

Summary
Deletion of the gene encoding the chromatin remodeler CHD1 is among the most common alterations in prostate cancer (PCa); however, the tumor suppressive functions of CHD1 and reasons for its tissue-specific loss remain undefined. We demonstrated that CHD1 occupied prostate-specific enhancers enriched for the androgen receptor (AR) and lineage-specific cofactors. Upon CHD1 loss, the AR cistrome was redistributed in patterns consistent with the oncogenic AR cistrome in PCa samples and drove tumor formation in the murine prostate. Notably, this cistrome shift was associated with a unique AR transcriptional signature enriched for pro-oncogenic pathways unique to this tumor subclass. Collectively, these data credential CHD1 as a tumor suppressor in the prostate that constrains AR binding/function to limit tumor progression.
Keywords: Prostate cancer, Androgen Receptor, Prostate cancer subclass, AR, CHD1, epigenetics, HOXB13, Chromatin remodeling, Cistrome reprogramming, Interactome
Graphical Abstract
Introduction
Precise regulation of chromatin signaling is essential for virtually all cellular processes, and chromatin dysregulation has recently emerged as a hallmark of cancer (Kadoch et al., 2013; Shen and Laird, 2013). In prostate cancer (PCa), alterations in epigenetic modifiers and chromatin remodelers are prevalent across the spectrum of the disease (Barbieri et al., 2012; Cancer Genome Atlas Research, 2015; Grasso et al., 2012; Kumar et al., 2011; Taylor et al., 2010). The most common of these is recurrent deletion of CHD1, encoding chromodomain helicase DNA-binding protein 1 (CHD1), that occurs in approximately 15% of primary human PCa (Barbieri et al., 2012; Barbieri and Tomlins, 2014; Cancer Genome Atlas Research, 2015; Kumar et al., 2011; Liu et al., 2012; Wedge et al., 2018). In normal tissue, CHD1 is ubiquitously expressed and functions as an ATP-dependent chromatin remodeler, localizing to promoters of actively transcribed genes marked by trimethylation of histone H3K4 (H3K4me3) and initiates nucleosome turnover to facilitate transcriptional initiation (de Dieuleveult et al., 2016; Flanagan et al., 2005; Lusser et al., 2005; Morettini et al., 2011; Siggens et al., 2015; Simic et al., 2003; Skene et al., 2014). Phenotypically, germline disruption of Chdl in mice is embryonically lethal, and it is required for maximally efficient transcription, cellular differentiation, and growth (Baumgart et al., 2017; Gaspar-Maia et al., 2009; Guzman-Ayala et al., 2015; Koh et al., 2015; Piatti et al., 2015). However, how disruption of these functions contributes to neoplastic formation within the prostate remains undefined.
Results
Chdl limits prostate tumorigenesis in vivo
Previous reports have demonstrated that homozygous deletion of CHD1 is frequently observed in PCa (Barbieri et al., 2012; Barbieri and Tomlins, 2014; Cancer Genome Atlas Research, 2015; Taylor et al., 2010), which we validated in our patient cohort (Figure 1A). Importantly, loss of CHD1 was largely restricted to tumors of prostate lineage (Figure 1A), rarely deleted in other cancer types (Cancer Genome Atlas Research, 2015), suggesting prostate-specific tumor suppressive functions for CHD1. We therefore crossed mice carrying a Chdl floxed allele (Chd1f/f) with Pb-Cre4 mice (Wu et al., 2001) to specifically delete Chdl in prostate epithelial cells to define the impact of its loss on prostate pathobiology (Figure 1B). At 1 year, there were no observed differences in histopathology, AR expression, or glandular structure due to Chdl homozygous loss (Figure 1C; Figure S1A). This is consistent with previous reports of CHD1 function in prostate tissue (Shenoy et al., 2017), and suggest that, like other well-established drivers of PCa in human disease such as ERG (Chen et al., 2013) and ETV1 (Baena et al., 2013), deregulation of Chdl alone is insufficient to drive tumorigenesis in the mouse prostate.
Figure 1.

Genomic loss of Chdl drives progression of prostate carcinoma in vivo. (A) CHD1 homozygous deletions in the TCGA database (The Cancer Genome Atlas Research, 2015) (Histogram) and Weill Cornell Institute for Precision Medicine database (pie charts) for all available samples ((Pauli et al., 2017) and in preparation). (B) Schematic of inducible Chdl knockout in the murine prostate. (C) H&E staining of prostates from Chd1+/+ and Chd1f/f mice in a Pb-Cre background at 1 year (50 μm scalebar). (D) Schematic of castration-testosterone re-supplementation in Chd1+/+ (n = 4) and Chd1f/f (n = 6) mice (top) and quantification of Ki67 staining after testosterone (T-pellet) re-supplementation (bottom) (50 μm scalebar). Data represent +/− SEM. (E) H&E staining of murine prostates at 1 year (50 μm scalebar). (F) Quantification of pathological features per genotype at 1 year. (G) Prostatic mass from 1 year old mice (sum of all lobes). Boxes represent the mean and interquartile range, with min and max values indicated. See also Figure S1.
Given the importance of androgen signaling in this tissue, we next characterized the impact of Chdl loss on androgen driven regrowth of prostate tissue after castration. Mice were castrated for 2 weeks prior to subcutaneous implantation of testosterone pellets. Following an additional 2 weeks for prostatic regrowth, tissues were assessed for pathological phenotypes (Figure 1D; Figure S1B-C). Both Chdl+/+ and Pb-Cre;Chdlf/f mice responded similarly to castration, exhibiting prostate regression, diminished gland size, and cytoplasmic staining of AR (Figure S1B). In response to androgen re-stimulation, both genotypes were capable of regenerating luminal structures with strong nuclear AR staining (Figure S1B); however, Chdl deficient mice showed increased proliferation (Ki67 staining) in regenerated epithelium (Figure 1D, Figure S1C). These data demonstrate that deletion of Chdl in prostatic tissue may lead to neoplastic phenotypes that are androgen-dependent.
PTEN is a well-characterized tumor suppressor in PCa, and acceleration of neoplastic phenotypes in Pten deleted mouse prostates have been considered key “base models” to examine the tumor suppressive functions of other genomic alterations (Grabowska et al., 2014). In order to define the impact of Chdl loss in promoting/accelerating pathological phenotypes we crossed Chdlf/f mice with mice in a conditional Pten deleted background. At one year, while heterozygous loss of Pten alone has a minimal phenotype in the murine prostate (Trotman et al., 2003; Wang et al., 2003), Pb-Cre4;Chdlf/f;Ptenf/+ demonstrated a high penetrance of focal high-grade prostatic intraepithelial neoplasia (HG-PIN) (Figure 1E–1F), with high levels of AR and elevated proliferative indices (Figure S1D–S1E). Furthermore, while homozygous loss of Pten alone in the murine prostate resulted in diffuse HG-PIN (Trotman et al., 2003), prostate lesions in Pb-Cre4;Chdlf/f;Ptenf/f frequently progressed to invasive carcinoma by 1 year of age (Figure 1E–1G). Histologically, these prostates were generally poorly differentiated, showed elevated proliferative indices, and remained AR positive (Figure 1E; Figure S1E–S1F). Together, these results demonstrate that Chdl loss cooperates with established drivers of PCa to promote tumor development in vivo, and provide evidence to credential Chdl as a bona-fide tumor suppressor in prostate tissue.
CHD1 localizes to the promoters of actively transcribed genes in PCa models
To uncover the mechanisms responsible for the tumor suppressive functions of CHD1, the cistrome of CHD1 and transcriptionally active (H3K4me3) and repressive (H3K27me3) histone marks were defined in AR positive PCa cells (Figure 2A; Figure S2A–S2B). Consistent with its known function, CHD1 peaks frequently overlapped with H3K4me3, and largely independent of the repressive H3K27me3 cistrome (Figure 2A–2B). These binding patterns were consistent across ~21,000 Ref-Seq genes, where CHD1 co-localized near the promoters of actively transcribed genes marked by H3K4me3, independent of H3K27me3 (Figure 2C–2E). CHD1 binding was enriched (along with H3K4me3) near the +1 nucleosome position (Figure 2D), and positively correlated with elevated transcript abundance (Figure 2F). These findings suggest that the canonical, promoter-specific role of CHD1 is conserved in prostate models and indicate that the mechanisms underlying the tumor suppressive action of CHD1 are likely independent of these functions.
Figure 2.

CHD1 co-localizes to the promoters of actively transcribed genes in PCa models. (A) Overlap of CHD1, H3K4me3, and H3K27me3 ChIP Seq peaks from LNCaP cells under androgen proficient conditions. (B) Representative binding of each factor on chromatin. (C) Heatmap of binding patterns for each factor from the Transcriptional Start Site (TSS) to the Transcriptional End Site (TES) of ~21000 RefSeq genes (gene body scaled to 3 kb, 500 bp before/after TSS/TES). (D) Average signal of H3K4me3 and CHD1 centered at the TSS of co-bound genes (normalized by RPKM). (E) Signal of CHD1 at all H3K4me3 or H3K27me3 peaks. (F) Ranked normalized RNA Seq transcripts obtained from androgen proficient conditions (GSE43785 (Chen et al., 2015)) ordered from most abundant (top) to least abundant (bottom), and the signal of CHD1, H3K4me3, and H3K27me3 are plotted for each gene centered around the TSS. 2-tailed spearman correlations are reported for each comparison; p < 0.0001 for all correlations. See also Figure S2.
