sections, which were stained with hematoxylin and eosin (H&E) to assess BM cellularity. Immunohistochemistry (IHC) was performed for CD3 (clone 2GV6) and CD34 (clone QBEnd/10; both from Ventana Medical Systems, Inc, Tucson, AZ) using a Ventana Benchmark Ultra automated staining instrument according to the manufacturer’s recommendations. The percentages of CD3‐positive cells and CD34‐positive blasts in the core biopsies were quantified by counting the number of CD3‐positive cells or CD34‐positive cells divided by the total cells counted in 10 microscopic fields at ×200 magnification using a ×20 objective for each case to arrive at the average percentage of CD3‐positive cells or CD34‐positive blasts per medium‐power field (MPF). To arrive at a measurement of absolute numbers of T cells per case, H&E‐stained and IHC biopsy sections were captured at ×200 magnification using an Olympus BX41 microscope with a ×20 objective, a DP72 Olympus camera, and Olympus CellSens Entry software (Olympus Corporation, Tokyo, Japan). Images representing single MPFs at ×200 magnification from 5 BM biopsies that had 100% cellularity were used to calculate the average number of hematopoietic cells contained in an MPF of BM with 100% cellularity (4500 cells per MPF with 100% cellularity). On the basis of Olympus software measurements, an MPF at ×200 magnification represented 350,000 μm2. Absolute T‐cell infiltrates from healthy donors (HDs) and patients with AML were calculated by multiplying the percentage CD3‐positive cells with the overall cellularity of the samples in each biopsy multiplied by the average number of cells contained in an MPF with 100% cellularity. Formula: (X number of CD3‐positive cells/100 cells = % CD3‐positive cells) * ([cellularity * 350,000 μm2]/1 MPF) * (1 MPF/350,000 μm2) * (4500 cells/1 MPF) = number of CD3‐positive cells/1 MPF.
Immunophenotyping of Lymphocytes and Blasts
Between March 2015 and May 2017, we performed 17‐color, multiparameter flow cytometry (MFC) on BMAs and PB mononuclear cells (PBMCs) from 107 patients with AML (39 with newly diagnosed AML, 68 with relapsed AML) and on 8 BMAs from HDs. These analyses were performed on freshly collected BMAs and PBMCs (within 12 hours of collection) by The University of Texas MD Anderson Cancer Center Immunotherapy Platform using MFC panels previously validated at the Immunotherapy Platform.20 HD BMAs and PBMCs were obtained from Lonza Walkersville Inc (Walkersville, MD) and Key Biologics (Memphis, TN). All patients provided written, informed consent. All research was performed in accordance with the Declaration of Helsinki and with The University of Texas MD Anderson Cancer Center Institutional Review Board guidelines.
PBMCs or BMAs from patients and HDs were collected in Vacutainer or Cell Preparation Tubes containing sodium heparin (BD Vacutainer, Franklin Lakes, NJ). Mononuclear cells were isolated from PB or BMAs by centrifuging the Cell Preparation Tubes at 2000 revolutions per minute (863g) for 15 minutes at room temperature. Samples were diluted 1:5 with phosphate‐buffered saline (PBS) and layered over 10 mL of Ficoll. The mixture was centrifuged at 2000 revolutions per minute (863g) for 20 minutes at room temperature with no brakes. The interface cells were then harvested and washed twice with PBS containing 10% fetal calf serum at 500g and 450g for 10 minutes, respectively. Mononuclear cells were resuspended in PBS, and MFC was performed using fluorescence‐conjugated monoclonal antibodies (Supporting Table 1). Cells were acquired using a Fortessa cell analyzer (BD Biosciences, Heidelberg, Germany), and the analysis was performed using FlowJo software (Tree Star, Ashland, OR).
We evaluated the expression of clinically actionable inhibitory checkpoint receptors (PD1, CTLA4, lymphocyte‐activation gene 3 [LAG3], TIM3) and activating checkpoint receptors (glucocorticoid‐induced tumor necrosis factor receptor‐related protein [GITR], OX40, 41BB [a type 2 transmembrane glycoprotein receptor], inducible T‐cell costimulatory [ICOS]) on the following T‐cell subsets: CD4‐positive T‐effector (Teff) cells were defined as CD3‐positive/CD4‐positive/CD127lo‐positive/forkhead box P3 (FoxP3)‐negative; CD4‐positive Tregs were defined as CD3‐positive/CD4‐positive/CD127‐negative/FoxP3‐positive; and CD8‐positive cells were defined as CD3‐positive/CD8‐positive in BMAs and PBMCs from 107 patients with AML. AML blasts were assessed for the 41BB ligand (41BBL), B7‐1, B7‐2, the ICOS ligand (ICOSL), galectin 9, PD‐L1, PD‐L2, and the OX40 ligand (OX40L). Eight BMAs isolated from HDs were used as controls for T‐cell subsets and the expression of checkpoint receptors on total CD3‐positive populations and on each T‐cell subsets.
A next‐generation sequencing‐based analysis for the detection of somatic mutations in the coding sequences of 28 myeloid‐associated genes was performed on DNA extracted from the BMAs. The methodology of our mutation analysis panel and coverage by genes has been previously published21 (Supporting Table 2).
We correlated the distribution of T‐cell subsets and the expression of immune checkpoint receptors on T‐cell subsets and the distribution of ligands on blasts with each patient’s age, karyotype, and baseline next‐generation sequencing for somatic mutations, including specifically tumor protein p53 (TP53), DNA methylation proteins (DNA methyltransferase 3α [DNMTα], isocitrate dehydrogenase 1[IDH1], IDH2, Tet methylcytosine dioxygenase 2 [TET2]), and Fms‐related tyrosine kinase 3 (FLT3).
Statistical Analysis
The compared groups were not of equal size and did not follow a normal distribution; therefore, differences between groups >2 were calculated using Kruskal‐Wallis 1‐way analyses of variance, and differences between groups of 2 were calculated using 2‐tailed Mann‐Whitney tests. Regression analyses and column dot plots were generated in Prism (GraphPad Software Inc, San Diego, CA). Data was reported as medians, and significance was defined as P < .05. For each subset of data, we evaluated the association between survival and each T‐cell subset and their individual checkpoint expression through a univariate Cox proportional‐hazards model.
