

Abstract
(N)-Methanocarba ([3.1.0]bicyclohexyl) adenosines and corresponding ribosides were synthesized to identify novel A1 adenosine receptor (A1AR) agonists for CNS or peripheral applications. Human and mouse AR binding was determined to assess the constrained ring system’s A1AR compatibility. N6-Dicyclobutylmethyl ribose agonist (9, MRS7469, >2000-fold selective for A1AR) and known truncated N6-dicyclopropylmethyl methanocarba 7 (MRS5474) were drug-like. The pure diastereoisomer of known riboside 4 displayed high hA1AR selectivity. Methanocarba modification reduced A1AR selectivity of N6-dicyclopropylmethyl and endo-norbornyl-adenosines, but increased ribavirin selectivity. Most analogues tested (ip.) were inactive or weak in inducing mouse hypothermia, despite mA1AR full agonism and variable mA3AR efficacy, but strong hypothermia by 9 depended on A1AR, which reflects CNS activity (determined using A1AR or A3AR null mice). Conserved hA1AR interactions were preserved in modeling of 9 and methanocarba equivalent 24 (~400-fold A1AR-selective). Thus, we identified, and characterized in vivo, ribose and methanocarba nucleosides, including with A1AR-enhancing N6-dicyclobutylmethyl-adenine and 1,2,4-triazole-3-carboxamide (40, MRS7451) nucleobases.
Keywords: adenosine receptor, G protein-coupled receptor, nucleosides, molecular modeling, hypothermia, structure activity relationship
Graphical Abstract
Introduction
The A1 adenosine receptor (A1AR) is coupled to the inhibition of adenylyl cyclase through the Gi protein and also acts on ion channels and MAP kinases.1,2 A1AR agonists are desired for their antiarrhythmic, antilipolytic, antinociceptive, cerebroprotective, cardioprotective, antidepressant, sleep-enhancing and antiseizure properties.1–7 Beneficial peripheral effects of A1AR agonists include protection in acute and chronic pain8,9 or as antilipolysis.10 We recently described the induction of mouse hypothermia through brain A1AR activation that is independent of peripheral A3AR activation.30 Pharmacologically-induced hypothermia has potential clinical applications, such as cerebroprotection in brain ischemia, but most A1AR agonists are unsuitable as clinical candidates because of their cardiovascular effects.1,4,11–13 Thus, there is a need for novel A1AR agonists that are well-tolerated for a variety of clinical applications, but with diminished side effects.
The first A1AR X-ray crystallographic structures, in complex with antagonists but not agonists, were reported and show promise to enable structure-based drug design (SBDD).14,15 More recently, a high resolution structure of an agonist-bound A1AR containing a Gi protein was determined using cryo-electron microscopy (cryo-EM).16 However, most potent A1AR agonists so far have been discovered through extensive empirical probing of their structure activity relationship (SAR), for example, leading to nucleosides 1–7 (Chart 1).1,2 Thus, adenosine derivatives variously substituted at the N6, 5′ and C2 positions have been reported as potent and selective A1AR agonists. Widely used modifications of adenosine to increase A1AR selectivity consist of introducing an N6-phenyl or an N6-cycloalkyl group, such as N6-cyclopentyl (1, 2, 5 and 6), N6-cyclohexyl or N6-endo-norbornyl (3, 4).8,17–19 (2S)-N6-(endo-norbornyl)-adenosine 3 is among the most selective mono-substituted agonists of A1AR.15 Recently, additional N6-bicycloalkyl derivatives containing heteroatoms were reported to be highly A1AR-selective agonists.20 Other commonly used modifications in AR agonists, such as 5′-Cl (4) and 5′-N-alkylamides (6), but not extended or bulky C2 substituents, are compatible with N6-cycloalkyl groups for retention of A1AR affinity.21–23 Alternative 5′ substituents, such as aryl thioethers, e.g. 5, and other ethers, were used to provide partial agonist activity at the A1AR, especially as applied to antiarrhythmic agents.1,24 The SAR of 5′-deoxyadenosine 5′-oxadiazole or 5′-tetrazole derivatives led to A1AR-selective agonists.1,18,19,25 However, the C8 position of adenosine was amenable to only limited derivatization with the retention of AR binding affinity, leading to partial A1AR agonists and A3AR antagonists.26,27
Chart 1.

Structures of reference AR agonists. All nucleosides shown are A1AR-selective,1,2 except 6, which is a mixed A1AR/A3AR agonist.32
Compound 4 is often used as a highly A1AR-selective agonist with a 12,000-fold window of selectivity in mouse (m) A1AR vs. mA3AR binding, which was shown to induce hypothermia via the A1AR.28–30 When administered peripherally in the mouse, it passes the blood-brain barrier (BBB) sufficiently to activate a central mA1AR, which lowered core body temperature (Tb). Furthermore, at intraperitoneal (ip.) doses up to 3 mg/kg, 4 did not cause intense activation of the peripheral mast cell mA3AR, as seen with other A1AR agonists. In addition to its high A1AR binding selectivity, this in vivo selectivity of 4 might be partly due to a possible lower A3AR efficacy, typical of adenosine derivatives with large N6 groups.17 Other A1AR agonists, such as 1a, although 2400-fold selective in mA1AR binding, did not achieve in vivo agonist selectivity due to activation of a peripheral mA3AR, consistent with its full efficacy at the human (h) A3AR.17 The greater propensity of AR agonists to activate mA3AR rather than brain A1AR after peripheral administration relates to the low BBB penetration of adenosine derivatives. This characteristic is partially a function of the multiple hydroxyl groups that reduce diffusion across biological membranes.31
The North (N)-methanocarba ([3.1.0]bicyclohexyl) modification increases the potency and selectivity of adenosine derivatives at the A3AR and tends to maintain affinity at the A1AR, but not A2AAR.32 We previously introduced a moderately selective hA1AR full agonist 7 that has a N6-dicyclopropylmethyl group, (N)-methanocarba substitution of ribose and only two hydroxyl groups due to 4′-truncation.33 This compound has demonstrated antiseizure and antidepressant activity.6,33 Here, we enlarged the pharmacological characterization of 7 and demonstrated its favorable absorption, distribution, metabolism, excretion and toxicological (ADME-tox) properties, suggesting further derivatization to expand the range of useful A1AR agonists.
Thus, we introduced novel ribose and (N)-methanocarba adenosine derivatives with the goal of enhancing their A1AR selectivity. One objective was to extend the SAR of (N)-methanocarba adenosine derivatives in the direction of A1AR in two species (h and m). We examined if it is feasible to overcome the tendency of this rigid ring system to produce A3AR selectivity, by appropriately substituting other positions. Previously, A1AR-enhancing N6-cycloalkyl groups were combined with the A3AR-enhancing (N)-methanocarba ring system to produce an agonist 6 of mixed A1AR/A3AR selectivity.32 Further modification could tune the selectivity, e.g. as seen in certain adenosine derivatives where a 2-chloro substitution increased hA1AR selectivity32 or affinity.34
Results
We have modified the 5′, C2 and other positions, and compared the receptor binding of some (N)-methanocarba nucleoside analogues directly with the corresponding ribosides. We have modeled the interaction of several novel A1AR-selective N6-dicyclobutylmethyl agonists at the hA1AR. We examined many of these A1AR agonists in an in vivo assay of hypothermia in the mouse, which can delineate peripheral A3AR and central A1AR components.30
Selection of target nucleosides
The nucleoside analogues examined in AR binding assays are shown in Table 1. Based on known AR agonists (e.g. 1–8, 14–18, 21, 23 and 38), related adenine substitutions were made in novel nucleosides in both the ribose (e.g. 9–13) and (N)-methanocarba (e.g. 22, 24–31 and 32–37, 39, 40) series. Adenosine derivatives with N6-cycloalkyl (such as N6-endo-norbornyl) and symmetric N6-dicycloalkylmethyl (N6-dicyclopropylmethyl and larger) groups were included as a potential means of enhancing selectivity for the A1AR. N6-endo-Norbornyl derivative 13 is a pure diastereoisomer of 4, corresponding to the predicted preferred stereochemistry for A1AR recognition from an earlier rA1AR study.28 The inclusion of N6-dicyclopropylmethyl analogues was based on the moderate A1AR selectivity of 7 and 8.17,33,34 In addition to known N6-dicyclopropylmethyl and N6-dicyclopentylmethyl substituents,33,34 here we introduced the first N6-(dicyclobutylmethyl)-adenosine analogues (9–12, 19, 24–28 and 35) to be evaluated at ARs. We also compared 2-Cl and 2-H on the adenine moiety.
Table 1.
Structures and binding affinitiesa of AR ligands in the species indicated, including reference compounds 1 - 7 and 15 - 18. R3 = OH, R4 = NHCH3, Y = N, unless noted.
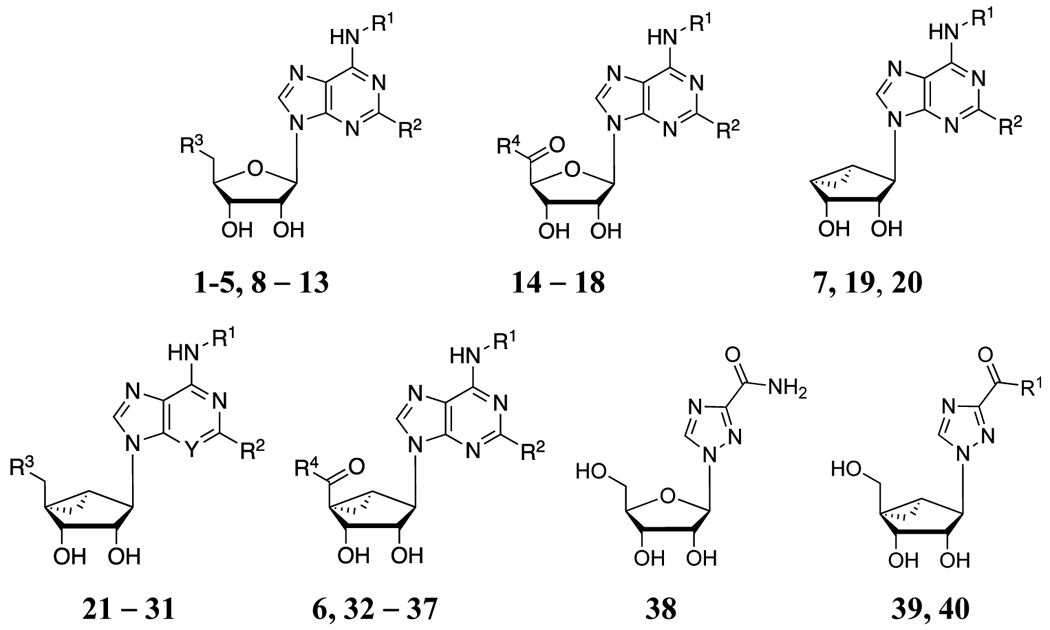 | ||||||
|---|---|---|---|---|---|---|
| Compd. (other changes) |
R1 | R2 | A1AR % inhibition or Ki (nM)a |
A2AAR % inhibitiona |
A3AR % inhibition or Ki (nM)a |
Off-target, receptor (h, unless noted) (Ki, μM)a |
| Ribosides, 5′-OH or 5′-Cl | ||||||
| 1ac,d |  |
H | 2.3 (h), 0.22±0.01 (m) |
794 (h), 808±89 (m) |
72±12 (h), 534±14 (m) |
ND |
| 2c,d |  |
Cl | 0.83 (h), 0.21±0.10 (m) |
2270 (h), 988 (m) |
38±6 (h), 17±5 (m) |
ND |
| 5f (R3 = S-(2-F)-Ph) | 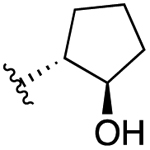 |
H | 12 (h) | >10,000 (h) | >1000 (h) | ND |
| 8b | 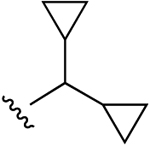 |
H | 0.85±0.27 (h), 0.8 (r) |
1470±380 (h), 1370 (r) |
41.3±5.3 (h) | none |
| 9 | 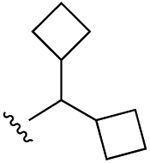 |
H | 2.14±0.52 (h), 0.37±0.02 (m) |
3550±440 (h) | 10,600±1400 (h), 897±45 (m) |
DAT 1.78, NET 4.82 |
| 10 (R3 = Cl) | 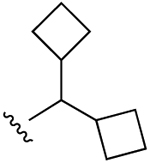 |
H | 4.90±0.87 (h), 0.71±0.12 (m) |
5200i (h) | 16,600±2000 (h), 2110±50 (m) |
TSPO 3.98, σ1 0.69, σ2 0.662 |
| 11 (R3 = SCH2CH3) | 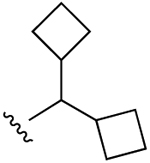 |
H | 63.4±12.1 (h), 3.58±0.01 (m) |
30±5% (h) | 6220±1480 (h), 718±150 (m) |
TSPO (90% inhib.j) σ1 0.626, σ2 0.642 |
| 12 | 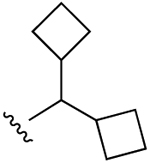 |
Cl | 17.8±8.7 (h), 0.65±0.06 (m) |
2550±540 (h) | 13,200±2500 (h), 653±85 (m) |
5HT2C 1.45 |
| 3c,d | 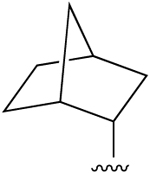 |
H | 0.38±0.19 (h), 0.34 (r), 0.14±0.02 (m) |
>10,000 (h), 477 (r) |
915±299 (h), 282 (r), 424±41 (m) |
ND |
| 4c (R3 = Cl) |
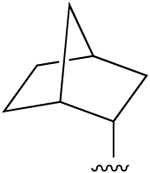 (±) (±) |
H | 0.51 (h), 0.20±0.01 (m) |
1340 (h), 3990±360 (m) |
1290 (h), 2410±330 (m) |
ND |
| 13c (R3 = Cl) | 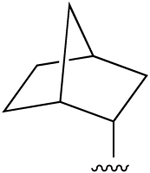 |
H | 0.76±0.42 (h) | 2050±570 (h) | 355±117 (h), 1560±140 (m) |
σ1 0.484, σ2 0.432, 5HT7 1.24 |
| Ribosides, 4′-carbonyl derivatives | ||||||
| 14 (R4 = OH) | H | H | 24±4% (h) | 11±4% (h) | 32±9% (h) | BZP (r) 7.34, σ1 1.48, σ2 0.796 |
| 15d (R3 = NH-Me) | H | H | 36.7±9.4 (h), 84 (r) |
466±95 (h), 67 (r) |
24.4±7.9 (h), 63 (r) |
none |
| 16c,d,h (R4 = NH-Et) | H | H | 6.8±2.4 (h),g 3.00±0.10 (h), 0.45±0.13 (m), 63 (r) |