The CHD1 interactome is enriched for nuclear receptor cofactors
To further characterize the function of CHD1 in prostate tissue, the chromatin-bound interactome of CHD1 was defined using Rapid Immunoprecipitation and Mass-spectrometry of Endogenous Peptides (RIME) (Mohammed et al., 2016; Stelloo et al., 2018a), with the interactome of H4K4me3 used as a promoter-specific control. Consistent with the overlap in their cistromes, 97 out of 293 (~33%) of the CHD1 interactome was shared with that of H3K4me3 and was enriched for proteins known to localize to promoters (e.g., POLR2A and POLR2B) (Table S1; Table S2). Importantly, a large portion of the CHD1 interactome was unique to CHD1, and was enriched for transcriptional regulators reported to affect nuclear receptor function (Figure 3A; Table S1; Table S2) (Gottlieb et al., 2012; Mooslehner et al., 2012; Stelloo et al., 2018a). While the CHD1 interactome did not contain nuclear receptors directly, the RIME-derived interactome of AR significantly overlapped with that of CHD1 (Figure 3A–3B), sharing 45% (33/73) of its chromatin-bound cofactors with CHD1 (Figure 3B). Consistent with this observation, Proximity Ligation Assays (PLA) confirmed that CHD1 and AR associate in nuclear foci on chromatin in an androgen-dependent manner (Figure 3C). These observations define the promoter-independent interactome of CHD1 and implicate CHD1 as a potential regulator of lineage-specific nuclear receptor activity.
Figure 3.

The CHD1 interactome is enriched for AR cofactors. (A) RIME was performed for H3K4me3 and CHD1 in androgen proficient conditions. Signal to noise ratios of significantly enriched peptides from H3K4me3 were subtracted from those of CHD1 and plotted by biological replicate. Orange: peptides unique to H3K4me3; Blue: unique to CHD1; Purple: common. Green: overlap with known AR interactors (Gottlieb et al., 2012; Mooslehner et al., 2012). (B) STRING analysis of AR (Stelloo et al., 2018a) and CHD1 interactomes in LNCaP cells. (C) Proximity Ligation Assay (PLA) for AR and CHD1 in LNCaP cells (left) and quantification of PLA signal from a single plane, plotted as the number of detected foci per cell (right). One-way Anova, Tukey’s multiple comparisons test: ** p < 0.01, **** p < 0.00001. Data represent +/− SEM. Scalebar 20 μm. See also Table S1–S2.
CHD1 colocalizes to enhancers enriched for AR and its cofactors
Nuclear receptors (including AR) bind enhancers across the genome at intronic and intergenic sites, largely independent of promoters (Kininis and Kraus, 2008). While the canonical functions of CHD1 have been described at promoters, its interactome further suggests a role in regulating nuclear receptor activity at enhancers. To define promoter-independent roles of CHD1, we annotated the CHD1 and H3K4me3 cistromes in prostate models. Approximately 20% of all CHD1 peaks were shared with H3K4me3, which localized at or near the promoters of actively transcribed genes (Figure 4A). Notably, ~80% of the CHD1 cistrome was found to be independent of direct H3K4me3 binding; these regions were heavily enriched for intronic/intergenic DNA and lacked promoter-specific regions (Figure 4A). Although no specific motifs were highly enriched across all CHD1 peaks (Figure S3), an unbiased motif analysis of CHD1 promoter-independent peaks revealed enrichment of motifs implicated in AR signaling (e.g. Androgen Response Element-ARE, FOXA1, and HOXB13) (Figure 4B). Furthermore, these enhancer-specific binding events were highly concordant with those of the AR cistrome, as ~40% of all high confidence AR peaks overlapped with that of CHD1, independent of H3K4me3 (Figure 4C). Strikingly, similar patterns of peak overlap were also seen between CHD1 and several established regulators of AR function, including FOXA1 (Zhao et al., 2016), HOXB13 (Pomerantz et al., 2015), and ETV1 (Chen et al., 2013) (Figure 4D-Top). Previous work characterizing the landscape flanking chromatin-bound AR described a bimodal distribution of nucleosome density defined by the active enhancer mark H3K4me2 that undergo active chromatin remodeling upon AR activation (He et al., 2010). As CHD1 has been shown to colocalize with H3K4me2 (Flanagan et al., 2005; Sims et al., 2005), we next analyzed the profile of CHD1 around AR and cofactors for evidence of nucleosome remodeling. Within a 2 kb window, the binding profile of CHD1 density around AR, FOXA1, HOXB13, and ETV1 was consistent with the bimodal nucleosome distribution exhibited by AR-bound, enhancer-associated chromatin (Figure 4D-Bottom). Together, these data define an enhancer-specific role for CHD1 in the prostate and nominate CHD1 as a context dependent chromatin remodeler for a subset of AR binding sites.
Figure 4.

CHD1 colocalizes to enhancers enriched for AR and its cofactors. (A) Overlap of CHD1 and H3K4me3 ChIP Seq peaks in LNCaP cells under androgen proficient conditions (top) and annotation of the common and unique peaks for H3K4me3 and CHD1 plotted as a percentage of all peaks (bottom). (B) Motif analysis of CHD1 peaks binned into promoter/TSS or intronic/intergenic categories (200 bp window). (C) Overlap of CHD1, AR, and H3K4me3 ChIP Seq peaks (top) and snapshot of AR, CHD1, and H3K4me3 signal at the KLK3 locus (bottom). (D) Overlap between CHD1 and AR, HOXB13 (Pomerantz et al., 2015), ETV1 (Chen et al., 2013), or FOXA1 (Zhao et al., 2016) ChIP Seq peaks in LNCaP cells under androgen-proficient conditions (top) and normalized average signal profile of CHD1 and each transcription factor at the peak center (bottom). See also Figure S3.
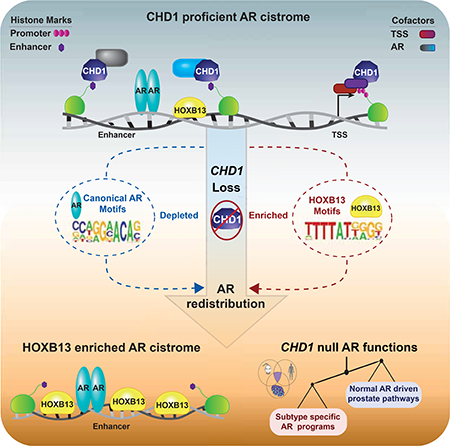
CHD1 loss redistributes the AR cistrome to HOXB13 enriched sites
In normal prostate epithelial tissue, AR serves to limit cell growth and promote luminal differentiation (Litvinov et al., 2003). During cancer pathogenesis the AR cistrome is expanded and driven towards sites that govern oncogenic programs (Pomerantz et al., 2015). However, the underlying mechanisms responsible for such reprogramming remain incompletely understood. Given the role of CHD1 at AR-associated enhancers, we hypothesized that CHD1 loss would alter AR signaling in prostate cells. Accordingly, we deleted CHD1 via CRISPR in the LNCaP line (sgCHD1) and validated complete loss of CHD1 protein (Figure 5A; Figure S4A). In contrast to other cell types (Guzman-Ayala et al., 2015; Koh et al., 2015; Piatti et al., 2015), deletion of CHD1 did not impair cell growth or tumorigenicity in a prostate-specific background (Figure S4B–S4G). Importantly, these cells remained highly AR positive and sensitive to androgen withdrawal and AR antagonists (Figure 5A; Figure S4B–S4C), demonstrating they accurately recapitulate critical features of primary disease. Furthermore, while low dose androgen was sufficient to engage the cell cycle in both parental and sgCHD1 cells, sgCHD1 cells were resistant to the growth inhibitory effects of high dose androgen (greater than 1 nM) (Figure 5B; Figure S4H). It has been postulated that this growth suppressive phenotype (Gao et al., 2016; Roediger et al., 2014), is due to a potential restoration of a “normal-like” AR transcriptional program (Gao et al., 2016). This suggests that CHD1 deficient prostate cells are compromised in their ability to engage this growth-suppressive AR cistrome, consistent with the proliferative phenotype seen in Chdl null regenerating prostates (Figure 1D; S1A–S1C).
Figure 5.

AR binding is redistributed in the absence of CHD1. (A) Immunoblot (top) and immunofluorescence (bottom) of LNCaP CRISPR-Cas9 cell lines with Ctrl or CHD1 specific sgRNA. Scale bar 20 μm. (B) Growth curve of sgCtrl (left) and sgCHD1 cells (right) in response to increasing doses of dihydrotestosterone (DHT). Data represents +/− SEM. (C) ChIP Seq of differential AR binding between sgCtrl and sgCHD1. (D) RPKM normalized AR signal (sgCtrl signal subtracted from sgCHD1 signal) at sites enriched or depleted with CHD1 loss (left) and snapshots of sgCHD1 enriched and depleted AR peaks (right). (E) AR ChIP Seq from (GSE70079 (Pomerantz et al., 2015)) patient tumors centered at sgCHD1 enriched and depleted sites. (F) H3K27ac ChIP Seq from annotated primary PCa tumors (ERR3 59744 (Kron et al., 2017)) binned into CHD1 null and CHD1 WT categories. Average signal is plotted at sgCHD1 enriched and depleted sites. See also Figures S4 and S5.
