

Abstract
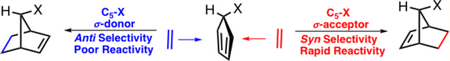
The reactivities and π-facial stereoselectivities of Diels–Alder reactions of 5-substituted cyclopentadienes were studied using density functional theory. Burnell and co-workers previously showed that the π-facial selectivities result from the energies required to distort the reactants into the transition state geometries. We have discovered the origins of these distortions. C5–X σ-donors predistort the cyclopentadiene into an envelope conformation that maximizes the stabilizing hyperconjugative interaction between the C5–X σ-bond and the diene π-system. This envelope conformation geometrically resembles the anti transition state. To minimize the destabilizing effect of negative hyperconjugation, C5–X σ-acceptors predistort in the opposite direction toward an envelope geometry that resembles the syn transition state. We now show how hyperconjugative effects of the C5–X substituent influence the stereoselectivities and have developed a unified model rationalizing the stereoselectivities and reactivities of 5-substituted cyclopentadiene Diels–Alder reactions.
Graphical Abstract
INTRODUCTION
Cyclopentadiene is generally more reactive than other cyclic dienes in the Diels–Alder reaction.1 Substitution at the 5-position of the cyclopentadiene can alter the reactivity, and perhaps surprisingly, in the normal-electron demand Diels– Alder reaction, electron-withdrawing substituents increase the reactivity! We have shown how this arises by destabilization of the cyclopentadiene.2 When the substituent at the 5-position is a σ-acceptor, negative hyperconjugation results in the cyclo-pentadiene having pseudo 4π electron antiaromatic character and accelerated reactivity.
5-Substituted cyclopentadienes (C5–X) are facially asymmetric and, depending on the substituent, will react on either the syn or anti face of the cyclopentadiene with regard to the C5–X substituent. As shown in Scheme 1, the π-facial stereoselectivity of the cycloaddition is considered syn when the dienophile (Y = Z) reacts on the same face of the C5–X substituent, whereas addition to the face opposite of the C5–X substituent is considered anti. Winstein and Woodward reported the first contrasteric (syn) Diels–Alder reaction of 5-acetoxycyclopentadiene (C5–OAc) with ethylene (Scheme 2) during their seminal studies on 7-norbornenyl cations.3
scheme 1.
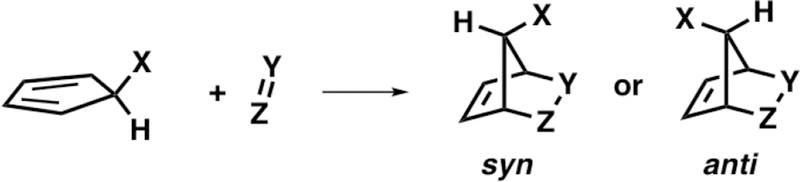
Syn and Anti Diels–Alder π-Facial Selectivity to a C5–X Cyclopentadiene with the X═Y Dienophile
Scheme 2.
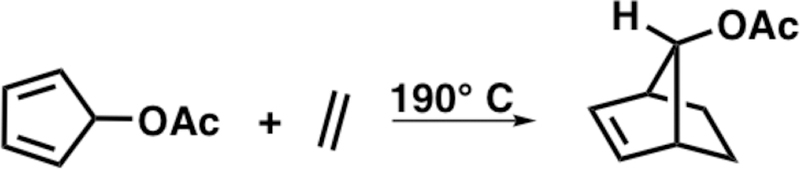
Reaction of C5–OAc with Ethylene Exclusively Forms the Syn Adduct3
Similar constrasteric cycloadditions have since been reported. Scheme 3 shows the π-facial selectivity in the Diels–Alder reactions of C5–F, C5–Cl, and C5–Br with dimethyl acetylenedicarboxylate (DMAD).4,5 C5–F reacts with syn π-facial stereoselectivity; C5–Cl forms a mixture of syn and anti adducts; and C5–Br reacts with anti π-facial stereoselectivity.
Scheme 3.
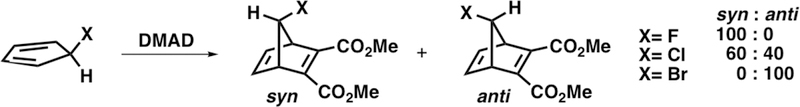
Syn and Anti π-Facial Stereoselectivity in the Diels–Alder Reactions of C5–F, C5–Cl, and C5–Br with DMAD4,5
Control of π-facial selectivity has been used in the synthesis of aconitine alkaloids. Scheme 4 shows how the reaction between 5-methoxycyclopentadiene 1 and cyclopropene 2 proceeds predominantly with syn selectivity to yield the desired intermediate 3a in the David Gin synthesis of neofinaconitine.6 The late David Gin visited our group in early 2011, and he brought the subject of π-facial stereoselectivity in cyclo-pentadienes to our attention. We have worked on this problem since then and now offer a comprehensive explanation for the reactivities and stereoselectivities of Diels–Alder reactions involving 5-substituted cyclopentadienes.
Scheme 4.
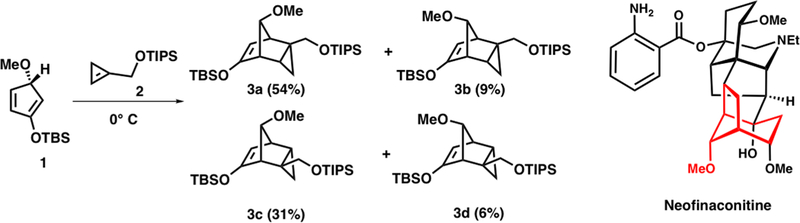
Gin’s Exploitation of π-Facial Stereoselectivity in the Total Synthesis of Neofinaconitine6
Many explanations have been offered, and it seems that the origin of π-facial stereoselectivity in 5-substituted cyclo-pentadiene cycloadditions remains unsettled.7 Cieplak proposed that the stereoselectivity of a number of nucleophilic reactions can be explained through the hyperconjugative stabilization of an incipient σ*-bond by an antiperiplanar donor σ-bond in the transition state.8 Fallis and Macaulay applied the Cieplak effect to Diels–Alder reactions of 5-substituted cyclopentadienes in order to rationalize the syn and anti π-facial stereoselectivity.9 They proposed that the cycloaddition occurs anti to the C5–X bond that is the better σ-donor. Scheme 5 shows the proposed σC5-X–σ* hyperconjugative interaction of the antiperiplanar C5–X bond with the incipient bonds. Anti stereoselectivity is predicted when the C5–X substituent is a stronger σ-donor than the hydrogen atom of the C5–H bond, while a C5–X substituent that is a worse σ-donor is predicted to give syn selectivity. This explanation does not, however, explain why anti C5-X σ-donors slow down, instead of accelerate, reactivity.