Results
T‐Cell Infiltration in BM Biopsies Was Comparable Between Patients With AML and Healthy Donors
We did not observe a significant difference in the percentage of CD3‐positive cells in BM biopsies from 13 patients who had relapsed/refractory AML (mean age ± standard deviation, 52 ± 14.9 years) and 14 age‐matched HDs (mean age ± standard deviation, 54 ± 13.6 years) (Fig. 1B, Supporting Table 3). When adjusting for overall cellularity as described above (see Materials and Methods), we also did not observe any significant difference in the calculated absolute CD3‐positive cell infiltration in per MPF at ×200 magnification (P = .14).
Figure 1.
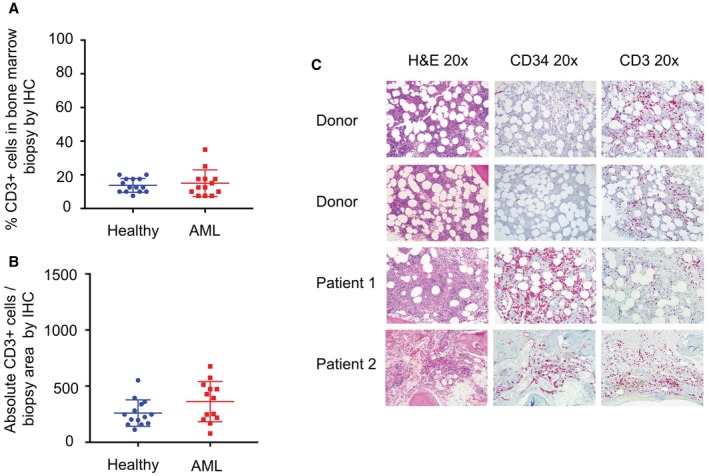
Immunochemistry (IHC) was used to quantify T‐cell infiltration in bone marrow biopsies from healthy donors and from patients with acute myeloid leukemia (AML). (A) No significant difference was observed in the percentage of cluster of differentiation 3 (CD3)‐positive (CD3+) cells per medium‐power field (MPF) in bone marrow biopsies from patients with AML (blue circles) and from age‐matched, healthy donors (red squares). (B) There also was no difference in the absolute CD3‐positive cell infiltration per MPF (at 200 magnification; calculated by multiplying the average percentage of CD3‐positive cells infiltrating the biopsy per MPF by the cellularity of each sample) and the number of average total hematopoietic cells in a bone marrow that had 100% cellularity (see Materials and Methods). (C) Representative photomicrographs of bone marrow biopsies are from 2 healthy donors and 2 patients with AML. H&E indicates hematoxylin and eosin staining.
Representative photomicrographs of BM biopsies stained in H&E and further stained for CD34 in blasts and for CD3 in T cells are provided in Figure 1C. The overall cellularity and CD3‐positive cell percentage, as well as age, for all 14 HDs and 13 patients with AML are listed in Supporting Table 3.
Checkpoint Expression by T Cells in BMAs Versus PBMCs
In total, 107 patients with AML (39 with newly diagnosed AML, 68 with relapsed AML) and 8 HDs were evaluated by MFC. A representative MFC gating strategy is illustrated in Figure 2A. We observed that our data from PB was quite discrepant from the BMA findings. We believed that focusing on the microenvironment in the BM was more appropriate and more likely to accurately reflect the immune status of the disease, as has been demonstrated on numerous occasions in solid tumor studies done on T cells isolated from the tumor site, rather than from PB. We have included the data comparing BM with PB among patients in Supporting Figure 1 and Supporting Table 4. Henceforth, our current discussions focus on the BMA findings, because we believe that T cells from the tumor microenvironment (in this case, from BMAs) would provide more representative information regarding changes in the immune system among patients with AML.
Figure 2.
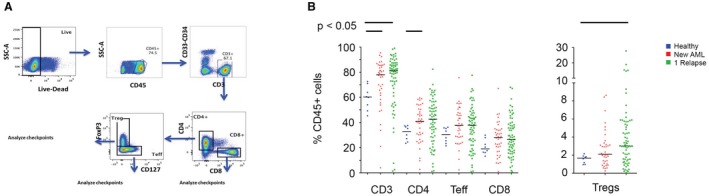
T‐cell subset distribution is illustrated in bone marrow aspirates from healthy donors, patients with newly diagnosed acute myeloid leukemia (AML), and patients with relapsed AML. (A) The flow‐cytometry gating strategy is illustrated. CD indicates cluster of differentiation; FoxP3, forkhead box P3; SCC‐A, side scatter area; Teff, Teff cells; Tregs, regulatory T cells. (B) T‐cell subsets are compared between healthy donors (blue circles), patients with newly diagnosed AML (red triangles), and patients with relapsed AML (green squares).When gating on CD45‐positive (CD43+) cells, there is an increase in the frequency of total T cells, CD4‐positive Teff cells, and Tregs in bone marrow aspirates from patients with AML compared with the aspirates from healthy donors.
Increased Frequency of Tregs in BMAs From Patients With AML
We did not observe a significant difference in the CD3‐positive cell subset as a percentage of cellularity or as the absolute CD3‐positive cell infiltration per MPF by BM IHC evaluation in age‐matched patients with AML and HDs, as noted above. All further MFC analyses are based on the population of interest: the nonleukemic, CD45hi gated BMA population. Among the CD45hi BMA population, the CD3‐positive cell subset was significantly more frequent in BMAs from patients with AML (HDs vs new AML vs relapsed AML: 60.3% vs 78% vs 81.1%, respectively; P = .02), but this was not observed in the total CD4‐positive T‐cell subset (HD vs new AML vs relapsed AML: 32.3% vs 40.8% vs 42.6%, respectively; P = .1), the CD4‐positive Teff cell subset (HD vs new AML vs relapsed AML: 30.5% vs 37.7% vs 37.8%, respectively; P = .2) or the CD8‐positive T‐cell subset (HD vs new AML vs relapsed AML: 19.1% vs 27.9% vs 26.4%, respectively; P = .3) (Fig. 2B).