Next, we directly assessed the impact of CHD1 loss on genome-wide localization of AR in sgCHD1 models. While loss of CHD1 did not impair the ability of AR to associate with chromatin globally, we detected roughly 21400 AR peaks that were differentially bound in the absence of CHD1 (Figure 5C; Figure S5A–S5C). These peaks were ~ equally distributed between enhanced (11193) and diminished (10278) AR binding, with nearly identical distribution across all genomic annotation classes except promoters, where enriched peaks were less abundant (1.6% vs 5.3%) (Figure 5C–5D; Figure S5D). To determine the relevance of this CHD1 null AR cistrome to human tumors, we examined AR occupancy at these sites in primary human PCa (n = 13) and normal prostate (n = 7) samples (Pomerantz et al., 2015). The tumor-associated AR cistrome mirrored that of the CHD1 null model, demonstrating elevated AR signal at enriched as compared to depleted sites (Figure 5E). This distinction was unique to the tumor cistrome of AR; the signal was universally weaker and no differences were observed between enriched or depleted sites in normal prostate tissue (Figure S5E). Similarly, epigenetic marks of active enhancers, H3K27ac, (Kron et al., 2017) were elevated at AR enriched sites in CHD1 null tumors compared with wild-type (WT) tumors, further supporting the relevance and subclass specificity of these sites (Figure 5F; Figure S5F–S5G). Thus, these data implicate CHD1 as a regulator of nuclear receptor occupancy on chromatin and identify a clinically relevant AR cistrome in CHD1 deficient prostate cells.
To more completely define the mechanistic basis of AR rewiring by CHD1 deletion, we analyzed the CHD1 null AR cistrome for motif enrichment. Within a 200 bp window, the HOXB13 motif was frequently observed within the enriched AR peaks, while AR Half-sites (a weaker AR binding motif containing only half of the canonical ARE) were significantly depleted (Figure 6A; Table S3). This motif pattern was highly consistent with the AR cistrome of human tumors, which demonstrated a shift away from mainly canonical ARE motifs and expanded to sites enriched for oncogenic AR cofactors (e.g. HOXB13) (Pomerantz et al., 2015) (Figures S6A, S6B). Other factors known to drive alternative AR binding patterns (e.g. FOXA1) were equally present in both datasets, suggesting that HOXB13 activity may underlie the divergent AR cistrome observed upon CHD1 loss. Indeed, HOXB13 occupancy at AR elevated sites was enriched in both human PCa tumors and LNCaP models (Pomerantz et al., 2015) (Figure 6B), validating that the motifs identified are actively bound HOXB13 sites. In order to determine the sufficiency of HOXB13 deregulation in promoting CHD1 null phenotypes, we next interrogated the AR cistrome in non-tumorigenic LHSAR prostate cells in the presence and absence of exogenous HOXB13 expression (Pomerantz et al., 2015). In control cells, AR was more frequently bound at sgCHD1 depleted sites, consistent with the maintenance of a normal AR program (Figure 6C-Left). However, upon HOXB13 induction, AR was redistributed to sgCHD1 enriched sites, phenocopying AR binding in the absence of CHD1 (Figure 6C-Right). To uncover how CHD1 regulates these phenotypes, we analyzed the motif enrichment of HOXB13 peaks overlapping with or independent of the CHD1 cistrome. Interestingly, co-bound peaks more frequently contained canonical AREs whereas CHD1 independent HOXB13 peaks were enriched for HOX and GATA motifs (Figure S6C). These CHD1 insensitive peaks contained nearly 1/3 of all AR peaks enriched in CHD1 null cells and were largely independent of the AR depleted sites within the same model (Figure S6C). Collectively these data suggest that CHD1 localizes to chromatin containing canonical AR binding sites, and upon its loss, AR is redistributed to regions enriched for other cofactors (e.g. HOXB13) that are insensitive to CHD1 activity. These data implicate HOXB13 as a driver of the CHD1 null AR cistrome.
Figure 6.

AR is enriched at HOXB13 sites in CHD1 null tumors. (A) Heatmap of differential motif enrichment between sgCHD1 enriched and depleted sites (left) and histogram of the average incidence of the HOXB13 (top) and AR-Halfsite (bottom) motifs from the peak center (right). (B) ChIP-seq for HOXB13 derived from LNCaP and primary human PCa (GSE70079 (Pomerantz et al., 2015)), was plotted at sgCHD1 enriched and depleted AR sites. (C) Average ChIP-seq signal for AR in control (left) and HOXB13 expressing (right) LHSAR cells at sgCHD1 enriched and depleted AR sites (GSE70079 (Pomerantz et al., 2015)). (D) AR ChIP Seq peaks from primary human prostate tumors (Stelloo et al., 2018b) binned into CHD1 deficient and CHD1 WT categories, merged, and assessed for overlap. (E) De novo motif analysis for CHD1 deficient tumor peaks (compared to CHD1 WT tumors) and enriched (blue) and de-enriched (red) motifs plotted according to rank and p value. Values next to motifs represent the best match motif score (out of 1). Gray indicates motifs with less significant p values. (F) ATAC-seq in the sgCHD1 isogenic model +/− 4 hr of DHT. 20k peaks/condition were randomly assessed for ARE and HOXB13 motif enrichment 100x. Plotted are the number of ARE (top) and HOXB13 (bottom) motifs per 20k peaks (left) and histogram of mean ARE and HOXB13 motif enrichments under DHT conditions (right). Data represent +/− SEM. (G) Immunoblot of LNCaP cells expressing HOXB13 or vector (left) and cell growth in androgen-depleted media in response to DHT (bottom). Data represent +/− SEM. See also Figure S6 and Table S3.
To validate the relevance of this shift in motif preference in human PCa samples, we analyzed the AR cistromes of primary PCa samples annotated for CHD1 genomic status (Stelloo et al., 2018b). Collectively, the number of AR binding sites was nearly equal between CHD1 deficient and CHD1 WT tumors (39745 vs. 36420) and a majority of these AR peaks were common between the two groups (Figure 6D), consistent with the AR cistromes in our isogenic LNCaP models of CHD1 loss (Figure 5C; S5B). Importantly, however, differential motif analyses of these collective peak sets uncovered a significant enrichment for HOXB13 and GATA, and a depletion of canonical ARE and AR-Halfsite motifs in CHD1 deficient tumors (Figure 6E). This pattern is highly consistent with the motif enrichment patterns uncovered in our preclinical CHD1 null models (Figure 6A) and provides compelling evidence to nominate CHD1 as a clinically relevant regulator of the AR cistrome.
To define how CHD1-regulated changes in chromatin accessibility relate to differential AR binding patterns across the genome, we performed ATAC-seq in the presence and absence of androgen in our isogenic models of CHD1 loss. As expected, ARE motifs were largely absent from ATAC peaks under androgen deprived conditions (Figure 6F). Upon androgen stimulation, AREs were enriched 2.7 fold in control cells, but only ~1.8 fold in the sgCHDl line (Figure 6F), consistent with changes in the AR cistrome upon CHD1 deletion. Parallel analyses of the HOXB13 motif showed an unexpected depletion (~25%) in control cells post-androgen stimulation (Figure 6F), with similar effects observed in ChIP seq datasets derived from prostate-specific active enhancer marks (H3K27ac and H3K4me2), as well as HOXB13 itself (Figure S6D–S6E). Importantly, sgCHD1 cells showed no significant reduction of HOXB13 motifs in ATAC peaks post-androgen stimulation (Figure 6F). Collectively, the motif enrichment patterns of CHD1 null open chromatin peaks mirrored those observed in the AR cistrome upon CHD1 deletion, suggesting that CHD1 is required for optimal chromatin remodeling of a subset of ARE and HOXB13 sites in response to androgen. These observations highlight a mechanism of AR rewiring in PCa and suggest that CHD1 is critical to maintain a chromatin state required for normal AR function.
Finally, to determine if HOXB13 deregulation was sufficient to phenocopy the CHD1 regulated growth effects in prostate cells, we overexpressed HOXB13 and assessed the impacts on androgen-driven growth. Therein, induction of HOXB13 was sufficient to phenocopy the biological effects of CHD1 deletion, promoting resistance to high dose androgens in a manner similar to CHD1 loss (Figure 6G). Together, these analyses implicate HOXB13 as a major driver of the CHD1 null AR cistrome, and suggest that CHD1 functions as a tumor suppressor by limiting binding of AR to canonical sites.