2.2±0.6 (h),g 35.0±14.0 (h), 12 (r) |
35±12 (h), 14.1±6.8 (m), 113 (r) |
none |
| 17d (R4 = NH-cPr) | H | H | 1.90±0.60 (h), 6.4 (r) |
50±10 (h), 13.4 (r) |
180±50 (h), 1600 (r) |
H1 7.88, σ1 4.12 (gp), σ2 4.35 |
| 18d,h (R4 = NH-(CH2)2OH) | H | H | 12.8±3.1 (h) | 505±30 (h) | 9450±1760 (h) | ND |
| (N)-Methanocarba, 4′-truncated | ||||||
| 7c | 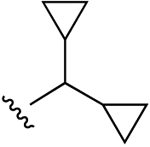 |
Cl | 47.9±10.5 (h), 5.20±0.05 (m) |
3950±410 (h), 34±9% (m) |
470±15 (h), 1060±250 (m) |
5HT2B 0.641, 5HT2C 1.85 |
| 19 | 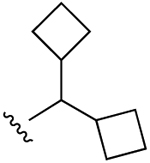 |
Cl | 961±639 (h) | 46±6% (h) | 822±449 (h), 385±52 (m) |
5HT2B 2.90, 5HT2C 8.32, α2A 6.96, TSPO 2.38 |
| 20b | 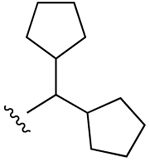 |
Cl | 34±3% (h) | 13±3% (h) | 48±2% (h) | 5HT2C 2.92, σ1 5.09, σ2 2.01 |
| (N)-Methanocarba, 5′-OH or 5′-Cl | ||||||
| 21b | H | Cl | 105±63 (h), 273 (r)b |
3420±270 (h), 1910 (r)b | 353±54 (h)d | none |
| 22 (R3= Cl) | H | Cl | 7.61±0.73 (h) | 1750±290 (h) | 253±148 (h) | σ1 1.97, σ2 2.55 |
| 23b | 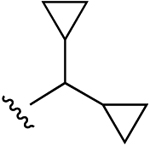 |
Cl | 39±17 (h), 0.71±0.06 (m) |
2200 (h), 41±9% (m) |
1600±210 (h), 1030±40 (m) |
5HT2B 0.641, 5HT2C 1.85, DAT 4.75 |
| 24 | 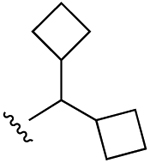 |
H | 22.7±3.0 (h), 0.53±0.19 (m) |
34±1% (h) |
51±6% (h), 918±21 (m) |
BZP (r) 4.08, 5HT2B 0.214, 5HT2C 1.32, σ1 1.79, σ2 0.753 |
| 25 | 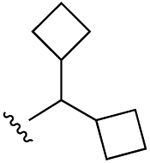 |
Cl | 8.96±1.02 (h), 2.47±0.26 (m) |
55±5% (h), 26±3% (m) |
25±2% (h), 612±58 (m) |
5HT2B 0.153, 5HT2C 0.238, M5 3.00, DAT 4.75 TSPO 2.93 |
| 26 (R3 = Cl) | 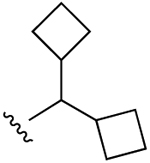 |
H | 32.7±12.4 (h), 1.05±0.19 (m) |
38±3% (h) |
49±4% (h), 934±18 (m) |
5HT2B 2.01, α2A 6.80, σ1 1.50, σ2 1.17, TSPO, 2.02 |
| 27 (R3 = Cl) | 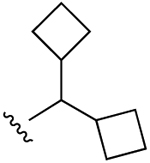 |
Cl | 120±23 (h), 11.2±0.8 (m) |
17±24% (h), 20±3% (m) |
3820±1820 (h), 1560±60 (m) |
5HT2B 1.52, 5HT2C 1.75, H2 2.60, σ2 0.349, TSPO, 2.89 |
| 28 (R3 = S-(2-F)-Ph) | 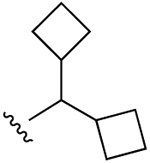 |
Cl |
45±4% (h), 2490±280 (m) |
22±5% (h), 20±9% (m) |
9750±4030 (h), 7440±660 (m) |
5HT2B 0.334, 5HT2C 1.46, σ2 0.583 |
| 29 | 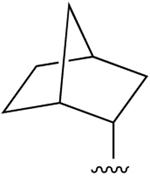 |
Cl | 23.6±5.2 (h), 1.05±0.03 (m) |
4260i (h), 15±2% (m) |
288±54 (h), 574±23 (m) |
5HT2B 0.472, NET 5.74 |
| 30 (Y = CH) | 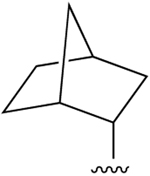 |
Cl | 240±19 (h) | 29±3% (h) | 145±74 (h) | none |
| 31 (R3 = Cl) | 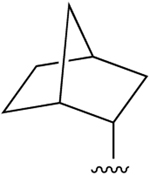 |
Cl | 44.8±1.3 (h), 1.86±0.10 (m) |
54±8% (h), 15±1% (m) |
456±201 (h), 503±13 (m) |
none |
| (N)-Methanocarba, 5′-carbonyl | ||||||
| 32b (R4 = O-Et) | 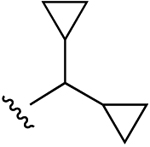 |
Cl | 360±74 (h) | 1570±180 (h) | 236±41 (h) | 5HT2B 0.015, 5HT2C 0.054, TSPO 2.50 |
| 33 (R4 = O-Et, 2′,3′-C(CH3)2) | 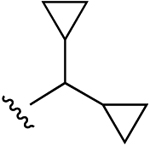 |
Cl | 49±9% (h) | 15±2% (h) | 41±6% (h) | 5HT5A 8.69, H2 6.38 |
| 34b (R4 = NH-Me) | 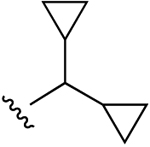 |
Cl | 110±14 (h) | 4320±870 (h) | 34±11 (h) | 5HT2B 0.023, 5HT2C 0.749 |
| 35 (R4 = O-Et) | 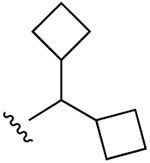 |
Cl | 73.8±14.2 (h) | 57±12% (h) | 1160±300 (h) | 5HT2B 0.097, 5HT2C 0.089, M3 5.90, σ2 2.02 |
| 6e (R4 = NH-Me) | 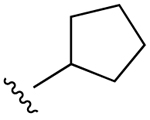 |
Cl | 18.3±6.3 (h), 0.68±0.02 (m) |
3250±300 (h) | 3.7±0.9 (h), 5.8±1.6 (r), 3.46±0.13 (m) |
5HT2B 0.012, σ1 1.55 |
| 36 (R4 = NH-cPr) | 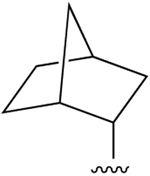 |
Cl | 1.22±0.05 (h), 0.45±0.03 (m) |
520±119 (h), 2650±560 (m) |
59.0±17.8 (h), 29.5±0.6 (m) |
5HT2B 1.57 |
| 37 (R4 = NH-(CH2)2OH) | 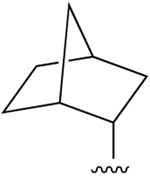 |
Cl | 17.6±5.3 (h), 1.56±0.09 (m) |
5400i (h), 23±1% (m) |
127±29 (h), 447±43 (m) |
5HT2B 0.718, KOR, 2.63 |
| Ribavirin analogues | ||||||
| 38g | - | - | 7410±100 (h), 4430±460 (m) |
23±6% (h) |
16±5% (h), 0% (m) |
ND |
| 39 | OCH3 | - |
6±4% (h), 2990±80 (m) |
14±5% (h) |
22±4% (h), 15±2% (m) |
β3 1.42, H3 4.52, σ2 1.72 |
| 40 | NH2 | - | 495±35 (h), 25.2±2.8 (m) |
11±3% (h) |
41±6% (h), 47±2% (m) |
5HT2B 1.91, 5HT2C 2.35, DAT 6.61, σ2 2.09 |
Binding in membranes of CHO or HEK293 (A2A only) cells stably expressing one of three hAR subtypes, unless noted (n = 3–5). The binding affinity for hA1, A2A and A3ARs was expressed as Ki values using agonists [3H]N6-R-phenylisopropyladenosine 51, [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine 52, or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide 53, respectively. A percent in italics refers to inhibition of binding at 10 μM. Nonspecific binding was determined using adenosine 5′-N-ethyluronamide 54 (10 μM). Values are expressed as the mean ± SEM (n = 3, unless noted). Ki values were calculated as reported.42 Off-target interactions determined by the PDSP. Receptor abbreviations are defined in the Supporting Information. Gp, guinea pig.
Data from Jacobson et al.32
Functional EC50 (nM) at hA2BAR: 16, 140±19; 18, 948±38.44
n = 1.
Percent inhibition at 10 μM.
ND, not determined.
On the ribose or pseudoribose moiety, adenosine-like 4′-CH2OH analogues, 4′-CH2Cl analogues, 4′-truncated analogues and 5′-N-alkyluronamides were included. Monosubstituted adenosine 5′-N-alkyluronamides 15–18 are known AR agonists upon which various A3AR-selective (N)-methanocarba analogues were based,32,35 although their selectivity across species has not been fully characterized.17,36 Small 5′-N-alkyluronamides of adenosine, 15 and 16, are potent but nonselective AR agonists. Certain ribose analogues with additional 5′-amide modifications were previously noted to increase hA1AR selectivity, including the 5′-N-cyclopropyl 17 and 5′-N-(2-hydroxyethyl) 18 uronamide derivatives of adenosine.23 The same 5′ substituents were incorporated into (N)-methanocarba derivatives 36 and 37, but their A1AR selectivity was reduced. 5′-Ester derivatives 32, 33 (2′,3′-protected intermediate) and 35 were also included among these prospective A1AR agonists, although ethyl ester 32 and the corresponding methyl ester displayed only partial agonism at the A3AR.34 Nevertheless, at the hA1AR 5′-ethyl ester 32 was already shown to be a full agonist (89% of maximal efficacy (Emax) of 1), therefore making esters relevant to the study of A1AR agonists.34 Although alkyl esters in general have limited in vivo half-lives, an (N)-methanocarba nucleoside 5′-ester derivative, similar to 32, was shown to be stable with respect to hydrolysis in vivo and in liver microsomes.34
Several modified nucleobases were studied to expand the range of A1AR agonists. Among simple adenosine derivatives, a 3-deaza modification was found to increase hA1AR selectivity.36 Therefore, we prepared compound 30, the 3-deaza analogue of 29, to see if this selectivity is also enhanced in the methanocarba series. One group of analogues containing a nonadenine nucleobase (a 1,2,4-triazole) was additionally included. The antiviral therapeutic nucleoside ribavirin 38 was reported to be a weak, but possibly selective agonist (Ki 86 μM at bovine A1AR; 7.4 μM at hA1AR).36,37 Therefore, we prepared two (N)-methanocarba analogues of ribavirin, i.e. methyl ester 39 and its precise equivalent, carboxamide 40.
Synthesis of target nucleosides
The nucleoside derivatives were prepared as shown in Schemes 1 and 2 and Schemes S1 – S3 (Supporting information) from known starting nucleoside intermediates.32,34 The route to (N)-methanocarba derivatives substituted at the N6 and C2 positions with typical A1AR-enhancing groups and at the 5′ position with an aryl thioether is shown in Scheme 1 and closely parallels previous syntheses.34 After functional group replacement at the 6-position and deprotection at the 5′ position, intermediates 60–62 were either fully deprotected, to give 24, 25 and 29, or substituted with 5′-chloro to give subsequently 26, 27 and 31 or progressed to the 5′-thioether 28. Scheme 2 shows the synthesis of ribose derivatives 9–12 containing the novel N6-dicyclobutylmethyl group and substituted at the 5′ position with an alkyl thioether. Starting with tribenzoyl-protected nucleoside precursors 67 and 68, synthesized as reported,38,39 nucleophilic substitution at the 6 position with dicyclobutylmethylamine was followed by protecting group replacement to yield 2′,3′-isopropylidene intermediate 71, which was subjected sequentially to 5′-Cl substitution, thiol substitution and isopropylidene deprotection to yield 10 and 11. Alternatively, compound 9 was prepared in one step from 6-chloropurine-9-riboside using published methods.28
Scheme 1.

Synthesis of (N)-methanocarba derivatives bearing typical A1AR-enhancing groups at the N6 and C2 positions with and optionally substituted at the 5′ position with chloro or an aryl thioether. Reagents and conditions: (i) R1NH2, DIPEA, 2-propanol, rt; (ii) TBAF, THF, rt; (iii) 10% TFA, MeOH, 70 oC; (iv) SOCl2, pyridine, CH3CN, −5 oC-rt (v) 2-F-PhSH, NaH, DMF, 0 oC-rt.
Scheme 2.

Synthesis of ribose derivatives containing the novel N6-dicyclobutylmethyl group and optionally substituted at the 5′ position with chloro or an alkyl thioether. Reagents and conditions: (i) dicyclobutylmethyl-amine, DIPEA, 2-propanol, rt; (ii) NH3/MeOH, rt; (iii) 2,2-dimethoxy propane, p-TSA, acetone, rt; (iv) SOCl2, pyridine, CH3CN, 0 oC-rt; (v) EtSNa, DMF, rt; (vi) 10% TFA, MeOH, 70 oC.
Scheme S1 (Supporting information) shows the route to (N)-methanocarba derivatives containing an N6-endo-norbonyl group. The synthesis of additional (N)-methanocarba or 5′-ester derivatives, including those bearing alternative nucleobases are shown in Schemes S2 and S3 (3-deaza-adenine in A or 1,2,4-triazole in B).
In vitro pharmacological characterization
The nucleoside analogues were examined in radioligand binding assays (Table 1) at four hARs and two or three mARs as previously described.30,33,40 In cases where there was only ~50% A3AR binding inhibition at 10 μM, an estimated Ki of ~10 μM was used in approximating the selectivity ratio. The hA2BAR affinities of selected compounds were measured in a radioligand binding assay using [3H]54,81 and most analogues tested showed <50% binding inhibition at 10 μM. Values at hA2BAR were (Ki, μM or % at 10 μM): 9, 8.62 μM; 19, 24%; 35, 38%; 38, 39%.
Comparison of ribose 5′-N-alkyluronamides.
In the ribose series, 5′-N-alkyluronamides (14–18) displayed variable AR selectivity, and several of these groups were later applied to (N)-methanocarba analogues. The 5′-N-cyclopropyluronamide 17 was notably 95-fold hA1AR-selective compared to hA3AR.44 Thus, for achieving A1AR selectivity in 5′-N-alkyluronamides, neither the commonly applied 5′-N-methyl- or 5′-N-ethyl-uronamide was optimal. At the hA3AR the 5′-N-methyl-uronamide 15 was best suited for optimizing affinity and selectivity, similar to previous results with the rat (r) ARs.45 In contrast, the corresponding carboxylic acid 14 was inactive or only weakly active in hAR binding, suggesting that a negative charge in that polar ribose-binding region of the AR binding site is not tolerated.
Introduction of other ribose modifications.