There was a significantly higher frequency of Tregs (HD vs new AML vs relapsed AML: 1.7% vs 2.1% vs 3%, respectively; P = .02) in patients with AML. It is important to emphasize that these median values do not reflect the skewed distribution for Tregs in BMAs from patients with AML noted on plotting Treg values for individual patients (Fig. 2B). When dividing patients with relapsed AML into first relapse versus >1 relapse, we observed a trend toward a decreased frequency of CD3‐positive cells, CD4‐positive Teff cells, and Tregs, but not CD8‐positive T cells, in patients with AML who had >1 relapse (Supporting Fig. 2).
Checkpoint Receptors PD1, OX40, and ICOS Are Expressed on T Cells in BMAs From Patients With Newly Diagnosed and Relapsed AML
When analyzing the expression of immune checkpoint markers on different T‐cell subsets, we observed that PD1 and OX40 emerged as checkpoints of interest. There was an increased frequency of PD1‐positive and OX40‐positive T cells in BMAs from patients with AML compared with HDs. PD1‐positive/CD8‐positive T cells were significantly less frequent in BMAs from HDs (12.1%) than in BMAs from patients with newly diagnosed AML (27.3%), first relapsed AML (25.5%), and >1 relapsed AML (34.7%)(P < .01). A similar but less pronounced trend was observed for the frequency of OX40‐positive/CD8‐positive T cells (HD vs new AML vs first relapsed AML vs >1 relapsed AML: 0.6%, 0.9%, 2.3%, and 2.1%, respectively; P = .08) (Fig. 3A).
Figure 3.
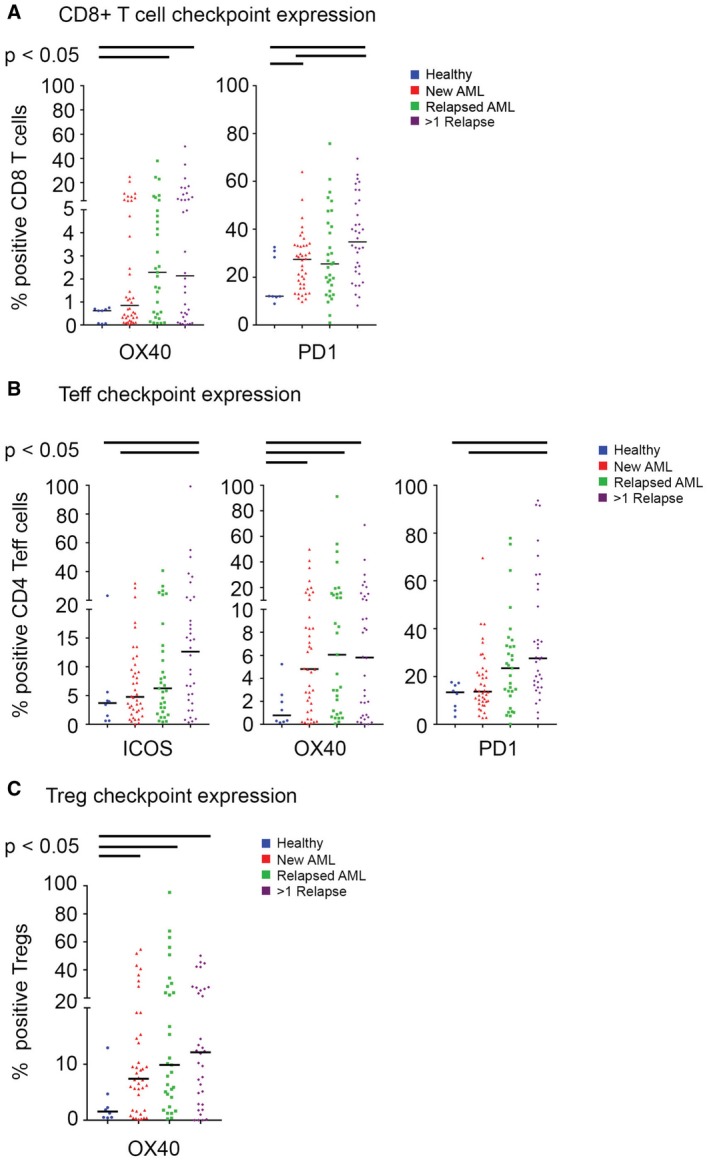
The expression of immune checkpoints (programmed cell death protein 1 [PD1], OX40, inducible T‐cell costimulatory [ICOS]) is illustrated on T‐cell subsets in bone marrow aspirates from healthy donors (blue circles) and from patients with newly diagnosed AML (red triangles), first relapsed AML (green squares), and multiple relapsed AML (purple diamonds). (A) OX40‐positive and PD1‐positive cluster of differentiation 8 (CD8)‐positive (CD8+) T cells; (B) ICOS‐positive, OX40‐positive, and PD1‐positive/CD4+ T‐effector (Teff) cells; and (C) OX40‐positive T‐regulatory cells (Tregs) were noted more frequently in bone marrow aspirates from patients with AML compared with the aspirates from healthy donors.
The frequency of PD1‐positive/CD4‐positive Teff cells in BMAs was significantly greater in BMAs from patients with AML than in those from HDs (HD vs new AML vs first relapsed AML vs >1 relapsed AML: 13.4%, 13.7%, 25.3% and 27.6%, respectively; P < .01) (Fig. 3B). The frequency of OX40‐positive/CD4‐positive T cells also was significantly greater in BMAs from patients with AML (HD vs new AML vs first relapsed AML vs >1 relapsed AML: 0.6%, 4.8%, 6.1% and 5.8%, respectively; P < .05). We also observed a higher frequency of ICOS‐positive/CD4‐positive Teff cells in AML BMAs (HD vs new AML vs first relapsed AML vs >1 relapsed AML: 3.7% vs 4.8% vs 6.3% vs 12.7%, respectively; P = .04).
When assessing checkpoint expression on Tregs, we only noted a significant increase in the frequency of OX40‐positive Tregs in BMAs from patients with AML (HD vs new AML vs first relapsed AML vs >1 relapsed AML: 1.6% vs 7.4% vs 9.9% vs 12.1%, respectively; P = .04) (Fig. 3C). The expression of individual immune checkpoint receptors on each T‐cell subset is illustrated in Supporting Figure 3. We did not observe any clinically meaningful associations between survival and the distribution of T‐cell subsets or the expression of individual immune checkpoint receptors on each T‐cell subset (Supporting Table 5).