Loss of CHD1 drives a unique AR transcriptome associated with activation of oncogenic pathways
In order to define transcriptional programs impacted by CHD1 deletion, we conduced Gene Set Enrichment Analysis of pathway Hallmarks using RNA-Seq data derived from 3 independent models of CHD1 deficiency, including human LNCaP sgCHD1 lines, Cre-inducible Chd1f/f murine organoids (Figure S7A–S7B), and CHD1 null primary PCa samples (The Cancer Genome Atlas-TCGA) (Cancer Genome Atlas Research, 2015). Interestingly, the canonical Androgen Response was the most significantly de-enriched pathway among all 3 datasets (Figure 7A). This result is consistent with the proposed role of CHD1 loss in deregulating conventional AR signaling networks, and demonstrate that the chromatin context of nuclear receptor signaling is a critical factor in defining downstream transcriptional programs.
Figure 7.

Loss of CHD1 drives a subtype specific AR transcriptome associated with activation of oncogenic pathways. (A) 3D representation of GSEA Hallmark enrichment for 3 independent datasets deficient in CHD1. Normalized Enrichment Scores (NES) for all hallmarks for each CHD1 null model (vs. WT counterpart) are plotted on the X (LNCaP), Y (murine organoid), and Z (CHD1 null human tumors) axes. Hallmarks with consistent de-enrichment (blue) or enrichment (red) are highlighted (boxes). Gray dots represent inconsistent enrichment patterns. Individual NES’s for each highlighted hallmark and dataset are shown. (B) TCGA RNA-seq data was annotated for the status of ERG, ETS, and CHD1 and AR score plotted for each tumor. The box shows interquartile range with median (solid line) and min and max (whiskers) indicated. (C) Z-scores of all genes in the AR score were averaged per subclass and plotted by rank. Highlighted: The 3 top (red) and bottom (blue) genes contributing to the CHD1 null AR score. (D) RNA-seq from sgCtrl and sgCHDl cells stimulated with androgen for 0, 3, or 8 hr. Normalized z-scores of AR score genes for each model. Diminished and elevated genes have a p value of < 0.05 and fold change cutoff of 1.3x. (E) Ranked normalized z-scores of AR score genes from the sgCHD1 model. Average z-scores from TCGA tumor subclasses and sgCtrl were used to cluster datasets based on the sgCHD1 ranked list. (F) AR signature of sgCHD1 cells. Adjusted p value < 0.05. (G) NES’s for each subclass were generated for the upregulated (orange up arrows) and downregulated (purple down arrows) genes of the sgCHDl AR signature. The downregulated NES was subtracted from the upregulated NES for each genotype. (H) Metascape analysis of the sgCHDl AR signature. Red: Relevant to PCa. Blue: Consistent with normal prostate function. Purple: Common. See also Figure S7 and Table S4.
An additional metric to assess global AR function is via the AR-score (Hieronymus et al., 2006), a well-established 20-gene signature that, unlike the AR Response signature, is used to assess the level of AR transcriptional activation rather than downstream biological pathways. Thus, we next used the AR-score to assess the level of AR activation in CHD1 deficient human tumors in order to determine if the de-enrichment of the Hallmark AR Response in CHD1 null models could be attributed to diminished AR activity. Contrary to the AR Response signature, AR activity by this metric was significantly higher in CHD1 null tumors as compared to subtypes driven by ERG or ETS factors (Figure 7B). Collectively, these results suggest that AR activity is not simply impaired in CHD1 deficient tumors, but rather that this subtype engages a unique, AR-dependent transcriptome.
To begin defining the specific AR transcriptome in CHD1 null tumors, we analyzed the expression patterns of the genes comprising the AR score across subclasses of PCa. Consistent with the findings above, we uncovered unique patterns of expression among subclasses (Figure 7C). While ERG and ETS AR signatures were relatively concordant, an inverted expression pattern was detected when compared to CHD1 null tumors, further implicating CHD1 loss in driving alternative AR programs (Figure 7C). To directly assess the impact of CHD1 loss on AR driven transcription, we stimulated sgCtrl and sgCHD1 models with dihydrotestosterone (DHT) and examined gene expression at 3 and 8 hr. Therein, deletion of CHD1 led to the diminished induction and increased expression of 35% and 10% of AR score genes, respectively (Figure 7D). Hierarchal clustering of cell line and tumor samples based upon ranked AR-scores showed that the sgCHD1 signature mirrored that of CHD1 null tumors and was sufficient to segregate CHD1 deficient samples from CHD1 proficient samples (Figure 7E). Similar results were also achieved by using a TCGA derived signature of CHD1 loss (Figure S7C). These findings describe subtype specific AR transcriptional regulation in PCa and further nominate CHD1 loss as a critical regulator of subtype-specific AR programs.
While the established AR-score is a broadly effective metric to estimate AR activity, the finding of distinct, subtype-specific AR transcriptional programs suggests context-dependent regulation of downstream pathways. Therefore, we generated a gene expression signature specific to sgCHD1 cells after AR stimulation with DHT. Compared to control cells, 514 genes were downregulated and 605 were upregulated in CHD1 null cells upon androgen stimulation (Figure 7F; Table S4). AR binding peaks attributed to transcriptional induction of genes in sgCHD1 cells were also enriched for HOXB13 and GATA motifs, consistent with CHD1 dependent changes in the AR cistrome (Figure S7D). We next tested the subtype specificity of this AR signature in human PCa tumors. Strikingly, this ranked-gene signature was highly concordant only in CHD1 deficient tumors and was inconsistent or discordant in other PCa subclasses (Figure 7G). An analysis of molecular pathways controlled by this AR program revealed an enrichment of oncogenic pathways relevant to PCa, and a de-enrichment of molecular processes associated with canonical prostate function (Figure 7H). Notably, the KEGG Pathway “Prostate Cancer” was both enriched and de-enriched in the CHD1 null AR signature, further highlighting the importance of chromatin context in defining cancer-associated networks. Combined, we conclude that CHD1 loss drives expression of a subtype-specific AR transcriptome, with activation of oncogenic pathways and diversion away from normal prostatic functions.
Discussion
The CHD1 gene resides at one of the most commonly deleted loci in primary PCa and is uniquely restricted to this tumor type. Our data provide evidence that CHD1 deficiency can drive tumorigenesis in vivo, helping to credential CHD1 as a bona fide tumor suppressor. Mechanistically, a promoter-independent function for CHD1 was defined at prostate-specific enhancers, where CHD1 complexed with a number of nuclear receptor-specific cofactors and was coenriched on chromatin with AR. Loss of CHD1 resulted in a redistribution of AR to sites enriched for HOXB13, which was associated with subtype-specific changes to the AR transcriptome. This AR signature was observed only in CHD1 null PCa samples and is consistent with engagement of pro-oncogenic pathways. These data provide critical insight into the context-dependent tumor suppressive functions of CHD1 and, more broadly, implicate deficiencies in chromatin remodeling as drivers of prostate pathogenesis.
Reprogramming of the AR cistrome is a hallmark of PCa; however, the underlying mechanisms driving such events have yet to be fully elucidated. Recent work has demonstrated that FOXA1 and HOXB13 motifs are the most tightly associated with the oncogenic AR cistrome, and deregulation of these two pioneer factors is sufficient to reprogram AR in normal cells (Pomerantz et al., 2015). Consistent with these observations, we found that CHD1 was required to remodel local chromatin for a subset of AREs, potentially varying the available pool of unbound AR and therefore altering conventional signaling patterns of this nuclear receptor. Consequently, with CHD1 loss we found that AR more frequently localized to accessible DNA enriched for HOXB13, which did not require CHD1 for accessibility. This colocalization on DNA was associated with a maintenance of open chromatin at androgen repressed HOXB13 sites, which has previously been suggested to play a pivotal role in AR reprogramming during tumorigenesis (Pomerantz et al., 2015). As such, deregulation of HOXB13 signaling underlies the redistribution of the AR cistrome in the absence of CHD1, providing mechanistic insight into the prostate-specific tumor suppressor functions of this chromatin remodeler.
Disruption of the HOXB13 pathway can be mediated through a variety of mechanisms and has emerged as a key event during prostate pathogenesis (Pomerantz et al., 2015). Interestingly, these alterations are frequently observed in the context of CHD1 deficiency, suggesting that disruption of these pathways converge to drive oncogenic transcriptional networks. For example, CHD1 deletion frequently co-occurs with SPOP mutations in PCa (Barbieri et al., 2012; Barbieri and Tomlins, 2014; Cancer Genome Atlas Research, 2015), which are reported to stabilize of a number of key AR cofactors, including HOXB13 (Blattner et al., 2017; Dai et al., 2017; Geng et al., 2013; Groner et al., 2016). Additionally, germline HOXB13 variants (G84E) have been associated with an increased risk for PCa development (Ewing et al., 2012), and are largely independent of ETS fusions events (similar to CHD1 loss) (Lotan et al., 2017). Together, these events may combine to affect the context of AR-driven transcription to drive PCa progression.
These findings offer insight into subclass-specific events that drive alternative AR programs critical to prostate pathogenesis. More broadly, this work credentials chromatin remodeling deficiencies as key events in the deregulation of lineage-specific programming and highlights the importance of chromatin architecture in oncogenic transcriptional regulation. Beyond PCa, these findings suggest that tissue-specific deficiencies in other chromatin remodelers may play similar roles in transcription factor reprogramming and offer alternative therapeutic opportunities to therapeutically target these tumors.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Christopher E. Barbieri (chb9074@med.cornell.edu).