Riboside and (N)-methanocarba derivatives were initially compared with the known N6-endo-norbornyl (3, 4, 13, 29–31, 36 and 37) substitution. In general, for this bulky N6 group, the selectivity for hA1AR or mA1AR compared to A3AR was greater among riboside derivatives than among similar methanocarba derivatives. For example, the mA1AR selectivity for N6-endo-norbornyl derivatives was 3000–12,000 fold in the ribose series and 70–500 fold in the methanocarba series, and the hA1AR selectivity showed an even greater divergence in each case. Each of the following three N6-dicycloalkylmethyl ribose analogues bound with similar or higher hA1AR selectivity (fold) compared to hA3AR than the equivalent methanocarba analogues: riboside 9 (4950) compared to methanocarba 24 (~400), 10 (3390) compared to 26 (~300), 12 (742) compared to 25 (>1000). Similarly, at the mA1AR, there tended to be lower selectivity (fold) compared to mA3AR in the methanocarba series: 9 (2420) compared to 24 (1730), 10 (2970) compared to 26 (890), 12 (1000) compared to 25 (248). Thus, among adenine derivatives examined, the ribose series generally resulted in comparable or greater A1AR selectivity.
The ribose hydroxyl groups favored A1AR selectivity. The N6-dicyclopropylmethyl analogues’ A1AR selectivity that was present in the ribose 8 and (N)-methanocarba 5′-OH 23 analogues was lost in the 5′-ester 32 and 5′-N-methyluronamide 34 series. As expected from structural information and previous SAR studies,45 the ribose 2′- and 3′-OH groups are important for AR recognition (cf. weakly binding isopropylidene derivatives 33).
Other ribose 5′ position modifications were compared. As with the known selective agonist 4, a 5′-deoxy-5′-Cl modification in ribose derivative 10 was conducive to high hA1AR (3390-fold) and mA1AR (2970-fold) selectivity compare to A3AR. The simple 5′-Cl analogue 22 exceeded the hA1AR affinity of the parent (N)-methanocarba 2-Cl-adenosine 21 by 14-fold with only minor effects on hA2AAR and hA3AR affinity, thereby increasing A1AR selectivity. However, in the 2-Cl methanocarba series with a large N6 group, both h and mA1AR selectivity was most pronounced in 5′-OH derivative 25, which displayed Ki < 10 nM at both h and mA1AR. The corresponding 5′-Cl derivative 27 was less potent and selective than 25 at the mA1AR (140-fold selective compared to mA3AR) and at the hA1AR (32-fold compared to hA3AR). An inactive methanocarba analogue 28 resembled known partial A1AR agonist 5 in having a large 5′-thioether. The A1AR selectivity of (N)-methanocarba derivatives 36 and 37 was reduced compared to the ribose series. Thus, a bulky 5′ position substitution was incompatible with a methanocarba modification using the criterion of A1AR affinity, and either a 4′-CH2OH or 5′-Cl group (depending on N6 substitution) was the best suited for maintaining A1AR selectivity in the methanocarba series.
Comparison of N6 substituents.
The N6-dicyclopropylmethyl group was incorporated into six analogues (7, 8, 23, 32–34). As previously reported,30,33 the N6-dicyclopropylmethyl group in the 4′-truncated methanocarba analogue 7 was associated with low (hA1AR) or high (mA1AR) selectivity.30 Among other N6-dicyclopropylmethyl analogues only 4′-CH2OH derivative 23 was highly mA1AR selective (1450-fold compared to mA3AR) but only moderately hA1AR selective (41-fold compared to hA3AR). However, the bulky N6-endo-norbornyl group was particularly suited for A1AR-selectivity in the ribose series. N6-endo-Norbornyl derivative 13 is a pure diastereoisomer of known agonist 4 and was 467-fold hA1AR-selective compared to hA3AR
In the (N)-methanocarba series, the 2-chloro-N6-endo-norbornyl derivatives 29 and 31 are selective at the mA1AR (550- and 270-fold, respectively, compared to mA3AR), but only slightly selective at the hA1AR (12- and 10-fold, respectively, compared to hA3AR). Thus, the high A1AR selectivity across several species associated with the N6-endo-norbornyl group in 5′-OH analogue 3 and 5′-Cl riboside 4 (±, a diastereomeric mixture)17 was generalizable to the methanocarba series.
In the series of 4′-truncated cycloalkyl homologues of methanocarba derivative 7, the corresponding N6-dicyclopentylmethyl analogue 20 was inactive in binding at the hARs.33 The intermediate N6-dicyclobutylmethyl homologue 19 was weak and nonselective. Thus, in this truncated methanocarba series, the N6-dicyclopropylmethyl group of 7 provided the highest A1AR affinity.
However, when a 4′-CH2OH was present, the preferred ring size differed from the truncated series. The novel N6-dicyclobutylmethyl ribose derivative 9 (2-H-adenine) was highly potent in A1AR binding. Compound 9 was 2400-fold mA1AR selective compared to the mA3AR; thus, it displayed equivalent selectivity and nearly the same mA1AR affinity (Ki 0.37 nM) as reference compound 1a, while at the hA1AR 9 was ~5000-fold and 1660-fold selective compared to hA3AR and hA2AAR, respectively. The affinity of 9 at hA2BAR was also very weak. Thus, the N6-dicyclobutylmethyl group was identified as promoting multispecies A1AR selectivity in adenosine derivatives.
Nucleobase modifications.
Only two variations at the adenine C2 position, H and Cl, were compared. With a 2-Cl present, the corresponding N6-dicyclobutylmethyl riboside 12 was 1000-fold mA1AR selective compared to mA3AR, but with slightly lower affinity at the mA1AR than its 2-H analogue 9. Similarly, at the hA1AR, the presence of 2-Cl led to reduced selectivity. Compound 12 bound at the hA1AR with 8-fold lower affinity than 9. Thus, in this series, 2-Cl was not advantageous for achieving A1AR selectivity over A3AR.
The 3-deaza-adenine methanocarba analogue 30 (5′-OH) was balanced in hA1AR/hA3AR affinity, i.e. the A1AR, but not the A3AR affinity was reduced by an order of magnitude by the loss of N3 when compared to its adenine equivalent 29. Thus, it was not advantageous to omit N3 for achieving hA1AR selectivity in the methanocarba series. Other deaza-adenine modifications were not examined.
With an alternative nucleobase, i.e. 1,2,4-triazole-3-carboxamide, the methanocarba analogue 40 was ~400-fold selective for the mA1AR and showed a 15-fold greater hA1AR affinity compared to known riboside 38. Thus, while the methanocarba modifcation reduced A1AR selectivity in the adenine series, with this alternative nucleobase, mA1AR selectivity was increased.
Functional assays of selected derivatives.
Truncated nucleoside 7 was previously shown to be a full agonist at the hA1AR in inhibition of cAMP accumlation.33 Here we demonstrated that it is a full agonist at the rA1AR expressed in CHO cells with an EC50 of 6.65±0.32 nM in stimulation of [35S]GTPγS binding (Figure 1A, Emax = 119% of 1 μM 16), determined as reported.41 In comparison, 2 displayed an EC50 of 1.65±0.47 nM, Emax = 103%. At the hA3AR, compound 7 proved to be a partial agonist with an EC50 value of 1.37 μM with ~half the Emax of 16 (Figure 1B). Functional assays using [35S]GTPγS binding evaluated mA1AR and mA3AR agonism (Table 2). All nucleosides tested were full agonists at mA1AR, with N6-endo-norbornyl (N)-methanocarba derivative 31 displaying a maximal 123% efficacy compared to standard 16. Compound 24 was confirmed to be a full mA3AR agonist at 10 μM with Emax of 92±3%, compared to 16. Compound 9 displayed 70% Emax at 10 μM, while other nucleosides (12, 24, 29 and 31) were partial mA3AR agonists with Emax of only 31 – 63%.
Figure 1.



A. Functional activity of agonists 2 and 7, in stimulation of guanine nucleotide binding at the rA1AR (recombinant A1AR membrane preparations from CHO-K1 cells, Perkin Elmer, compared to 2) B. Effects of agonists 7 and 16 in inhibition of cAMP accumulation at hA3AR (in A3AR-expressing CHO cells, treated with 10 μM forskolin, compared to 16). 100% value defined as effect of 1 μM 16. Also, functional assays at the hA1AR are shown for several derivatives (EC50 or IC50 in nM): stimulation of [35S]GTPγS binding (C, 9, 28.0±9.0; 16, 0.12±0.05; 40, 758±175); inhibition of forskolin-stimulated cAMP production (D, 9, 0.14; 40, 87); β-arrestin2 recruitment (E, 9, 209±90; 16, 5.03±2.84; 40, 2460±800).
Table 2.
Functional activity of selected nucleoside derivatives (%Emax at 10 μM) at mA1AR and mA3AR in a guanine nucleotide binding assay.
| Compound (MW, cLogPa) |
Binding Ki (and selectivityb) for mA1AR | Emax, mA1AR, %c | Emax, mA3AR, %c |
|---|---|---|---|
|
9
(389, −1.10) |
0.37 nM (2420) | 101±7 | 70±9 |
|
12 (424, −0.39) |
0.65 nM (1000) | 90±19 | 63±5 |
|
24
(400, −1.62) |
0.53 nM (1730) | 97±11 | 92±3 |
|
29
(406, 1.18) |
1.05 nM (547) | 104±6 | 62±5 |
|
31 (424, 2.38) |
1.86 nM (270) | 123±16 | 50±11 |
|
40
(254, −4.52) |
25.2 nM (~400) | ND | 31±3 |
Calculated using ChemDrawProfessional, v. 16.0.
Fold, compared to mA3AR (Data from Table 1).
Mean (effect of 10 μM of test compound as a % of 10 μM 16) ± SEM in increasing AR-mediated [35S]GTPγS binding, n = 3–6.
ND, not determined.
Several compounds were compared in functional assays at the hA1AR: stimulation of [35S]GTPγS binding; inhibition of forskolin-stimulated cAMP production; β-arrestin2 recruitment (Fig. 1C–E).41 Varying agonist efficacies are shown for 9, 38 and 40 in comparison to standard agonist 16. The (N)-methanocarba analogue 40 appears to be more potent and efficacious than the parent antiviral drug 38.
Off-target interactions.
Table 1 shows other weak (μM) interactions detected for various nucleosides, at serotonin receptors, histamine receptors, other G protein-coupled receptors (GPCRs), sigma receptors and transporters. Key agonists 9 and 40 had several off-target interactions at transporters and receptors with Ki values > 1 μM, i.e. considerably weaker than their mA3AR affinity. However, previously identified32 mixed agonist 6 bound to the 5HT2C serotonin receptor with a Ki of 12 nM. Therefore, minimizing the off-target activity of the present series at 5HT2BR and other GPCRs, determined by the NIMH Psychoactive Drug Screening Program (PDSP, Figure S1, Supporting information),56 was an important consideration in the SAR optimization. Broad screening data (DiscoverX) of truncated methanocarba derivative 7 (at 10 μM) showed that it is not promiscuous, as it interacts only at the A3AR and not in other GPCR assays (Table S1, Supporting information) except for the 5HT2B serotonin receptor (5HT2BR), in agreement with our previous report.34 In a kinase screen, only two weak hits at 10 μM were observed for 7 among 403 kinases (Figure S2, Supporting information).
In vivo biological characterization and ADME-tox studies
Selected analogues (9, 10, 12, 24–27, 29, 31 and 40, Table 3) were examined for the ability to induce hypothermia in the mouse when administered ip. and were compared with published data in the same assay for compounds 1a, 4 and 7.30 With 1a and 7, the peripheral A3AR agonist hypothermic effect predominated, while 4 had a clear central A1AR agonist effect.30 Compound 9 produced hypothermia and hypoactivity at 1 or 3 mg/kg i.p in wild-type mice. As expected,30 hypothermia was generally accompanied by reduced activity for all compounds tested. There was a loss of 9-induced hypothermia in A1AR KO and in A1AR/A3AR double KO mice, but not in A3AR KO mice (Figure 2A–E). Thus, the hypothermia induced by 9 was dependent on A1AR, without a contribution from A3AR. 5′-Chloro derivative 31 at up to 3 mg/kg ip. did not significantly induce hypothermia in WT mice (Table 3). Furthermore, compounds 10, 12, 24–27, and 40 (at 3 or 10 mg/kg, ip.) also had no pronounced effect on Tb (Figure S3, Supporting information). In addition, there was a hypothermic effect in response to (N)-methanocarba 5′-OH analogue 29 (3 mg/kg, ip.). The hypothermia induced by 29 was slightly attenuated in mice lacking A1AR, more diminished in mice without A3AR, and completely lost in mice missing both A1AR and A3AR (Figure 2F–J). These results suggest that 29 caused hypothermia via both central A1AR and peripheral A3AR.30 The nucleoside effects on locomotor activity depression usually paralleled the effects in hypothermia (Table 3). In Figure 3, dose response curves for hypothermia and locomotor depression induced by 9 are compared.
Table 3.
Hypothermia and locomotor activity parameters in wild-type C57BL/6J mice.
| Compound | Dose (mg/kg) | Tb (°C) | Tb P value | Activity (counts) | Activity P value | ARs causing hypothermia |
|---|---|---|---|---|---|---|
| Vehicle | 36.3±0.8 | 15.5±5.5 | ||||
| 1a | 1 | 32.4±0.5 | <0.0001 | 1.5±0.5 | 0.0078 | A3AR > A1AR30 |
| 4 | 3 | 32.2±1.3 | 0.0014 | 2.8±1.6 | 0.0055 | A1AR > A3AR30 |
| 7 | 3 | 33.4±1.7 | <0.0001 | 5.5±4.1 | <0.0001 | A3AR >> A1AR30 |
| 9 | 3 | 33.0±0.7 | <0.0001 | 6.6±2.3 | <0.0001 | A1AR selective (Figure 2) |
| 10 | 10 | 35.8±0.1 | ns | 20.5±4.6 | ns | |
| 12 | 3 | 35.5±0.8 | 0.047 | 13.5±3.5 | ns | |
| 24 | 3 | 36.2±0.4 | ns | 18.1±6.8 | ns | |
| 25 | 10 | 36.6±0.2 | ns | 18.4±3.2 | ns | |
| 26 | 3 | 35.9±0.5 | ns | 14.8±1.7 | 0.041 | |
| 27 | 10 | 36.5±0.6 | ns | 18.9±10.2 | ns | |
| 29 | 3 | 33.4±0.7 | <0.0001 | 10.0±4.3 | 0.046 | A3AR > A1AR (Figure 2) |
| 31 | 3 | 36.6±0.5 | 0.055 | 16.8±3.6 | 0.040 | |
| 40 | 10 | 35.7±0.7 | ns | 19.1±5.3 | ns |
Compounds were dosed ip. Tb and activity were measured by telemetry and the averages from dosing to 60 min were calculated. P value >0.05 was considered not significant (ns).
Data are for the highest dose used and are mean ±SD from 3 to 22 mice/group (usually 5–7).
P values are unpaired t-Test vs vehicle-treated controls assayed simultaneously.
Vehicle data are mean ±SD from all (n=83) vehicle treated mice.
Figure 2.

Effects of nucleoside derivatives on Tb in WT (C57BL/6J male) or AR-KO mice. Compound 9 (A-E) or 29 (F-J), both at 3 mg/kg, ip., and vehicle were dosed in wild-type mice (WT, A, F), or mice lacking A1AR (B, G), A3AR (C, H), or both A1AR and A3AR (D, I). E, J. Tb during the first 60 min after dosing, mean ±SEM, N =4 to 12/group. t-Test (2-tailed, paired or unpaired as appropriate) P values for vehicle vs compound, within genotype.