Scheme 5.
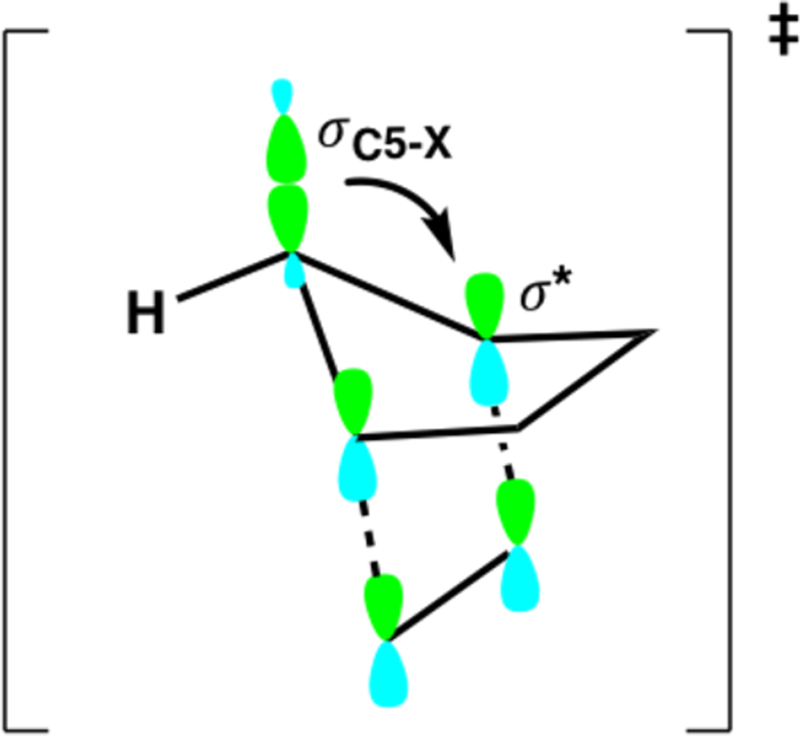
Hyperconjugative Stabilization of the Incipient σ* Bonds by the Antiperiplanar C5–X σ-Bond (Cieplak Effect)
Inagaki, Fujimoto, and Fukui explained the π-facial stereoselectivity of 5-substituted cyclopentadienes with orbital mixing.10 They proposed that orbital mixing between the nonbonding orbital (n) of the C5–X substituent, the diene π-HOMO, and the σ-orbitals of the diene carbon framework determines the π-facial selectivity. When the π-HOMO lies higher in energy than the nonbonding orbital of the C5–X substituent, the mixing results in an increase in the amplitude of the C1 and C4 p-orbitals on the syn face of the 5-substituted cyclopentadiene (Scheme 6). Conversely, when the π-HOMO lies lower in energy than the nonbonding orbital of the C5–X substituent, the mixing increases the amplitude of the C1 and C4 p-orbitals on the anti face of the 5-substituted cyclo-pentadiene. The dienophile then reacts on the face of the cyclopentadiene with the largest p-orbital amplitude at the C1 and C4 positions.
Scheme 6.
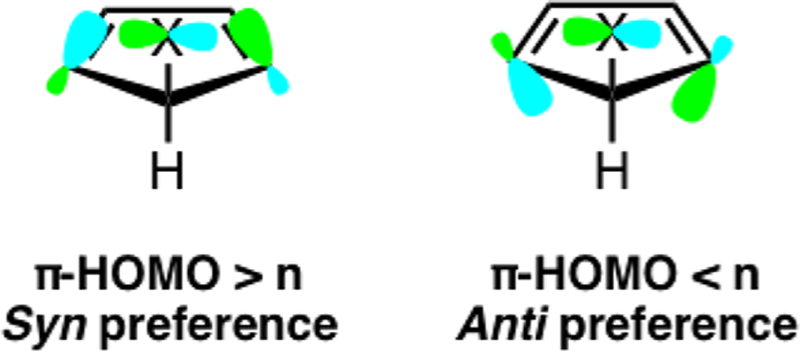
Deformation of the π-HOMO from Orbital Mixing Proposed by Inagaki, Fujimoto, and Fukui
Burnell and co-workers studied computationally the π-facial selectivities of C5–X cyclopentadienes.11,12 They amassed extensive computational evidence to show that the π-facial stereoselectivity is controlled by the energy required to deform the diene into the transition state geometry. They associated the deformation with the change of the C1–C5–X angle between the ground and transition state geometries.
We have recently reported cyclopentadienes as potential bioorthogonal reactants13 and showed how the C5–X substituent has a very large effect on the Diels–Alder reactivity.2 Understanding the reactivity and stereoselectivity trends in 5-substituted cyclopentadienes will be of further value in the development of new bioorthogonal cyclo-pentadienes. To better understand how the reactivity and the syn and anti π-facial stereoselectivity trends relate to the properties of the C5–X substituent, we have investigated a wide scope of C5–X cyclopentadienes (Scheme 7) with the distortion/interaction–activation strain model.14
Scheme 7.