Experimental model and subject details
Mouse breeding, genotyping, and tissue processing
All mouse studies were approved by Weill Cornell Medicine (WCM) Institutional Care and Use Committee under protocol 2015–0022. Chdl-Exonl6-f/f mice were previously published (Guzman-Ayala et al., 2015) and were obtained from the Jackson Laboratories repository (strain Chdltm1c(KOMP)Rsan). For prostate-specific deletion of Chdl alone or in combination with Pten, Chdlff mice were crossed with the previously described Pb-Cre4;Ptenf/+ mice (Trotman et al., 2003; Wang et al., 2003). Only Pb-Cre4-positive male mice were used for Pb-Cre4 crosses. All described mice are in a C57BL/6 background. Genotyping was performed and confirmed by TransNetYX. Whole murine prostates were micro-dissected from mice at the indicated age, imaged, weighed, and then fixed overnight in 4% PFA. Sections were then transferred to 70% EtOH solution and paraffin embedded, sectioned, and stained by the Weill Cornell Medicine Translational Research Program Core. For genotyping primers see Table S5.
Murine organoid generation and growth
Prostates from the mice of the given genotype were harvested between 3–4 months of age and processed and grown as previously described (Drost et al., 2016) to generate both 3D and 2D organoid cultures. Where applicable, removal of the floxed allele was achieved through transient viral infection of 2D organoids using adenoviral expressing CRE or empty vector (Vector Labs Cat #1774 and 1660). After 72 hr, cells were propagated in 3D or 2D culture and assessed for CHD1 loss via immunoblot and IF.
Murine pathology review
All sections were reviewed by a board-certified genitourinary pathologist with expertise in human and murine models of prostate cancer (B.D.R). Reviews were performed blinded to age and genotype.
Human cell lines
LNCaP cells were purchased from the American Type Culture Collection (ATCC- Item # CRL-1740), grown on poly-L-lysine coated plates in 5% Fetal Bovine Serum (FBS) containing RPMI-1640, and incubated at 37°C and 5% CO2. Cells were passaged twice weekly or once cultures reached 80% confluency. All 2D and 3D cultures were assessed for mycoplasma monthly via the highly sensitive PCR based kit from ABM (Cat #ABM G238). Where applicable, cell line identity was validated yearly though the Human STR profiling cell authentication service provided by ATCC.
CRISPR model generation
LNCaP cells (< passage 15) were transfected with All-In-One pLentiCRISPR v2/sgRNA plasmids containing either control or CHD1 specific sgRNAs (ACCCAGAATCATCATCCGAC or TTCTGATCCGCTATTAGATG) purchased from GenScript. Cells were selected with puromycin until resistant populations emerged, and then assayed for CHD1 expression via immunoblot.
Transient models
LNCaP cells were transfected with Ctrl or Flag-HOXB13 expressing constructs (GeneCopoeia Cat# EX-M0909-M13, EX-NEG-M03) using the Xfect transfection system (Clontech Cat # 631317). Six hr post transfection, cells were plated in 96 well plates and assed for growth using Incucyte Zoom technology.
Method Detail
Murine Castration-testosterone re-supplementation
Mice of the given genotypes were aged to 3–4 months before surgical castration. Two weeks post castration, 15 mg testosterone pellets (A-151 Innovative Research of America) were implanted subcutaneously into all mice and prostates harvested for analysis 2 weeks thereafter.
Immunofluorescence
Cells were plated and grown on coverslips for 48–72 hr then fixed for 15 min with 4% PFA at room temperature. Cells were washed twice with PBS and then permeabilized with 0.1% Triton-X100 in PBS for 10 min. Following an additional PBS wash, cells were blocked with 10% goat serum and 0.5% BSA in PBS for 30 min at room temperature. Primary antibody was applied at the specified dilution in blocking solution overnight at 4°C. The following morning, cells were washed twice with PBS and incubated with the appropriate fluorescent secondary antibody (in blocking solution) for 30 min in the dark. Coverslips were then washed three times with PBS, mounted with Prolong Gold antifade mount solution with DAPI (Thermofisher Cat # P36931), and visualized using a fluorescent microscope.
Growth assays
Five thousand cells were plated in a 96 well plate in phenol-red free RPMI-1640 containing 5% charcoal dextran treated serum (CDT). Cells were allowed to attach overnight and were treated with varying concentrations of DHT (in EtOH). Growth was monitored and calculated using Incucyte software. The average of 4 images per well were plotted in biological triplicate for each cell line and each condition.
ChIP and ChIP Sequencing
LNCaP cells were grown in RPMI-1640 supplemented with 5% FBS and stimulated with 10 nM DHT for 3 hr. Twenty million cells per replicate were fixed using 1% formaldehyde for 10 min at 24°C, quenched for 5 min with 0.125 M glycine, and stored at −80°C until use. Samples were thawed on ice and processed for ChIP as previously described (McNair et al., 2018). Briefly, fixed pellets were lysed in 1% SDS lysis buffer, sonicated for 27 cycles (30s on/off) in a temperature controlled Bioruptor 300 to obtain a size range of 250–400 bp. Samples were spun at 15000 RPM for 20 min to remove debris, and individual samples were incubated with Protein A/antibody conjugated beads overnight rocking at 4°C. Samples were washed 6 times with increasing salt buffers and DNA eluted at 65°C overnight. All samples were treated with RNAse A for 30 min at 37°C followed by Proteinase K at 65°C for 1 hr. DNA was purified using phenol chloroform and individual ChIP samples verified by q-PCR. ChIP sequencing libraries (20 ng DNA per sample) were constructed using the KAPA Hyper Prep Kit (Illuminia-Kapa biosystems Cat # KK8502, NimbleGen SeqCap Adapter Kit A- Roche Cat # 07 141 530 001) according to the manufacturer’s instructions. Libraries were assessed for quality, purity, and size using DNA High Sensitivity Bioanalyzer chips (Aglient Technology Cat #5067–4626), and libraries passing quality control (equal size distribution between 250–400 bp, no adapter contamination peaks, no degradation peaks) were quantified using the Library Quantification Kit from Illiumina (Kapa Biosystems KK4854). Libraries were pooled to a final concentration of 10nM and sequenced using the Illumina HiSeq 4000.
Quantitative PCR
Quantitative PCR (Q-PCR) was performed using 4% of purified ChIP DNA per reaction. Reactions were conducted in technical triplicate and quantified as previously described (Augello et al., 2013). Standard error was calculated for each locus and condition, with statistical differences defined as p < 0.05 using a student’s T test within Prism software. For primer details see Table S5.
Immunoblot
Cells were harvested under the described conditions and lysed in RIPA buffer. 20–30 μg of total cell lysate were loaded onto poly-acrylamide gels, transferred to nitro-cellulose membranes, and probed for the indicated targets as previously described (Augello et al., 2013). For antibody use and details see Table S6.
Chromatin Tethering
Isogenic models of CHD1 loss were processed for AR tethered chromatin under androgen proficient conditions as previously described (52). Briefly, 250000 cells were washed with Buffer A and lysed with 100 μL of Buffer B on ice. Cells were then spun at 20000 RPM for 30 min to isolate soluble from chromatin fractions. Chromatin fractions were resuspended in 200 uL of Buffer B/1 × SDS-lysis buffer, with soluble fractions diluted to 200 μL using the same buffer. All samples were sonicated in a Bioruptor 3000 for 5 cycles on high (30s on/off) and boiled at 100°C for 5 min. Samples were then run on a polyacrylamide gel for analysis.
Xenograft Studies
6–8 weeks old athymic nude male mice were subcutaneously injected on the flank with 2 million cells suspended in a 1:1 ratio of PBS to Matrigel in a volume of 100 μL Tumors were monitored for growth twice weekly, and volume measured using electronic calipers. Mice were sacrificed 2 months post-injection, or when the tumor reached 800 mm3.
Growth Curves
Isogenic cell line models were plated in phenol red-free RPMI-1640 supplemented with 5% charcoal dextran treated (CDT) serum (Hyclone Cat # SH30068.03HI) (7000 cells per well in a 96 well plate). Cells were allowed to attach overnight and subsequently treated with increasing concentrations of DHT or 0.1% vehicle (EtOH). Treatments were conducted in biological triplicate and proliferation over time was monitored and calculated using IncuCyte Zoom technology/software.
Proximity Ligation Assay
LNCaP cells were grown on Poly-L-Lysine coated coverslips in androgen proficient, or androgen deficient conditions for 72 hr. Cells were then fixed and permeabilized as per the immunofluorescence protocol above. PLA was then carried out using the Duolink PLA kit (Sigma Aldrich Cat# DUO92101) following the manufacturer’s instructions and the indicated antibodies (Table S6). Nine images were taken for each PLA condition at 20x and 40x magnification in a single plane. Signal was then quantified for each image at 20x as number of foci/cell using Image J software.