Figure 3.

Effect of compound 9 (ip) on body temperature and locomotor activity in wild-type C57BL/6J mice.
The pharmacokinetics of 9 (3 mg/kg, p.o.) in the male rat (Table S2, Figure S4, Supporting information) demonstrated a maximal concentration of 121±44 ng/mL, which corresponds to 310 nM. The oral bioavailability was 31–43 %F, depending on the dose (1 – 10 mg/kg), and the oral half-life in plasma was 0.9 – 1.3 h. Thus, peripheral concentrations of 9 in the hypothermia tests are expected to be above its A1AR Ki value for hours after dosing.
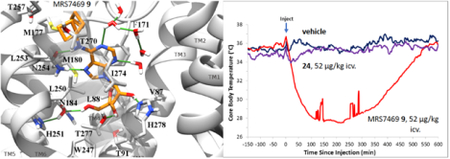
A compound that failed to lower Tb when administered ip., 24, produced intense hypothermia when given intracerebroventricularly (icv., Figure S3C,D, Supporting information). Both compounds 9 and 24 administered by an icv. route caused robust hypothermia with Tb dropping to ~24 °C (5 μg injected, which is equivalent to 0.157 mg/kg). This indicated that compound 24 can cause hypothermia centrally but lacks sufficient concentration in the brain when administered peripherally. In the second experiment, with doses of 1.67 μg/mouse (= 0.052 mg/kg), 9 induced deep hypothermia lasting for ~10 h while 24 reduced Tb only slightly.
As we reported, compound 7 displayed the optimal A1AR selectivity in the series of 4′-truncated N6-dicycloalkylmethyl analogues.33 The pharmacokinetics of 7 was determined in the mouse, demonstrating elimination t1/2 values of 2.5 to 4 h following ip. (3, 15, or 30 mg/kg) administration and an elimination t1/2 of 5 to 11 h with oral administration (dose-dependent; 1, 3, 10 mg/kg) (Figures S5 and S6, Supporting information). The bioavailability was calculated to be 98 and 124 %F for 1 and 3 mg/kg p.o., respectively. Thus, compound 7 has high oral bioavailability. Other ADME-tox measurements (Table S3, Supporting information) also demonstrated drug-like properties of 7: stable in simulated intestinal fluid and human liver microsomes, 51% remaining in simulated gastric fluid (120 min), IC50 at hCYP P450 enzymes (2C9, 2D6, 3A4) >25 μM and CACO-2 cell efflux ratio of 0.91 (i.e. no efflux). Plasma protein binding was 90% (rat) and 94% (mouse).
The cardiovascular effects of 7 and prototypical A1AR agonist 2 were examined preliminarily in the anesthetized rat (Figure S7, Supporting information), although heart rate is known to be lowered by anesthesia. A high dose of 7 (3 mg/kg, ip.) displayed modest effects on heart rate (slow onset of reduction) and blood pressure (a modest, transient rise). Although not directly comparable, a low dose of 2 (0.02 mg/kg, iv.) produced a sustained rise in blood pressure.
The pharmacokinetics of 7 in the monkey was also determined following oral and iv. administration (Figure S8, Supporting information). After consuming (in a food treat) compound 7 (1 or 3 mg/kg body weight), female rhesus monkeys (awake and fed) displayed no adverse reaction up to 4 h post-dosing. The pharmacokinetics of 7 was then determined in awake male cynomolgus monkeys following iv. (0.5 mg/kg) and p.o. (1.0 mg/kg) administration. The bioavailability was estimated as 24%, and there were no clinical observations noted throughout the one-day observation period. Thus, this A1AR agonist was well tolerated in non-human primates.
Beyond the 4′-truncated series, the highly A1AR-selective agonist 9 was examined in other ADME-tox tests (Supporting information). The ligand efficiency (LE) of 9 is a favorable 0.43, and other parameters predict drug-likeness. 9 showed no inhibition against 1A2, 2C19, 2D6, 3A4 CYP isozymes and weak inhibition at the 2C9 isozyme. It was nontoxic in HEP-G2 hepatocytes, displayed a hERG IC50 of >30 μM (fluorescence polarization assay) and was highly soluble in aqueous medium containing 1% DMSO at H 7.4 (>193 μg/kg, Supporting information). The compound showed high permeability in CACO-2 cells, with a low efflux ratio of 3.68. It was completely stable in simulated gastric and intestinal fluids and in plasma of three species (h, m and rat), but showed medium clearance in liver microsomes of the same species.
Another factor to consider in light of the in vivo activity profiles is that the 2-Cl group, as in 12, decreases the polarity (cLogP 9 = −1.1; cLogP 12 = −0.39) and might increase plasma protein binding. If so, this would reduce the free fraction of drug and influence its ability to induce mast cell degranulation and to freely diffuse into the CNS. Therefore, the plasma protein binding of 9 and five other analogues (Table 4) was determined in three species (m, r, h). The analogue 40 of ribavirin was estimated to be largely free in plasma, unlike other analogues. Curiously, substituting ribose 9 with (N)-methanocarba 24 tended to increase the free fraction. Conversely, substituting a chlorine atom at the 2 position (12 vs. 9, for m and r) or 5′ position (31 vs. 29, for all three species) decreased the free fraction. In mouse plasma the free fractions of 24 (~28%) was greater than for 9 (~21%). Therefore, a comparison of plasma protein binding did not explain the inactivity of 24 in inducing hypothermia.
Table 4.
Plasma protein binding at 10 μM in three species, expressed as unbound percent.a
| Compound | human, % freeb |
mouse, % freeb |
rat, % freeb |
|---|---|---|---|
| 9 | 6.05±1.60 | 20.6±1.2 | 14.6±0.01 |
| 12 | 5.81±0.39 | 4.74±0.67 | 4.64±0.06 |
| 24 | 14.6±1.3 | 27.5+1.7 | 17.8±1.2 |
| 29 | 9.75±2.03 | 7.68±0.39 | 6.59±0.49 |
| 31 | 4.04±0.60 | 2.95±0.84 | 2.53±0.45 |
| 40 | c | c | c |
Procedure is in the Supporting information. Verapamil and warfarin served as positive controls.
Mean ± SD.
Estimated >80% free.
Molecular Modeling
Although we previously based hA1AR homology modeling on an agonist-bound hA2AAR, we sought a closer template for this study.46 The current hA1AR antagonist-bound X-ray structures were found to be unsuitable as templates for agonist docking by us and others.29 Thus, modeling hA1AR interactions was based on the recently reported cryo-electron microscopic (cryo-EM) structure.16 We modeled the hA1AR interactions of two selective A1AR agonists: riboside 9 and its methanocarba equivalent 24.
No water molecules were resolved in the cryo-EM structure of hA1AR, but in the adenosine-bound hA2AAR X-ray structure (2YDO59) a water molecule mediates a H-bond network involving the adenosine 5′-OH group, the conserved His250 (6.52, His251 in hA1AR) and Asn181 (5.43, Asn184 in hA1AR). Assuming that a water molecule could have the same role in hA1AR, this molecule was copied to the hA1AR-adenosine complex and minimized to result in formation of a similar H-bond network with 5′-OH, His251(6.52) and Asn184(5.43).
The binding pocket entrance in the cryo-EM hA1AR structure is hindered by some bulky amino acid side chains, which interfere with computational docking of N6-substituted adenosine derivatives. For this reason, Glu172 (EL2), Met177 (5.37), Thr257 (6.58), Lys265 (EL3) and Thr270 (7.34) were temporarily mutated into Ala residues before performing docking simulations. After obtaining a docking pose for compound 9, the aforementioned residues were retro-mutated restoring the original coordinates and were minimized together with the docked ligand. To assess the stability of the proposed binding mode and to observe its temporal evolution, the complex obtained after molecular docking was subjected to MD simulations. Moreover, the methanocarba equivalent of compound 9, compound 24, was superposed by flexible alignment to compound 9’s selected pose, to allow comparison of the two analogs.
To better reproduce the planarity of the N6 nitrogen atom during MD, the dihedral angle C5-C6-N6-C1 (Figure S9) parameters were optimized by fitting the QM Potential Energy Surface (PES) obtained by a dihedral angle scan. 30 ns MD simulations were run (in triplicate) for both the compounds, and the trajectories were analyzed after aligning the protein to the Cα carbon atoms of the initial receptor structure.
Both compounds appeared stabilized in the suggested binding mode, as indicated by an average heavy atom root-mean-square deviation (RMSD) of 1.6 Å in the case of compound 9 and of 1.7 Å in the case of compound 24. Moreover, both compounds’ RMSD and ligand-receptor interaction energies tended to plateau (Fig. SI10–11, A-B) indicating both geometrical and energetical stability. The ligand was anchored to the receptor by a pattern of interactions that are typical for the AR family, involving His251 (6.52), Asn254 (6.55), Thr277 (7.41), His278 (7.42) (Fig. 4, panel A-C; Fig. SI10–11, panel D). The side chain amide of Asn254 (6.55) was engaged in a bidentate H-bond with each ligand, involving N6H as H-bond donor and N7 as acceptor, which was maintained during each entire simulation. The ribosyl 2′-OH group of 9 transiently interacted with His278 (7.42), while this direct contact was less prominent for 24. In the starting adenosine-bound conformation of hA1AR, the hydroxyl group of Thr277 (7.41) pointed far away from 3′-OH group, contrary to the residue at equivalent position of hA2AAR, Ser277 (7.41), which interacts with the 3′ group of ribose through a H-bond. However, a rotation of Thr277 was observed during the MD simulations, together with the formation of a H-bond with 3′-OH group of both 9 and 24. Another key residue was His251 (6.52), which interacts through a direct or water-mediated H-bonding with the 5′-OH group of each compound.
Figure 4.

Conformation of compounds 9 (panel A-B) and 24 (panel C-D) bound to hA1AR (PDB ID: 6D9H) in the last frame of a 30 ns MD simulation. Compounds 9 and 24 are respectively rendered by orange and green sticks. In panels A and C the receptor is represented by a gray ribbon (TM7 transparent to allow the visualization of the pose), with significative residues rendered by gray sticks. Water molecules within 3 Å from the ligands are reported and the pattern of H-bonds is highlighted by green lines. Panels B and D report a different view of the complexes, with the binding site surface colored according to residue hydrophobic character (from purple to cyan for hydrophobic to hydrophilic contributions).
Another important contribution to the stabilization of the bound state was provided by the conserved π- π interaction between Phe171 (EL2) and the aromatic purine base over time. The two cyclobutyl substituents fit into two clefts, respectively between EL2, TM5 and TM6 in one case, and TM6 and TM7 in the other. The character of the two clefts is mainly hydrophobic, with one cyclobutyl surrounded (4 Å distance) by Phe171 (EL2), Glu172 (EL2), Met177 (5.37), Met180 (5.40), Asn254 (6.55) and the other by Leu250 (6.51), Leu253 (6.54), Thr257 (6.58), Lys265 (EL3), Thr270 (7.34), Ile274 (7.38) (Fig. 4, panel B-D, Fig. SI10–11, panel C).
During simulation of 9, an approach between Glu172 (EL2) and Lys265 (EL3), which in the cryo-EM structure are ~7 Å apart, is observed, resulting in a water-mediated interaction. This resembles the interaction between A2AAR Glu169 (EL2) and His264 (EL3) reported in many crystal structures,75 which is known to have a role in decreasing the dissociation rate of A2AAR ligands,76 hinting at a similar effect in hA1AR.
Discussion
Additional AR agonists that display A1AR selectivity in vivo and are well tolerated are still needed. Often the dose range of an agonist to produce a desired effect of A1AR activation, such as antiseizure activity, without toxic side effects is narrow.4,33 Few A1AR agonists are suitable for dose escalation in vivo due to their cardiovascular effects,1,4,10 but we have shown that (N)-methanocarba derivative 7 is an exception to this usual caveat of A1AR agonists, being non-lethal in the mouse at 60 mg/kg. Compound 7 within the 4′-truncated series achieved the optimal A1AR binding selectivity, as other N6-dicycloalkyl groups failed to increase A1AR affinity.33 We have further characterized its A1AR full agonism and its weak A3AR partial agonism. 7 is relatively stable and well tolerated in vitro and in vivo, in rodents and nonhuman primates. Along with 7 as a structural lead in this series, we patterned some of the target molecules on the endo-norbornyl analogue 4, a highly selective A1AR agonist.
Unlike 4, which has a racemic substituent as the N6 group, compounds 3, 13, 29–31, 36 and 37 are stereochemically pure in the form that is more potent in rA1AR binding,28 rather than being a diastereomeric mixture. This defined endo-norbornyl group stereochemistry is distinctly advantageous for pharmacological studies. Another N6 group present in many analogues, dicyclobutylmethyl, has never before appeared in published reports on adenosine agonists. However, it is an important molecular lead toward the refinement of selective A1AR agonists that are well tolerated in vivo.
In a comparison of non-truncated ribose and methanocarba derivatives, we found that A1AR-selectivity is more generally associated with the former series. Furthermore, N6-dicyclobutylmethyl derivatives were identified as novel A1AR agonists. Two ribose analogues, 5′-OH 9 and 5′-Cl-5′-deoxy 10 derivatives, containing the N6-dicyclobutylmethyl group were found to be >2000-fold selective for the mA1AR compared to the mA3AR. Furthermore, 9 and 10 are highly selective for the hA1AR compared to other hAR subtypes. Our efforts to identify methanocarba derivatives that display a clear preference for the A1AR did not reach the degree of selectivity seen in the ribose series. Nevertheless, N6-dicyclobutylmethyl derivatives 24–26 were selective at both hA1AR and mA1AR (≥200-fold selective compared to A3AR). In the series of methanocarba N6-dicycloalkylmethyl analogues, the previously reported N6-dicyclopropylmethyl analogue 23 was less selective at the hA1AR than the higher homologue 25 but more selective at the mA1AR.
Focusing on nucleosides 9 and 24, we have suggested a hypothetical binding mode through MD simulations. Both compounds are stabilized by a network of conserved interactions typical of both the adenosine-bound A2AAR X-ray crystal structure and cryo-EM A1AR. The π-π interaction with Phe171 (EL2) as well as a H-bond network involving His251 (6.52), Thr277 (7.41), His278 (7.42) and Asn254 (6.55) and engaging respectively the ribosyl 2′, 3′, and 5′-OH groups, the exocyclic N6H and the endocyclic N7 nitrogen atom are maintained during the simulations. The two cyclobutyl substituents insert into two hydrophobic clefts and consequently bury receptor hydrophobic surfaces that contribute to binding through a hydrophobic effect.