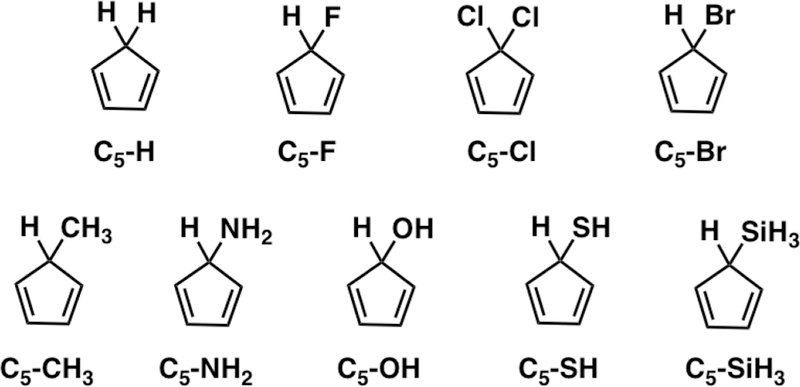
C5–X Cyclopentadienes Studied in This Work
COMPUTATIONAL METHODS
All calculations were performed with Gaussian 09.15 Geometry optimizations and frequency calculations were calculated with the M06–2X16 functional and the 6–31G(d) basis set. The M06–2X functional has been found to accurately reproduce experimental trends in the reactivity and selectivity of Diels–Alder reactions.17 Normal mode analysis of each structure verified that each stationary point is either a first-order saddle point or an energy minimum. Single-point energies were computed using the 6–311++G(d,p) basis set. The distortion/interaction model was applied to the transition state structures to disect the activation energies into the distortion and interaction energy components. The procedure to carry out this analysis procedure has been reviewed recently.14
RESULTS AND DISCUSSION
The anti and syn transition structures and the activation free energies (ΔG⧧) for the Diels–Alder reactions of the 5-substituted cyclopentadienes with ethylene are shown in Figure 1. Syn π-facial stereoselectivity is favored when the C5 substituent is F, OH, NH2, or Cl. Poor π-facial selectivity is predicted when the substituent is Br, SH, or Me. Anti π-facial stereoselectivity is favored when the C5–X substituent is SiH3. The activation free energies of the syn and anti transition states range from 24 to 38 and from 29 to 33 kcal/mol, respectively. There is a correlation between the electronegativity of the C5– X substituent and the activation barriers, as observed earlier by Burnell.11,12 Electron-withdrawing substituents accelerate the reactivity, and electron-donating substituents decrease the reactivity, with a range in activation energies of 10 kcal/mol.
Figure 1.
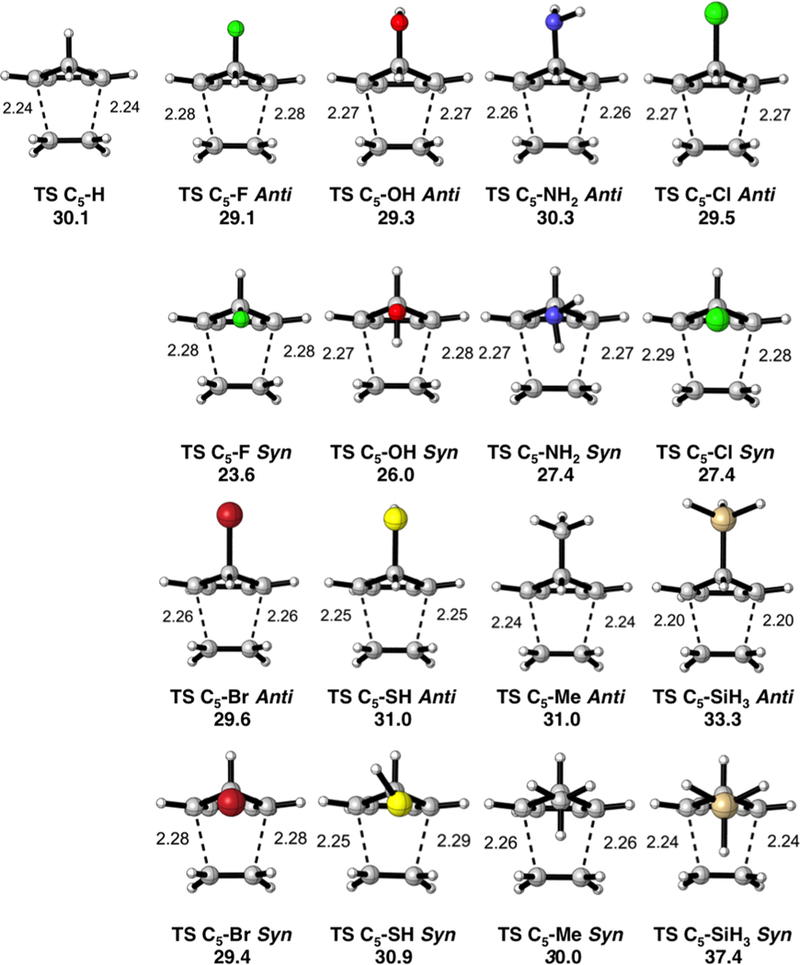
Transition state structures with forming bond lengths reported in Å and activation free energies (ΔG⧧) in kcal/mol for the syn and anti Diels–Alder reactions of the 5-substituted cyclopentadienes with ethylene.
Extensive computational and experimental studies by the Schleyer group on the aromaticity of cyclic π-systems with one saturated linkage (cyclopropene, cyclopentadiene, and cyclo-heptatriene) show that the substituents at the saturated linkage contribute to the π-electron count as pseudo π-donors or π-acceptors via hyperconjugative interactions with the π-system.18 The effect of the C5–X substituent on the stability on the cyclopentadiene was estimated here with the isodesmic equation shown in Figure 2. This isodesmic equation measures the aromatic stabilization enthalpy (HASE) of the cyclo-pentadiene relative to nonconjugated cyclopentadienes for which cyclic electron delocalization of the π-bonds is not possible. A positive reaction enthalpy in the isodesmic equation indicates that cyclic delocalization of the π-electrons via hyperconjugation is stabilizing. The weak hyperconjugative donors, C5–H and C5–Me, are stabilized by 2–3 kcal/mol, which arises mostly from the favorable π-conjugation. Silyl substitution further stabilizes the cyclopentadiene to 7.4 kcal/mol, whereas fluorine substitution destabilizes the cyclo-pentadiene to −3.4 kcal/mol in the isodesmic equation. When the C5–X substituent is a σ-acceptor, the hyper-conjugative π–σ*C5-X interaction destabilizes the cyclopenta-diene by giving it pseudo 4π electron antiaromatic character. When the C5–X substituent is a σ-donor, positive hyperconjugation stabilizes the cyclopentadiene by giving it pseudo 6π electron aromatic character. The aromatic stabilization energies influence the activation and reaction energies.2
Figure 2.