Quantification and statistical analysis
ChIP Sequencing Analysis
Human cell line reads (FASTQ files) were validated for quality using FastQC software (Version 0.11.7), and single end reads with a score > 29 were aligned to the hg19 human reference genome using Bowtie2 (v2.2.9) software (default parameters). SAM files were converted to BAM format, sorted, PCR duplicates removed, ENCODE blacklist regions removed, and indexed using Samtools software (v1.7). Duplicate BAM files were combined to generate RPKM normalized bigwig files for each factor using Deeptools (v3.0) (Ramirez et al., 2014) which were then used to generate subsequent heatmaps and binding profiles (Deeptools v3.0). Previously aligned and normalized bigwig files from Pomerantz et. al. (Pomerantz et al., 2015) and Kron et al. (Kron et al., 2017) were downloaded and analyzed in their published format. H3K27ac dataset utilized only ERG negative tumors as to avoid driver effects specific to this subclass. When applicable, NCBI RefSeq gene annotation and locations were downloaded from the USCS genome browser (Karolchik et al., 2004), and assessed for factor binding enrichment using Deeptools (v3.0). Multiple transcriptional start sites were collapsed prior to analysis.
Peak calling
Peaks were called using MACS2 (Zhang et al., 2008) with a p value < 10−8 or q = 0.05 (replicates combined) using the narrow peak caller for all datasets except H3K27me3 which used the broad calling option with q = 0.1. Peak overlap and Venn diagrams were generated using pybedtools and bedtools intersect function (Quinlan and Hall, 2010) and was defined as overlap ≥ 1 bp. Differential peak calling was determined from 2 biological replicates using the Diffbind R package (v3.7) with peak calls for individual replicates from MACS2 using q = 0.05, an FDR = 0.01, and the EdgeR default settings as previously described (Stark et. al. 2011, Bioconductor).
Motif Analysis
Motif analysis was conducted using Homer v4.8.3 (Heinz et al., 2010) using a 200 bp window around the center of each peak. Motif density around peaks was calculated using Homer and JASPER definitions of the conserved motif. To determine motif enrichment between datasets with similar peak numbers, peak sets of the control were used as background (−bg flag in findmotifsgenome.pl function). For analyses where the number of peaks differed greatly between conditions, 20000 peaks were randomly selected using bedtools sample function and assessed for known JASPER motif enrichment using Homer software (annotatepeaks.pl) run 100 times for each condition/replicate. For enhancer specific assessment of motif enrichment, peaks localized to promoters/TSS were removed prior to analysis. Where indicated, motif enrichment was validated using pScan (default parameters) (Zambelli et al., 2013). Unless otherwise noted, significant motif enrichment was considered only if the p value was ≤ 1−20.
Motif analysis of primary human prostate tumors
Copy number analysis of primary human prostate cancers for the CHD1 locus was performed as described in (Stelloo et al., 2018b). AR peaks from these tumors (Stelloo et al., 2018b) were binned into CHD1 deficient (homozygous and heterozygous deletions) and CHD1 WT. ERG high tumors were eliminated from this analysis to avoid observations driven by the dominant ERG signal. Thirteen CHD1 deficient and 17 CHD1 WT tumors (minimum 1000 peaks) were utilized for motif enrichment. Peaks from each genotype were merged to generate a CHD1 null and CHD1 WT peak sets. Differential de novo motif analysis was determined for each peak set using the opposing dataset as background using Homer software (v4.1.0) findmotifsgenome.pl function (−bg flag) using the hg19 genome build and a 200 bp window flanking the center of each peak.
Genomic region annotation
Cis-regulatory element analysis was performed using CEAS v1.0.2 (Shin et al., 2009) in conjunction with the Homer annotatepeaks.pl function and the hg19 genome annotation.
ATAC Seq
LNCaP isogenic models of CHD1 loss were starved of androgen for 72 hr and treated with either vehicle (0.1% EtOH) or DHT (10 nM) for 4 hr. Cells were then fixed using 4% PFA, washed in PBS 2x, and 50000 cells/condition were submitted in biological duplicate to the Dana Farber Center for Functional Cancer Epigenetics for ATAC Seq library preparation, sequencing, processing, and analysis using the Omni-ATAC protocol as previously described (Corces et al., 2017). Briefly, 105 cells were resuspended in 1 mL of cold ATAC-seq resuspension buffer (RSB; 10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2 in water). Cells were centrifuged at 500 g for 5 min in a pre-chilled (4°C) fixed-angle centrifuge, and the supernatant was carefully aspirated. Cell pellets were resuspended in 50 gL RSB containing 0.1% NP40, 0.1% Tween-20, and 0.01% digitonin by pipetting up and down three times. Cell lysis reaction was incubated on ice for 3 min. After lysis, 1 mL RSB containing 0.1% Tween-20 (without NP40 or digitonin) was added, and the tubes were inverted to mix. Nuclei were then centrifuged for 10 min at 500 g in a pre-chilled fixed-angle centrifuge. Supernatant was removed and nuclei were resuspended in 50 gL of transposition mix (Corces et al., 2017) (2.5 gL transposase26 (100 nM final), 16.5 gL PBS, 0.5 gL 1% digitonin, 0.5 gL 10% Tween-20, and 5 gL water) by pipetting up and down six times. Transposition reactions were incubated at 37°C for 30 min in a thermomixer with shaking at 1000 rpm. Reactions were cleaned up with QIAquick PCR spin columns. Library quantitation and number of amplification cycles was determined as described (Buenrostro et al., 2015). After sequencing on a NextSeq 500 per manufacturer instructions, ChiLin pipeline 2.0.0 was used for quality control and pre-processing (Qin et al., 2016). This includes Burrows-Wheeler Aligner (BWA) for read mapping (Li and Durbin, 2009), Model-based Analysis of ChIP-Seq (MACS) as a peak caller (Zhang et al., 2008), and DESeq2 for differential peak analysis (Love et al., 2014). For ATAC peak motif enrichment, peak sets between replicates were merged prior to analysis.
RNA-seq analysis
Growth Conditions- Isogenic LNCaP cells were grown in androgen deprived media for 72 hr and then stimulated with 1nM DHT (or vehicle) for 3 and 8 hr in biological triplicate. Murine organoids were grown in 2D in complete prostate organoid media for 48 hr and harvested for RNA in biological triplicate. Human cell lines and mouse organoids were prepared for RNA sequencing using TruSeq RNA Library Preparation Kit v2. RNA integrity was verified using the Agilent Bioanalyzer 2100 (Agilent Technologies). Cell lines samples were then sequenced with the HiSeq 2500 to generate 2×51-bp paired-end reads, and organoids samples were sequenced with 2×151-bp paired-end reads. Human cell line reads (FASTQ files) were mapped to the human reference genome sequence (hg19) using STAR v2.4.0j (Dobin et al., 2013), and mouse organoid reads were mapped to the mouse reference genome sequence (mm10). The resulting BAM files were subsequently converted into mapped-read format (MRF) using RSEQtools (v2.1) (Habegger et al., 2011). The read count of each gene was calculated via HTSeq (Anders et al., 2015) using GENCODE as reference gene annotation set. Quantification of gene expression was performed via RSEQtools (v2.1), and expression levels (RPKM) were estimated by counting all nucleotides mapped to each gene and were normalized by the total number of mapped nucleotides (per million) and the gene length (per kb). The genomic status and AR output score of TCGA samples were downloaded from TCGA study (Cancer Genome Atlas Research, 2015). The AR output score of our samples were calculated by following the similar strategy as TCGA study (Cancer Genome Atlas Research, 2015). Specifically, the AR output score was derived from the mRNA expression of 20 genes that were experimentally validated AR transcriptional targets from LNCaP cell line (Hieronymus et al., 2006). Here the Z-score for the expression of each gene in each sample was calculated and the AR score for each sample was then computed as the sum of the Z-scores of 20 AR signaling genes. Heatmap and hierarchical clustering were performed via using correlation distance and Ward’s method. GSEA (Subramanian et al., 2005) was performed using JAVA program and run in pre-ranked mode to identify enriched signatures. We used the gene sets in the Molecular Signature Database (MSigDB) (Subramanian et al., 2005). The GSEA plot, normalized enrichment score and q -values were derived from GSEA output for each MSigDB hallmark signature. Differentially expression analyses were performed using DESeq2 (v1.20.0) (Love et al., 2014) based on the gene read count data. Multiple-hypothesis testing was considered by using Benjamini-Hochberg (BH; FDR) correction. The CHD1 null AR signature from the LNCaP isogenic model of CHD1 loss was generated by first defining all differentially expressed genes in the sgCHDl line between vehicle and 8 hr DHT treatment using an FDR of 0.01. These androgen regulated genes were then assessed for changes between the sgCtrl and sgCHDl lines at the 8 hr DHT timepoint using an adjusted p value of 0.05 as a significance threshold.
Generation of CHD1 null transcriptional signature from TCGA
By following a similar strategy (Blattner et al., 2017), we developed the CHD1 deletion transcriptional signature that included 282 genes differentially expressed between CHD1 deleted and WT samples from TCGA prostate cancer RNA-seq data. The low-expressed genes (mean RSEM < 1) were filtered prior to analysis. Specifically, we identified significantly differentially expressed genes by comparing CHD1 deletion and WT cases as determined from genomic analyses among TCGA samples lacking ETS family gene fusions (ERG, ETV1, ETV4 and FLI1), using Wilcoxon rank-sum test and controlled for false discovery using Benjamini-Hochberg adjustment (FDR < 1×10−5). Heatmap and hierarchical clustering were performed via using correlation distance and Ward’s method on TCGA and LNCaP model.