Alternative nucleobases might be suitable for potent and selective A1AR agonists.36 The human plasma concentrations achieved with clinical doses of ribavirin 38 are equal to or greater than its Ki value at the hA1AR.47 Interestingly, coffee consumption was found to ameliorate some ribavirin side effects, including fatigue.48,49 The AR antagonism associated with ingested caffeine doses raises the possibility that AR activation might be one mechanism responsible for a subset of the numerous side effects of ribavirin.48 Dyspnea is associated with ribavirin use, and this is a known consequence of peripheral A1AR activation.50
The 1,2,4-triazole-3-carboxamide analogue 40 illustrates a means of circumventing the tendency of the (N)-methanocarba modification toward high A3AR affinity by using an alternative nucleobase. In fact, compared to 38, its A1AR affinity is greatly enhanced, to achieve ~400-fold mA1AR selectivity compared to mA3AR. However, not all non-adenine nucleobases have this enhancing effect, as was shown with the 1,3-dialkylxanthine-7-ribosides, which display only a small rA1AR enhancement upon replacement of ribose with (N)-methanocarba (Ki 24 μM for the 1,3-di-n-butyl analogue).63
In the (N)-methanocarba series, conversion of 5′-OH to 5′-Cl either slightly (26 vs. 24) or greatly reduced (27 vs. 25) the A1AR-affinity. A comparison of 21 and 22 indicated a beneficial effect of 5′-Cl substitution in a simple (N)-methanocarba derivative. Also, a 3-deaza modification greatly reduced A1AR affinity (30 vs. 29), but also reduced off-target interactions.
As reported, either central mA1AR or robust peripheral mast cell mA3AR activation can lower body temperature.30 We have focused on 9 as a novel lead, and we demonstrated its ability to induce centrally-mediated A1AR hypothermia when administered ip. in the mouse, which presumably requires it to cross the BBB. Peripheral A3AR activation by 9 also enhances A1AR-induced hypothermia, but the A3AR is not required to lower Tb. Similarly, compound 29 activated both receptors to greatly reduce Tb, although its A1AR effect was less pronounced than for 9. However, compounds 10, 24–27 and 31 did not significantly lower Tb in WT mice, although all were considerably mA1AR-selective. Therefore, these compounds failed to activate either central A1AR or peripheral mast cell A3AR when administered peripherally. The lack of an A1AR effect to cause hypothermia is consistent with low brain entry of the nucleosides.31 However, the lack of a peripheral A3AR effect was surprising and might be explained by a low agonist efficacy at this receptor. As noted, there is reason to expect that 4 is not a full agonist at the A3AR, which might contribute to its favoring the central A1AR effect vs. the peripheral A3AR mast cell effect in Carlin et al.30 Other data on adenosine derivatives with large N6 groups show that they tend to be partial agonists or antagonists at the A3AR, while fully activating the A1AR.17 This could be an important factor in the overall selectivity of 4 as seen in vivo, in addition to its 10,000-fold binding selectivity for mA1AR vs mA3AR. However, several compounds that are similar to 9 in efficacy measurements (Table 2) did not lower Tb. It is worth noting that the mA3AR functional assay in Table 2 might overestimate the intrinsic efficacy of the nucleosides at that subtype, because it utilized HEK cells overexpressing the receptor. Also, the nucleoside concentration (10 μM) used to determine Emax in the guanine nucleotide binding assay is most likely higher than the in vivo plasma concentration achieved for each nucleoside, based on the rat pharmacokinetic data for 9. Thus, many compounds might lack robust activation of mA3AR receptor in vivo.
We also have to consider a pharmacokinetic explanation for the lack of hypothermia of certain analogues, such as 10. Since many nucleosides do not appreciably cross the BBB,31 it is not surprising that most analogues administered ip. did not elicit an A1AR-mediated hypothermia. There is no reason to expect that these compounds are rapidly metabolized or excreted in the mouse, as the relative stability of 9 was shown in the rat. The molecular weights of 4 and its dicyclobutylmethyl analogue 10 are roughly equivalent, so mass alone is not a factor, although shape might be a factor in the BBB permeability.51 There is no direct correlation between ability to induce hypothermia and molecular weight or cLogP of any of the nucleosides in Table 2.
The moderate in vivo potency of 9 to induce hypothermia in the mouse is not comparable to high potency of relatively nonselective AR agonists, such as 16 and N6-(R-phenylisopropyl)-adenosine.52–54 A possible explanation might result from robust activation of multiple AR subtypes and/or less hindered distribution in the body by 16 and its congeners.
High plasma protein binding might also account for the reduced ability of a given nucleoside to enter the brain.80 Although these novel nucleosides have closely related structures, knowing the free fraction of each nucleoside would help determine the available, free concentration of agonist to diffuse across the BBB and to act on the mA3AR in mast cells. However, the data did not provide a plausible explanation for the different effects in hypothermia. A comparison of the hypothermic effects of 9 and 24, administered either ip. or icv., indicated a greatly reduced ability of 24 to cross the BBB compared to 9.
Conclusions
(N)-Methanocarba adenosine derivatives and corresponding ribosides were synthesized and compared pharmacologically to identify novel A1AR agonists for CNS or peripheral applications. The N6-dicyclobutylmethyl ribose analogue 9 and the previously reported truncated N6-dicyclopropylmethyl (N)-methanocarba mA1AR-selective agonist 7 were drug-like. In the methanocarba series, appropriate adenine or pseudoribose functionalization, or use of an alternative ribavirin nucleobase could provide selectively binding A1AR agonists. Nevertheless, A1AR selectivity, such as in stereochemically unambiguous N6-endo-norbornyl derivatives, was more consistently achieved with a ribose ring. Hypothermia induced by BBB-penetrant 9 was primarily driven by the A1AR, which is evidence that the nucleoside is CNS-active. Other nucleosides that selectively bind at A1AR and are full agonists did not induce hypothermia, indicating insufficient brain concentrations. However, it was surprising that only a few of these compounds elicited an A3AR-mediated hypothermia. We modeled hA1AR interactions of dicyclobutylmethyl derivatives 9 and 24 with MD, and conserved interactions were preserved. However, we have not studied the cardiovascular effects of these A1AR agonists. These novel A1AR agonists can now be evaluated in other A1AR central and peripheral actions, such as in pain relief or inhibition of lipolysis.
Experimental section
Chemical synthesis
Materials and instrumentation
Most of the reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Other reagents were purchased from Small Molecules, Inc. (Hoboken, NJ), Anichem (North Brunswick, NJ), PharmaBlock, Inc. (Sunnyvale, CA), Frontier Scientific (Logan, UT) and Enamine (Monmouth Jct., NJ). 1H NMR spectra were obtained with a Bruker 400 spectrometer using CDCl3, CD3OD and DMSO as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ 0.00) for CDCl3 and water (δ 3.30) for CD3OD. NMR spectra were collected with a Bruker AV spectrometer equipped with a z-gradient [1H, 13C, 15N]-cryoprobe. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Sigma-Aldrich. Purity of the final nucleoside derivatives was checked using a Hewlett−Packard 1100 HPLC equipped with a Zorbax SB-Aq 5 μm analytical column (50 × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 5 mM TBAP (tetrabutylammonium dihydrogen phosphate) −CH3CN from 80:20 to 0:100 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity by HPLC analysis (detection at 254 nm). Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless noted. Observed mass accuracies are those expected based on known instrument performance as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
(2R,3R,4R,5R)-2-(6-((Dicyclobutylmethyl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol (9)
Saturated methanolic ammonia solution (25 mL) was added to compound 69 (425 mg, 0.60 mmol) and the solution stirred overnight at temperature in a sealed tube. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 25:1) to give compound 9 (220 mg, 93%) as colorless powder. 1H NMR (CD3OD, 400 MHz) δ 8.28 (s, 1H), 8.20 (s, 1H), 5.97 (d, J = 6.4 Hz, 1H), 4.77 (t, J = 5.6 Hz, 1H), 4.41 (t, J = 7.2 Hz, 1H), 4.34 (d, J = 4.8 Hz, 1H), 4.19 (d, J = 6.0 Hz, 1H), 3.92 (d, J1 = 2.4 Hz, J2 = 12.4 Hz, 1H), 3.78 (d, J1 = 2.4 Hz, J2 = 12.4 Hz, 1H), 2.55–2.51 (m, 2H), 2.02–1.83 (m, 10H), 1.79–1.75 (m, 2H). HRMS calc. C19H28N5O4 (M + H)+: 390.2141; found 390.2144.
(2S,3S,4R,5R)-2-(Chloromethyl)-5-(6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (10)
Compound 10 (89%) was prepared from compound 72 following the same method as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.27 (s, 1H), 8.24 (s, 1H), 6.05 (d, J = 5.2 Hz, 1H), 4.79 (d, J = 5.2 Hz, 1H), 4.41–4.39 (m, 2H), 4.31–4.27 (d, J1 = 4.8 Hz, J2 = 5.2 Hz, 1H), 3.97 (d, J1 = 5.2 Hz, J2 = 6.8 Hz, 1H), 3.87 (d, J1 = 5.2 Hz, J2 = 6.8 Hz, 1H), 2.57–2.50 (m, 2H), 2.01–1.83 (m, 10H), 1.79–1.75 (m, 2H). HRMS calc. C19H27N5O4Cl (M + H)+: 408.1802; found 408.1800.
(2R,3R,4S,5S)-2-(6-((Dicyclobutylmethyl)amino)-9H-purin-9-yl)-5-((ethylthio)methyl)tetrahydrofuran-3,4-diol (11)
Compound 11 (91%) was prepared from compound 73 following the same method as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.30 (s, 1H), 8.23 (s, 1H), 6.02 (d, J = 5.2 Hz, 1H), 4.78 (t, J = 6.0 Hz, 1H), 4.39–4.33 (m, 2H), 4.24–4.21 (m, 1H), 3.01 (d, J1 = 5.6 Hz, J2 = 8.8 Hz, 1H), 2.93 (d, J1 = 5.6 Hz, J2 = 8.8 Hz, 1H), 2.63–2.48 (m, 4H), 2.01–1.83 (m, 10H), 1.76–1.75 (m, 2H), 1.22 (t, J = 7.6 Hz, 3H). HRMS calc. C21H32N5O3S (M + H)+: 434.2226; found 434.2232.
(2R,3R,4R,5R)-2-(2-Chloro-6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol (12)
Compound 12 (91%) was prepared from compound 70 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 8.25 (s, 1H), 5.92 (d, J = 6.0 Hz, 1H), 4.70 (t, J = 5.2 Hz, 1H), 4.40–4.32 (m, 2H), 4.17 (d, J = 6.8 Hz, 1H), 3.92 (d, J1 = 2.4 Hz, J2 = 10.0 Hz, 1H), 3.78 (d, J1 = 2.4 Hz, J2 = 10.0 Hz, 1H), 2.55–2.47 (m, 2H), 2.01–1.84 (m, 10H), 1.77–1.76 (m, 2H). HRMS calc. C19H27N5O4Cl (M + H)+: 424.1752; found 424.1750.
(1R,2R,3S,4R,5S)-4-(6-((Dicyclobutylmethyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (24)
A solution of compound 60 (30 mg, 0.02 mmol) in methanol (3 mL) and 10% TFA in water (3 mL) was heated at 70 °C for 6hrs. Solvent was evaporated under vacuum and the residue was purified on flash silica gel silica chromatography (CH2Cl2:MeOH = 25:1) to give compound 24 (23 mg, 86%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.47 (s, 1H), 8.22 (s, 1H), 4.82 (d, J = 6.4 Hz, 1H), 4.40 (t, J = 6.8 Hz, 1H), 4.31 (d, J = 11.6 Hz, 1H), 3.91 (d, J = 6.4 Hz, 1H), 3.33 (d, J = 11.6 Hz, 1H), 2.54–2.50 (m, 2H), 1.98–1.83 (m, 10H), 1.76–1.75 (m, 2H), 1.68–1.65 (m, 1H), 1.54 (t, J = 5.2 Hz, 1H), 0.78–0.75 (m, 1H). HRMS calc. C21H30N5O3 (M + H)+: 400.2349; found 400.2356.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (25)
Compound 25 (87%) was prepared from compound 61 following the same method as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.81 (s, 1H), 4.79 (d, J = 6.8 Hz, 1H), 4.37 (d, J = 8.0 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.90 (d, J = 6.4 Hz, 1H), 2.53–2.47 (m, 2H), 1.99–1.88 (m, 10H), 1.84–1.82 (m, 2H), 1.65–1.62 (m, 1H), 1.56 (d, J = 4.8 Hz, 1H), 0.78–0.75 (m, 1H). HRMS calc. C21H29N5O3Cl (M + H)+: 434.1959; found 434.1964.
(1S,2R,3S,4R,5S)-1-(Chloromethyl)-4-(6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-2,3-diol (26)
Compound 26 (88%) was prepared from compound 63 following the same method as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.36 (s, 1H), 8.25 (s, 1H), 4.83 (d, J = 6.0 Hz, 1H), 4.40 (t, J = 7.2 Hz, 1H), 4.33 (d, J = 11.6 Hz, 1H), 4.01 (d, J = 6.4 Hz, 1H), 3.65 (d, J = 11.6 Hz, 1H), 2.54–2.51 (m, 2H), 1.99–1.83 (m, 10H), 1.79–1.75 (m, 4H), 0.99–0.95 (m, 1H). HRMS calc. C21H29N5O2Cl (M + H)+: 418.2010; found 418.2016.
(1S,2R,3S,4R,5S)-4-(2-Chloro-6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)-1-(chloromethyl)bicyclo[3.1.0]hexane-2,3-diol (27)
Compound 27 (89%) was prepared from compound 64 following the same procedure as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.29 (s, 1H), 4.83–4.80 (m, 2H), 4.38 (t, J = 8.0 Hz, 1H), 4.31 (d, J = 11.6 Hz, 1H), 3.99 (d, J = 6.8 Hz, 1H), 3.69 (d, J = 11.6 Hz, 1H), 2.53–2.47 (m, 2H), 1.99–1.88 (m, 10H), 1.84–1.74 (m, 4H), 0.99–0.95 (m, 1H). HRMS calc. C21H28N5O2Cl2 (M + H)+: 452.1620; found 452.1624.
(1S,2R,3S,4R,5S)-4-(2-Chloro-6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)-1-(((2-fluorophenyl)thio)methyl)bicyclo[3.1.0]hexane-2,3-diol (28)
Compound 28 (90%) was prepared from compound 66 following the same procedure as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 1H), 7.49–7.45 (m, 1H), 7.20–7.15 (m, 1H), 7.07–7.02 (m, 2H), 4.84 (d, J = 6.0 Hz, 1H), 4.71 (s, 1H), 4.38 (t, J = 7.6 Hz, 1H), 4.05 (d, J = 6.8 Hz, 1H), 3.69 (d, J = 13.6 Hz, 1H), 3.31 (d, J = 13.6 Hz, 1H), 2.54–2.48 (m, 2H), 2.02–1.89 (m, 10H), 1.84–1.77 (m, 2H), 1.59–1.54 (m, 2H), 0.91–0.87 (m, 1H). HRMS calc. C27H32N5O2ClSF (M + H)+: 544.1949; found 544.1951.
(1R,2R,3S,4R,5S)-4-(6-(((1R,2S,4S)-Bicyclo[2.2.1]heptan-2-yl)amino)-2-chloro-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (29)
Compound 29 (84%) was prepared from compound 62 following the same method as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.77 (d, J = 6.8 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 4.30 (br s, 1H), 3.88 (d, J = 6.8 Hz, 1H), 3.38 (d, J = 11.6 Hz, 1H), 2.35–2.34 (m, 2H), 1.93–1.87 (m, 1H), 1.64–1.53 (m, 5H), 1.49–1.34 (m, 2H), 1.27–1.21 (m, 2H), 0.78–0.74 (m, 1H). HRMS calc. C19H25N5O3Cl (M + H)+: 406.1646; found 406.1647.