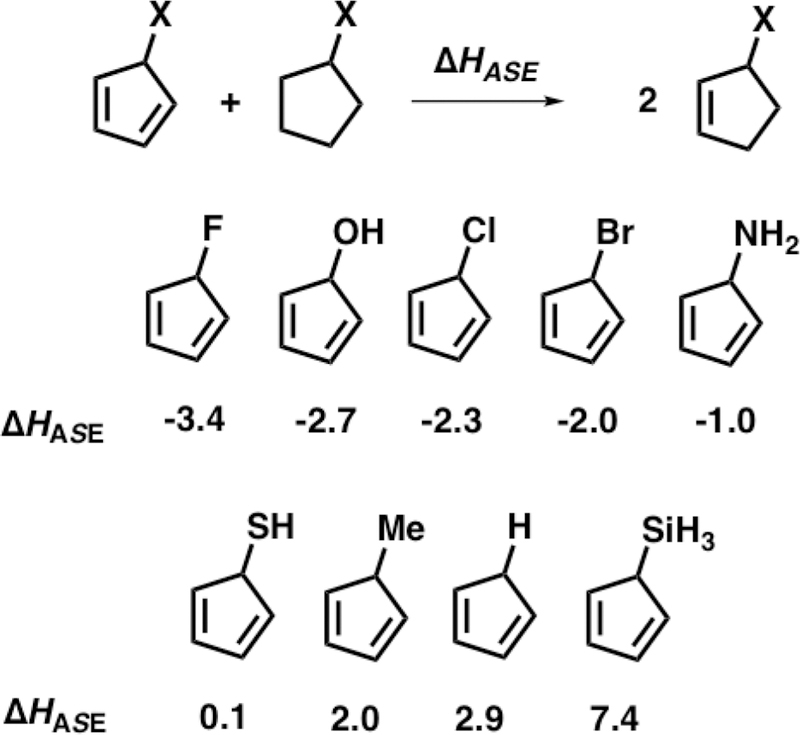
Isodesmic equation (specifically hypohomodesmotic)19 and calculated aromatic stabilization enthalpies of the cyclopentadienes.
Figure 3a shows a plot of ΔH of reaction for the reactions of the C5–X cyclopentadienes vs the aromatic stabilization energy (ΔHASE) of the diene. The linear correlation suggests that the exothermicities of these cycloadditions are related to the stabilities of the C5–X cyclopentadienes. The norbornene π-bond donates into the σ*C5-X bond of substituent anti to the norbornene π-bond.20 The syn adducts are more stable than the anti adduct with the exception of C5–SiH3. The syn preference becomes increasingly favored as the C5–X substituent becomes a stronger σ-acceptor, a result of a more stabilizing π–σ*C5-X interaction.
Figure 3.
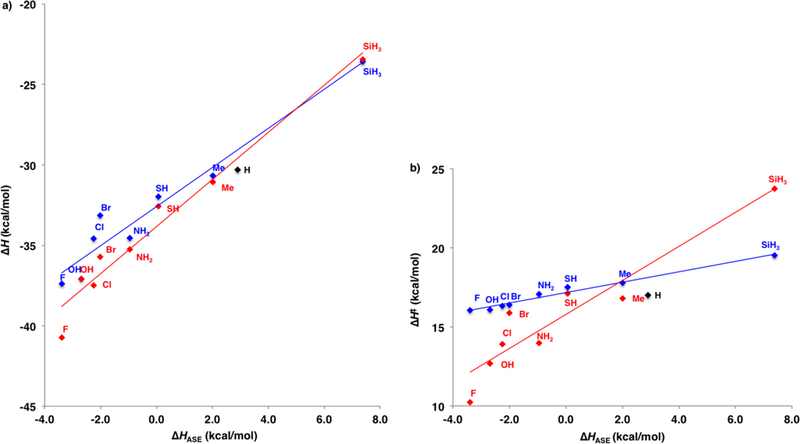
Plots of the reaction enthalpies (a) and activation enthalpies (b) against the calculated aromatic stabilization enthalpies. Syn: red, (a) ΔH = 1.2ΔHASE – 33, r2 = 0.95; (b) ΔH⧧ = 1.1ΔHASE + 16, r2 = 0.90. Anti: blue, (a) ΔH = 1.5ΔHASE – 34, r2 = 0.95; (b) ΔH⧧ = 0.33ΔHASE + 17, r2 = 0.98.
Figure 3b shows a plot of the ΔH⧧ for the C5–X cycloadditions with ethylene against the diene aromatic stabilization energies. Here the correlations are quite different for the syn and anti reactions with slopes of 1.1 and 0.33, respectively. The syn reactions are clearly favored for electron-withdrawing substituents, while the silyl-substituted cyclo-pentadiene reacts with a strong preference for the formation of the anti adduct.
We applied the distortion/interaction-activation strain analysis14 in order to understand the origins of the syn and anti π-facial stereoselectivity in Diels–Alder reactions of 5-substituted cyclopentadienes. This analysis dissects the electronic activation energies in the distortion and interaction energies of the reaction. The distortion energy (ΔEd) is the energy required to deform the reactants into the corresponding transition structures, and the interaction energy (ΔEi) comprises of the interactions that occur between the diene and dienophile as they approach each other and adopt the geometries of the transition state. Figure 4 shows a plot of the stereoselectivity measured as the difference in the anti and syn electronic activation energies (ΔE⧧(syn) – ΔE⧧(anti)) with the difference in the distortion (ΔE⧧(syn) – ΔE⧧(anti)) required to achieve the anti and syn transition states.
Figure 4.