RIME analysis
Cells were prepared as per the ChIP protocol above and processed for RIME as previously described (Mohammed et al., 2016). Peptides were desalted on hand-packed C18 STAGE tip columns (Rappsilber et al., 2003). Eluted peptides were dried down in a centrifugal evaporator, reconstituted in 5% formic acid and analyzed by nanospray LC-MS/MS on an Orbitrap Fusion mass spectrometer (ThermoFisher). Peptides were separated by reverse-phase HPLC on a hand-packed column (packed with 40 cm of 1.8 pm, 120 Ä pores, Sepax GP-C18, Sepax Technologies, Newark, DE) using a 85 min gradient of 5–27% buffer B (ACN, 0.1% FA) at a 350 nl/min. Peptides were detected using a Top20 method. For each cycle, one full MS scan of m/z = 375–1400 was acquired in the Orbitrap at a resolution of 120000 at m/z with AGC target = 5×105. Each full scan was followed by the selection of up to 20 of the most intense ions for CID and MS/MS analysis in the linear ion trap. Selected ions were excluded from further analysis for 30 s. Ions with charge 1+ or unassigned were also rejected. Maximum ion accumulation times were 100 ms for each full MS scan and 35 ms for MS/MS scans.
MS2 spectra were searched using SEQUEST (v28 revision 13) against a composite database containing all Swiss-Prot reviewed human protein sequences (20193 target sequences downloaded from www.uniprot.org on March 18, 2016) and their reversed complement, using the following parameters: a precursor mass tolerance of +/− 20 ppm; 1.0 Da product ion mass tolerance; tryptic digestion; up to two missed cleavages; static modifications of carbamidomethylation on cysteine (+57.0214), and a dynamic modification of methionine oxidation (+15.9949). Peptide spectral matches were filtered to 1% FDR using the target-decoy strategy (Elias and Gygi, 2007) combined with linear discriminant analysis (LDA) using SEQUEST scoring parameters including Xcorr, ΔCn’, precursor mass error, and charge state (Huttlin et al., 2010).
Two independent biological replicates were processed for each IP, the log signal to noise ratios (S/N) from IgG samples were subtracted from each IP condition, and those with a score > 0.2 and a minimum of 6 peptides between replicates were included for further processing. These data were then analyzed using the Crapome database with isotype matched IgG and Crapome controls (Mellacheruvu et al., 2013), and only peptides with a SAINT score > 0.9 (out of 1) were included in downstream analyses.
Ki67 quantification
Three independent, representative images were taken at 20x magnification for each mouse prostate. The percentage of Ki67 positive epithelial cells was calculated for each image (at least 500 total cells per image) and the mean and standard error of all images for each genotype was determined. Normal distribution of the data was confirmed using D’Agostino-Pearson omnibus normality test prior to a student’s 2-sided t-test which was to determine statistical differences between the two genotypes. For the castration-restimulation experiments, Ki67 counts are representative of differences observed in the anterior prostate gland.
Tumor-xenograft volume quantification
Volume for each xenograft was calculated using electronic calipers (w*l*h - mm3) and plotted 10 weeks post-injection. The difference in the mean volume between xenograft lines was determined using a 2-sided students t-test (p value = 0.803).
Xenograft-tumor take quantification
Formation of palpable tumor formation was performed twice weekly and reported for each xenograft line. Tumor-free survival was calculated using both the logrank Mantel-Cox and Gehan-Breslow-Wilcoxon test. Curves were not statistically different using either test with p values of 0.61 and 0.82, respectively.
PLA quantification
A minimum of 75 cells per condition were quantified in a single plane for detectable foci using image J. The number of foci per cell per condition is reported. Differences in the mean number of foci between conditions was determined using One-way ANOVA and Turkey’s multiple comparisons test.
Growth Curves
Biological triplicates for each cell line and condition were assessed for confluency using Incucyte software every 4 hr for the indicated duration. The mean and standard error of each time point is plotted. Differences in growth in response DHT/enzalutamide were determined via EC50 calculations 100 hr post treatment. The individual biological triplicates (and calculated standard error) for each dose and cell line were used as input, and differences in EC50 calculated using non-linear regression software, least squared (ordinary) fit, and the Extra sum-of-squares F test (p value < 0.05) to determine statistical differences between EC50 values.
String analyses
Peptides significantly enriched in the CHD1 RIME dataset were analyzed for overlap with that of the previously identified AR RIME interactome (Stelloo et al., 2018a). Common proteins were uploaded to the STRING database (STRING-db.org v10.5) and assessed for potential direct and indirect interactions.
Data and software availability:
RNA-Seq data has been deposited in GEO under the accession GSE117429
ChIP-Seq data has been deposited in GEO under the accession GSE117430
Mass-Spec data has been deposited in PRIDE under the accession PXD010468
ATAC-Seq data has been deposited in GEO under the accession GSE123333
Supplementary Material
Table S1: Raw RIME data, related to Figure 3.
Table S2: Significantly enriched proteins in RIME datasets, related to Figure 3.
Table S3: Pscan motif analysis of sgCHD1 enriched and depleted peaks, related to Figure 6.
Table S4: Differentially expressed genes with DHT in sgCtrl and sgCHD1 cells, related to Figure 7.
Highlights:
CHD1 acts as a prostate-specific tumor suppressor in vivo.
CHD1 regulates AR occupancy at a subset of lineage specific enhancers.
Loss of CHD1 redistributes the AR cistrome to favor oncogenic AR-driven pathways.
Significance.
Deletion of the CHD1 locus occurs in approximately 15% of primary prostate cancer (PCa) cases, and is uniquely restricted to tumors of this tissue type. Our data uncover an enrichment of CHD1 at lineage-specific enhancers, which is required to constrain oncogenic androgen receptor (AR) binding and limit prostate neoplasia in vivo. The discovery that CHD1 can regulate nuclear receptor activity on chromatin not only reveals a promoter-independent role for this nucleosome remodeler, but also credentials CHD1 as a bona-fide tumor-suppressor within prostate tissue. These findings provide needed insight into the underlying mechanisms and pathways of subclass-specific PCa development and more broadly, highlight the importance of deregulation of chromatin architecture in the development and progression of human cancer.
Acknowledgements
We are grateful to the prostate cancer patients and families who contributed to this research. We thank The Cancer Genome Atlas Research Network (TCGA) and cBioPortal. We thank the WCM Genomics and Epigenomics Core Facilities, the Biospecimen and Pathology and Computational and Biostatistics Cores of the WCM SPORE in Prostate Cancer, and the Center for Functional Cancer Epigenetics at The Dana Farber Cancer Institute. Special thanks to O. Krijgsman for providing computational pipelines and to C. McNair and J. Franciosi for valuable feedback and technical assistance. This work was supported by: the NCI (WCM SPORE in Prostate Cancer, P50CA211024-01 C.E.B., K08CA187417-01, C.E.B., R37CA215040, C.E.B.), a Urology Care Foundation Rising Star in Urology Research Award (C.E.B.), Damon Runyon Cancer Research Foundation MetLife Foundation Family Clinical Investigator Award (C.E.B.), Department of Defense Postdoctoral Fellowship (M.A.A.), and the Prostate Cancer Foundation (C.E.B., M.A.A.)-Prostate Cancer Foundation Young Investigator award (M.A.A), the Dutch Cancer Society/ Alpe d’HuZes (10084- W.Z), and The Movember Foundation (NKI01- W.Z).