(1S,2R,3S,4R,5S)-4-(6-(((1R,2S,4S)-Bicyclo[2.2.1]heptan-2-yl)amino)-2-chloro-9H-purin-9-yl)-1-(chloromethyl)bicyclo[3.1.0]hexane-2,3-diol (31)
Compound 31 (93%) was prepared from compound 65 following the same procedure as for compound 24. 1H NMR (CD3OD, 400 MHz) δ 8.28 (s, 1H), 4.81–4.79 (m, 2H), 4.31 (d, J = 11.6 Hz, 1H), 4.03 (br s, 1H), 3.97 (d, J = 6.8 Hz, 1H), 3.68 (d, J = 11.6 Hz, 1H), 2.38–2.32 (m, 2H), 1.92–1.87 (m, 1H), 1.82–1.79 (m, 1H), 1.75 (t, J = 4.8 Hz, 1H), 1.61–1.55 (m, 3H), 1.48–1.34 (m, 2H), 1.30–1.23 (m, 2H), 0.98–0.96 (m, 1H). HRMS calc. C19H24N5O2Cl2 (M + H)+: 424.1307; found 424.1314.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-N-(dicyclobutylmethyl)-9H-purin-6-amine (57)
Dicyclobutylmethylamine (302 mg, 2.17 mmol) and DIPEA (0.75 mL, 4.37 mmol) were added to a solution of compound 55 (250 mg, 0.43 mmol) in 2-propanol and the solution heated at 75 °C overnight. Solvent was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (hexane:ethyl acetate = 3:1) to give compound 57 (267 mg, 91%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.36 (s, 1H), 8.19 (s, 1H), 7.67–7.644 (m, 4H), 7.44–7.33 (m, 6H), 5.40 (d, J = 7.2 Hz, 1H), 5.04 (s, 1H), 4.72 (d, J = 6.8 Hz, 1H), 4.40 (t, J = 7.2 Hz, 1H), 4.23 (d, J = 10.8 Hz, 1H), (d, J = 10.8 Hz, 1H), 2.54–2.50 (m, 2H), 1.99–1.88 (m, 10H), 1.76–1.75 (m 2H), 1.66–1.65 (m, 1H), 1.51 (s, 3H), 1.25 (s, 3H), 1.16 (t, J = 4.8 Hz, 1H), 1.10 (s, 9H), 0.92–0.88 (m, 1H). HRMS calc. C40H52N5O3Si (M + H)+: 678.3839; found 678.3832.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-(dicyclobutylmethyl)-9H-purin-6-amine (58)
Compound 58 (87%) was prepared from compound 56 following the same method as for compound 57.1H NMR (CD3OD, 400 MHz) δ 8.27 (s, 1H), 7.66 (d, J = 6.8 Hz, 4H), 7.44–7.31 (m, 6H), 5.35 (d, J = 7.2 Hz, 1H), 4.95 (s, 1H), 4.71 (d, J = 7.2 Hz, 1H), 4.39 (t, J = 8.0 Hz, 1H), 4.23 (d, J = 6.4 Hz, 1H), 3.76 (d, J = 10.8 Hz, 1H), 2.54–2.50 (m, 2H), 2.01–1.89 (m, 10H), 1.83–1.76 (m, 2H), 1.63–1.60 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.14–1.11 (m, 1H), 1.08 (s, 9H), 0.98–0.93 (m, 1H). HRMS calc. C40H51N5O3SiCl (M + H)+: 712.3450; found 712.3445.
N-((1R,2S,4S)-Bicyclo[2.2.1]heptan-2-yl)-9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-9H-purin-6-amine (59)
(1R, 2S, 4S)-Bicyclo[2.2.1]nonane (306 mg, 2.75 mmol) and triethylamine (0.76 mL, 5.51 mmol) were added to a solution of compound 56 (336 mg, 0.55 mmol) in methanol and the solution stirred at room temperature overnight. Solvent was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (hexane:ethyl acetate = 4:1) to give compound 59 (319 mg, 82%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.26 (s, 1H), 7.66–7.64 (m, 4H), 7.43–7.32 (m, 6H), 5.34 (d, J = 6.8 Hz, 1H), 4.95 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 4.22 (d, J = 6.8 Hz, 1H), 4.03 (br s, 1H), 3.76 (d, J = 6.8 Hz, 1H), 2.36–2.34 (m, 2H), 1.94–1.87 (m, 1H), 1.62–1.59 (m, 4H), 1.52 (s, 3H), 1.48–1.36 (m, 2H), 1.29–1.24 (m, 5H), 1.12 (t, J = 5.2 Hz, 1H), 1.09 (s, 9H), 0.98–0.94 (m, 1H). HRMS calc. C38H47N5O3SiCl (M + H)+: 684.3137; found 684.3143.
((3aR,3bR,4aS,5R,5aS)-5-(6-((Dicyclobutylmethyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (60)
TBAF (0.6 mL, 1M solution in THF) was added to a solution of compound 57 (267 mg, 0.39 mmol) in THF and the solution stirred for 1 h at room temperature. Solvent was evaporated and the residue was purified on silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give compound 60 (161 mg, 93%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.29 (s, 1H), 8.21 (s, 1H), 5.43 (d, J = 7.2 Hz, 1H), 5.03 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 4.40 (t, J = 6.4 Hz, 1H), 4.11 (d, J = 10.8 Hz, 1H), 3.46 (d, J = 10.8 Hz, 1H), 2.55–2.51 (m, 2H), 1.98–1.82 (m, 10H), 1.79–1.76 (m, 3H), 1.53 (s, 3H), 1.25 (s, 3H), 1.19 (t, J = 5.2 Hz, 1H), 1.00–0.96 (m, 1H). HRMS calc. C24H34N5O3 (M + H)+: 440.2662; found 440.2662.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (61)
Compound 61 (89%) was prepared from compound 58 following the same method as for compound 60. 1H NMR (CD3OD, 400 MHz) δ 8.22 (s, 1H), 5.39 (d, J = 7.2 Hz, 1H), 4.95 (s, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.38 (t, J = 8.0 Hz, 1H), 4.01 (d, J = 11.6 Hz, 1H), 3.63 (d, J = 11.6 Hz, 1H), 2.53–2.47 (m, 2H), 1.99–1.84 (m, 10H), 1.78–1.69 (m, 3H), 1.52 (s, 3H), 1.26 (s, 3H), 1.15 (t, J = 4.8 Hz, 1H), 1.00–0.97 (m, 1H). HRMS calc. C24H33N5O3Cl (M + H)+: 474.2272; found 474.2274.
((3aR,3bR,4aS,5R,5aS)-5-(6-(((1R,2S,4S)-Bicyclo[2.2.1]heptan-2-yl)amino)-2-chloro-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (62)
Compound 62 (91%) was prepared from compound 59 following the same method as for compound 60. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.38 (d, J = 6.4 Hz, 1H), 4.95 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 4.00–3.97 (m, 2H), 3.62 (d, J = 12.0 Hz, 1H), 2.35–2.33 (m, 2H), 1.99–1.87 (m, 1H), 1.71–1.67 (m, 1H), 1.61–1.58 (m, 3H), 1.52 (s, 3H), 1.48–1.33 (m, 2H), 1.31–1.22 (m, 5H), 1.14 (t, J = 4.8 Hz, 1H), 0.99–0.95 (m, 1H). HRMS calc. C22H29N5O3Cl (M + H)+: 446.1959; found 446.1953.
9-((3aR,3bS,4aS,5R,5aS)-3b-(Chloromethyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-N-(dicyclobutylmethyl)-9H-purin-6-amine (63)
Thionyl chloride (0.048 mL, 0.39 mL) was added dropwise to a solution of compound 60 (58 mg, 0.13 mmol) in dry CH3CN at −5 °C followed by pyridine (32 L, 0.39 mmol) and the solution stirred for 30 min in the same condition. Then the reaction mixture was brought to room temperature and stirred overnight. Water was added into the reaction and the reaction mixture was neutralized with 1M NaHCO3 solution. The aqueous layer was extracted with CH2Cl2 (3 times), dried over Na2SO4, filtered and evaporated. The residue was purified by flash silica gel column chromatography (hexane:ethyl acetate = 2:1) to afford the compound 63 (37 mg, 62%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.39 (s, 1H), 8.37 (s, 1H), 5.41 (d, J = 7.2 Hz, 1H), 5.09 (s, 1H), 4.83 (d, J = 6.8 Hz, 1H), 4.32 (t, J = 6.8 Hz, 1H), 3.93 (d, J = 10.4 Hz, 1H), 2.64–2.60 (m, 2H), 2.03–1.85 (m, 10H), 1.80–1.78 (m, 3H), 1.54 (s, 3H), 1.36 (t, J = 5.2 Hz, 1H), 1.27 (s, 3H), 1.19–1.15 (m, 1H). HRMS calc. C24H33N5O2Cl (M + H)+: 458.2323; found 458.2322.
2-Chloro-9-((3aR,3bS,4aS,5R,5aS)-3b-(chloromethyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-N-(dicyclobutylmethyl)-9H-purin-6-amine (64)
Compound 64 (65%) was prepared from compound 61 following the same method as for compound 63. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 5.38 (d, J = 7.2 Hz, 1H), 4.94 (s, 1H), 4.78 (d, J = 6.8 Hz, 1H), 4.37 (t, J = 8.0 Hz, 1H), 4.15 (d, J = 11.6 Hz, 1H), 3.79 (d, J = 11.6 Hz, 1H), 2.55–2.49 (m, 2H), 1.99–1.86 (m, 10H), 1.84–1.74 (m, 3H), 1.53 (s, 3H), 1.30–1.27 (m, 4H), 1.14–1.10 (m, 1H). HRMS calc. C24H32N5O2Cl (M + H)+: 492.1933; found 492.1935.
N-((1R,2S,4S)-Bicyclo[2.2.1]heptan-2-yl)-2-chloro-9-((3aR,3bS,4aS,5R,5aS)-3b-(chloromethyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-9H-purin-6-amine (65)
Compound 65 (63%) was prepared from compound 62 following the same method as for compound 63. 1H NMR (CD3OD, 400 MHz) δ 8.14 (s, 1H), 5.36 (d, J = 7.2 Hz, 1H), 4.94 (s, 1H), 4.77 (d, J = 7.2 Hz, 1H), 4.13–4.02 (m, 2H), 3.80 (d, J = 11.6 Hz, 1H), 2.35–2.33 (m, 2H), 1.92–1.86 (m, 1H), 1.81–1.77 (m, 1H), 1.64–1.61 (m, 2H), 1.53 (s, 3H), 1.47–1.33 (m, 2H), 1.30–1.23 (m, 7H), 1.14–1.10 (m, 1H). HRMS calc. C22H28N5O2Cl2 (M + H)+: 464.1620; found 464.1614.
2-Chloro-N-(dicyclobutylmethyl)-9-((3aR,3bS,4aS,5R,5aS)-3b-(((2-fluorophenyl)thio)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-9H-purin-6-amine (66)
NaH (5.6 mg, 0.23 mmol) was added portion wise to an ice-cold solution of 2-fluoro-thiophenol (38 mg, 0.07 mmol) in dry DMF (1 mL). After hydrogen evolution completed, compound 64 (29 mg, 0.05 mmol) was added in dry DMF (0.5 mL) and the solution stirred overnight at room temperature. The reaction mixture was quenched with water, and the aqueous layer was extracted with ethyl acetate (3 times), dried over Na2SO4, filtered and evaporated. The residue was purified on flash silica gel column chromatography (hexane:ethyl acetate = 1:1) to give compound 66 (29 mg, 85%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.06 (s, 1H), 7.52–7.48 (m, 1H), 7.21–7.16 (m, 1H), 7.06–7.00 (m, 2H), 5.36 (d, J = 6.8 Hz, 1H), 4.81 (s, 1H), 4.79 (d, J = 6.8 Hz, 1H), 4.40 (t, J = 6.8 Hz, 1H), 3.66 (d, J = 12.0 Hz, 1H), 3.29 (d, J = 12.0 Hz, 1H), 2.53–2.50 (m, 2H), 2.02–1.84 (m, 10H), 1.78–1.76 (m, 2H), 1.58–1.52 (m, 1H), 1.52 (s, 3H), 1.26 (m, 3H), 1.11 (t, J = 5.2 Hz, 1H), 0.97–0.93 (m, 1H). HRMS calc. C30H36N5O2ClSF (M + H)+: 584.2262; found 584.2271.
(2R,3R,4R,5R)-2-((Benzoyloxy)methyl)-5-(6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diyl dibenzoate (69)
Compound 69 (91%) was prepared from compound 67 following the same method as for compound 57. 1H NMR (CD3OD, 400 MHz) δ 8.26 (s, 1H), 8.08 (s, 1H), 8.06–7.93 (m, 6H), 7.64–7.57 (m, 3H), 7.48–7.37 (m, 6H), 6.54–6.49 (m, 2H), 6.36 (t, J = 5.6 Hz, 1H), 4.92–4.91 (m, 2H), 4.73 (dd, J1 = 3.2 Hz, J2 = 8.0 Hz, 1H), 4.37 (br s, 1H), 2.55–2.49 (m, 2H), 1.97–1.85 (m, 10H), 1.83–1.76 (m, 2H). HRMS calc. C40H40N5O7 (M + H)+: 702.2928; found 702.2919.
(2R,3R,4R,5R)-2-((Benzoyloxy)methyl)-5-(2-chloro-6-((dicyclobutylmethyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diyl dibenzoate (70)
Compound 70 (89%) was prepared from compound 68 following the same method as for compound 57. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 8.04–7.94 (m, 6H), 7.63–7.57 (m, 3H), 7.46–7.37 (m, 6H), 6.50 (d, J = 4.0 Hz, 1H), 6.35–6.28 (m, 2H), 4.75–4.71 (m, 1H), 4.36 (t, J = 8.0 Hz, 1H), 2.51–2.46 (m, 2H), 1.98–1.87 (m, 10H), 1.82–1.75 (m, 2H). HRMS calc. C40H39N5O7Cl (M + H)+: 736.2538; found 736.2526.
((3aR,4R,6R,6aR)-6-(6-((Dicyclobutylmethyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methanol (71)
2,2-Dimethoxypropane (0.31 mL, 2.57 mmol) and p-toluenesulfonic acid (97 mg, 0.51 mmol) were added to a solution of compound 9 (200 mg, 0.51 mmol) in acetone (10 mL) and stirred for 5 h at room temperature. Reaction mixture was neutralized with NaHCO3, filtered and evaporated. The residue was purified on flash silica gel column chromatography (hexane:ethyl acetate = 1:2) to give compound 71 (211 mg, 96%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 2H), 5.38 (d, J= 6.8 Hz, 1H), 4.98 (s, 1H), 4.73 (d, J = 6.8 Hz, 1H), 4.43–4.33 (m, 2H), 3.84–3.67 (m, 2H), 2.08–1.98 (m, 2H), 1.96– 1.72 (m,10H), 1.73–1.69 (m, 2H), 1.52 (s, 3H), 1.26 (s, 3H). HRMS calc. C22H32N5O4 (M + H)+: 430.2454; found 430.2450.