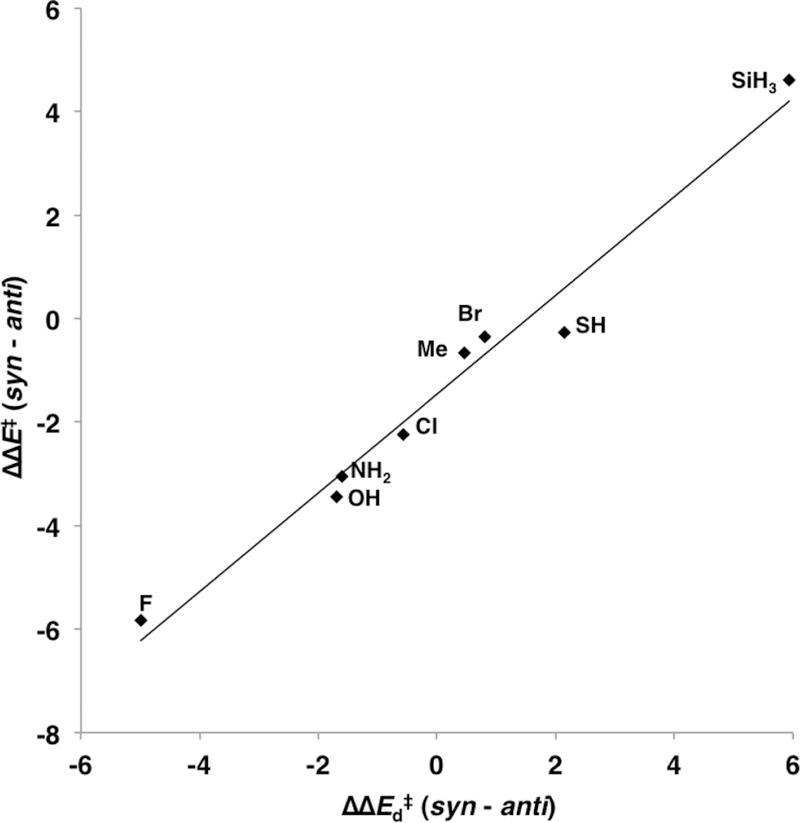
Plot of π-facial selectivity (ΔΔE⧧(syn–anti)) against the differences in the distortion energies (ΔΔEd⧧(syn–anti)) (ΔΔE⧧ = 0.95ΔΔEd⧧ – 1.5, r2 = 0.98).
The excellent linear correlation suggests that the π-facial selectivity results from differences in the energies required to distort the reactants into the syn and anti geometries, as originally proposed by Burnell.11,12 We also performed the distortion/interaction-activation strain analysis along the intrinsic reaction coordinate defined by the length of the forming C–C bonds. These plots are provided in the Supporting Information. When the forming bond lengths of the stereoisomers in the transition state are similar, as observed in these reactions, performing the distortion/interaction-activation strain analysis at the TS and along the IRC leads to the same conclusion, that the syn and anti stereoselectivity is distortion controlled.
As shown in Figure 5, the cyclopentadiene (C5–H) ground state is planar. The electronic nature of the C5–X substituent predistorts the cyclopentadiene into an envelope geometry. The angle θenv is defined as the angle at which the C5 atom of the cyclopentadiene puckers above or below the plane of the cyclopentadiene. The value of θenv is negative when the C5 atom extends below the plane and positive when it extends above the plane of the cyclopentadiene. When C5–X is a σ-donor, the C5 atom distorts above the plane of the cyclopentadiene. This distortion aligns the C5–X bond with the cyclopentadiene π-system to maximize the stabilizing hyperconjugation interaction that induces hyperconjugative aromaticity.2,18 The C5 atom in C5–SiH3 is distorted 3° above the plane of the cyclopentadiene. When C5–X is a σ-acceptor, the C5 atom distorts below the plane of the cyclopentadiene to minimize the overlap of the diene π-system with the σ*C5-X and reduce the destabilizing effect of the hyperconjugative aromaticity. For σ-acceptors, C5–F, C5–OH, and C5–Cl, the C5 atom is predistorted 2 to 4° below the plane of the cyclopentadiene ring. The poor σ-donors/acceptors, C5–Br, C5–CH3, C5–SH, and C5–NH2, are nearly planar with the C5 atom predistorted less than 2° relative to the plane of the cyclopentadiene.
Figure 5.
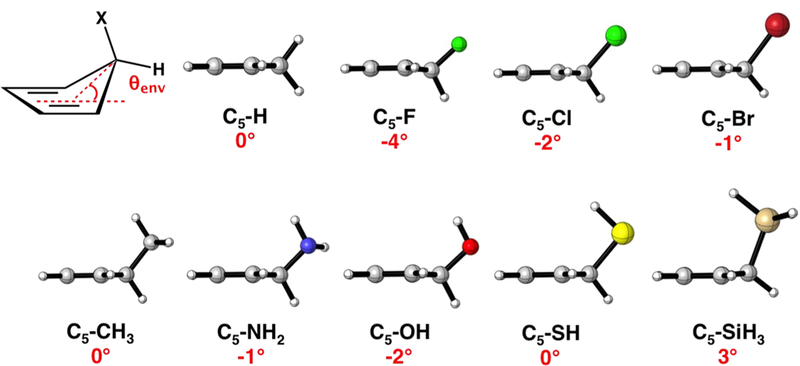
Optimized M06–2X/6–31G(d) ground state geometries of the C5–X cyclopentadienes with θenv, the angle measuring the out-of-plane distortion of the C5 atom, reported in degrees.
In the syn and anti transition structures θenv ranges from −18° to −19° and from 16° to 19°, respectively. Figure 6 shows θenv for the syn and anti Diels–Alder reactions of C5–F, C5–Br, and C5–SiH3 with ethylene. For the syn and anti reactions of C5–F, the syn reactions require a −14° change about θenv and a change of 21° to achieve the anti transitiond state geometry. To achieve the syn and anti transition state geometries, θenv in C5–Br distorts 17° and −18°, respectively. For C5–SiH3, the change about θenv to achieve the syn and anti transition state geometries is 16° and −21° from the ground state geometry, respectively.
Figure 6.