TIM3 and LAG3 Are Coexpressed More Frequently With PD1 on T Cells Isolated From AML BMAs
Previous results have indicated that PD1‐positive/TIM3‐positive (double‐positive) T cells represent highly exhausted T cells within the immune microenvironment in murine models of AML and are associated with an earlier relapse after ASCT in patients with AML.17, 22 Coexpression of PD1 with either TIM3 or LAG3 on T cells also has been associated with immune exhaustion in patients with solid cancers.23 We assessed the frequency of each T‐cell subset that co‐expressed PD1 with TIM3 or LAG3 in BMAs from HDs and patients with AML.
When we compared the PD1‐positive/TIM3‐positive expression gated on each T‐cell subset, we observed a trend toward increased frequency of PD1‐positive/TIM3‐positive/CD8‐positive T cells in BMAs from patients with AML (HD vs new AML vs relapsed AML: 0.75%, 1.36%, and 1.72%, respectively; P = .09) and PD1‐positive/TIM3‐positive/CD4‐positive Teff cells (HD vs new AML vs relapsed AML: 0.75, 2.2%, and 2.8%, respectively; P = .16), but not for PD1‐positive/TIM3‐positive Tregs (HD vs new AML vs relapsed AML: 0.75%, 1.6%, and 1.9%, respectively; P = .35) (Fig. 4A). We observed a similar pattern for PD1‐positive/LAG3‐positive/CD8‐positive T cells (HD vs new AML vs relapsed AML: 2.71%, 4.69%, and 8.98%, respectively; P < .01) and for PD1‐positive/LAG3‐positive/CD4‐positive Teff cells (HD vs new AML vs relapsed AML: 2.71%, 14.3%, and 13.5%, respectively; P = .05), but not for PD1‐positive/LAG3‐positive Tregs (HD vs new AML vs relapsed AML: 2.72%, 0.71%, and 3.06%, respectively; P = .15) (Fig. 4B). However, the median frequencies of the double‐positive cells in the BMAs from HDs and from patients with AML do not truly reflect the bimodal distributions of the double‐positive cells we noted in different T‐cell subsets, and subgroups of patients appeared to have markedly higher frequencies of double‐positive T‐cell subsets in their BMAs (Fig. 4).
Figure 4.
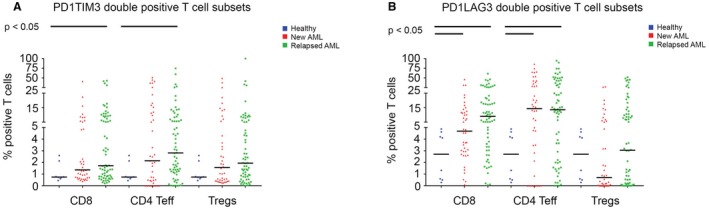
The frequency of programed cell death 1 (PD1)/T‐cell immunoglobulin and mucin‐domain containing‐3(TIM3) (PD1TIM3) double‐positive T cells and of PD1/lymphocyte‐activation gene 3 (LAG3) (PD1LAG3) double‐positive T cells is illustrated in bone marrow aspirates from healthy donors (blue circles), patients with newly diagnosed acute myeloid leukemia (AML) (red triangles), and patients with relapsed AML (green squares). (A) There is an increased frequency of PD1/TIM3 double‐positive cluster of differentiation 8 (CD8)‐positive T cells and PD1/TIM3 double‐positive CD4‐positive T effector (Teff) cells in bone marrow aspirates from patients with AML compared with aspirates from healthy donors. (B) There is an increased frequency of PD1/LAG3 double‐positive CD8‐positive and CD4‐positive Teff cells in bone marrow aspirates from patients with AML compared with aspirates from healthy donors.
Age, Mutational Profile, and Karyotypic Status Influence the Composition of T‐Cell Subsets and Immune‐Checkpoint Ligand Expression on AML Blasts
We assessed for correlations between molecular mutations, karyotype, BM blasts, and age in patients with AML with the frequencies of each T‐cell subset, the expression of individual immune checkpoint receptors on each T‐cell subset, and the expression of immune checkpoint ligands on AML blasts. We observed that BM blasts were more frequently positive for PD‐L1 (6.95% vs 12.5%; P = .05) and for 41BBL (5.16% vs 13.65%; P = .01) in patients who had TP53‐mutated AML (Fig. 5). There also was a trend toward increased frequency of PD‐L1–positive blasts in BMAs from patients who had AML with complex cytogenetics (6.2% vs 10%; P = .09). We also assessed the percentage of pretherapy BM blasts (both as a continuous variable and as a binomial variable using blast cutoffs of 20%, 30%, and 50%) and did not observe any correlation between the BM blast burden and BM Tregs, CD3‐positive cells, or CD8‐positive cells in patients with either new or relapsed AML.
Figure 5.
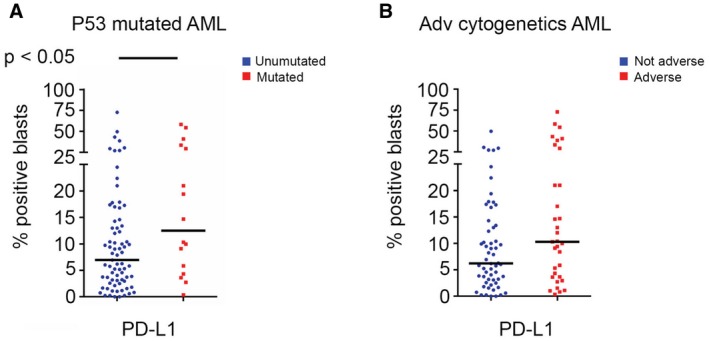
Programmed cell death 1 ligand (PD‐L1) expression is illustrated according to the percentage of positive blasts in patients with acute myeloid leukemia (AML). (A) PD‐L1–positive blasts were noted more frequently in bone marrow aspirates from patients who had tumor protein 53 (TP53)‐mutated AML versus those who had non‐TP53–mutated AML. (B) PD‐L1–positive blasts are compared between patients who had AML with adverse (Adv) cytogenetics versus those who had nonadverse cytogenetics.