9-((3aR,4R,6S,6aS)-6-(Chloromethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-N-(dicyclobutylmethyl)-9H-purin-6-amine (72)
Compound 72 (67%) was prepared from compound 71 following the same method as for compound 63. 1H NMR (CD3OD, 400 MHz) δ 8.25 (s, 2H), 6.24 (d, J = 2.4 Hz, 1H), 5.50 (d, J1 = 2.4 Hz, J2 = 4.4 Hz, 1H), 5.16 (d, J1 = 2.4 Hz, J2 = 6.0 Hz, 1H), 4.45–4.37 (m, 2H), 3.84 (d, J1 = 4.4 Hz, J2 = 7.2 Hz, 1H), 3.70 (d, J1 = 4.4 Hz, J2 = 7.2 Hz, 1H), 2.54–2.50 (m, 2H), 2.02–1.83 (m, 10H), 1.76–1.75 (m, 2H), 1.61 (s, 3H), 1.40 (s, 3H). HRMS calc. C22H31N5O3Cl (M + H)+: 448.2115; found 448.2113.
N-(Dicyclobutylmethyl)-9-((3aR,4R,6S,6aS)-6-((ethylthio)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (73)
Sodium thioethoxide (27.8 mg, 0.33 mmol) was added to a solution of compound 72 (37 mg, 0.08 mmol) in dry DMF (1 mL) at 0 °C and the solution stirred at room temperature for 1 h. The reaction mixture was quenched with water, and the aqueous layer was extracted with ethyl acetate (3 times), dried over Na2SO4, filtered and evaporated. The residue was purified on flash silica gel column chromatography (hexane:ethyl acetate = 2:1) to give compound 73 (34 mg, 87%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.26 (s, 1H), 8.25 (s, 1H), 6.19 (d, J = 2.4 Hz, 1H), 5.56 (d, J1 = 1.6 Hz, J2 = 4.8 Hz, 1H), 5.08 (m, 1H), 4.39–4.26 (m, 2H), 2.82–2.80 (m, 2H), 2.55–2.46 (m, 4H), 2.01–1.83 (m, 10H), 1.77–1.75 (m, 2H), 1.60 (s, 3H), 1.40 (s, 3H), 1.14 (t, J = 7.2 Hz, 3H). HRMS calc. C24H36N5O3S (M + H)+: 474.2539; found 474.2541.
Pharmacological characterization
Functional assay at rA1AR was performed by GVK Biosciences, Hyderbad, India (Study No: 050–13-IVP). rA1AR membranes (Perkin Elmer Rat A1: 6110511400UA) were pretreated with adenosine deaminase at 1U/mL. 20 μl of 1 nM [35S]GTPγS was added to all wells of a 96-well plate. 20 μl of 10 μM GDP, 20 μl of membrane preparations (5 μg membranes/well) and 20 μl of PVT SPA (PolyVinyl Toluene Scintillation Proximity Assay) beads (1 μg/well) were added, and the total assay volume was made up to 100 μl by adding assay buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 1 mM MgCl2). The reaction mixture was incubated for 60 min at room temperature. Post-incubation, the plate was centrifuged at 2000 rpm for 10 min, and radioactivity was measured.
Functional assays at hA1AR, mA1AR and mA3AR were performed as described.41 In the hA1AR-mediated stimulation of [35S]GTPγS binding and inhibition of forskolin-stimulated cAMP production, an antagonist of the A2BAR (8-[4-[4-(4-chlorophenzyl)piperazide-1-sulfonyl)phenyl]]-1-propylxanthine, 1 μM) was added to prevent activation of the endogenous receptor in HEK cells.
Body temperature.
All animal experiments performed in the manuscript were conducted in compliance with institutional guidelines. Tb and activity were measured in male C57BL/6J mice and A1AR, A3AR, and A1AR/A3AR deficient mice continuously by telemetry as previously described.30 The following compounds (vehicle) were administered ip.: compounds 4, 10, 29 and 31 (10% DMSO); compound 1a (saline); compound 7 (10% DMSO/27% PEG400); compounds 9, 12, 24, 25, 26, 27 and 40 (15% DMSO/15% Kolliphor EL/70% saline).
Central infusions:
Mice were anesthetized with ketamine/xylazine (80/10 mg/kg, ip.). Sterile guide cannulas (5.25 mm, 26 gauge, Plastics One, Roanoke, VA) were unilaterally implanted into the lateral ventricle (coordinates relative to bregma: −0.34 mm anterior, 1.0 mm lateral, and +1.7 mm ventral) and fixed with dental cement (Parkell, Edgewood, NY). Compounds in 5 μl were infused (0.5 μl/min) through a 33-gauge cannula protruding 0.5 mm past the tip of the guide cannula using PE-50 tubing fitted to a 5 μl syringe (Hamilton, Reno, NV).
Molecular Modeling
Ligand-Protein Complex Preparation
The cryo-EM structure of A1AR was retrieved from the Protein Data Bank (PDB ID: 6D9H)55 and prepared using the Protein Preparation Wizard tool57 of the Schrödinger suite (Maestro 2018–1).58 His78/251/278 were protonated on the Nδ nitrogen (named HSD according to the CHARMM nomenclature), while His264 was protonated on Nε (HSE). The Gi protein was removed from the complex before proceeding with the simulations.
Residues Glu172 (EL2), Met177 (5.37), Thr257 (6.58), Lys265 (EL3) and Thr270 (7.34) were mutated into alanines, and the resulting receptor was used for docking simulations. A water molecule was copied from the A2AAR 2YDO59 structure (RESID 2017) and minimized with OPLS360 force field. Compound 9 was docked to the receptor using Glide61,62 docking software of the Schrödinger suite,58 with XP scoring function63 and constraining the compound core to the adenosine scaffold. Glu172 (EL2), Met177 (5.37), Thr257 (6.58), Lys265 (EL3) and Thr270 (7.34) were restored from the cryo-EM structure and finally minimized in presence of the docked compound with OPLS3 force field.60 The conformation of compound 24 was obtained by flexible alignment to compound 9 selected pose.
Molecular Dynamics
The HTMD (version 5.2.0)64 module was used to prepare the systems for MD simulations. An 80×80 Å 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayer was obtained using the VMD Membrane Plugin tool.65 After computing the orientation of the A1AR structure in a membrane through the Positioning of Proteins in Membrane (PPM) web server,66 the ligand-protein complexes were embedded into the membrane leaflet and the overlapping lipids (within 1.3 Å) were removed. The systems were solvated with TIP3P67 water molecules, resulting in a simulation box of 80×80×120 Å, and Na+/Cl− counter-ions were added to neutralize the system in a 0.154 M concentration. The simulations were run using the ACEMD68 MD engine with CHARMM3669,70 force field for protein, lipids, water and ions, and CGenFF (3.0.1)71,72 for the ligands. Missing ligands parameters were assigned by analogy using the ParamChem73 web service (version 1.0.0), with few modifications: adenosine parameters were used for the adenine moiety, and the dihedral angle C5-C6-N6-C1 (Figure S9) was optimized by means of the HTMD64 Parametrize tool, using QM calculations as target data (Hartree-Fock (HF) theory and 6–31G* basis set) and Psi4 code (version 1.2.1).74
The systems were subjected to an equilibration phase involving 5000 conjugate-gradient minimization steps and 40 ns MD simulation in the NPT ensemble, by applying initial positional harmonic restraints to ligand and protein atoms (0.8 kcal mol−1 Å−2 for ligand atoms, 0.85 kcal mol−1 Å−2 for Cα carbon atoms, and 0.4 kcal mol−1 Å−2 for the other protein atoms), linearly reduced in the last 20 ns. A Berendsen barostat (relaxation time 800 fs) was used to maintain the pressure at 1 atm. The equilibrated systems were subjected to three 30 ns MD simulations in the NVT ensemble each. In both equilibration and productive simulations, the temperature was maintained at 310K through a Langevin thermostat (with damping constant 1 ps−1 and 0.1 ps−1 in the case of equilibration and production, respectively). A timestep of 2 fs was employed and the M-SHAKE75 algorithm was used to constrain bonds containing hydrogen atoms. A cutoff of 9 Å was used for non-bonded interactions, with a switching distance of 7.5 Å. The Particle Mesh Ewald (PME)76 method was used to compute long-range electrostatic interactions beyond the non-bonded cutoff, with grid spacing of 1Å.
Trajectory Analysis
Trajectories were aligned by superposing protein Cα carbon atoms to the initial conformation and were analyzed in terms of ligand heavy atoms RMSD, ligand-protein electrostatic and van der Waals interactions, number of H-bonds and number of contacts between protein and ligand. Analysis were performed through an in-house Tcl script employing VMD62 text commands. Ligand-protein electrostatic and van der Waals interactions were computed with NAMD.77 All the data were plotted using the Gnuplot (version 5.0) software78 and figures were produced using Chimera.79
Supplementary Material
Acknowledgments:
We acknowledge support from the NIH Intramural Research Program (NIDDK, ZIA DK031117–28 and ZIA DK075063; NIA AG000356) and the National Institutes of Health (NHLBI R01 grant HL133589). We thank John Lloyd (NIDDK) for mass spectral determinations and Robert O’Connor (NIDDK) for NMR spectra. We thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health’s Psychoactive Drug Screening Program (Contract # HHSN-271-2008-00025-C) for screening data.
Abbreviations:
- AR
adenosine receptor
- HEK 293
human embryonic kidney 293
- DAT
the dopamine transporter
- DMF
dimethylformamide
- DIPEA
diisopropylethylamine
- GPCR
G protein-coupled receptor
- HRMS
high resolution mass spectrometry
- MD
molecular dynamics
- NET
the norepinephrine transporter
- PDSP
NIMH Psychoactive Drug Screening Program
- RMSD
root-mean-square deviation
- SAR
structure activity relationship
- SERT
the serotonin transporter
- Tb
body temperature
- TBAF
tetrabutylammonium fluoride
- TBAP
tetrabutylammonium dihydrogenphosphate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TM
transmembrane
- TSPO
the translocator protein
Footnotes
Supporting Information Available:
Chemical synthesis schemes S1–S3 and associated procedures; PDSP and DiscoverX screening results; ADME-tox studies of 7 and 9; in vivo characterization of various nucleosides, including hypothermia measurements; molecular modeling methods and results; MD simulation videos of receptor-bound 9 and 24; PDB files containing the coordinates of A1AR, ligand (9 or 24) and water molecules within 3 Å from the ligand, extracted from the last frame of MD simulations, Molecular Formula Strings.
References
- 1.a) Kiesman WF; Elzein E; Zablocki J A1 Adenosine receptor antagonists, agonists, and allosteric enhancers. Handb. Exp. Pharmacol 2009, 193, 25–58. [DOI] [PubMed] [Google Scholar]; b) Schenone S; Brullo C; Musumeci F; Bruno O; Botta M A1 receptors ligands: past, present and future trends. Curr. Top. Med. Chem 2010, 10, 878–901. [DOI] [PubMed] [Google Scholar]; c) Giorgi I; Nieri P Adenosine A1 modulators: a patent update (2008 to present). Exp. Opin. Therap. Patents 2013, 23, 1109–1121. [DOI] [PubMed] [Google Scholar]
- 2.Gao ZG; Tosh DK; Jain S; Yu J; Suresh RR; Jacobson KA Chapter 4. A1 adenosine receptor agonists, antagonists and allosteric modulators In: The Adenosine Receptors Varani K. (ed.). Springer, 2018, 34, 59–89, DOI: 10.1007/978-3-319-90808-3_4. [DOI] [Google Scholar]
- 3.Luongo L; Petrelli R; Gatta L; Giordano C; Guida F; Vita P; Franchetti P; Grifantini M; Novellis VD; Cappellacci L; Maione S 5′-Chloro-5′-deoxy-(±)-ENBA, a potent and selective adenosine A1 receptor agonist, alleviates neuropathic pain in mice through functional glial and microglial changes without affecting motor or cardiovascular functions. Molecules 2012, 17, 13712–13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knutsen LJS; Lau J; Petersen H; Thomsen C; Weis JU; Shalmi M; Judge ME; Hansen AJ; Sheardown MJ N-Substituted adenosines as novel neuroprotective A1 agonists with diminished hypotensive effects. J. Med. Chem 1999, 42, 3463–3477 [DOI] [PubMed] [Google Scholar]
- 5.Schulte G; Sommerschild H; Yang J; Tokuno S; Goiny M; Lövdahl C; Johansson B; Fredholm BB; Valen G Adenosine A1 receptors are necessary for protection of the murine heart by remote, delayed adaptation to ischaemia. Acta Physiol. Scand 2004, 182, 133–143. [DOI] [PubMed] [Google Scholar]
- 6.Serchov T; Clement HW; Schwartz MK; Iasevoli F; Tosh DK; Idzko M; Jacobson KA; de Bartolomeis A; Normann C; Biber K; van Calker D Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of Homer1a. Neuron 2015, 87, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Boison D Adenosine as a neuromodulator in neurological diseases. Curr. Opin. Pharmacol 2008, 8, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Greene RW; Bjorness TE; Suzuki A The adenosine-mediated, neuronal-glial, homeostatic sleep response. Curr. Opin. Neurobiol 2017, 44, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrelli R; Scortichini M; Kachler S; Boccella S; Cerchia C; Torquati I; Del Bello F; Salvemini D; Novellino E; Luongo L; Maione S; Jacobson K; Lavecchia A; Klotz KN; Cappellacci L Exploring the role of N6-substituents in potent dual acting 5′-C-ethyl-tetrazolyl-adenosine derivatives: synthesis, binding, functional assays and antinociceptive effects in mice. J. Med. Chem 2017, 60, 4327–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawynok J Adenosine receptor targets for pain. Neuroscience 2016, 338:1–18. [DOI] [PubMed] [Google Scholar]
- 10.a) Tozzi M; Novak I Purinergic receptors in adipose tissue as potential targets in metabolic disorders. Front Pharmacol. 2017, 8, 878. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Merkel LA; Hawkins ED; Colussi DJ; Greenland BD; Smits GJ; Perrone MH; Cox BF Cardiovascular and antilipolytic effects of the adenosine agonist GR79236. Pharmacology 1995, 51, 224–236. [DOI] [PubMed] [Google Scholar]
- 11.Mor M; Shalev A; Dror S; Pikovsky O; Beharier O; Moran A; Katz A; Etzion Y INO-8875, a highly-selective A1 adenosine receptor agonist: Evaluation of chronotropic, dromotropic and hemodynamic effects in rats. J. Pharmacol. Exp. Therap 2010, Retrieved from http://jpet.aspetjournals.org/content/early/2010/05/21/jpet.110.169383.abstract [DOI] [PubMed] [Google Scholar]
- 12.Tupone D; Madden CJ; Morrison SF Central activation of the A1 adenosine receptor (A1AR) induces a hypothermic, torpor-like state in the rat. J. Neurosci 2013, 33, 14512–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jinka TR; Combs VM; Drew KL Translating drug-induced hibernation to therapeutic hypothermia. ACS Chem. Neurosci 2015, 6, 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glukhova A; Thal DM; Nguyen AT; Vecchio EA; Jörg M; Scammells PJ; May LT; Sexton PM; Christopoulos A Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877.e13. [DOI] [PubMed] [Google Scholar]
- 15.Cheng RKY; Segala E; Robertson N; Deflorian F; Doré AS; Errey JC; Fiez-Vandal C; Marshall FH; Cooke RM Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 2017, 25, 1275–1285. [DOI] [PubMed] [Google Scholar]