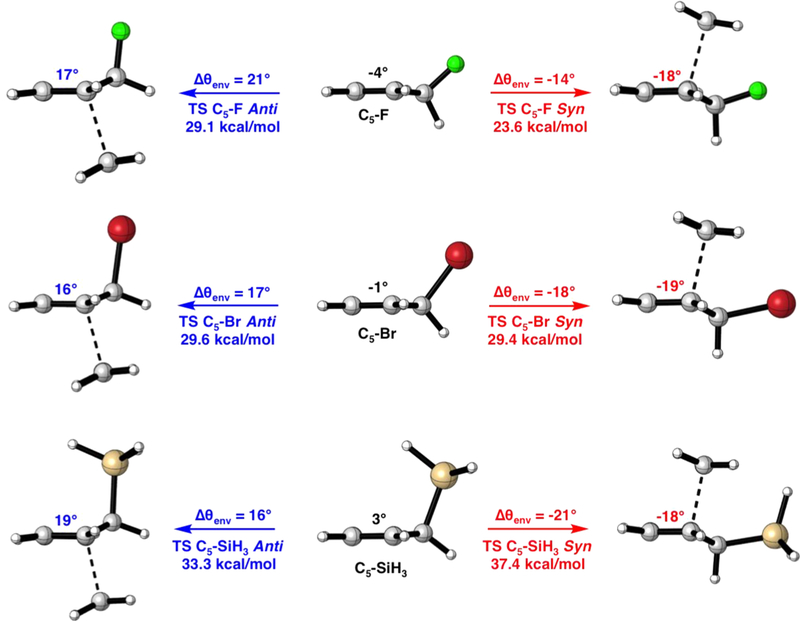
Ground and syn and anti transition state structures of C5–F, C5–Br, and C5–SiH3 with θenv shown in degrees.
The stereoselectivity of C5–X cyclopentadiene Diels–Alder reactions is determined by the distortion energies, which are related to how the C5–X cyclopentadiene is predistorted in the ground state. Figure 7a shows a strong linear correlation when the difference in the diene distortion energies of the syn and anti transition states is plotted against the difference in the cyclopentadiene envelope angle, θenv, required to achieve the syn and anti transition state geometries. The transition state that requires less of a change in θenv is the stereoselectively favored reaction. When the substituent is a σ-donor, the ground state is predistorted into an envelope geometry that resembles and favors the anti transition state, whereas σ-acceptors predistort the cyclopentadiene ground state into an envelope geometry that resembles and favors the syn transition state.
Figure 7.
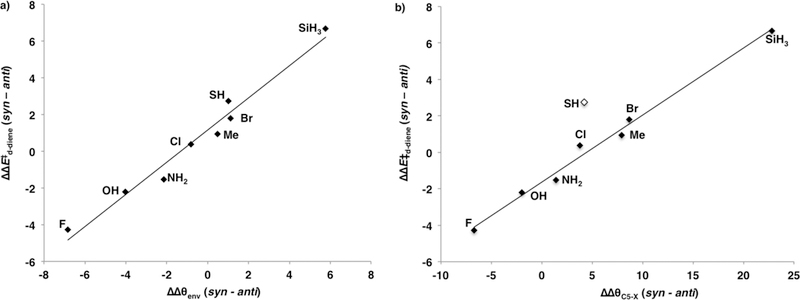
(a) Plot of differences in the diene distortion energies (ΔΔEd-diene⧧(syn–anti)) against the change in the envelope angle required to achieve the syn and anti transition state geometries ΔΔθenv(syn–anti) (ΔΔEd-diene⧧ = 0.88ΔΔθenv + 1.2, r2 = 0.97). (b) Plot of differences in the diene distortion energies (ΔΔEd-diene⧧(syn–anti)) against the change in the bending of the C5-X bond required to achieve the syn and anti transition state geometries (ΔΔEd-diene⧧ = 0.37ΔΔθC5-X – 1.6, r2 = 0.99).
We have also considered the contribution of the bending of the C5–X bond to the distortion energy as proposed by Burnell.12 The C5–X (θC5-X) bond angle is measured relative to the plane of the cyclopentadiene defined by the C1C4C5 atoms. Figure 7b shows a plot of the differences in the diene distortion energies against the difference in the bending of the C5–X (θC5-X) bond from the plane of the cyclopentadiene between the syn and anti transition state. There is a strong linear correlation between the diene distortion and the bending of the C5–X bond from the plane of the diene with the exception of C5–SH, which is an outlier in the distribution.
The x-intercept shows that for diene distortion of the syn and anti transition states to be equal (ΔΔEd-diene⧧ = 0) an additional 5° distortion of the C5–X bond toward the anti transition state is required about the C5–X bond. Figure 7a shows the plot of the difference in the diene distortion energies against the difference in θenv between the syn and anti transition states. From the x-intercept there is only a 1° difference between the envelope geometry of the syn and anti transition states when the distortion energy of the syn and anti transition states is equal (ΔΔEd-diene⧧ = 0).
The diffculty of distorting a bond is related to the strength of the bending force constants. Table 1 summarizes the computed force constants associated with the bending of the C5–X bond and the C5 carbon relative to the plane of the cyclopentadiene in the C5–X cyclopentadiene ground states. The force constants for the bending of the C5–X bonds range from 0.032 to 0.097 millidynes/Å and are significantly lower than the out-of-plane bending force constants associated with the out-of-plane bending of the C5 carbon atom, which ranges from 0.56 to 1.20 millidynes/Å. The bending of the C5–X alkyl bonds contributes less to the diene distortion energies of the syn and anti transition states than the distortion associated with the bending of the C5 carbon from the plane of the cyclopentadiene and as a result has less influence on the stereoselectivity.
Table 1.
Force Constants Computed at the M06–2X/6–31G(d) Level of Theory for Bending of the C5–X Bonds and for the out-of-Plane Motion of the C5 Atom from the Plane of the Cyclopentadiene
| C5–X | bending of C5–X bond (mDyne/Å) |
out-of-plane bending of C5 atom (mDyne/Å) |
|---|---|---|
| C5–F | 0.097 | 1.20 |
| C5–Cl | 0.064 | 1.12 |
| C5–Br | 0.047 | 0.80 |
| C5–CH3 | 0.047 | 0.56 |
| C5–NH2 | 0.049 | 0.79 |
| C5–OH | 0.066 | 0.97 |
| C5–SH | 0.048 | 1.20 |
| C –SiH | 0.032 | 0.84 |
The strength of the hyperconjugation interaction between the π-system and the C5–X substituent determines the extent of the predistortion. The electronegativity of the C5–X substituent correlates with the envelope angle θenv in the ground state geometries of the C5–X cyclopentadienes (Figure 8a) and is a useful way to predict the π-facial stereoselectivity in Diels–Alder reactions of C5–X cyclopentadiene with ethylene (Figure 8b). As the C5–X substituent becomes a stronger σ-acceptor it predistorts increasingly toward the envelope conformation of the syn transition state and becomes increasingly selective for the syn reaction.