We observed a decreased frequency of CD4‐positive Teff cells (32.2% vs 41.8%; P = .03) in BMAs from patients who had AML with adverse cytogenetics. There was a higher frequency in the total CD4‐positive T‐cell subset (44.6% vs 36.2%; P = .03) in BMAs from patients with AML that harbored somatic mutations in certain DNA methylation pathways (DNMT3A, IDH1, IDH2, TET2), driven by an increase in the frequency of the CD4‐positive Teff‐cell subset (43.2% vs 32%; P < .01). We did not observe any other significant differences in the composition of the BMA T‐cell subsets by age, cytogenetic subgroup, or somatic myeloid‐associated mutations. A detailed distribution of all evaluated checkpoint ligands on AML blasts in BMA by mutation, cytogenetics, and age is provided in Supporting Figure 4; and distribution by the same variables for T‐cell subsets and individual checkpoint receptor expression on each T‐cell subset are provided in Supporting Figure 5.
Discussion
Previous investigators have evaluated the function of T cells in patients with AML.24, 25 PB‐derived and BM‐derived T cells did not appear to be functionally impaired in patients with AML compared with HD controls on CD3/CD28 activation studies.24, 26 The current study is 1 of the first to broadly examine T‐cell frequencies, T‐cell subset distribution, and the expression of checkpoint receptors in a large population of patients with AML (N = 107) to better understand the T‐cell and immune checkpoint landscape of AML, to trigger additional research in this area, and to identify ideal targets and scenarios for the introduction of immune checkpoint blockade and T‐cell–harnessing strategies in patients with AML. The novelty of this study lies in the size of our study population and in the number of markers interrogated in the analyses. We realize and accept that this is not a comprehensive summary of the entire immune biology, including the impact of tumor microenvironmental factors, such as myeloid‐derived suppressor cells, mesenchymal stem cells, monocytes, and dendritic cells. Such studies are ongoing in multiple institutions.
We used IHC on BM slides rather than bead counting on BMAs to quantitatively compare T‐cell infiltration in BM samples from age‐matched HDs and from patients with AML, because IHC allowed us to accurately calculate both the cellularity of the sample as well as the percentage of T cells per MPF. IHC on BM slides also was less likely to be impacted by hemodilution with blood than BMAs. We did not observe any difference in the percentage of CD3‐positive cells per MPF or in the absolute number of CD3‐positive cells per MPF in the BM biopsies of age‐matched HDs and patients with AML. Indeed, these findings, although apparently counterintuitive, are in line with previously published data demonstrating that the absolute T‐cell numbers in PB were either within the same range in patients with AML and in HDs27 or were increased in patients with AML compared with HDs.28 We could not identify any prior studies that specifically compared T‐cell distribution or multiple checkpoint receptor expression in BM samples from HDs with samples from patients who had AML. Our data indicating preservation of the T‐cell population in AML BM is important and is in line with the recently demonstrated clinical efficacy of therapies that depend on a preserved BM T‐cell population to generate response, such as bispecific antibodies, immune checkpoint inhibitors, and autologous chimeric antigen receptor therapies.9, 11, 29, 30
When we assessed for how this population of T cells was divided into different subsets by MFC gating on CD45hi, nonleukemic cells, we observed that the frequency of CD8‐positive/CD4‐positive Teff‐cell and Treg subsets in BMAs from patients with AML were higher compared with those from HDs. Consistent with previously published data from solid tumors,31 our findings demonstrate that the distribution of T‐cell subsets and the expression of immune checkpoint receptors in PB were not representative of the findings in BM among patients with AML. This suggests that BMAs/BM biopsy samples, and not PB samples, should be used for monitoring immune markers in immune‐based clinical trials. We noted an increased frequency of OX40‐positive and PD1‐positive/CD8‐positive T cells and CD4‐positive Teff cells in BMAs from patients with AML compared with BMAs from HDs. OX40 also was expressed more frequently on Tregs in BMAs from patients with AML.
PD1, CTLA4, TIM3, and LAG3 are co‐inhibitory receptors that, when activated, dampen the function of Teff cells. Blockage of these receptors by antagonist antibodies enhances T‐cell function by “removing the brakes.” OX40, ICOS, GITR, and 41BB are costimulatory receptors that, when activated, enhance Teff‐cell activation. Activation of these receptors by agonist antibodies enhances T‐cell function by “pressing the accelerator.” The expression of OX40, PD1, TIM3, and LAG3 by T cells in the BM from patients with AML, as noted in the current study, suggests that antigen‐experienced T cells are infiltrating the AML BM. Increased ICOS expression on CD4‐positive and CD8‐positive cells in patients with relapsed AML further indicates that BM infiltration activates T cells, because ICOS is a marker of activation.
Several of these immune markers are clinically targetable with available or emerging agents. OX40 and PD1 are expressed by T cells after CD28 activation and are being evaluated as therapeutic targets in clinical trials or already have been approved in solid and hematologic malignancies.32, 33, 34, 35, 36, 37 Targeting PD1 in combination with other checkpoints, particularly CTLA4, may be attractive because of the potential for synergy with dual checkpoint blockade, as has been demonstrated clinically in melanoma and renal cell carcinoma.38, 39, 40 Dual checkpoint inhibitor therapy with PD1 and CTLA4 inhibition before ASCT (NCT02397720) and after ASCT (NCT03600155 and NCT01822509) in relapsed and high‐risk AML, and with PD1 and TIM3 inhibition (NCT03066648) in relapsed AML, are being evaluated in clinical trials for patients with AML. There is preclinical41, 42 and early clinical43 evidence that signaling through OX40 promotes CD4‐positive and CD8‐positive T‐cell survival while concomitantly inhibiting the differentiation and function of Tregs by preventing FoxP3 expression,44, 45, 46 making OX40 an attractive therapeutic target.43, 47 OX40 was differentially expressed in relapsed/new AML compared with HDs and demonstrated a bimodal distribution, with significantly higher expression of OX40 on CD8‐positive T cells and Tregs in some patients, suggesting that these patients may benefit most from OX40 agonists. A clinical trial evaluating the OX40 agonist PF‐04518600 as a single agent and in various combinations with hypomethylating agents, PD1 antibodies, and 41BB agonists in patients with AML is ongoing (NCT03390296).