- 16.Draper-Joyce CJ; Khoshouei M; Thal DM; Liang Y-L; Nguyen ATN; Furness SGB; Venugopal H; Baltos JA; Plitzko JM; Danev R; Baumeister W; May LT; Wootten D; Sexton PM; Glukhova A; Christopoulos A Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nature, 2018, 558, 559–563 [DOI] [PubMed] [Google Scholar]
- 17.Gao ZG; Blaustein J; Gross AS; Melman N; Jacobson KA N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem. Pharmacol 2003, 65, 1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrelli R; Torquati I; Kachler S; Luongo L; Maione S; Franchetti P; Grifantini M; Novellino E; Lavecchia A; Klotz K-N; Cappellacci L 5′-C-Ethyl-tetrazolyl-N6-substituted adenosine and 2-chloro-adenosine derivatives as highly potent dual acting A1 adenosine receptor agonists and A3 adenosine receptor antagonists. J. Med. Chem 2015, 58, 2560–2566. [DOI] [PubMed] [Google Scholar]
- 19.Petrelli R; Scortichini M; Belardo C; Boccella S; Luongo L; Capone F; Kachler S; Vita P; Del Bello F; Sabatino Maione S; Lavecchia A; Klotz KN; Cappellacci L Structure-based design, synthesis, and in vivo antinociceptive effects of selective A1 adenosine receptor agonists. J. Med. Chem 2018, 61, 305–318. [DOI] [PubMed] [Google Scholar]
- 20.Knight A; Hemmings JL; Winfield I; Leuenberger M; Frattini E; Frenguelli BG; Dowell SJ; Lochner M; Ladds G Discovery of novel adenosine receptor agonists that exhibit subtype selectivity. J. Med. Chem 2016, 59, 947–964. [DOI] [PubMed] [Google Scholar]
- 21.Olsson RA; Kusachi S; Thompson RD; Ukena D; Padgett W; Daly JW N6-substituted N-alkyladenosine-5′-uronamides: bifunctional ligands having recognition groups for A1 and A2 adenosine receptors. J. Med. Chem 1986, 29, 1683–1689. [DOI] [PubMed] [Google Scholar]
- 22.Elzein E Kalla R; Li X; Perry T; Marquart T; Micklatcher M; Li Y; Wu Y; Zeng D; Zablocki JA N6-Cycloalkyl-2-substituted adenosine derivatives as selective, high affinity adenosine A1 receptor agonists. Bioorg. Med. Chem. Lett 2007, 17, 161–166. [DOI] [PubMed] [Google Scholar]
- 23.Tosh DK; Phan K; Gao ZG; Gakh A; Xu F; Deflorian F; Abagyan R; Stevens RC; Jacobson KA; Katritch V Optimization of adenosine 5′-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment-based searching. J. Med. Chem 2012, 55, 4297–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison CF; Elzein E; Jiang B; Ibrahim PN; Marquart T; Palle V; Shenk KD; Varkhedkar V; Maa T; Wu L; Wu Y; Zeng D; Fong I; Lustig D; Leung K; Zablocki JA Structure-affinity relationships of 5′-aromatic ethers and 5′-aromatic sulfides as partial A1 adenosine agonists, potential supraventricular antiarrhythmic agents. Bioorg. Med. Chem. Lett 2004, 14, 3793–3797. [DOI] [PubMed] [Google Scholar]
- 25.Bountra C; Hyafil F; Kirilovsky JE Use of Adenosine Derivatives For Treating Dyslipidemia, Obesity, Cardiovascular Risk Factors, Metabolic Syndrome, Polycystic Ovary Syndrome, NIDDM, 2005, patent WO 2005053712 A1. [Google Scholar]
- 26.Roelen H; Veldman N; Spek AL; von Frijtag Drabbe Künzel J; Mathôt RAA; IJzerman AP N6,C8-Disubstituted adenosine derivatives as partial agonists for adenosine A1 receptors. J. Med. Chem 1996, 39, 1463–1471. [DOI] [PubMed] [Google Scholar]
- 27.Volpini R; Costanzi S; Lambertucci C; Vittori S; Klotz KN; Lorenzen A; and Cristalli G Introduction of alkynyl chains on C-8 of adenosine led to very selective antagonists of the A3 adenosine receptor. Bioorg. Med. Chem. Lett 2001, 11, 1931–1934. [DOI] [PubMed] [Google Scholar]
- 28.Trivedi BK; Bridges AJ; Patt WC; Priebe SR; Bruns RF N6-Bicycloalkyladenosines with unusually high potency and selectivity for the adenosine A1 receptor. J. Med. Chem 1989, 32, 8–11. [DOI] [PubMed] [Google Scholar]
- 29.Franchetti P; Cappellacci L; Vita P; Petrelli R; Lavecchia A; Kachler S; et al. N6-Cycloalkyl- and N6-bicycloalkyl-C5′(C2′)-modified adenosine derivatives as high-affinity and selective agonists at the human A1 adenosine receptor with antinociceptive effects in mice. J. Med. Chem 2009, 52, 2393–2406. [DOI] [PubMed] [Google Scholar]
- 30.Carlin JL; Jain S; Gizewski E; Wan TC; Tosh DK; Xiao C; Auchampach JA; Jacobson KA; Gavrilova O; Reitman ML Hypothermia in mouse is caused by adenosine A1 and A3 receptor agonists and AMP via three distinct mechanisms. Neuropharmacology 2017, 114, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaddelee MP; Read KD; Cleypool CG; IJzerman AP; Danhof M; de Boer AG Brain penetration of synthetic adenosine A1 receptor agonists in situ: role of the rENT1 nucleoside transporter and binding to blood constituents. Eur. J. Pharm. Sci 2005, 24, 59–66. [DOI] [PubMed] [Google Scholar]
- 32.(a) Jacobson KA; Gao ZG; Tchilibon S; Duong HT; Joshi BV; Sonin D; Liang BT Semirational design of (N)-methanocarba nucleosides as dual acting A1 and A3 adenosine receptor agonists: Novel prototypes for cardioprotection. J. Med. Chem 2005, 48, 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tosh DK; Deflorian F; Phan K; Gao ZG; Wan TC; Gizewski E; Auchampach JA; Jacobson KA Structure-guided design of A3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem 2012, 55, 4847–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.a) Tosh DK; Paoletta S; Deflorian F; Phan K; Moss SM; Gao ZG; Jiang X; Jacobson KA Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: Receptor docking and potent anticonvulsant activity. J. Med. Chem 2012, 55, 8075–8090. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tosh DK; Phan K; Deflorian F; Wei Q; Gao ZG; Jacobson KA Truncated (N)-methanocarba nucleosides as A1 adenosine receptor agonists and partial agonists: Overcoming lack of a recognition element. ACS Med. Chem. Lett 2011, 2, 626–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tosh DK; Ciancetta A; Warnick E; Crane S; Gao ZG; Jacobson KA Structure-based scaffold repurposing for G protein-coupled receptors: Transformation of adenosine derivatives into 5HT2B/5HT2C serotonin receptor antagonists. J. Med. Chem 2016, 59, 11006–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller CE; Jacobson KA Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta - Biomembranes 2011, 1808, 1290–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodríguez D; Chakraborty S; Warnick E; Crane S; Gao ZG; O’Connor RO; Jacobson KA; Carlsson J Structure-based screening of uncharted chemical space for atypical adenosine receptor agonists. ACS Chem. Biol 2016, 11, 2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.a) Franchetti P; Cappellacci L; Grifantini M; Senatore G; Martini C; Luccacchini A Tiazofurin analogues as selective agonists of A1 adenosine receptors. Res. Commun. Mol. Pathol. Pharmacol 1995, 87, 103–105. [Google Scholar]; b) Janać B; Pešić V; Peković S; Rakić L; Stojiljković M Different effects of adenosine A1 agonist ribavirin on amphetamine-induced total locomotor and stereotypic activities in rats. Ann. N.Y. Acad. Sci 2005, 1048, 396–399. [DOI] [PubMed] [Google Scholar]
- 38.Schaeffer HJ; Thomas HJ Synthesis of potential anticancer agents. XIV. Ribosides of 2,6-disubstituted purines. J. Am. Chem. Soc 1958, 80, 3738–3742. [Google Scholar]
- 39.Zelli R; Zeinyeh W; Haudecoeur R; Alliot J; Boucherle B; Callebaut I; Décout J-L A one-pot synthesis of highly functionalized purines. Org. Lett 2017, 19, 6360–6363. [DOI] [PubMed] [Google Scholar]
- 40.Wan TC; Kreckler LM; Van Orman J; Auchampach JA Pharmacological characterization of recombinant mouse adenosine receptors expressed in HEK 293 cells Abstract, 4th International Symposium of Nucleosides and Nucleotides, Chapel Hill, NC, June 9–11th, 2004. [Google Scholar]
- 41.Du L; Gao ZG; Paoletta S; Wan TC; Barbour S; van Veldhoven JP; IJzerman AP; Jacobson KA; Auchampach JA Species differences and mechanism of action of A3 adenosine receptor allosteric modulators. Purinergic Signalling 2018, 14, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng YC; Prusoff WH Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 43.van der Hoeven D; Wan TC; Gizewski ET; Kreckler LM; Maas JE; Van Orman J; Ravid K; Auchampach JA A role for the low-affinity A2B adenosine receptor in regulating superoxide generation by murine neutrophils. J. Pharmacol. Exp. Therap 2011, 338, 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobson KA; Ohno M; Duong HT; Kim SK; Tchilibon S; Cesnek M; Holy A; Gao ZG A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chemistry and Biology 2005, 12, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallo-Rodriguez C; Ji X-D; Melman N; Siegman BD; Sanders LH; Orlina J; Fischer B; Pu Q-L; Olah ME; van Galen PJM; Stiles GL; Jacobson KA Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J. Med. Chem 1994, 37, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu F; Wu H; Katritch V; Han GW; Jacobson KA; Gao ZG; Cherezov V; Stevens RC Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindahl K; Stahle L; Bruchfeld A; Schvarcz R High-dose ribavirin in combination with standard dose peginterferon for treatment of patients with chronic hepatitis C. Hepatology 2005, 41, 275–279. [DOI] [PubMed] [Google Scholar]
- 48.Carrieri MP; Cohen J; Salmon-Ceron D; Winnock M Coffee consumption and reduced self-reported side effects in HIV-HCV co-infected patients during PEG-IFN and ribavirin treatment: results from ANRS CO13 HEPAVIH. J. Hepatol 2012, 56, 745–747. [DOI] [PubMed] [Google Scholar]
- 49.Freedman N; Curto T; Lindsay K; Wright E; Sinha R; Everhart J; HALT-C Trial Group. Coffee consumption is associated with response to peginterferon and ribavirin therapy in patients with chronic hepatitis C. Gastroenterology 2011, 140, 1961–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong JL; Ho CY; Kwong K; Lee LY Activation of pulmonary C fibers by adenosine in anaesthetized rats: role of adenosine A1 receptors. J. Physiol. (Lond) 1998, 508, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van de Waterbeemd H; Camenisch G; Folkers G; Chretien JR; Raevsky OA Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors, J. Drug Targeting 1998, 6, 151–165. [DOI] [PubMed] [Google Scholar]
- 52.Yang JN; Tiselius C; Dare E; Johansson B; Valen G; Fredholm BB Sex differences in mouse heart rate and body temperature and in their regulation by adenosine A1 receptors. Acta Physiol 2007, 190, 63–75. [DOI] [PubMed] [Google Scholar]
- 53.Yang JN; Wang Y; Garcia-Roves PM; Bjornholm M; Fredholm BB Adenosine A3 receptors regulate heart rate, motor activity and body temperature. Acta Physiol 2010, 199, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carlin JL; Jain S; Duroux R; Suresh RR; Xiao C; Auchampach JA; Jacobson KA; Gavrilova O; Reitman ML Activation of adenosine A2A or A2B receptors causes hypothermia in mice. Neuropharmacology 2018, 139, 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin Q; Fan S; Zhang Y; Xu M; Zhang H; Yang Y; Lee AP; Woltering JM; Ravi V; Gunter HM; Luo W; Gao Z; Lim ZW; Qin G; Schneider RF; Wang X; Xiong P; Li G; Wang K; Min J; Zhang C; Qiu Y; Bai J; He W; Bian C; Zhang X; Shan D; Qu H; Sun Y; Gao Q; Huang L; Shi Q; Meyer A; Venkatesh B Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nature 2016, 540, 395–399.27974754 [Google Scholar]
- 56.Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madhavi Sastry G; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 58.Maestro; Schrödinger, LLC: New York, 2018. [Google Scholar]
- 59.Lebon G; Warne T; Edwards PC; Bennett K; Langmead CJ; Leslie AGW; Tate CG Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- 61.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 62.Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 63.Ravi G; Lee K; Ji X. -d.; Kim HS; Soltysiak KA; Marquez VE; Jacobson KA Synthesis and purine receptor affinity of 6-oxopurine nucleosides and nucleotides containing (N)methanocarba-pseudoribose rings. Bioorg. Med. Chem. Lett 2001, 11, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doerr S; Harvey MJ; Noé F; De Fabritiis G HTMD: High-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput 2016, 12, 1845–1852. [DOI] [PubMed] [Google Scholar]
- 65.Humphrey W; Dalke A; Schulten K VMD - Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 66.Lomize MA; Pogozheva ID; Joo H; Mosberg HI; Lomize AL OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 2012, 40, D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 68.Harvey M; Giupponi G; De Fabritiis G ACEMD: Accelerated molecular dynamics simulations in the microseconds timescale. J. Chem. Theory Comput 2009, 5, 1632–1639. [DOI] [PubMed] [Google Scholar]
- 69.Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi1 and chi2 dihedral angles. J. Chem. Theory Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vanommeslaeghe K; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vanommeslaeghe K; Raman EP; MacKerell AD Jr. Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 2012, 52,3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.http://cgenff.paramchem.org, accessed 06/2018
- 74.Parrish RM; Burns LA; Smith DGA; Simmonett AC; DePrince AE; Hohenstein EG; Bozkaya U; Sokolov AY; Di Remigio R; Richard RM; Gonthier JF; James AM; McAlexander HR; Kumar A; Saitow M; Wang X; Pritchard BP; Verma P; Schaefer HF; Patkowski K; King RA; Valeev EF; Evangelista FA; Turney JM; Crawford TD; Sherrill CD Psi4 1.1: An open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J. Chem. Theory Comput 2017, 13, 3185–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kräutler V; Van Gunsteren WF; Hünenberger PH A Fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem 2001, 22, 501–508. [Google Scholar]
- 76.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A. Smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- 77.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Williams T; Kelley C Gnuplot 5.0, http://www.gnuplot.info, accessed August 15, 2018. [Google Scholar]
- 79.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 80.Waterhouse RN Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol 2003, 5, 376–389. [DOI] [PubMed] [Google Scholar]
- 81.Guo M; Gao Z-G; Tyler R; Stodden T; Wang G-J; Wiers C; Fowler J; Rice KC; Jacobson KA; Kim SW; Volkow N Preclinical evaluation of the first adenosine A1 receptor partial agonist radioligand for positron emission tomography (PET) imaging. J. Med. Chem 2018, 61, 9966–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.