Figure 8.
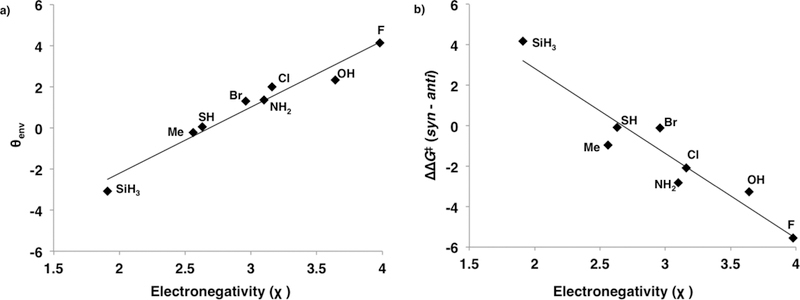
(a) Plot of θenv in the ground state of the C5–X cyclopentadienes against the electronegativity of the C5–X substituent (θenv = 3.2χ – 8.7, r2 = 0.96). (b) Plot of π-facial selectivity against the electronegativity of the C5–X substituent (ΔΔG⧧ = −4.2χ + 11.2, r2 = 0.90).
The influence of the dienophile on the π-facial stereoselectivity was investigated by calculating the syn and anti Diels–Alder stereoselectivities for the concerted reactions with maleic anhydride (MA), tetracyanoethylene (TCNE), 1,2,4-triazoline-3,5-dione (TAD), and acetylene. Figure 9 summa-rizes the syn and anti π-facial stereoselectivity of the C5–X cyclopentadienes with these dienophiles. For C5–Br, C5–SH, and C5–Me, which are poor σ donor/acceptors, the π-facial stereoselectivity can be influenced by interactions between the C5–X substituent and the dienophile. Steric interactions destabilize the syn transition state when TCNE is the dienophile, and anti π-facial selectivity becomes favored for C5–Cl, C5–Br, C5–SH, and C5–Me. Lone pair repulsions between the nitrogens of TAD with the halogen lone pair on the Cl and Br destabilize the syn transition states and result in poor stereoselectivity for the Diels–Alder reaction of C5–Cl and anti stereoselectivity in the reaction of C–Br with TAD. The predistortion of the C–F, C – ground 5 OH, and C52 states toward the syn transition state geometry is significant enough that the destabilizing interactions between the C5–X substituent and the dienophile in the syn transition state do not overrule the syn selectivity of these dienes. The Diels–Alder reaction of C5–SiH3 strongly favors the anti cycloaddition with all of the studied dienophiles.
Figure 9.
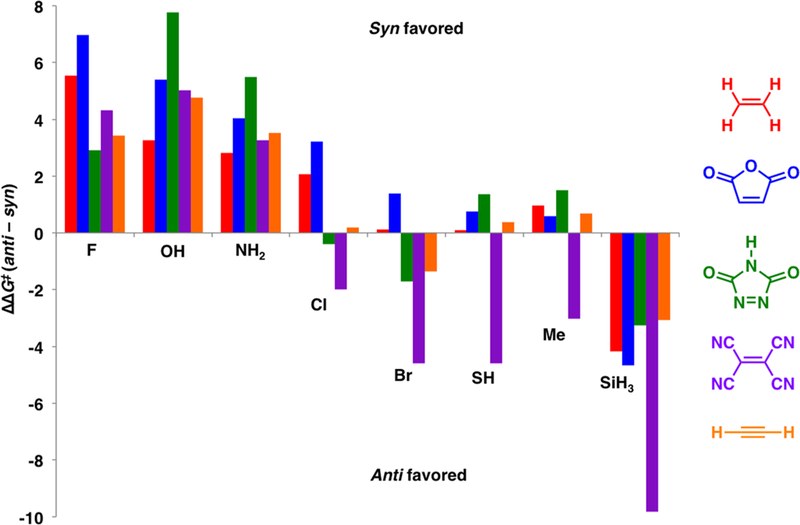
Histogram showing the syn and anti π-facial stereoselectivity in the Diels–Alder reactions of the C5–X cyclopentadienes with ethylene (red), maleic anhydride (blue), 1,2,4-triazoline-3,5-dione (green), tetracyanoethylene (purple), and acetylene (orange).
CONCLUSION
The π-facial selectivity of C5–X cyclopentadienes is distortion controlled. When the C5–X substituent is a strong σ-acceptor (X = F, OH, and NH2) the cyclopentadiene adopts an envelope geometry that minimizes the destabilizing π–σ*C5-X hyperconjugative interaction that provides the cyclopentadiene with antiaromatic character. This distortion causes the cyclopentadiene to resemble the envelope geometry of the syn transition and lessens the distortion energy required of the syn cycloaddition. Conversely, when the C5–X substituent is a σ-donor (X = SiH3) the C5 atom distorts to maximize the effect of the stabilizing hyperconjugative interaction that provides the cyclopentadiene with aromatic character. This distortion of the ground state causes the cyclopentadiene to resemble the envelope geometry of the anti transition state, and anti π-facial selectivity is favored. When the C5–X substituent is a poor σ-acceptor/donor (X = Cl, Br, SH, and Me), the π-facial selectivity is sensitive to the nature of the dienophile.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Science Foundation (CHE-1361104 and CHE-1764328) the National Institute of Health, National Institute of General Medical Science (R01 GM109078), for financial support of this research. Computer time was provided by the UCLA Institute for Digital Research and Education (IDRE).
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b02537.