PD1‐positive/TIM3‐positive and PD1‐positive/LAG3‐positive cell populations have been associated with immune exhaustion and relapse post‐ASCT in patients with AML17, 22 and appear to annotate an antigen‐experienced population in patients with AML.23 Together, these data suggest that a proportion of the T cells that infiltrate the AML BM may be doing so as part of an endogenous immune response against leukemia. This double‐positive T‐cell population was particularly evident in the BMAs from patients with multiply relapsed AML, likely because of advanced immune exhaustion from persistent antigen stimulation or multiple prior AML therapies.17, 22 Although blocking PD1 and TIM3 has demonstrated promise in animal models of AML,22, 30 to date, there is no clinical evidence that TIM3 blockade can enhance or rescue resistance to PD1 blockade in patients with solid and hematologic malignancies. On the basis of our experience, we believe that there are 2 populations of patients with AML: those who have an inflamed/exhausted immune microenvironment and those who do not, as depicted in Figure 4 by the presence of 2 populations of exhausted T cells that coexpress LAG3 or TIM3 with PD1 on the surface of their Teff cells in some patients with AML, but not in others. It is likely that patients with a higher frequency of double‐positive T cells are those who are less likely to respond to single immune checkpoint inhibition, because they probably have immune exhaustion at multiple levels, which may be difficult to overcome with PD1 and/or CTLA‐4 therapy alone. Such “exhausted” patients either may not be ideal candidates for T‐cell harnessing immunotherapies or, alternatively, may require dual checkpoint blockade or dual immune modality therapy to overcome immune exhaustion.
We observed that the frequency of Tregs in BMAs from patients with AML was significantly higher than that in BMAs from HDs. The frequency of Tregs in AML BMAs progressively increased with the number of relapses. We also noted that the frequency of PD‐L1–positive blasts was higher in patients who had TP53‐mutated AML compared with those who had TP53 wild‐type AML. It is known that TP53 loss induces PD‐L1 expression indirectly, because p53 induces microRNA‐34 (miR‐34) expression and miR‐34 binds to the 3′‐untranslated region of PD‐L1 to inhibit PD‐L1 expression,48 In a study that targeted miR‐34 in a syngeneic nonsmall cell lung cancer model, the authors demonstrated that p53 loss induced PD‐L1 expression and that restoring miR‐34 restored immunogenicity by reducing PD‐L1 expression, with resulting CD8‐positive T‐cell infiltration and increased circulating interferon‐γ.48 Likewise, eliminating miR‐34 resulted in PD‐L1 expression in AML.49 We believe that this may be a possible explanation for the increased PD‐L1 in patients with TP53‐mutated AML. P53 is known to induce the expression of ERAP1 (endoplasmic reticulum aminopeptidase 1), a key protein involved in antigen processing, as well as major histocompatibility class I; and cells infected by HPV are known to be resistant to interferon signaling.50 These are key mechanisms that have been associated with acquired resistance to immune checkpoint blockade in patients. It remains to be determined in clinical trials whether or not the increased expression of PD‐L1 in patients with TP53‐mutated AML will translate into higher sensitivity and better responses to PD1/PD‐L1–based therapies.
To overcome the multilayered immune suppression observed seen in AML, patients may need combinations of immune checkpoint antibodies, checkpoint antibodies with BiTE (bi‐specific T‐cell engager) antibodies or HMAs, or strategies that include external supplementation with activated T cells, such as chimeric antigen receptor (CAR)‐T cells. One such strategy that has been evaluated in the clinic with early encouraging results is combining HMAs with immune checkpoint inhibitors in patients who have MDS/AML.11, 12, 51
Our current study has limitations. This was a set of 107 nonselected patients who received different modalities of treatment, such as HMA‐based, cytotoxic, targeted therapies and investigational therapies. The study included frontline and relapsed patients with AML. Therefore, the presence or absence of MRD during and after therapy varied based on the treatment modality received by individual patients. Furthermore, the immune profiling was only done at 1 time point, and not sequentially, in these patients. Because of these Treg factors, an assessment for correlations between Treg frequency and MRD status in these patients was not possible. Another limitation of our study was that the data focused on checkpoint receptor and ligand expression on T lymphocytes and AML blasts in BMAs from patients with AML. Possible contributions by natural killer cells and other myeloid subsets, such as macrophages, monocytes, myeloid‐derived suppressor cells, or other components of the tumor microenvironment, were not analyzed for this report, but these efforts are underway in our institution and in other groups using mass cytometry approaches. We evaluated how the distribution of T‐cell subsets, the expression of individual checkpoint receptors on total CD3‐positive and T‐cell subsets, and the expression of individual ligands on AML blasts could have an impact on the survival of patients, and we did not identify any clinically meaningful associations. This result must be interpreted cautiously, because our data only provide a snapshot in time without longitudinal follow‐up on these patients. Therefore, we cannot conclusively determine whether the increased frequency of Tregs or PD1/TIM3 double‐positive T‐cell subsets facilitated an AML relapse or indeed represented secondary changes in T‐cell biology as a consequence of the relapse. In addition, the patients received various different therapies on different clinical trials ongoing at our institution in that timeframe. Thus, as we learn more about the associations between the immune microenvironment and AML biology, the information gained may help guide treatments and potentially allow for the personalized selection of immune checkpoint pathways to target in a given patient with AML.
Funding Support
This work was supported in part by The University of Texas MD Anderson Cancer Center Leukemia Support Grant (CA016672), The University of Texas MD Anderson Cancer Center Dick Clark Leukemia Specialized Programs of Research Excellence (SPORE) grant (CA100632), the National Institutes of Health (T32 grant CA009666), the Charif Souki Cancer Research Fund, the Intramural Research Program of the National Heart, Lung, and Blood Institute of the National Institutes of Health, and generous philanthropic contributions to The MD Anderson Moon Shots Program.