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson PD, McKissic SA, Logan M, Roh M, Franco ΟE, Wang J, Doubinskaia I, van der Meer R, Hayward SW, Eischen CM, et al. (2012). Nkx3.1 and Myc crossregulate shared target genes in mouse and human prostate tumorigenesis. J Clin Invest 122, 1907–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, Magee MS, Frigo DE, Wilder-Romans K, Shilkrut M, Han S, et al. (2013). Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest 123, 493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, et al. (2013). ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev 27, 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. (2012). Exorne sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 44, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, and Tomlins SA (2014). The prostate cancer genome: perspectives and potential. Urol Oncol 32, 53 e15–22. [DOI] [PubMed] [Google Scholar]
- Baumgart SJ, Najafova Z, Hossan T, Xie W, Nagarajan S, Kari V, Ditzel N, Kassem M, and Johnsen SA (2017). CHD1 regulates cell fate determination by activation of differentiation-induced genes. Nucleic Acids Res 45, 7722–7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, Gao D, Nataraj S, Deonarine LD, Augello MA, Sailer V, et al. (2017). SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 31, 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang ΗY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chi P, Rockowitz S, laquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, et al. (2013). ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med 19, 1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Lan X, Thomas-Ahner JM, Wu D, Liu X, Ye Z, Wang L, Sunkel B, Grenade C, Chen J, et al. (2015). Agonist and antagonist switch DNA motifs recognized by human androgen receptor in prostate cancer. EMBO J 34, 502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corees MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Gan W, L¡ X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, et al. (2017). Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 23, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Dieuleveult M, Yen K, Hmitou I, Depaux A, Boussouar F, Bou Dargham D, Jounier S, Humbertclaude H, Ribierre F, Baulard C, et al. (2016). Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature 530, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, and Clevers H (2016). Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc 11, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein Identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, et al. (2012). Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med 366, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, and Khorasanizadeh S (2005). Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438,1181–1185. [DOI] [PubMed] [Google Scholar]
- Gao S, Gao Y, He ΗH, Han D, Han W, Avery A, Macoska JA, Liu X, Chen S, Ma F, et al. (2016). Androgen Receptor Tumor Suppressor Function Is Mediated by Recruitment of Retinoblastoma Protein. Cell Rep 17, 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho-Santos J, McManus ΜT, Plath K, Meshorer E, and Ramalho-Santos M (2009). Chdl regulates open chromatin and pluripotency of embryonic stem cells. Nature 460, 863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, et al. (2013). Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A 110, 6997–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, and Trifiro M (2012). The androgen receptor gene mutations database: 2012 update. Hum Mutat 33, 887–894. [DOI] [PubMed] [Google Scholar]
- Grabowska MM, DeGraff DJ, Yu X, Jin RJ, Chen Z, Borowsky AD, and Matusik RJ (2014). Mouse models of prostate cancer: picking the best model for the question. Cancer Metastasis Rev 33, 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. (2012). The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D, Houtman R, Cato ACB, Tschopp P, Gu L, et al. (2016). TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 29, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Ayala M, Sachs M, Koh FM, Onodera C, Bulut-Karslioglu A, Lin CJ, Wong P, Nitta R, Song JS, and Ramalho-Santos M (2015). Chdl is essential for the high transcriptional output and rapid growth of the mouse epiblast. Development 142, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habegger L, Sboner A, Gianoulis TA, Rozowsky J, Agarwal A, Snyder M, and Gerstein M (2011). RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics 27, 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelett DJ, Rhie SK, Gaddis M, Yan C, Lakeland DL, Coetzee SG, Ellipse G.-O. N. c., Practical c., Henderson BE, Noushmehr H, et al. (2014). Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet 10, el004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He ΗH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. (2010). Nucleosome dynamics define transcriptional enhancers. Nat Genet 42, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, et al. (2006). Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10, 321–330. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski ΜP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ΜE, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussier D, and Kent WJ (2004). The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32, D493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kininis M, and Kraus WL (2008). A global view of transcriptional regulation by nuclear receptors: gene expression, factor localization, and DNA sequence analysis. Nucl Recept Signal 6, e005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh FM, Lizama CO, Wong P, Hawkins JS, Zovein AC, and Ramalho-Santos M (2015). Emergence of hematopoietic stem and progenitor cells involves a Chdl-dependent increase in total nascent transcription. Proc Natl Acad Sci U S A 112, E1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krön KJ, Murison A, Zhou S, Huang V, Yamaguchi TN, Shiah YJ, Fraser M, van der Kwast T, Boutros PC, Bristow RG, and Lupien M (2017). TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat Genet 49, 1336–1345. [DOI] [PubMed] [Google Scholar]
- Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, Coleman I, Ng SB, Salipante SJ, Rieder MJ, et al. (2011). Exorne sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A 108, 17087–17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L¡ H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinov IV, De Marzo AM, and Isaacs JT (2003). Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab 88, 2972–2982. [DOI] [PubMed] [Google Scholar]
- Liu W, Lindberg J, Sui G, Luo J, Egevad L, Li T, Xie C, Wan M, Kim ST, Wang Z, et al. (2012). Identification of novel CHDl-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene 31, 3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotan TL, Torres A, Zhang M, Tosoian JJ, Guedes LB, Fedor H, Hicks J, Ewing CM, Isaacs SD, Johng D, et al. (2017). Somatic molecular subtyping of prostate tumors from HOXB13 G84E carriers. Oncotarget 8, 22772–22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love ΜI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusser A, Urwin DL, and Kadonaga JT (2005). Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat Struct Mol Biol 12,160–166. [DOI] [PubMed] [Google Scholar]
- Magee JA, Chang LW, Stormo GD, and Milbrandt J (2006). Direct, androgen receptor- mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology 147, 590–598. [DOI] [PubMed] [Google Scholar]
- McNair C, Xu K, Mandigo AC, Benelli M, Leiby B, Rodrigues D, Lindberg J, Gronberg H, Crespo M, De Laere B, et al. (2018). Differential impact of RB status on E2F1 reprogramming in human cancer. J Clin Invest 128, 341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, Taylor C, Brown GD, Papachristou EK, Carroll JS, and D’Santos CS (2016). Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc 11, 316–326. [DOI] [PubMed] [Google Scholar]
- Mooslehner KA, Davies JD, and Hughes IA (2012). A cell model for conditional profiling of androgen-receptor-interacting proteins. Int J Endocrinol 2012, 381824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morettini S, Tribus M, Zeilner A, Sebald J, Campo-Fernandez B, Scheran G, Worle H, Podhraski V, Fyodorov DV, and Lusser A (2011). The chromodomains of CHD1 are critical for enzymatic activity but less important for chromatin localization. Nucleic Acids Res 39, 3103–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Soner A, Sailer V, Augello M, Puca L, Rosati R, et al. (2017). Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 7, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti P, Lim CY, Nat R, Villunger A, Geley S, Shue YT, Soratroi C, Moser M, and Lusser A (2015). Embryonic stem cell differentiation requires full length Chdl. Sei Rep 5, 8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Li F, Takeda DY, Lend R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, et al. (2015). The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet 47,1346–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, et al. (2016). ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, and Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75, 663–670. [DOI] [PubMed] [Google Scholar]
- Roediger J, Hessenkemper W, Bartsch S, Manvelyan M, Huettner SS, Liehr T, Esmaeili M, Foller S, Petersen I, Grimm MO, and Baniahmad A (2014). Supraphysiological androgen levels induce cellular senescence in human prostate cancer cells through the Src-Akt pathway. Mol Cancer 13, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Myers M, and Brown M (2002). Formation of the androgen receptor transcription complex. Mol Cell 9, 601–610. [DOI] [PubMed] [Google Scholar]
- Shen H, and Laird PW (2013). Interplay between the cancer genome and epigenome. Cell 153, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo W, Koh FM, Wang C, Zhang LZ, Wang Y, Gil V, et al. (2017). CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann Oncol 28,1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Liu T, Manrai AK, and Liu XS (2009). CEAS: cis-regulatory element annotation system. Bioinformatics 25, 2605–2606. [DOI] [PubMed] [Google Scholar]
- Siggens L, Cordeddu L, Ronnerblad M, Lennartsson A, and Ekwall K (2015). Transcription-coupled recruitment of human CHD1 and CHD2 influences chromatin accessibility and histone H3 and H3.3 occupancy at active chromatin regions. Epigenetics Chromatin 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simie R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, and Arndt KM (2003). Chromatin remodeling protein Chdl interacts with transcription elongation factors and localizes to transcribed genes. EMBO J 22,1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ 3rd, Chen CF, Santos-Rosa H, Kouzarides T, Patel SS, and Reinberg D (2005). Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J Biol Chem 280, 41789–41792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Hernandez AE, Groudine M, and Henikoff S (2014). The nucleosomal barrier to promoter escape by RNA polymerase II is overcome by the chromatin remodeler Chdl. Elite 3, e02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelloo S, Nevedomskaya E, Kim Y, Hoekman L, Bleijerveld OB, Mirza T, Wessels LFA, van Weerden WM, Altelaar AFM, Bergman AM, and Zwart W (2018a). Endogenous androgen receptor proteomic profiling reveals genomic subcomplex involved in prostate tumorigenesis. Oncogene 37, 313–322. [DOI] [PubMed] [Google Scholar]
- Stelloo S, Nevedomskaya E, Kim Y, Schuurman K, Valle-Encinas E, Lobo J, Krijgsman O, Peeper DS, Chang SL, Feng FY, et al. (2018b). Integrative epigenetic taxonomy of primary prostate cancer. Nat Commun 9, 4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sei USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, et al. (2003). Pten dose dictates cancer progression in the prostate. PLoS Biol 1, E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, and Brown M (2007). A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, et al. (2003). Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221. [DOI] [PubMed] [Google Scholar]
- Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, Ghori M, Zamora J, Butler A, Whitaker H, Kote-Jarai Zv et al. (2018). Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet 50, 682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, Sangiorgi FO, Maxson RE, Sucov ΗM, and Roy-Burman P (2001). Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev 101, 61–69. [DOI] [PubMed] [Google Scholar]
- Zambelli F, Pesóle G, and Pavesi G (2013). PscanChIP: Finding over-represented transcription factor-binding site motifs and their correlations in sequences from ChIP-Seq experiments. Nucleic Acids Res 41, W535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JC, Fong KW, Jin HJ, Yang YA, Kim J, and Yu J (2016). FOXA1 acts upstream of GATA2 and AR in hormonal regulation of gene expression. Oncogene. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Raw RIME data, related to Figure 3.
Table S2: Significantly enriched proteins in RIME datasets, related to Figure 3.
Table S3: Pscan motif analysis of sgCHD1 enriched and depleted peaks, related to Figure 6.
Table S4: Differentially expressed genes with DHT in sgCtrl and sgCHD1 cells, related to Figure 7.