Cartesian coordinates and energies of all optimized structures and transition structures (PDF)
REFERENCES
- (1).Levandowski BJ; Houk KN Theoretical Analysis of Reactivity Patterns in Diels–Alder Reactions of Cyclopentadiene, Cyclohexadiene, and Cycloheptadiene with Symmetrical and Unsymmetrical Dienophiles. J. Org. Chem 2015, 80, 3530–3537. [DOI] [PubMed] [Google Scholar]
- (2).Levandowski BJ; Zou L; Houk KN Schleyer hyperconjugative aromaticity and Diels–Alder reactivity of 5-substituted cyclopentadienes. J. Comput. Chem 2016, 37, 117–123. [DOI] [PubMed] [Google Scholar]
- (3).Winstein S; Shatavsky M; Norton C; Woodward RB 7-Norbornenyl and 7-Norbornyl Cations. J. Am. Chem. Soc 1955, 77, 4183–4184. [Google Scholar]
- (4).McClinton MA; Sik VJ 5-Fluorocyclopentadiene: synthesis and utility. J. Chem. Soc., Perkin Trans. 1 1992, 15, 1891–1895. [Google Scholar]
- (5).Franck-Neumann M; Sedrati M Studies on the effect of remote substituents on stereoreactivity. III. Influence of direct electronic activation of the dipolarophilic double-bond on the course of diazoalkanes cycloaddition to 7-halonorbornadienes. Tetrahedron Lett 1983, 24, 1391–1394. [Google Scholar]
- (6).Shi Y; Wilmot JT; Nordstrøm LU; Tan DS; Gin DY Total Synthesis, Relay Synthesis, and Structural Confirmation of the C18-Norditerpenoid Alkaloid Neofinaconitine. J. Am. Chem. Soc 2013, 135, 14313–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ishida M; Inagaki S π-Facial Selectivity of Diels-Alder Reactions. Top. Curr. Chem 2009, 289, 183–218. [DOI] [PubMed] [Google Scholar]
- (8).Cieplak AS Inductive and Resonance Effects of Substituents on π-Face Selection. Chem. Rev 1999, 99, 1265–1336. [DOI] [PubMed] [Google Scholar]
- (9).Macaulay JB; Fallis AG Heteroatom-directed.pi.-facial diastereoselection in Diels-Alder cycloadditions of plane-nonsym-metric cyclopentadienes. J. Am. Chem. Soc 1990, 112, 1136–1144. [Google Scholar]
- (10).(a) Inagaki S; Fujimoto H; Fukui K Orbital Mixing Rule. J. Am. Chem. Soc 1976, 98, 4054–4061. [Google Scholar]; (b) Ishida M; Beniya Y; Inagaki S; Kato S Application of the orbital mixing rule to heteroatom-dependent.pi-facial stereoselectivity in the Diels-Alder reaction of 5-substituted 1,3-cyclopentadienes. J. Am. Chem. Soc 1990, 112, 8980–8982. [Google Scholar]; (c) Ishida M; Aoyama T; Beniya Y; Yamabe S; Kato S; Inagaki S π-Facial Selectivity in the Diels–Alder Reaction of 5-Substituted 1,3-Cyclopentadienes. Bull. Chem. Soc. Jpn 1993, 66, 3430–3439. [Google Scholar]
- (11).Xidos JD; Poirier RA; Pye CC; Burnell DJ An ab Initio Study of Facial Selectivity in the Diels–Alder Reaction. J. Org. Chem 1998, 63, 105–112. [DOI] [PubMed] [Google Scholar]
- (12).Xidos JD; Poirier RA; Burnell DJ A computational examination of Diels–Alder reactions with 1,3-cyclopentadienes bearing anionic and cationic substituents at C-5. Tetrahedron Lett 2000, 41, 995–998. [Google Scholar]
- (13).Levandowski BJ; Gamache RF; Murphy JM; Houk KN Readily Accessible Ambiphilic Cyclopentadienes for Bioorthogonal Labeling. J. Am. Chem. Soc 2018, 140 (20), 6426–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam MJ; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian, Inc: Wallingford CT, 2009. [Google Scholar]
- (16).Zhao Y; Truhlar DG The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, non-covalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- (17).(a) Pieniazek SN; Clemente FR; Houk KN Sources of error in DFT computations of C-C bond formation thermochemis-tries: π→σ transformations and error cancellation by DFT methods. Angew. Chem., Int. Ed 2008, 47, 7746–7749. [DOI] [PubMed] [Google Scholar]; (b) Lan Y; Zou L; Cao Y; Houk KN Computational Methods To Calculate Accurate Activation and Reaction Energies of 1,3-Dipolar Cycloadditions of 24 1,3-Dipoles. J. Phys. Chem. A 2011, 115, 13906–13920. [DOI] [PubMed] [Google Scholar]; (c) Levan-dowski BJ; Hamlin TA; Bickelhaupt FM; Houk KN Role of Orbital Interactions and Activation Strain (Distortion Energies) on Reactivities in the Normal and Inverse Electron-Demand Cyclo-additions of Strained and Unstrained Cycloalkenes. J. Org. Chem 2017, 82, 8668–8675. [DOI] [PubMed] [Google Scholar]; (d) Levandowski BJ; Hamlin TA; Helgeson RC; Bickelhaupt FM; Houk KN Origins of the Endo and Exo Selectivities in Cyclopropenone, Iminocyclopropene, and Triafulvene Diels–Alder Cycloadditions. J. Org. Chem 2018, 83 (6), 3164–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Nyulaśzi L; Schleyer P. v. R. Hyperconjugative π-Aromaticity: How To Make Cyclopentadiene Aromatic. J. Am. Chem. Soc 1999, 121, 6872–6875. [Google Scholar]; (b) Fernandez I; Wu JI; Schleyer P. v. R. J. Substituent Effects on Hyperconjugative Aromaticity and Antiaromaticity in Planar Cyclopolyenes. Org. Lett 2013, 15, 2990–2993. [DOI] [PubMed] [Google Scholar]; (c) Levandowski BJ; Houk KN Hyperconjugative, Secondary Orbital, Electrostatic, and Steric Effects on the Reactivities and Endo and Exo Stereoselectivities of Cyclopropene Diels–Alder Reactions. J. Am. Chem. Soc 2016, 138, 16731–16736. [DOI] [PubMed] [Google Scholar]
- (19).Wheeler SE; Houk KN; Schleyer P. v. R.; Allen WD A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc 2009, 131, 2547–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Adcock W; Angus DI; Lowe DA 19F and 13C NMR Study of Some Norborn-7-yl Fluoride Derivatives. Magn. Reson. Chem 1996, 34, 675–690. [Google Scholar]; (b) Levandowski BJ; Herath D; Gallup NM; Houk KN Origin of π-Facial Stereoselectivity in Thiophene 1-Oxide Cycloadditions. J. Org. Chem 2018, 83, 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.