Conflict of Interest Disclosures
Christopher S. Hourigan reports laboratory research funding from Merck and Sellas outside the submitted work. James P. Allison reports consulting fees from and stock ownership in Jounce, Kite Pharma, Neon, Amgen, Forty‐Seven, Apricity, Polaris, Marker Therapeutics, Codiak, BioAlta, ImaginAB, Tvardi Therapeutics, and TapImmune outside the submitted work; patents issued with The Regents of the University of California for “Blockade of Lymphocyte Down‐Regulation Associated With CTLA‐4 Signaling” (US5811097 A, US5855887 A, US6051227 A, and US7229628 B1), “Stimulation of T Cells Against Self Antigens Using CTLA‐4 Blocking Agents” (US20060034844 A1), “Diagnosis of Prostate Cancer with SPAS‐1 Cancer Antigen (US7704701 B2), and “Methods and Compositions for Localized Secretion of Anti‐CTLA‐4 Antibodies” (US9868961 B2); a patent issued with Biosante Pharmaceuticals and The Regents of the University of California for “Cancer Immunotherapy Compositions and Methods of Use” (US7919079 B2); a patient issued with the Icahn School of Medicine at Mount Sinai and Memorial Sloan Kettering Cancer Center for “Newcastle Disease Viruses and Uses Thereof” (US20160015760 A1); a patent issued with the Board of Regents of The University of Texas System and Memorial Sloan Kettering Cancer Center for “Combination Immunotherapy for the Treatment of Cancer (US9375475 B2); a patent issued with Albert Einstein College of Medicine Inc and the Sloan‐Kettering Institute for Cancer Research for “Antibodies to Human B7X for Treatment of Metastatic Cancer” (US9447186 B2); a patent issued for “SPAS‐1 Cancer Antigen (US20020150588 A1); a patent issued for “Compositions and Methods for Modulating Lymphocyte Activity (US20040175380 A1); a patent pending with The Regents of the University of California for “Stimulation of T Cells Against Self Antigens Using CTLA‐4 Blocking Agents” (US20090269353 A1); and a patent pending for “Multi‐Antigen Immunotherapy for Melanoma and Prostate Cancer.” Padmanee Sharma reports consulting fees from and stock ownership in Jounce, Neon, Constellation, Oncolytics, BioAlta, Forty‐Seven, Apricity, Polaris, Marker Therapeutics, and Codiak outside the submitted work and consulting fees from Kite Pharma, Pieris, Merck, and BioMx outside the submitted work. The remaining authors made no disclosures.
Author Contributions
Patrick Williams: Monitored the patients, curated the data, reviewed the data, performed the formal analysis, writing–initial draft, writing–review and comments/revisions, and approved the final version. Sreyashi Basu: Curated the data; reviewed the data; performed the flow‐cytometry, molecular, and cytogenetic analyses; performed the formal analysis; writing–initial draft; writing–review and comments/revisions; and approved the final version. Guillermo Garcia‐Manero: Treated patients, writing–review and comments/revisions, and approved the final version. Christopher S. Hourigan: Performed the immunohistochemistry on bone marrow biopsies and quantified the CD34 and CD3 infiltrates, writing–review and comments/revisions, and approved the final version. Karolyn A. Oetjen: Performed the immunohistochemistry on bone marrow biopsies and quantified the CD34 and CD3 infiltrates, writing–review and comments/revisions, and approved the final version. Jorge E. Cortes: Treated patients, writing–review and comments/revisions, and approved the final version. Farhad Ravandi: Treated patients, writing–review and comments/revisions, and approved the final version. Elias J. Jabbour: Treated patients, writing–review and comments/revisions, and approved the final version. Zainab Al‐Hamal: Curated the data; performed the flow‐cytometry, performed the formal analysis; writing–review and comments/revisions; and approved the final version. Marina Konopleva: Treated patients, writing–review and comments/revisions, and approved the final version. Jing Ning: Curated the data, performed the formal analysis, writing–review and comments/revisions, and approved the final version. Lianchun Xiao: Curated the data, performed the formal analysis, writing–review and comments/revisions, and approved the final version. Juliana Hidalgo Lopez: Curated the data; performed the flow‐cytometry, molecular, and cytogenetic analyses; performed the formal analysis; writing–review and comments/revisions; and approved the final version. Steve M. Kornblau: Treated patients, writing–review and comments/revisions, and approved the final version. Michael Andreeff: Treated patients, writing–review and comments/revisions, and approved the final version. Wilmer Flores: Curated the data, performed the formal analysis writing–review and comments/revisions, and approved the final version. Carlos Bueso‐Ramos: Curated the data; performed the flow‐cytometry, molecular, and cytogenetic analyses; performed the formal analysis; writing–review and comments/revisions; and approved the final version. Jorge Blando: curated the data, performed the formal analysis, writing–review and comments/revisions, and approved the final version. Pallavi Galera: Performed the immunohistochemistry on bone marrow biopsies and quantified the CD34 and CD3 infiltrates, writing–review and comments/revisions, and approved the final version. Katherine R. Calvo: Performed the immunohistochemistry on bone marrow biopsies and quantified the CD34 and CD3 infiltrates, writing–review and comments/revisions, and approved the final version. Gheath Al‐Atrash: Treated patients, writing–review and comments/revisions, and approved the final version. James P. Allison: Curated the data; performed the flow‐cytometry, molecular, and cytogenetic analyses; performed the formal analysis; writing–review and comments/revisions; and approved the final version. Hagop M. Kantarjian: Treated patients, writing–review and comments/revisions, and approved the final version. Padmanee Sharma: Curated the data; reviewed the data; performed the flow‐cytometry, performed the formal analysis; writing–initial draft; writing–review and comments/revisions; and approved the final version. Naval G. Daver: Treated patients, monitored the patients, curated the data, reviewed the data, performed the formal analysis, writing–initial draft, writing–review and comments/revisions, and approved the final version.
Supporting information
See editorial on pages 1410‐3, this issue.
Contributor Information
Padmanee Sharma, Email: padsharma@mdanderson.org.
Naval G. Daver, Email: ndaver@mdanderson.org.
References
- 1. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med. 2015;373:1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dubey C, Croft M, Swain SL. Naive and effector CD4 T cells differ in their requirements for T cell receptor versus costimulatory signals. J Immunol. 1996;157:3280‐3289. [PubMed] [Google Scholar]
- 4. Ansell SM, Lesokhin AM, Borrello I, et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Younes A, Santoro A, Shipp M, et al. Nivolumab for classical Hodgkin's lymphoma after failure of both autologous stem‐cell transplantation and brentuximab vedotin: a multicentre, multicohort, single‐arm phase 2 trial. Lancet Oncol. 2016;17:1283‐1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herbaux C, Gauthier J, Brice P, et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood. 2017;129:2471‐2478. [DOI] [PubMed] [Google Scholar]
- 7. Westin JR, Chu F, Zhang M, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open‐label, phase 2 trial. Lancet Oncol. 2014;15:69‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B‐cell lymphoma. Blood. 2017;130:267‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375:143‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Albring JC, Inselmann S, Sauer T, et al. PD‐1 checkpoint blockade in patients with relapsed AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2017;52:317‐320. [DOI] [PubMed] [Google Scholar]
- 11. Daver N, Basu S, Garcia‐Manero G, et al.Phase IB/II study of nivolumab in combination with 5‐azacytidine (Aza) in patients (pts) with relapsed acute myeloid leukemia (AML) [abstract S474]. Paper presented at: 22nd Congress of the European Hematology Association; June 22–25, 2017; Madrid, Spain.
- 12. Garcia‐Manero G, Daver N, Montalban‐Bravo G, et al.An update of a phase II study of nivolumab (Nivo) or ipilimumab (Ipi) with azacytidine in patients with previously treated or untreated myelodysplastic syndrome (MDS) [abstract S487]. Paper presented at: 22nd Congress of the European Hematology Association; June 22–25, 2017; Madrid, Spain.
- 13. Szczepanski MJ, Szajnik M, Czystowska M, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. 2009;15:3325‐3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Zheng J, Liu J, et al. Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur J Haematol. 2005;75:468‐476. [DOI] [PubMed] [Google Scholar]
- 15. Chen X, Liu S, Wang L, Zhang W, Ji Y, Ma X. Clinical significance of B7–H1 (PD‐L1) expression in human acute leukemia. Cancer Biol Ther. 2008;7:622‐627. [DOI] [PubMed] [Google Scholar]
- 16. Yang H, Bueso‐Ramos C, DiNardo C, et al. Expression of PD‐L1, PD‐L2, PD‐1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28:1280‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kong Y, Zhang J, Claxton DF, et al. PD‐1(hi)TIM‐3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation [serial online]. Blood Cancer J. 2015;5:e330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Q, Munger ME, Highfill SL, et al. Program death‐1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116:2484‐2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L, Gajewski TF, Kline J. PD‐1/PD‐L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545‐1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang C, Welsh JW, de Groot P, et al. Ipilimumab with stereotactic ablative radiation therapy: phase I results and immunologic correlates from peripheral T cells. Clin Cancer Res. 2017;23:1388‐1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luthra R, Patel KP, Reddy NG, et al. Next‐generation sequencing‐based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99:465‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Q, Munger ME, Veenstra RG, et al. Coexpression of Tim‐3 and PD‐1 identifies a CD8+ T‐cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117:4501‐4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gros A, Robbins PF, Yao X, et al. PD‐1 identifies the patient‐specific CD8(+) tumor‐reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246‐2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schnorfeil FM, Lichtenegger FS, Emmerig K, et al. T cells are functionally not impaired in AML: increased PD‐1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment [serial online]. J Hematol Oncol. 2015;8:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wendelbo O, Nesthus I, Sjo M, Paulsen K, Ernst P, Bruserud O. Functional characterization of T lymphocytes derived from patients with acute myelogenous leukemia and chemotherapy‐induced leukopenia. Cancer Immunol Immunother. 2004;53:740‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lamble A, Kosaka Y, Huang F, et al. Mass cytometry as a modality to identify candidates for immune checkpoint inhibitor therapy within acute myeloid leukemia [abstract]. Blood. 2016;128:2829. [Google Scholar]
- 27. Vidriales MB, Orfao A, Lopez‐Berges MC, et al. Lymphoid subsets in acute myeloid leukemias: increased number of cells with NK phenotype and normal T‐cell distribution. Ann Hematol. 1993;67:217‐222. [DOI] [PubMed] [Google Scholar]
- 28. Le Dieu R, Taussig DC, Ramsay AG, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. 2009;114:3909‐3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uy GL, Godwin J, Rettig MP, et al. Preliminary results of a phase 1 study of flotetuzumab, a CD123 X CD3 bispecific dart protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome [abstract]. Blood. 2017;130:637. [Google Scholar]
- 30. Budde L, Song JY, Kim Y, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123‐specific CAR T cells: a first‐in‐human clinical trial [abstract]. Blood. 2017;130:811. [Google Scholar]
- 31. Gros A, Parkhurst MR, Tran E, et al. Prospective identification of neoantigen‐specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22:433‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366:2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iwai Y, Terawaki S, Honjo T. PD‐1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133‐144. [DOI] [PubMed] [Google Scholar]
- 35. Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682‐687. [DOI] [PubMed] [Google Scholar]
- 36. Velu V, Titanji K, Zhu B, et al. Enhancing SIV‐specific immunity in vivo by PD‐1 blockade. Nature. 2009;458:206‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blank C, Brown I, Peterson AC, et al. PD‐L1/B7H‐1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140‐1145. [DOI] [PubMed] [Google Scholar]
- 38. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:1270‐1271. [DOI] [PubMed] [Google Scholar]
- 39. Wolchok JD, Chiarion‐Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal‐cell carcinoma. N Engl J Med. 2018;378:1277‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68:5206‐5215. [DOI] [PubMed] [Google Scholar]
- 42. Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Curti BD, Kovacsovics‐Bankowski M, Morris N, et al. OX40 is a potent immune‐stimulating target in late‐stage cancer patients. Cancer Res. 2013;73:7189‐7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zou W, Chen L. Inhibitory B7‐family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467‐477. [DOI] [PubMed] [Google Scholar]
- 45. Boussiotis VA. Molecular and biochemical aspects of the PD‐1 checkpoint pathway. N Engl J Med. 2016;375:1767‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T‐cell biology and immune disease. Immunol Rev. 2009;229:173–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Montler R, Bell RB, Thalhofer C, et al. OX40, PD‐1 and CTLA‐4 are selectively expressed on tumor‐infiltrating T cells in head and neck cancer [serial online]. Clin Transl Immunol. 2016;5(e70):2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cortez MA, Ivan C, Valdecanas D, et al. PDL1 regulation by p53 via miR‐34 [serial online]. J Natl Cancer Inst. 2016;108:djv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Li J, Dong K, et al. Tumor suppressor miR‐34a targets PD‐L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015;27:443–452. [DOI] [PubMed] [Google Scholar]
- 50. Hebner C, Beglin M, Laimins LA. Human papillomavirus E6 proteins mediate resistance to interferon‐induced growth arrest through inhibition of p53 acetylation. J Virol. 2007;81:12740‐12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daver N, Boddu P, Garcia‐Manero G, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia. 2018;32:1094‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials