

Abstract
The preparation and characterization of a series of di-, tri- and tetrasaccharide analogs of β-(1→3)-glucans is described in which each pyranoside ring is replaced by a 5-thiopyranosyl ring and each glycosidic oxygen by a thioether. These oligomeric 1,5-dithio-D-glucopyranose derivatives were shown to inhibit the staining of human neutrophils and of mouse macrophages by fluorescent anti-CR3 and anti-dectin-1 antibodies, respectively. The compounds were also demonstrated to stimulate phagocytosis and pinocytosis indicative of binding to the carbohydrate binding domains of complement receptor 3 (CR3) and dectin-1. Activity in all three assays was optimum at the level of the trisaccharide mimic suggesting that while the replacement of ethereal oxygens by thioethers results in greater affinity for the aromatic-lined hydrophobic binding pockets, the presence of multiple longer C-S bonds eventually results in a mismatch and a loss of affinity.
Graphical Abstract
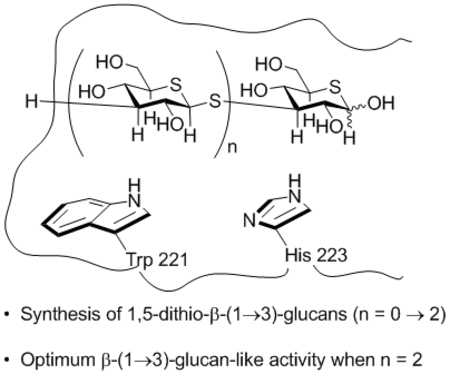
Introduction
The β-(1→3)-glucans (Figure 1) are widely–occurring natural immunomodulating agents, for which yeasts, seaweeds, fungi and grains are the most common sources.1–9 The immunostimulating properties of the β-(1→3)-glucans have resulted in their application as agents to enhance the natural immune system and to relieve side effects associated with chemotherapy. For example, schizophyllan and lentinan, fungal β-(1→3)-glucans with different molecular weight distributions and degrees of β-(1→6)-branching, are used in the treatment of uterine, stomach, colorectal and gastro-intestinal cancers.9–13 The β-(1→3)-glucans are also known to potentiate tumor-specific antibodies, to modulate the effects of radiation and photodynamic therapy14–17 to mitigate allergic rhinitis,18 regulate stress,9 to afford protection of the liver,19 and to protect from symptoms of Inflammatory Bowel Diesease.20–27
Figure 1.
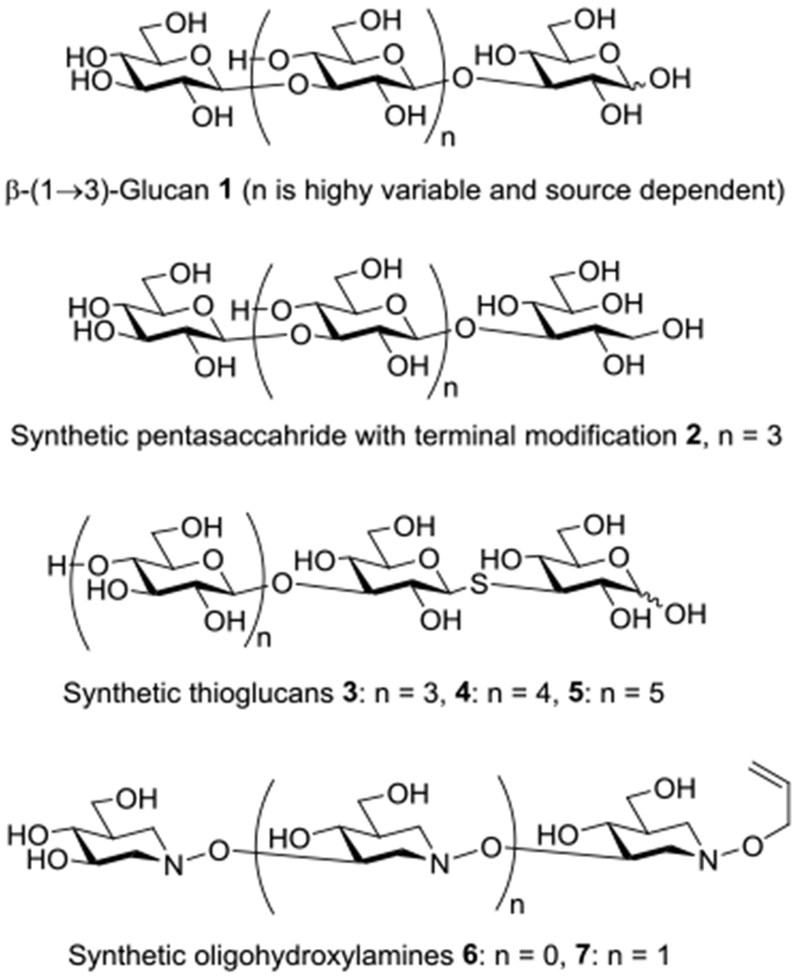
Structures of β-(1→3)-glucan and synthetic analogs
Despite their widespread availability in nature, the heterogeneity of natural isolates complicates the isolation of pure homogeneous glycoforms of β-(1→3)-glucans for biological studies and the establishment of structure activity relationships. Accordingly, much effort has been devoted to the chemical synthesis of β-(1→3)-glucans resulting in the preparation of multiple oligomers and evaluation of their immunomodulating properities.28–33
The immunostimulating properties of the β-(1→3)-glucans are considered to arise primarily from their affinity for the lectin regions of Complement Receptor 3 (CR3)8,9,34–41 and Dectin-142–44 to which their binding triggers a cascade of effects including phagocytosis.9 Studies with homogenous β-(1→3)-glucans obtained by controlled acidic hydrolysis and extensive purification demonstrated that the shortest β-(1→3)-glucan capable of detectable binding to recombinant murine Dectin-1 in a microarray format is the 10- or 11-mer.45 Subsequently a surface plasmon resonance-based assay revealed the heptasaccharide to be the minimum binding unit for recombinant murine Dectin-1.46 Earlier work with glucans isolated from yeast cell walls indicated that the interaction of pure β-glucans with monocyte glucan receptors (now recognized to be CR3) showed specificity for the linear chains of β-(1→3)-D-glucans, and that the heptasaccharide, subsequently revised to the tetrasaccharide, was the smallest β-(1→3)-glucan able to block the ability of monocytes to ingest zymosan (via CR3).9 With a series of homogeneous synthetic β-(1→3)-glucans it has been demonstrated that even the tetramer and especially the pentamer are sufficient to show immunostimulatory effects, such as the potentiation of phagocytosis, approaching those of phycarine, a β-(1→3)-glucan isolated from brown algae.29 Short synthetic β-(1→3)-glucans (penta- and hexamers) modified at the reducing end by the replacement of the terminal glucopyranose residue by its manno-stereoisomer, by a 4-deoxyglucopyranose moiety, and by gluco- and manno-configured glycitols (eg, 2) retain the ability to promote phagocytosis.30,47
X-Ray crystallographic studies of recombinant Dectin 1 reveal a shallow carbohydrate binding groove featuring a hydrophobic pocket lined by the side chains of Trp 221 and His 223.48 STD-NMR experiments revealed that laminarin, a natural β-(1→3)-glucan from brown algae with degree of polymerization from 18-31, binds to recombinant Dectin-1 through interaction of the α-faces of its terminal pyranose rings (at both the reducing and non-reducing ends) with the hydrophobic binding patch of the lectin domain (Figure 2).49,50 Interestingly however, in view of the immunostimulatory effects observed with penta- and hexamers,30,47 STD NMR experiments revealed little or no binding between a synthetic hexamer and recombinant CR3 and Dectin-1.49 More recent STD NMR studies revealed a synthetic β-(1→3)-glucan hexadecamer, but not a hexamer to bind to the lectin domain of Dectin-1.50 These observations are consistent with a relatively weak interaction between the lectin-binding domain and a short carbohydrate epitope, as is typical in carbohydrate-protein interactions,51–54 that is significantly enhanced by a multivalent effect arising in this case by the repeated presentation of the epitope in the form of the polymeric glucan. Surface-plasmon resonance (SPR) experiments are consistent with the binding of multiple Dectin-1 molecules to a single polymeric β-(1→3)-glucan backbone with affinity increasing in an additive fashion, i.e., through a multivalent interaction.46 The availability of defined synthetic β-(1→3)-glucans has also enabled their detailed conformational analysis by NMR spectroscopy.50,55
Figure 2.
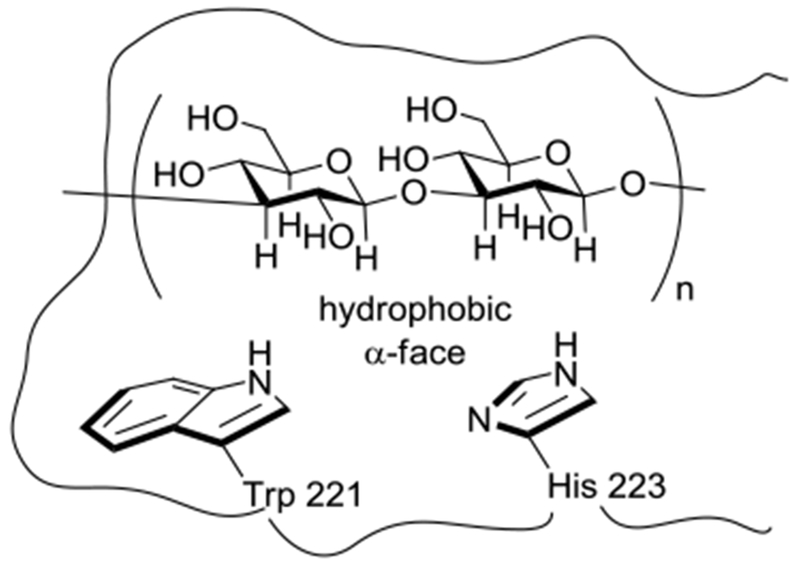
Schematic representation of the hydrophobic α-face of a disaccharide unit of a β-(1→3)-glucan in complex with the hydrophobic binding pocket of the Dectin-1 lectin domain
The identification of hydrophobic binding pockets in CR3 and Dectin-1 accepting small carbohydrate epitopes suggests that the ligand-receptor interaction may be enhanced by modification of the carbohydrate epitope. Efforts to approach the β-(1→3)-glycans in this manner were limited to the short synthetic β-(1→3)-glucans and their deoxy and 1-thia analogs 3-5 56,57 before our recent description of synthetic hydroxylamine-based glycan analogs 6 and 7 which show significant affinity for CR3 and Dectin-1.56
We hypothesized that the enhanced binding of the oligohydroxylamines relative to that of comparable length β-(1→3)-glucans is the result of the replacement of the C2-OH group in the repeat unit by a C-H bond and of the ring oxygen by a methylene group, both of which augment the hydrophobicity of the α-face of the repeat unit, and so enhance affinity for the hydrophobic binding pocket of the lectin domain in CR3 and Dectin-1 (Figure 3).57 The enhanced effects of thioglycosides 3-5 with respect to the corresponding simple glycosides, in addition to resistance to acidic and enzymatic hydrolysis,57 might also arise from enhanced interactions with proteins due to the replacement of the glycosidic oxygen by sulfur (Figure 3)58 as is well recognized and exploited in medicinal chemistry.59–61 Indeed, similar observations and hypotheses have been made by others albeit for different saccharide mimics in other contexts.62–67
Figure 3.
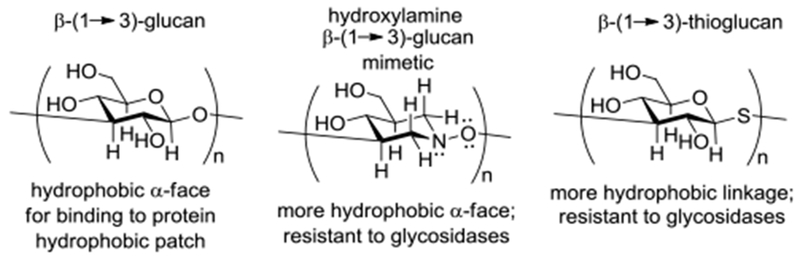
Hydrophobic α-faces of the β-(1→3)-glucan, oligomeric hydroxylamines 6 and 7, and thioglucans.
Building on these observations we designed a series of short β-(1→3)-glucan mimetics in which both the ring and glycosidic oxygens are replaced by sulfur and report here on the synthesis of the di-, tri-, and tetramers 8-10 (Figure 4). We also report that these short oligomeric molecules inhibit binding of fluorescent anti-CR3 and anti-Dectin-1 antibodies to human neutrophils and mouse macrophages, respectively, and stimulate phagocytosis and pinocytosis.
Figure 4.
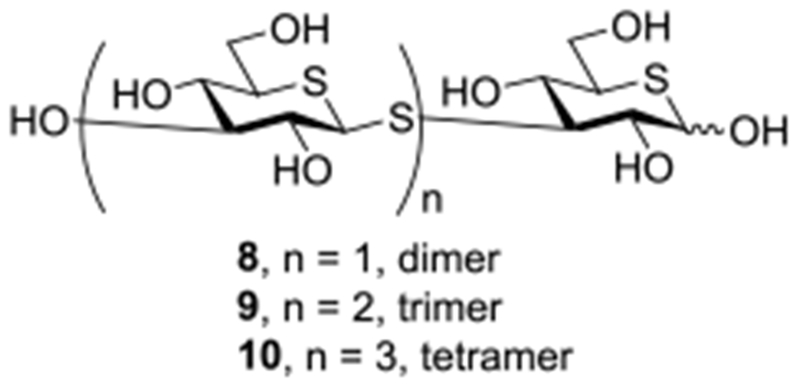
Di-, tri- and tetrameric dithiaglucose mimics 8-10 of β-(1→3)-glucans
Results and Discussion
Synthesis.
Synthetic efforts began with the large scale preparation of pentaacetyl 5-thia-D-glucopyranose essentially following the method of Whistler.68 Thus, 1,2;5,6-di-O-isopropylidene-D-glucofuranose was converted to 3-O-acetyl-1,2-O-isopropylidene-D-glucofuranose 11 by known methods69 and then exposed to a 10% molar excess of benzoyl chloride in dichloromethane at −30 °C in the presence of pyridine and 4-dimethylaminopyridine. In situ treatment of the resulting monobenzoate with methanesulfonyl chloride then gave the fully functionalized glucofuranose derivative 1270 in 83% yield (Scheme 1). Treatment with sodium methoxide in methanol, neutralization with Amberlyst IR-120 resin, and subsequent heating with thiourea next afforded the known 5,6-dideoxy-5,6-episulfide 1371 in 65% yield. Heating with potassium acetate in a mixture of acetic anhydride and acetic acid then provided the 3,5,6-triacetyl derivative of 1,2-O-isopropylidene-5-thia-D-glucofuranose 1472 in 43% yield. Finally, isopropylidene removal with aqueous acetic acid, followed by Zemplen deesterification, and peracetylation gave pentaacetyl 5-thia-D-glucopyranose 1572 in 60% yield (Scheme 1).
Scheme 1.
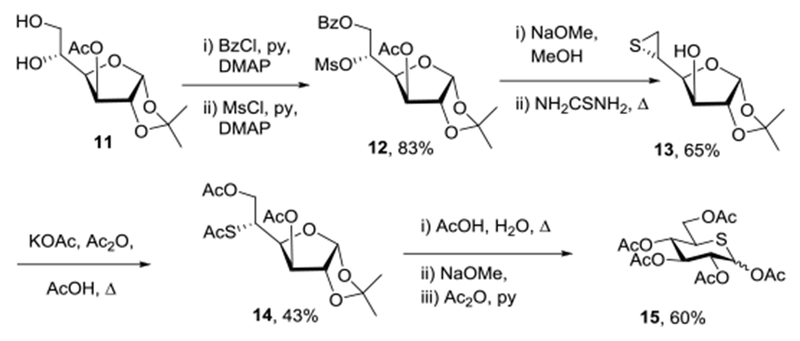
Synthesis of Pentaacetyl 5-Thia-D-glucopyranose 15
Adapting the approaches of Ferrières to short β-(1→3)-glucans73 and of Hindsgaul to thia-linked derivatives,74 Zemplen desesterification of 15 gave a crude preparation of 5-thia-D-glucose that was transformed in 73% overall yield to the diisopropylidene furanose derivative 1675 on stirring in anhydrous acetone with p-toluenesulfonic acid (Scheme 2). Swern oxidation followed by sodium borohydride reduction then afforded the allo-isomer 1775 in 57% yield. Finally, reaction with triflic anhydride and pyridine provided the triflate 18 in 85% yield in the form of a white solid (Scheme 2). A second aliquot of pentaacetyl 5-thia-D-glucopyranose 15 was treated with hydrogen bromide in acetic acid at 0 °C to give a crude preparation of the two anomers of tetraacetyl-5-thia-D-glucopyranosyl bromide which, on stirring with potassium thioacetate in dimethylformamide at 0 °C, gave 46% of pentaacetyl 1,5-dithia-β-D-glucopyranose 19 (Scheme 2).
Scheme 2.
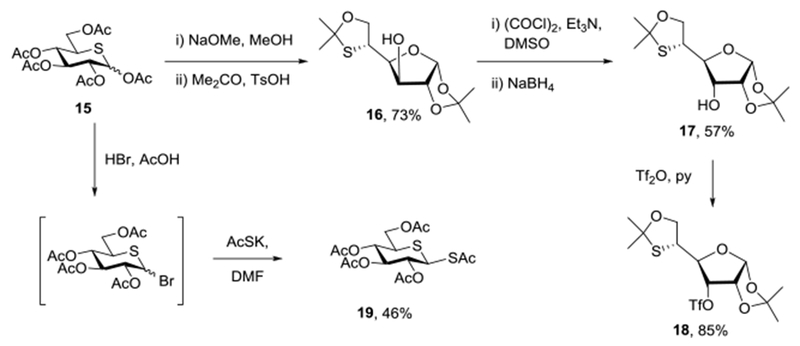
Synthesis of the Key Building Blocks 18 and 19
A mixture of triflate 18 with a 20 mol% excess of the β-diathia sugar 19 was stirred with diethylamine76,77 at 0 °C in DMF resulting in selective cleavage of the labile anomeric thioacetate, and alkylation of the resulting thiolate to give the disaccharide mimetic 20 in 86% yield (Scheme 3). As expected from the absence of mutarotation seen in other cyclic dithiahemiacetals,78 20 was isolated in the form of a single β-anomer about of the dithioglycosidic linkage. Zemplen deesterification then gave 21 quantitatively, and heating with Dowex-50 resin followed by acetylation gave the peracetyl laminaribiose derivative 22 in 91% yield (Scheme 3). Zemplen deesterification of 22 finally gave the target trithia-laminaribiose derivative 8 in 95 yield as a 4:1 α:β-anomeric mixture. Treatment of the peracetate 22 with hydrogen bromide in acetic acid72 afforded the corresponding anomeric bromide 23 in 73% yield, whose exposure to potassium thioacetate gave the β-thioacetate 24 in 89% yield. Selective cleavage of the thioacetate in 24 with diethylamine in the presence of triflate 18 then afforded the crystalline trisaccharide mimetic 25 in 64% isolated yield, that was converted to the peracetyl derivative 27 in 95% yield by acidic hydrolysis of the isopropylidene derivatives and acetylation. Finally, removal of the acetates with sodium methoxide gave the trisaccharide mimetic 9 in the form of a 4:1 α:β-mixture of anomers in quantitative fashion. Application of the sequence of hydrogen bromide in acetic acid and then potassium thioacetate to the peracetate 27 gave first the α-bromide 28 and then the β-thioacetate 29 in 43% and 90% yields, respectively. Subsequent treatment of 29 with diethylamine and triflate 18 afforded the tetrasaccahride mimetic 30 in 46% yield from which the esters were removed with sodium methoxide to give 31 quantitatively. Finally hydrolysis of the isopropylidene group with Dowex-50 resin water afforded the tetrasaccharide mimetic 10 in 85% yield as a 4:1 α:β anomeric mixture (Scheme 3).
Scheme 3.
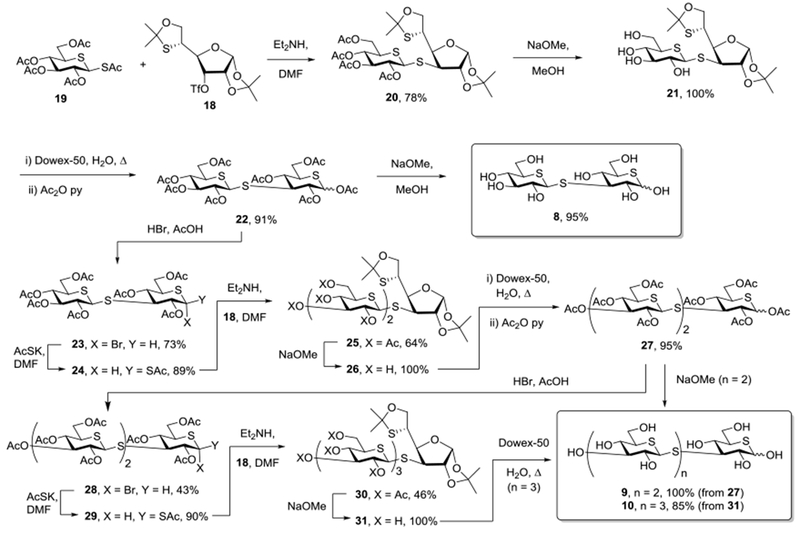
Synthesis of 1,5-Dithialaminaribose 8, triose 9, and tetraose 10
Evaluation of Binding to CR3 and Dectin-1 Receptors.
Glycan mimetics 8-10 were screened for their ability to inhibit anti-CR3 or anti-Dectin-1 fluorescein isothiocyanate (FITC)-conjugated antibody staining of human neutrophils and mouse macrophages, which is indicative of their affinity for CR3 and Dectin-1 (Table 1).37 For comparison purposes the previously reported performances of the di- and trimeric hydroxylamines 6 and 7 in the same assay are also presented in Table 1.
Table 1.
Percentage Inhibition of anti-CR3 and anti-Dectin-1-FITC Antibody Staining of Neutrophils and Macrophages by 0.1 μg.mL−1 Substrate.
| Cmpd | Oligomer No | % Inhibition of anti-CR3-FITC Staining of Human Neutrophilsa | % Inhibition of anti-Dectin-1-FITC Staining of Mouse Macrophagesa |
|---|---|---|---|
| 8 | dimer | 20.2 ± 1.7 | 30.9 ± 3.4 |
| 9 | trimer | 37.3 ± 3.2 | 42.2 ± 3.8 |
| 10 | tetramer | 21.5 ± 1.4 | 33.1 ± 2.9 |
| 6 | dimer | 26.4 ± 2.7 | 28.2 ± 2.9 |
| 7 | trimer | 34.2 ± 3.3 | 43.1 ± 3.5 |
Mean ± SD
Evaluation of phagocytic activity.
The ability of glycan mimetics 8-10 to stimulate phagocytosis of synthetic polymeric 2-hydroxyethyl methacrylate particles79 by human macrophage-like RAW 264 cells was also examined (Table 2) in comparison to the commercial highly purified yeast-derived insoluble Glucan #300.80 Again, for comparison purposes the phagocytic activity of the di- and trimeric hydroxylamines 6 and 7 are also presented in Table 2. In a comparable assay, 47.4 ± 5.2 and 58.3 ± 4.9% stimulation of phagocytosis was observed with laminaritetraose and pentaose, the smallest β-(1→3)-glucans for which data is available.29
Table 2.
Percentage Stimulation of Phagocytosis.
| Cmpd | Oligomer No | % Stimulation of Phagocytosis (Raw 264 macrophages, 10 μg/mL, 24 h)a |
|---|---|---|
| 8 | dimer | 8.7 ± 0.7 |
| 9 | trimer | 16.3 ± 0.9 |
| 10 | tetramer | 6.9 ± 0.3 |
| 6 | dimer | 7.8 ± 1.1 |
| 7 | trimer | 16.6 ± 2.0 |
| Glucan #300 | - | 37.5 ± 2.5 |
Mean ± SD; significant difference from PBS control (2.1 ± 0.2) at P < 0.05 level.
Finally, the ability of glycan mimetics 8-10 to stimulate pinocytosis, an important mechanism of cellular internalization, was determined in comparison to Glucan #300 by spectrophotometric measurement of neutral red dye accumulation by mouse macrophages on incubation with the dye (Table 3).81
Table 3.
Percentage Stimulation of Pinocytosis.
| Cmpd | Oligomer No | Uptake of Neutral red dye by mouse macrophages after 2 h, (ng/1 × 105 cells)a |
|---|---|---|
| 8 | dimer | 7.7 ± 1.61 |
| 9 | trimer | 12.8 ± 1.72 |
| 10 | tetramer | 3.37 ± 0.36 |
| Glucan #300 | - | 38.9 ± 2.81 |
Mean ± SD; significant difference from PBS control (1.77 ± 0.11) at P < 0.05 level.
Discussion.
Each of the di-, tri- and tetrameric forms of 1,5-dithia-D-glucopyranose 8-10 displayed the ability to inhibit staining of human neutrophils and mouse macrophages by fluorescent anti-CR3 and anti-Dectin-1 antibodies, respectively (Table 1) indicative of their binding to the carbohydrate binding domains of CR3 and Dectin-1. Notably, the greatest inhibition of staining by the fluorescent antibodies was observed with the trimer 9 and not with the longer tetramer 10. Further, the levels of inhibition of antibody staining by the dimer 8 and the trimer 9 were comparable to those observed previously with the di- and trimeric hydroxylamines 6 and 7, respectively. This pattern of activity is repeated in the stimulation of phagocytosis by 8-10 (Table 2), with the trimer 9 being the most active and the levels of activity comparable with those of the hydroxylamines of corresponding length. The most active compound, the trimer 9, was half as effective at stimulating phagocytosis as the yeast derived β-(1→3)-glucan Glucan #300, which was selected on the basis of its superior immunostimulating activities in comparative studies,80 and approximately one third as active as laminaritetraose – the smallest β-(1→3)glucan for which data is available.29 Finally, the same pattern was found for the stimulation of pinocytosis of neutral red dye by mouse macrophages (Table 3), with the trimer 9 being more active than either the dimer 8 or the tetramer 10, and approximately one third as active as the comparator Glucan #300.
Conclusions
As already demonstrated with the di- and trimeric hydroxylamines 6 and 7,56 the present study demonstrates that small molecule mimetics of β-(1→3)-glucans can be designed that display significant activity in the inhibition of staining of human neutrophils and mouse macrophages by fluorescent anti-CR3 and anti-Dectin-1 antibodies suggestive of binding to the carbohydrate binding domains of the respective proteins. The affinity for the carbohydrate binding domains of CR3 and Dectin-1 is reflected in the stimulation of phagocytosis and of pinocytosis by compounds 8-10. Unexpectedly, the trimer 9 is more active than either the dimer 8 or the tetramer 10 in each of the three assays conducted suggesting that, at least for the present series of glucan mimetics, there is little to be gained by preparing higher oligomers. This maximization of activity in the trimer 9 might be accounted for by a tradeoff between the greater affinity for the CR3 and Dectin-1 carbohydrate binding sites arising from the presence of the multiple thioethers on the positive side, and the accumulation of multiple long C-S bonds eventually causing a mismatch with the binding site on the negative side.
Experimental Section
General.
All reagents and solvents were purchased from commercial suppliers and were used without further purification unless otherwise stated. All reactions were performed under an argon atmosphere unless otherwise stated. Reactions were monitored by analytical thin-layer chromatography with pre-coated glass backed plates and visualized by UV absorption (254 nm) or by staining with a 5% solution of H2SO4 in MeOH or ceric ammonium molybdate solution (4.0 g ceric sulfate; 10 g ammonium molybdate; 40 mL H2SO4; 360 mL H2O) followed by heating. Optical rotations were measured with an automatic polarimeter in the solvent specified at 589 nm at 23 °C with a path length of 10 cm. 1H, 13C, HSQC, HMBC, COSY, and TOCSY NMR spectra were recorded at 400 or 600 MHz. High resolution mass spectra were recorded with a Walters LC/MS with an electrospray source coupled to a time-of-flight mass analyzer. Melting points were recorded with an electrothermal melting point apparatus.
General Procedure A: Coupling Reactions.
To a 1.0 M solution of a 1-S-acetyl-5-thio-β-D-glucopyranose 19 and triflate 18 (1.1 eq) in DMF was added diethylamine (2.5 eq) dropwise at 0 °C. The reaction mixture was stirred at room temperature until completion. The reaction mixture was diluted with ethyl acetate and washed with water, brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel chromatography.
General Procedure B: Deacetylation.
At 0 °C, sodium methoxide (0.2 eq) was added to a 0.1 M solution of substrate in anhydrous methanol, which was then stirred until completion. Amberlyst IR120 resin was added to neutralize the reaction. When the pH was neutral (monitored by pH paper), the resin was filtered off and the solution was concentrated in vacuo. Furanosyl systems were subjected to acidic hydrolysis according to general procedure C, whereas the pyranose forms were purified by Sephadex G-25 gel chromatography eluting with water.
General procedure C: Hydrolysis and Acetylation.
A solution of the substrate in deionized water (0.05 M) was treated with DOWEX-50WX2 hydrogen form resin (400 mg per 100 mg substrate) then stirred at 90 °C until the completion. The resin was filtered off and the solution was concentrated in vacuo. The crude mixture was taken up in pyridine (0.5 M), acetic anhydride (2 eq per OH) and 4-(dimethylamino)pyridine (0.1 eq) were added, and the reaction mixture was stirred at room temperature until the completion. The solution was concentrated in vacuo and purified by silica gel chromatography.
General Procedure D: Bromination.
To a solution of per-O-acetyl-5-thio-glucopyranose in anhydrous DCM (0.5 M) was added 33% HBr in acetic acid (7.0 eq) at 0 °C. The reaction mixture was kept at 5 °C for 12 h, then was diluted with DCM and quenched with ice cold aqueous NaHCO3. The organic layer was washed with water, and brine, dried with MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography.
General procedure E: Thioacylation.
At 0 °C, potassium thioacetate (1.5 eq) was added to a solution of the per-O-acetyl-5-thio-glucopyranosyl bromide in DMF (1.0 M) and the reaction mixture stirred 12 h at room temperature. After completion, the reaction mixture was diluted with ethyl acetate and washed with water, and brine, and dried with MgSO4. The reaction mixture was concentrated in vacuo and purified by silica gel chromatography.
3-O-Acetyl-5-O-methanesulfonyl-6-O-benzoyl-1,2-O-isopropylidene-α-D-glucofuranose (12).
To a stirred solution of compound 1169 (21.0 g, 0.08 mol) in DCM (500 mL, 0.16 M) was added pyridine (30.0 mL, 4.66 mol) and DMAP (0.98 g, 8.0 mmol). At −30 °C benzoyl chloride (10.1 mL, 0.09 mol) was added dropwise and the reaction mixture was stirred at −30 °C for 1 h. After completion, methanesulfonyl chloride (12.2 mL, 0.16 mol) was added to the reaction mixture at −30 °C. The reaction mixture was stirred at room temperature for 10 min before it was concentrated at 40 °C to remove DCM. After DCM was removed, the reaction mixture was stirred at room temperature until completion. The reaction mixture was diluted with DCM and quenched with aqueous NaHCO3. The solution was washed with water, and brine and dried over MgSO4. The reaction mixture concentrated in vacuo to give product 12 (29.5 g, 83%) as a white solid, whose spectral data are identical with literature.70
5,6-Anhydro-5,6-epithio-1,2-O-isopropylidene-α-D-glucofuranose (13).
At 0 °C, to a stirred solution of compound 12 (24 g, 0.05 mol) in anhydrous methanol (500 mL, 0.1 M) was added NaOMe (3.2 g, 0.06 mol) portionwise. The reaction mixture was stirred at 0 °C until the completion. Amberlyst IR 120 was added portionwise to neutralize the reaction mixture. The resin was filtered off and thiourea (7.6 g, 0.1 mol) was added to the reaction mixture which was then stirred at 80 °C for 3 h before it was concentrated in vacuo. The residue was dissolved in DCM and washed with water, and brine and dried over MgSO4 and concentrated in vacuo. The crude mixture was crystallized from ethyl acetate and hexane to give product 13 (7.1 g, 65%) as a white solid, mp 140-142 °C, with spectral data identical to the literature.71
3,6-Di-O-acetyl-5-S-acetyl-1,2-O-isopropylidene-α-D-glucofuranose (14).
A stirred solution of compound 13 (25.5 g, 0.12 mol) in a mixture of acetic anhydride (213 mL) and glacial acetic acid (43 mL) was treated with anhydrous potassium acetate (18.7 g, 0.18 mol) and stirred at 145 °C for 12 h. After the reaction mixture was cooled, it was poured into ice water and extracted with chloroform. The chloroform layer was washed with aqueous NaHCO3, and brine, dried over MgSO4, and concentrated in vacuo. The residue was decolorized with charcoal and recrystallized from ethanol to give 14 (18.1 g, 43%) as white crystals (mp 145-146 °C), with spectal data are identical to the literature.72
1,2,3,4,6-Penta-O-acetyl-5-thio-α,β-D-glucopyranose (15).
A solution of compound 14 (17.5 g, 0.05 mol) in acetic acid (100 mL) and water (100 mL) was stirred at 90 °C for 12 h, then concentrated in vacuo and co-evaporated with toluene (310 mL) to remove acetic acid. The residue was dissolved in anhydrous methanol (300 mL) and NaOMe (540 mg, 0.01 mol) was slowly added at 0 °C, followed by stirring at 0 °C for 4 h before Amberlyst IR120 resin was added to quench the reaction (monitored by pH paper). The resin was filtered off and the filtrate was concentrated in vacuo. The residue was dissolved in pyridine (100 mL) and acetic anhydride (46 mL, 0.5 mol) was added at 0 °C. The reaction mixture was stirred at room temperature for 12 h before it was concentrated in vacuo. The residue was purified by silica gel chromatography eluting ethyl acetate: hexane (1:4) to give 15 (11.5 g, 60%) as a colorless oil, with spectral data identical to the literature.72
1,2-O;5-S,6-O-Di-isopropylidene-5-thio-α-D-glucofuranose (16).
To a stirred solution of compound 15 (3.6 g, 8.8 mmol) in anhydrous methanol was added NaOMe (75 mg, 1.4 mmol). The reaction mixture was stirred at room temperature for 10 min before Amberlyst IR 120 resin was added to quench the reaction (monitored by pH paper). The resin was filtered off and the filtrate was concentrated in vacuo then was dissolved in anhydrous acetone (100 mL) and treated with p-toluenesulfonic acid (3.0 g, 17.4 mmol). The reaction mixture was stirred at room temperature for 12 h before aqueous NaHCO3 was added. The reaction mixture was diluted with ethyl acetate and washed with water, and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography with eluting with hexane: ethyl acetate (3:1) to give 16 (1.8 g, 74%) as a colorless oil with spectra identical to the literature.75
1,2-O;5-S,6-O-Di-isopropylidene-5-thio-α-D-allofuranose (17).
To a solution of dimethyl sulfoxide (5.6 mL, 0.08 mol) in DCM (22 mL) at −78 °C was added oxalyl chloride (3.4 mL, 0.04 mol) dropwise. The reaction mixture was stirred at −78 °C for 10 min before a solution of 16 (3.5 g, 12.6 mmol) in DCM (20 mL) was added dropwise. The reaction mixture was stirred at −78 °C for 1 h before triethylamine (12.6 mL, 0.09 mol) was added. Stirring was continued at −78 °C for 10 min before the reaction mixture was poured into ice water, extracted with ethyl acetate, washed with aqueous NaHCO3, and brine, dried over MgSO4 and concentrated in vacuo. The residue was dissolved in ethanol (70 mL), cooled to 0 °C, and treated with sodium borohydride (715 mg, 0.02 mol). After stirring at 0 °C for 30 min, the reaction mixture was quenched with acetone, diluted with DCM and washed with aqueous NaHCO3, and brine, and dried over MgSO4. The reaction mixture was concentrated in vacuo and purified by silica gel chromatography (eluent: hexane:ethyl acetate 3:1) to give 17 as a white solid (2.0 g, 57%) with spectral data identical to the literature.75
3-O-Trifluoromethanesulfonyl-1,2-O;5-S,6-O-di-isopropylidene-5-thio-α-D-allofuranose (18).
To a stirred solution of compound 17 (2.0 g, 7.2 mmol) and pyridine (5.8 mL, 72 mmol) at 0 °C was added trifluoromethanesulfonic anhydride (1.8 mL, 10.8 mmol) dropwise. The reaction mixture was stirred at 0 °C for 0.5 h before ice cold aqueous NaHCO3 was added. The reaction mixture was diluted in DCM, washed by water, and brine and dried over MgSO4. The reaction mixture was concentrated in vacuo and purified by silica gel chromatography (eluent: hexane:ethyl acetate 10:1) to give product 18 as a white solid (2.5 g, 85%). Mp 79-80 °C. [α]23D = +15.0° (c 0.6, CHCl3). 1H NMR (600 MHz, CDCl3) δ 5.78 (d, J1, 2 = 3.9 Hz, 1H, H1), 4.87 (dd, J3,4 = 7.6 Hz, J3,2 = 5.3 Hz, 1H, H3), 4.73 (t, J = 4.6 Hz, 1H, H2), 4.32 (t, J = 7.2 Hz, 1H, H4), 4.25 (dd, J6, 6’ = 10.3 Hz, J6, 5 = 2.5 Hz, 1H, H6), 4.13 (dd, J6, 6’ = 10.3 Hz, J6’, 5 = 5.8 Hz, 1H, H6’), 3.79 (m, 1H, H5), 1.69 (s, 3H), 1.60 (s, 3H), 1.56 (s, 3H), 1.36 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 118.3 (CF3), 114.2 (isopropylidene C), 103.8 (C1), 93.2 (isopropylidene C), 83.2 (C3), 79.0 (C4), 77.6 (C2), 70.6 (C6), 51.7 (C5), 30.7, 30.4, 26.8, 26.5 (isopropylidene CH3). HRMS m/z [M+Na]+ calcd for C13H19O7F3NaS2 431.0422, found 431.0424.
1-S-Acetyl-2,3,4,6-tetra-O-acetyl-1,5-dithio-β-D-glucopyranose (19).
Compound 15 (4.95 g, 12.2 mmol) was subjected to bromination according to general procedure D to give an α,β mixture of anomeric bromides. The crude product was subjected to thioacylation according to general procedure E to give the α,β mixture of anomeric thioacetates from which the β-isomer was isolated by silica gel chromatography eluting with hexane:ethyl acetate (3:1) to give 19 (2.36 g, 46%) as a colorless solid. Mp 121-122 °C. [α]23D = +66.1° (c 0.6, CHCl3). 1H NMR (600 MHz, CDCl3) δ 5.26 (dd, J4,5 = 10.7 Hz, J4,3 = 9.6 Hz, 1H, H4), 5.22 (dd, J2,1 = 11.0 Hz, J2,3 = 9.4 Hz, 1H, H2), 5.08 (t, J = 9.5 Hz, 1H, H3), 4.66 (d, J1,2 = 11.0 Hz, 1H, H1), 4.25 (dd, J6,6’ = 12.1 Hz, J6,5 = 5.4 Hz, 1H, H6), 4.09 (dd, J6’,6 = 12.1 Hz, J6’,5 = 3.2 Hz, 1H, H6’), 3.41 – 3.35 (m, 1H, H5), 2.35 (s, 3H, SCOCH3), 2.05 (s, 3H), 2.01 (s, 3H), 1.98 (s, 3H), 1.97 (s, 3H).13C NMR (151 MHz, CDCl3) δ 191.4 (SCOCH3), 170.5, 169.5, 169.34, 169.29 (OCOCH3), 74.4 (C3), 72.4 (C2), 71.5 (C4), 61.0 (C6), 44.9 (C1), 44.0 (C5), 30.5 (SCOCH3), 20.6, 20.5, 20.4, 20.4 (OCOCH3). HRMS m/z [M+Na]+ calcd for C16H22O9NaS2 445.0603, found 445.0605.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-1,2-O;5-S,6-O-di-isopropylidene-3,5-dideoxy-3,5-dithio-α-D-glucofuranose (20).
Compounds 18 (0.66 g, 1.5 mmol) and 19 (0.73 g, 1.8 mmol) were coupled according to general procedure A. The crude reaction mixture was subjected to silica gel chromatography (eluent: ethyl acetate:hexane 1:4) to give the product 20 (0.75 g, 78%) as a colorless solid. Mp 161-162 °C. [α]23D = +20.0° (c 0.6, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.85 (d, J1b, 2b = 3.6 Hz, 1H, H1b), 5.29 (dd, J4a, 5a = 10.7 Hz, J4a, 3a = 9.3 Hz, 1H, H4a), 5.13 (dd, J2a, 1a = 10.6 Hz, J2a, 3a = 9.4 Hz, 1H, H2a), 5.05 (t, J = 9.4 Hz, 1H, H3a), 4.69 (d, J2b, 1b = 3.6 Hz, 1H, H2b), 4.39 (dd, J4b, 5b = 10.5 Hz, J4b, 3b = 4.0 Hz, 1H, H4b), 4.34 – 4.26 (m, 2H, H6a&6b), 4.15 – 4.06 (m, 2H, H6’a&6’b), 3.98 (d, J1a, 2a = 10.6 Hz, 1H, H1a), 3.65 – 3.60 (m, 1H, H5b), 3.57 (d, J3b, 4b = 4.0 Hz, 1H, H3b), 3.23 – 3.18 (m, 1H, H5a), 2.07 (s, 3H), 2.06 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H), 1.66 (s, 3H), 1.59 (s, 3H), 1.53 (s, 3H), 1.34 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.5, 169.7, 169.3, 169.2 (OCOCH3), 112.3 (isopropylidene C), 105.3 (C1b), 92.7 (isopropylidene C), 86.0 (C2b), 81.4 (C4b), 74.5 (C3a), 73.7 (C2a), 72.0 (C6b), 71.6 (C4a), 61.0 (C6a), 53.4 (C3b), 50.6 (C5b), 47.9 (C1a), 44.6 (C5a), 31.6, 30.7, 26.7, 26.4 (isopropylidene CH3), 20.6, 20.6, 20.5, 20.4 (OCOCH3). HRMS m/z [M+Na]+ calcd for C26H38O12NaS3 661.1423, found 661.1423.
5-Thio-β-D-glucopyranosyl-(1→3)-1,2-O;5-S,6-O-di-isopropylidene-3,5-dideoxy-3,5-dithio-α-D-glucofuranose (21).
Compound 20 (800 mg, 1.2 mmol) was subjected to deacylation according to general procedure B to give the product 21 (590 mg, quantitative) as a colorless solid. [α]23D = +43.7° (c 1.4, MeOH).1H NMR (600 MHz, CD3OD) δ 5.87 (d, J1b, 2b = 3.6 Hz, 1H, H1b), 4.89 (d, J2b, 1b = 3.6 Hz, 1H, H2b), 4.36 (dd, J4b, 5b = 10.5 Hz, J4b, 3b = 3.9 Hz, 1H, H4b), 4.25 (dd, J6b, 6’b = 9.9 Hz, J6b, 5b = 2.2 Hz, 1H, H6b), 4.09 (dd, J6’b, 6b = 9.9 Hz, J6’b, 5b = 4.8 Hz, 1H, H6’b), 3.89 (dd, J6a,6’a = 11.4 Hz, J6a, 5a = 4.3 Hz, 1H, H6a), 3.82 (d, J1a, 2a = 10.2 Hz, 1H, H1a), 3.75 (dd, J6’a, 6a = 11.4 Hz, J6’a, 5a = 6.0 Hz, 1H, H6’a), 3.63 (d, J3b, 2b = 3.9 Hz, 1H, H3b), 3.62 – 3.60 (m, 1H, H5b) 3.53 (t, J = 9.5 Hz, 1H, H4a), 3.35 (dd, J2a, 1a = 10.2 Hz, J2a, 3a = 8.7 Hz, 1H, 3H, H2a), 3.16 (t, J = 8.8 Hz, 1H, H3a) 2.90 – 2.86 (m, 1H, H5a) 1.62 (s, 3H), 1.55 (s, 3H), 1.46 (s, 3H), 1.30 (s, 3H). 13C NMR (151 MHz, CD3OD) δ 111.6 (isopropylidene C), 105.4 (C1b), 91.9 (isopropylidene C), 85.6 (C2b), 81.1 (C4b), 79.4 (C3a), 76.3 (C2a), 74.8 (C4a), 71.6 (C6b), 61.5 (C6a), 52.2 (C3b), 50.5 (C5b), 49.6 (C5a), 49.2 (C1a), 30.7, 29.6, 25.5, 25.1 (isopropylidene CH3). HRMS m/z [M+Na]+ calcd for C18H30O8S3Na 493.1000, found 493.0999.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-α,β-D-glucopyranose (22).
Compound 21 (590 mg, 1.2 mmol) was deprotected according to general procedure C to give the product 22 (830 mg, 91%) as a 3:1 α:β mixture in the form of a colorless oil. 1H NMR (600 MHz, CDCl3) α isomer: δ 6.02 (d, J = 3.1 Hz, 1H, H1bα), 3.54–3.49 (m, 1H, H5bα), 2.20 (s, 3H), 2.15 (s, 3H), 2.12 (s, 3H), 2.08 (s, 3H), 2.05 (s, 3H), 1.98 (s, 3H), 1.97 (s, 3H), β isomer: 5.84 (d, J = 8.4Hz, 1H, H1bβ), 5.37 (dd, J1,2 = 9.6 Hz, J2,3 = 8.4 Hz, 1H, H2bβ), 3.20 – 3.17 (m, 1H, H5bβ), 3.05 (t, J = 10.1 Hz, 1H, H3bβ), 2.14 (s, 3H), 2.13 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H); common and overlapping signals: 5.31 – 5.21 (m, H4a & H2bα), 5.15 – 4.94 (m, H2a & H3a & H4bβ & H4bα), 4.32 – 4.22 (m, H6a & H6bα & H6’bβ), 4.18 – 4.13 (m, H6’a), 4.09 – 4.02 (m, H6’bα & H1a & H6bβ), 3.36 – 3.22 (m, H5a & H3bα), 2.01 – 2.00 (m, Acetyl CH3) . 13C NMR (151 MHz, CDCl3) δ 170.5, 169.7, 169.2, 169.1, 168.9 (Carbonyl C), 75.8 (C2b), 74.2 (C3a), 73.2 (C2a), 71.8 (C4a), 70.5 (C4b& C1b), 61.5 (C6b), 61.1 (C6a), 50.3 (C1a), 49.9 (C3b), 44.5 (C5a), 40.6 (C5b), 21.1, 20.8, 20.6, 20.5, 20.4, 20.3 (Acetyl CH3). HRMS m/z [M+Na]+ calcd for C28H38O16S3Na 749.1220, found 749.1223.
5-Thio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-α,β-D-glucopyranose (8).
Compound 22 (20 mg, 0.03 mmol) was subjected to deacetylation according to general procedure B to give 8 (10 mg, 95%) as a 4:1 α:β mixture in the form of a white solid. 1H NMR (600 MHz, D2O) α isomer: δ 4.86 (d, J = 3.0 Hz, 1H, H1bα), 3.12 – 3.09 (m, 1H, H5bα), 3.00 (t, J = 10.7 Hz, 1H, H3bα), β isomer: 4.60 (d, J = 8.9Hz, 1H, H1bβ), 2.94 – 2.91 (m, 1H, H5bβ), 2.67 (t, J = 10.4 Hz, 1H, H3bβ); common and overlapping signals: 3.82 – 3.62 (m, H2bα,2bβ & H6a,6’a & H6b,6’b), 3.95 (d, J = 10.4 Hz, H1a), 3.54 – 3.41 (m, H4a & H4bα & H4bβ), 3.34 (dd, J2,1 = 10.4 Hz, J2,3 = 9.8 Hz, H2a), 3.17 (t, J = 9.8Hz, H3a), 2.90–2.84 (m, H5a). 13C NMR (151 MHz, D2O) δ 77.8 (C3a), 77.2 (C2bβ) 76.2 (C2a), 75.0 (C1bβ) 74.6 (C2bα), 72.9 (C4a), 72.4 (C1bα), 70.8 (C4b), 60.7 (C6bβ), 60.5 (C6bα), 60.0 (C6a), 57.5 (C3bβ) 54.4 (C3bα), 48.59 (C1a), 48.56 (C5a), 48.1 (C5bβ), 43.4 (C5bα). HRMS m/z [M+Na]+ calcd for C12H22O8NaS3 413.0374, found 413.0374.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-α-D-glucopyranosyl bromide (23).
Compound 22 (246 mg, 0.34 mmol) was subjected to bromination according to general procedure D to give product 23 (184 mg, 73%) as a colorless oil. [α]23D = +112.0° (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 5.44 (d, J1b,2b = 3.4 Hz, 1H, H1b), 5.26 (dd, J4a,3a = 10.7 Hz, J4a,5a = 9.6 Hz, 1H, H4a), 5.09 (t, J = 10.7 Hz, 1H, H3a), 5.05 (t, J = 10.9 Hz, 1H, H2a), 5.00 (t, J = 9.5 Hz, 1H, H4a), 4.92 (dd, J2b,3b = 10.9 Hz, J2b,1b = 3.4 Hz, 1H, H2b), 4.36 (dd, J6b,6’b = 12.2 Hz, J6b,5b = 4.9 Hz, 1H, H6b), 4.25 (dd, J6a,6’a = 12.0 Hz, J6a,5a = 5.0 Hz, 1H, H6a), 4.13 (dd, J6a,6’a = 12.1 Hz, J6b,5b = 3.4 Hz, 1H, H6’a), 4.10 – 4.07 (m, 2H, H6’a & H1a), 3.62 – 3.59 (m, 1H, H5b), 3.37 (t, J = 10.9 Hz, 1H, H3b), 3.27– 3.34 (m, 1H, H5a), 2.20 (s, 3H), 2.15 (s, 3H), 2.06 (s, 4H), 2.03 (s, 3H), 1.99 (s, 3H), 1.97 (s, 3H), 1.96 (s, 3H) 13C NMR (151 MHz, CDCl3) δ 170.5, 169.7, 169.3, 169.2, 169.0, 76.7 (C2b), 74.3(C3a), 73.3 (C2a), 71.6 (C4a), 70.1 (C4b), 61.1 (C6a), 61.0 (C6b), 54.8 (C3b), 50.7 (C1a), 49.9 (C1b), 44.5 (C5a), 42.4 (C5b), 20.9, 20.8, 20.6, 20.5, 20.4, 20.3 (OCOCH3). HRMS m/z [M+Na]+ calcd for C26H35O14NaS3Br 769.0270, found 769.0269.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-1-S-acetyl-2,4,6-tri-O-acetyl-1,3,5-trideoxy-1,3,5-trithio-β-D-glucopyranose (24).
Compound 23 (67 mg, 0.09 mmol) was used according to general procedure E to give 24 (60 mg, 89%) as a colorless solid. [α]23D = +43.5° (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ 5.25 (dd, J = 10.7, 9.5 Hz, 1H, H4a), 5.21 (t, J = 10.7 Hz, 1H, H2b), 5.06 (dd, J = 11.0, 9.5 Hz, 1H, H2a), 4.95 (t, J = 10.7 Hz, 1H, H4b), 4.93 (t, J = 9.5 Hz, 1H, H3a), 4.57 (d, J = 10.7 Hz, 1H, H1b), 4.23 (dd, J = 12.0, 5.3 Hz, 1H, H6a), 4.18 (dd, J = 12.0, 5.6 Hz, 1H, H6b), 4.13 (dd, J = 12.0, 3.3 Hz, 1H, H6’a), 4.09 (dd, J = 12.0, 3.2 Hz, 1H, H6’b), 3.99 (d, J = 11.0 Hz, 1H, H1a), 3.34 (m, 1H, H5b), 3.16 (m, 1H, H5a), 3.01 (t, J = 10.8 Hz, 1H, H3b), 2.35 (s, 3H, SCOCH3), 2.12 (s, 3H), 2.10 (s, 3H), 2.06 (s, 3H), 2.02 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H), 1.95 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 191.9 (SCOCH3), 170.5, 170.4, 169.7, 169.3, 169.2, 169.1, 168.9 (OCOCH3), 75.2 (C2b), 74.4 (C3a), 72.9 (C2a), 71.6 (C4a), 70.2 (C4b), 61.6 (C6a), 61.0 (C6b), 55.1 (C3b), 50.5 (C1a), 47.2 (C5b), 45.8 (C1b), 44.6 (C5a), 30.5 (SCOCH3), 20.8, 20.8, 20.6, 20.6, 20.4, 20.4, 20.2 (OCOCH3). HRMS m/z [M+Na]+ calcd for C28H38O15NaS4 765.0991, found 765.0989.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-1,2-O;5-S,6-O-di-isopropylidene-3,5-dideoxy-3,5-dithio-α-D-glucofuranose (25).
Compounds 24 (615 mg, 0.83 mmol) and 18 (507 mg, 1.24 mmol) were subjected to coupling according to general procedure A to give 25 (510 mg, 64%) as white crystals. Mp 163–164 °C. [α]23D = +26.5° (c 0.4, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 5.87 (d, J1c,2c = 3.6 Hz, 1H, H1c), 5.26 (dd, J4a,5a = 10.7 Hz, J4a,3a = 9.6 Hz, 1H, H4a), 5.16 (t, J = 10.6 Hz, 1H, H2b), 5.08 (dd, J1a,2a = 11.0 Hz, J2a,3a = 9.5 Hz, 1H, H2a), 5.02 – 4.94 (m, 2H, H4b & H3a), 4.71 (d, J1c,2c = 3.6 Hz, 1H, H2c), 4.39 (dd, J4c,5c = 10.5 Hz, J3c,4c = 4.0 Hz, 1H, H4c), 4.32 (dd, J6c,5c = 10.0 Hz, J6c,6’c = 2.5 Hz, 1H, H6c), 4.25 (dd, J6a,6’a = 12.0 Hz, J6a,5a = 5.1 Hz, 1H, H6a), 4.22 (dd, J6b,6’b = 12.0 Hz, J6b,5b = 5.4 Hz, 1H, H6b), 4.15 – 4.11 (m, 2H, H6’a & H6’c), 4.09 (dd, J6b,6’b = 12.0 Hz, J6’b,5b = 3.3 Hz, 1H, H6’b), 4.00 (d, J1a,2a = 11.0 Hz, 1H, H1a), 3.83 (d, J1b,2b = 10.6 Hz, 1H, H1b), 3.64 (m, 1H, H5c), 3.55 (d, J3c, 4c = 4.0 Hz, 1H, H3c), 3.25 – 3.14 (m, 2H, H5a & H5b), 2.93 (t, J = 10.8 Hz, 1H, H3b), 2.20 (s, 3H), 2.16 (s, 3H), 2.13 (s, 3H), 2.08 (s, 3H), 2.05 (s, 3H), 2.00 (s, 3H), 1.97 (s, 3H), 1.66 (s, 3H), 1.60 (s, 3H), 1.53 (s, 3H), 1.35 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.6, 170.5, 169.8, 169.3, 169.2, 169.1, 168.8, 112.3 (isopropylidene C), 105.4 (C-1c), 92.7 (isopropylidene C), 86.1 (C2c), 81.4 (C4c), 76.2 (C2b), 74.3 (C3a), 72.6 (C2a), 72.0 (C6c), 71.9 (C4a), 70.3 (C4b), 61.7 (C6b), 61.2 (C6a), 55.3 (C3b), 53.6 (C3c), 50.7 (C1a), 50.6 (C5c), 50.1 (C1b), 47.0 (C5b), 44.4 (C5a), 31.6, 30.7, 26.7, 26.5 (Isopropylidene CH3), 21.0, 20.9, 20.6, 20.5, 20.4, 20.2 (Acetyl CH3). HRMS m/z [M+Na]+ calcd for C38H54O18NaS5 981.1811, found 981.1810.
5-Thio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-1,2-O;5-S,6-O-diisopropylidene-3,5-dideoxy-3,5-dithio-α-D-glucofuranose (26).
Compound 25 (568 mg, 0.59 mmol) was deacetylated according to general procedure B to give 26 (394 mg, quantitative) as a colorless solid. [α]23D = +17.2° (c 2.6, MeOH). 1H NMR (600 MHz, CD3OD) δ 5.90 (d, J1c,2c = 3.5 Hz, 1H, H1c), 4.92 (d, J2c,1c = 3.5 Hz, 1H, H2c), 4.36 (dd, J4c,5c = 10.5 Hz, J4c,3c = 3.9 Hz, 1H, H4c), 4.25 (dd, J6c,6’c = 10.0 Hz, J6c,5c = 2.1 Hz, 1H, H6c), 4.10 (dd, J6c,6’c = 9.9 Hz, J6’c,5c = 4.8 Hz, 1H, H6’c), 3.92 (d, J = 10.1 Hz, 1H, H1a), 3.91 – 3.85 (m, 3H, H1b, H6a, H6b), 3.82 (dd, J6b,6’b = 11.5 Hz, J6’b,5b = 5.6 Hz, 1H, H6’b), 3.73 (dd, J6’a,6a = 11.4 Hz, J6’a,5a = 6.0 Hz, 1H, H6’a), 3.69 (d, J3c,4c = 3.9 Hz, 1H, H3c), 3.64 – 3.61 (m, 1H, H5c), 3.55 – 3.47 (m, 2H, H4a & H4b), 3.47 – 3.39 (m, 2H, H2a & H2b), 3.19 (t, J = 8.9 Hz, 1H, H3a), 2.98 – 2.95 (m, 1H, H5a), 2.93 – 2.90 (m, 1H, H5b), 2.74 (t, J = 10.1 Hz, 1H, H3b), 1.62 (s, 3H), 1.56 (s, 3H), 1.46 (s, 3H), 1.32 (s, 3H), (isopropylidene CH3). 13C NMR (151 MHz, CD3OD) δ 111.6 (isopropylidene C), 105.5 (C1c), 91.9 (isopropylidene C), 85.5 (C2c), 81.2 (C4c), 79.0 (C3a), 77.9 (C2a), 76.2 (C2b), 74.4 (C4a), 73.0 (C4b), 71.6(C6c), 62.4 (C3b), 61.8 (C6b), 61.5 (C6a), 52.1(C3c), 51.6 (C5b), 51.4 (C1b), 50.5 (C5c), 49.9 (C5a), 49.5 (C1a), 30.7, 29.7, 25.6, 25.3 (CH3). HRMS m/z [M+Na]+ calcd for C24H40O11NaS5 687.1072, found 687.1073.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-1,2,4,6-tetra-O-acetyl-3,5-dideoxy-3,5-dithio-α,β-D-glucopyranose (27).
Compound 26 (394 mg, 0.59 mmol) was deprotected according to general procedure C to give 27 (586 mg, 95%) as a 3:1 α:β mixture in the form of a colorless solid. 1H NMR (600 MHz, CDCl3) δ α isomer: 6.03 (d, J1cα, 2cα = 3.1 Hz, 1H, H1cα), 3.56 – 3.48 (m, 1H, H5cα), 3.28 (t, J = 11.5 Hz, 1H, Η3cα), 2.20 (s, 3H), 2.14 (s, 3H), 2.01 (s, 3H), 2.00 (s, 3H), 1.97 (s, 3H), β isomer: 5.87 (d, J1cβ, 2cβ = 7.1 Hz, 1H, Η1cβ), 5.36 (t, J = 7.1Hz, 1H, H2cβ), 3.37 – 3.33 (m, 1H, H5cβ), 2.09 (s, 3H); common and overlapping signals: 5.28 – 5.23 (m, H4a & H2cα), 5.14 – 5.04 (m, H2b & H2a & H4cα & H4cβ), 4.99 – 4.93 (m, H4b & H3a), 4.30 – 4.08 (m, H6a,6’a & H6b,6’b & H6cα & H6cβ), 4.05 (dd, J6cβ, 6’cβ = 12.1 Hz, J6’cβ, 5cβ = 3.3 Hz, H6’cβ), 4.03 – 3.91 (m, H1b & H1a & Η6’cα), , 3.25 – 3.21 (m, H5b), 3.17– 3.05 (m, H5a & H3cβ), 2.93 – 2.86 (m, H3b), 2.17 – 2.11 (m, Acetyl CH3), 2.08 – 2.04 (m, Acetyl CH3), . 13C NMR (151 MHz, CDCl3) δ 170.57, 170.54, 170.42, 169.67, 169.23, 169.17, 169.14, 169.10, 168.99, 168.86, 168.72 (Carbonyl C), 76.2 (C2b), 75.5 (C2cα), 74.8 (C2cβ), 74.2 (C3a), 72.7 (C1cβ), 72.6 (C2a), 71.9 (C4a), 70.8 (C4cα), 70.5 (C1cα & C4b), 70.2 (C4cβ), 62.8 (C6cβ), 61.8 (C6cα), 61.5 (C6b), 61.1 (C6a), 51.6 (C1b), 50.8 (C1a), 50.0 (C3cα), 46.6 (C5b), 44.5 (C5a), 42.9 (C5cβ), 40.7 (C5cα), 21.1, 21.0, 20.9, 20.8, 20.7, 20.7, 20.6, 20.6, 20.5, 20.4, 20.2 (Acetyl CH3). HRMS m/z [M+Na]+ calcd for C40H54O22NaS5 1069.1608, found 1069.1613.
5-Thio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-α,β-D-glucopyranose (9).
Compound 27 (20 mg, 0.02 mmol) was deacetylated according to general procedure B to give 9 (11 mg, quantitative), 4:1 α:β mixture as a colorless solid. 1H NMR (600 MHz, D2O) δ α isomer: 4.85 (d, J = 2.9 Hz, 1H, H1cα), β isomer: 4.59 (d, J = 12.0 Hz, 1H, H1cβ); common and overlapping signals: 3.98 (t, J = 10.9 Hz, H1a&H1b), 3.83 – 3.64 (m, H6a,6’a&H6b,6’b&H6c,6’c), 3.58 – 3.42 (m, H2b&H4a&H4b&H4c), 3.35 (t, J = 9.0 Hz, H2a), 3.17 (t, J = 9.0 Hz, H3a), 3.14 – 3.10 (m, H5c), 3.05 (t, J = 10.7 Hz, H3c), 2.95– 2.91 (m, H5b), 2.90–2.85 (m, H5a), 2.73 (t, J = 10.4 Hz, H3b). 13C NMR (151 MHz, D2O) δ 77.9 (C3a), 77.3 (C2cβ), 76.1 (C2a&C2b), 75.1 (C1cβ), 74.7 (C2cα), 72.8 (C4a), 72.5 (C1cα), 70.8 (C4b), 70.7 (C4cβ), 70.6 (C4cα), 60.7 (C6cβ), 60.5 (C6cα), 60.4 (C6b), 60.0 (C6a), 59.2 (C3b), 57.6 (C3cβ), 54.5 (C3cα), 50.6 (C5b), 50.4 (C1b), 48.8 (C1a), 48.9 (C5a), 48.1 (C5cβ), 43.5 (C5cα). HRMS m/z [M+Na]+ calcd for C18H32O11NaS5 607.0446, found 607.0447
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-trideoxy-3,5-dithio-α-D-glucopyranosyl bromide (28).
Bromination was performed on compound 27 (53 mg, 0.05 mmol) according to general procedure D to give 28 (23 mg, 43%) as a colorless oil. [α]23D = +90.0° (c 0.6, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 5.47 (d, J = 3.4 Hz, 1H, H1c), 5.17 – 5.02 (m, 1H, H4a), 5.17 – 5.02 (m, 3H, H2a&H2b& H3a), 5.01 – 4.89 (m, 3H, H2c&H4b&H4c), 4.34 (dd, J6a,6’a = 12.2 Hz, J6a,5a = 5.1 Hz, 1H, H6a), 4.29 – 4.06 (m, 5H, H6’a&H6b&H6’b&H6c&H6’c), 4.00 (d, J = 10.7 Hz, 1H, H1b), 3.95 (d, J = 10.9 Hz, 1H, H1a), 3.67 – 3.58 (m, 1H, H5c), 3.42 (t, J = 10.9 Hz, 1H, H3c), 3.29 – 3.20 (m, 1H, H5b), 3.15 – 3.07 (m, 1H, H5a), 2.97 – 2.85 (m, 1H, H3b). 2.23 (s, 3H), 2.16 (s, 3H), 2.14 (s, 3H), 2.12 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H), 1.97 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.5, 170.4, 169.7, 169.3, 169.2, 169.1, 169.1, 168.9, 168.6 (Carbonyl C), 76.3 (C2c), 76.2 (C2b), 74.3 (C3a), 72.6 (C2a), 71.9 (C4a), 70.6 (C4c), 70.4 (C4b), 61.7 (C6c), 61.1 (C6b&C6a), 55.3 (C3b), 55.0 (C3c), 51.6 (C1b), 50.8 (C1a), 50.6 (C1c), 46.8 (C5b), 44.5 (C5a), 42.6 (C5c), 20.9, 20.8, 20.6, 20.5, 20.4, 20.2 (Acetyl CH3). HRMS m/z [M+Na]+ calcd for C37H47O25NaS3Br 1089.0650, found 1089.0653.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-1-S-acetyl-2,4,6-tri-O-acetyl-1,3,5-trideoxy-1,3,5-trithio-β-D-glucopyranose (29).
The bromide 28 (34 mg, 0.03mmol) was substituted by potassium thioacetate (5mg, 1.5 equiv) according to general procedure E to give 29 (30mg, 90%) as a colorless solid. [α]23D = +99.2° (c 0.6, CHCl3). 1H NMR (600 MHz, CDCl3) δ 5.25 (t, J = 10.9 Hz, 1H, H4a), 5.22 (t, J =10.6 Hz, 1H, H2c), 5.16 – 5.07 (m, 1H, H2b), 5.05 (dd, J2a,1a = 11.0 Hz, J2a,3a = 9.5 Hz, 1H, H2a), 5.00 – 4.93 (m, 4H, H3a&H4b&H4c), 4.60 (d, J1c,2c = 10.6 Hz, 1H, H1c), 4.28 – 4.09 (m, 6H, H6a,6’a & H6b,6’b& H6c, 6’c), 3.93 (d, J1a, 2a = 11.0 Hz, 1H, H1a), 3.90 (d, J1b, 2b = 10.6 Hz, 1H, H1b) , 3.37 – 3.34 (m, 1H, H5c), 3.16 – 3.13 (m, 1H, H5b), 3.11 – 3.07 (m, 1H, H5a), 3.03 (t, J = 10.7 Hz, 1H, H3c), 2.88 – 2.80 (m, 1H, H3b), 2.38 (s, 3H), 2.13 (s, 6H), 2.12 (s, 6H), 2.06 (s, 6H), 2.04 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H), 1.97 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 192.0, 170.5, 170.4, 169.7, 169.3, 169.1, 168.5 (Carbonyl C), 76.1 (C2c), 75.3 (C2b), 74.3 (C3a), 72.5 (C2a), 71.8 (C4a), 70.5 (C4c), 70.4 (C4b) 61.7 (C6c), 61.7 (C6b), 61.1 (C6a), 55.3 (C3b), 54.9 (C3c), 52.4 (C1b), 50.8 (C1a), 47.2 (C5c), 46.8 (C5b), 45.7 (C1c), 44.5 (C5a), 30.5 (S-Acetyl CH3), 20.8, 20.7, 20.60, 20.5, 20.4, 20.2 (Acetyl CH3). HRMS m/z [M+Na]+ calcd for C40H54O21NaS6 1085.1380, found 1085.1383.
2,3,4,6-Tetra-O-acetyl-5-thio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-2,4,6-tri-O-acetyl-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-1,2-O-5-S,6-O-di-isopropylidene-3,5-dideoxy-3,5-dithio-α-D-glucofuranose (30).
Compounds 29 (510 mg, 0.48 mmol) and 18 (490 mg, 0.72 mmol) were coupled together according to general procedure A to give product 30 (283 mg, 46%) as white crystals. Mp 139–140 °C. [α]23D = +31.6° (c 0.3, CHCl3). 1H NMR (600 MHz, CDCl3) δ 5.88 (d, J1d,2d = 3.6 Hz, 1H, H1d), 5.27 (t, J = 10.1 Hz, 1H, H4a), 5.17 (t, J = 10.6 Hz, 1H, H2c), 5.13 – 5.08 (m, 1H, H2b), 5.05 (t, J = 10.3 Hz, 1H, H2a), 5.01 – 4.94 (m, 3H, H3a&H4b&H4c), 4.70 (d, J2d,1d = 3.6 Hz, 1H, H2d), 4.40 (dd, J4d,5d = 10.5 Hz, J4d,3d = 4.0 Hz, 1H, H4d), 4.33 (dd, J6d,6’d = 10.1 Hz, J6d,5d = 2.4 Hz, 1H, H6d), 4.29 – 4.18 (m, 3H, H6a&H6b&H6c), 4.17 – 4.07 (m, 4H, H6’a&H6’b&H6’c&H6’d), 3.94 (d, J1a,2a = 11.1 Hz, 1H, H1a), 3.90 (d, J1b,2b = 10.3 Hz, 1H, H1b), 3.84 (d, J1c,2c = 10.6 Hz, 1H, H1c), 3.67 – 3.62 (m, 1H, H5d), 3.57 (d, J3d,4d = 4.0 Hz, 1H, H3d), 3.20 –3.15 (m, 2H, H5b&H5c), 3.11 – 3.08 (m, 1H, H5a), 2.95 (t, J = 10.7 Hz, 1H, H3c), 2.88–2.82 (m, 1H, H3b), 2.21 (s, 3H), 2.14 (s, 3H), 2.12 (s, 3H), 2.11 (s, 3H), 2.06 (s, 6H), 2.05 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H), 1.97 (s, 3H), 1.67 (s, 3H), 1.60 (s, 3H), 1.53 (s, 3H), 1.35 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.6, 170.4, 169.7, 169.2, 169.1, 168.7, 168.3, 112.3 (isopropylidene C), 105.4 (C1d), 92.7 (isopropylidene C), 86.1 (C2d), 81.4 (C4d), 76.2 (C2c), 75.7 (C2b), 74.2 (C3a), 72.5 (C2a), 72.0 (C6d), 71.8 (C4a), 70.6 (C4c) 70.4 (C4b), 61.8 (C6c), 61.7 (C6b), 61.0 (C6a), 55.3 (C3b), 54.9 (C3c), 53.5 (C3d), 52.5 (C1b), 50.8 (C1a), 50.6 (C5d), 50.0 (C1c), 47.0 (C5c), 46.6 (C5b), 44.5 (C5a), 31.6, 30.7, 26.7, 26.5 (Isopropylidene CH3), 21.0, 20.9, 20.8, 20.7, 20.6, 20.5, 20.4, 20.2 (Acetyl CH3). HRMS m/z [M+Na]+ calcd for C50H70O24NaS7 1301.2200, found 1301.2205.
5-Thio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-1,2-O-5-S,6-O-di-isopropylidene-3,5-dideoxy-3,5-dithio-α-D-glucofuranose (31).
Compound 30 (150 mg, 0.12 mmol) was deacetylated according to general procedure B to give 31 (100 mg, quantitative) as a colorless solid. [α]23D = +18.9° (c 0.5, MeOH). 1H NMR (600 MHz, CD3OD) δ 5.88 (d, J1d,2d = 3.6 Hz, 1H, H1d), 4.90 (d, J2d,1d = 3.6 Hz, 1H, H2d), 4.37 (dd, J4d,5d = 10.5 Hz, J4d,3d = 4.0 Hz, 1H, H4d), 4.25 (dd, J = 10.0, 1.8 Hz, 1H, H6d), 4.10 (dd, J = 8.0, 4.9, 1.6 Hz, 1H, H6’d), 4.00 (d, J = 10.0 Hz, 1H, H1b) 3.97 (d, J = 10.4 Hz, 1H, H1a), 3.94 – 3.86 (m, 4H, H1c&H6a&H6b&H6c), 3.81 – 3.72 (m, 2H, H6’a&H6’b), 3.72 – 3.67 (m, H6’c1H), 3.66 (d, J = 4.0 Hz, 1H, H3d), 3.65 – 3.60 (m, 1H, H5d), 3.56 – 3.43 (m, 5H, H4a&H4b&H4c&H2b&H2c), 3.38 (t, J = 10.4 Hz, 1H, H2a), 3.17 (t, J = 8.9 Hz, 1H, H3a), 3.02 – 2.88 (m, 3H, H5a&H5b&H5c), 2.85 – 2.69 (m, 2H, H3b&H3c), 1.62 (s, 3H), 1.56 (s, 3H), 1.46 (s, 3H), 1.31 (s, 3H). 13C NMR (151 MHz, CD3OD) δ 111.6 (isopropylidene C), 105.4 (C1d), 92.0 (isopropylidene C), 85.6 (C2d), 81.1 (C4d), 78.6 (C3a), 77.5 (C2a), 77.0 (C2b), 75.8 (C2c), 73.7 (C4a), 71.9 (C4b), 71.8 (C4c), 71.6 (C6d), 61.8 (C3b), 61.2 (C6c), 61.1 (C3c), 60.9 (C6a&C6b), 52.5 (C3d), 51.7 (C5b), 51.4 (C5c), 51.3 (C1b), 51.1 (C1c), 50.5 (C5d), 49.6 (C5a&C1a), 30.7, 29.6, 25.5, 25.2 (isopropylidene-CH3). HRMS m/z [M+Na]+ calcd for C30H50O14NaS7 881.1143, found 881.1141.
5-Thio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-β-D-glucopyranosyl-(1→3)-3,5-dideoxy-3,5-dithio-α,β-D-glucopyranose (10).
Compound 31 (100 mg, 0.12 mmol) was deprotected according to general procedure C to give product 10 (78 mg, 85%) as 4:1 α:β mixture in the form of a white solid. 1H NMR (600 MHz, D2O) α isomer: δ 4.86 (d, J = 3.0 Hz, 1H, H1dα), β isomer: 4.58 (d, J = 8.8Hz, 1H, H1dβ); commona and overlapping signals: 4.02 – 3.90 (m, H1a& H1b& H1c), 3.86 – 3.59 (m, 9.0H, H6a,6’a & H6b,6’b & H6c,6’c & H6d,6’d & H2d), 3.57 – 3.37 (m, H4a & H4b & H4c & H4d, H2b & H2c), 3.31 (t, J = 9.0 Hz H2a), 3.13 (t, J = 9.0 Hz, H3a), 3.10 – 3.06 (m, H5d), 3.01 (t, J = 10.6 Hz, H3d), 2.96 – 2.81 (m, H5a & H5b & H5c), 2.76 – 2.66 (m, H3b & H3c). 13C NMR (151 MHz, D2O) δ 77.9 (C3a), 77.3 (C2dβ), 76.2 (C2a&C2b&C2c), 75.1 (C1dβ), 74.7 (C2dα), 72.9 (C4a), 72.5 (C1dα), 70.9 (C4b), 70.8 (C4dβ), 70.6 (C4c&C4dα), 60.6 (C6b & C6c), 60.5 (C6a), 60.0 (C3c), 59.3 (C3b), 59.2 (C3dβ), 54.5 (C3dα), 50.7 (C1b) 50.6 (C5b & C5c), 50.4 (C1c), 48.8 (C1a), 48.6 (C5a), 48.1 (C5dβ), 43.5 (C5dα). HRMS m/z [M+Na]+ calcd for C24H42O14NaS7 801.0517, found 801.0518.
Inhibition of anti-CR3-FITC antibody staining of human neutrophils and of anti-Dectin 1-FITC antibody staining of mouse macrophages.
For fluorescent staining, anti-CR3-FITC antibodies (MN-41 donated by Drs. Allison Eddy and Alfred Michael of the University of Minnesota, Minneapolis, MN, and rat anti Mouse Dectin-1 antibody labeled with FITC (purchased from AbD Serotec, Raleigh, NC) were employed. Either human neutrophils or mouse peritoneal macrophages were incubated with 0.1 μg.mL−1 of tested samples for 0.5 h on ice and washed. Subsequently, the cells were stained with antibodies on ice using standard techniques. After centrifugation of cells through a 3 mL cushion of 12% BSA in PBS, the cells were re-suspended in PBS containing 1% BSA and 10 mM sodium azide. Cell cytometry was performed with a Becton Dickinson-LSRII instrument. The inhibition of CR3 receptor and Dectin-1 receptor staining was calculated as described.37
Stimulation of phagocytosis.
The technique employing phagocytosis of synthetic polymeric microspheres was described earlier.82 8287Human cells (cell line RAW 264) were incubated in vitro with 10 μg.mL−1 of tested samples for 24 h at 37 °C. After washing, 0.05 mL of 2-hydroxyethyl methacrylate particles (HEMA; 5×108/mL) was added. The test tubes were incubated at 37 °C for 1 h, with intermittent shaking. Smears were stained with Wright stain. Cells with three or more HEMA particles were considered positive. The insoluble glucan Glucan #300 used as comparison standard was obtained from Yeast-derived insoluble Glucan #300 (>85% dry w/w basis) was purchased from Transfer Point (Columbia, SC, USA). This glucan contains 96% carbohydrates and 2.1% proteins. Neutral sugar analysis confirmed 91.3% glucose and 8% mannose.
Stimulation of pinocytosis.
Stimulation of pinocytosis was determined spectrophotometrically as described.81
Supplementary Material
Acknowledgments.
We thank the NIH (GM62160) for support of this work and acknowledge the NSF (MRI-084043) for funds in support of the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b01645.
Copies of the 1H and 13C NMR spectra of all new compounds (PDF)
References
- (1).Williams DL; W. LD; Ensley HE In Toxicology of 1,3-Beta-Glucans Young S-H, Castranova V, Eds.; CRC Press: Boca Raton, 2005, p 1–34. [Google Scholar]
- (2).Tsoni SV; Brown GD β-Glucans and Dectin 1. Annal New York Acad. Sci 2008, 1143, 41–60. [DOI] [PubMed] [Google Scholar]
- (3).Barsanti L; Passarelli V; Evangelista V; Frassanito AM; Gualtieri P Chemistry, Physico-Chemistry and Applications Linked to Biological Activities of β-Glucans. Nat. Prod. Rep. 2011, 28, 457–466. [DOI] [PubMed] [Google Scholar]
- (4).Chlubnova I; Sylla B; Nugier-Chauvin C; Daniellou R; Legentil L; Kralova B; Ferrières V Natural Glycans and Glycoconjugates as Immunomodulating Agents. Nat. Prod. Rep. 2011, 28, 937–952. [DOI] [PubMed] [Google Scholar]
- (5).Sze DM-Y; Chan GC-F Effects of Beta-Glucans on Different Immune Cell Populations and Cancers Recent Trends Med. Plants Res. 2012, 62, 179–196. [Google Scholar]
- (6).Vannucci L; Krizan J; Sima P; Stakheev D; Caja F; Rajsiglova L; Horak V; Saieh M Immunostimulatory Properties and Antitumor Activities of Glucans. Int. J. Oncology 2013, 43, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Synytsya A; Novak M Structural Diversity of Fungal Glucans. Carbohydr. Polym. 2013, 92, 792–809. [DOI] [PubMed] [Google Scholar]
- (8).Bohn JA; BeMiller JN β-D-(1,3)-Glucans as Biological Response Modifiers: A Review of Structure-Functional Activity Relationships. Carbohydr. Polym. 1995, 28, 3–14. [Google Scholar]
- (9).Legentil L; Paris F; Ballet C; Trouvelot S; Daire X; Vetvicka V; Ferrieres V Molecular Interactions of β-(1→3)-Glucans with Their Receptors. Molecules 2015, 20, 9745–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Matsuoka H; Seo Y; Wakasugi H; Saito T; Tomoda H Lentinan Potentiates Immunity and Prolongs the Survival Time of Some Patients. Anticancer Res. 1997, 17, 2751–2755. [PubMed] [Google Scholar]
- (11).Chihara G Preclinical Evaluation of Lentinan in Animal Models. Adv. Exp. Med. Biol. 1983, 166, 189–197. [DOI] [PubMed] [Google Scholar]
- (12).Ina K; Furuta R; Kataoka T; Kayukawa S; Yoshida T; Miwa T; Yamamura Y; Takeuchi Y Lentinan Prolonged Survival in Patients with Gastric Cancer Receiving S-1-Based Chemotherapy. World J. Clin. Oncol 2011, 2, 339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fujimoto S Clinical Efficacies of Schizophyllan (SPG) on Advanced Gastric Cancer. Nippon Geka Gakkai Zasshi 1989, 90, 1447–1450. [PubMed] [Google Scholar]
- (14).Xiang D; Sharma VR; Freter CE; Yan J Anti-Tumor Monoclonal Antibodies in Conjunction with β-Glucans: A Novel Anti-Cancer Immunotherapy. Curr. Med. Chem. 2012, 19, 4298–4305. [DOI] [PubMed] [Google Scholar]
- (15).Descroix K; Ferrieres V; Jamois F; Yvin J-C; Plusquellec D Recent Progress in the Field of β-(1,3)-Glucans and New Applications. Minirev. Med. Chem 2006, 6, 1341–1349. [DOI] [PubMed] [Google Scholar]
- (16).Krosl G; Korbelik M Potentiation of Photodynamic Therpay by Immunotherapy; The Effect of Schizophyllan. Cancer Lett. 1994, 84, 43–49. [DOI] [PubMed] [Google Scholar]
- (17).Okamura K; Suzuki M; Chihara T; Fujiwara A; Fukuda T; Goto S; Ichinohe K; Jimi S; Kasamatsu T; Kawai N; Mizuguchi K; Mori S; Nakano H; Noda K; Sekiba K; Suzuki K; Suzuki T; Takahashi K; Takeuchi K; Takeuchi S; Yajima A; Ogawa N Clinical Evaluation of Schizophyllan Combined with Irradiation in Patients with Cervical Cancer: A Randomized Controlled Study. Cancer 1986, 58, 865–872. [DOI] [PubMed] [Google Scholar]
- (18).Kirmaz C; Bayrak P; Yilmaz O; Yuksel H Effects of Glucan Treatment on the Th1/Th2 Balance in Patients with Allergic Rhinitis: A Double-Blind Placebo-Controlled Study. Eur. Cytokine Netw. 2005, 16, 128–134. [PubMed] [Google Scholar]
- (19).Horvathova E; Eckl P; Bresgen N; Slamenova D Evaluation of Genotoxic and Cytotoxic Effects of H2O2 and DMNQ on Freshly Isolated Rat Hepatocytes; Protective Effects of Carboxymethyl Chitin-Glucan Neuro. Endocrinol. Lett 2008, 29, 644–648. [PubMed] [Google Scholar]
- (20).Nosal’ova V; Bobek P; Cerna S; Galbavy S; Stvrtina S Effects of Pleuran (β-Glucan Isolated from Pleurotus ostreatus) on Experimental Colitis in Rats. Physiol. Res 2001, 50, 575–581. [PubMed] [Google Scholar]
- (21).Wilczak J; Blaszczyk K; Kamola D; Gajewska M; Harasym JP; Jalosinska M; Gudej S; Suchecka D; Oczkowski M; Gromadzka-Ostrowska J The Effect of Low or High Molecular Weight Oat beta-Glucans on the Inflammatory and Oxidative Stress Status in the Colon of Rats with LPS-Induced Enteritis. Food & Function 2015, 6, 590–603. [DOI] [PubMed] [Google Scholar]
- (22).Lavi I; Levinson D; Peri I; Nimri L; Hadar Y; Schwartz B Orally Administered Glucans from the Edible Mushroom Pleurotus pulmonarius Reduce Acute Inflammation in Dextran Sulfate Sodium-Induced Experimental Colitis. Br. J. Nutr. 2010, 103, 393–402. [DOI] [PubMed] [Google Scholar]
- (23).Lee K-H; Park M; Ji K-Y; Lee H-Y; Jang J-H; Yoon IJ; Oh S-S; Kim S-M; Jeong YH; Yun C-H; Kim M-K; Lee IY; Choi H-R; Ko K.-s.; Kang H-S Bacterial β-(1,3)-Glucan Prevents DSS-Induced IBD by Restoring the Reduced Population of Regulatory T Cells. Immunobiol 2014, 219, 802–812. [DOI] [PubMed] [Google Scholar]
- (24).Nishitani Y; Zhang L; Yoshida M; Azuma T; Kanazawa K; Hashimoto T; Mizuno M Intestinal Anti-Inflammatory Activity of Lentinan: Influence on IL-8 and TNFR1 Expression in Intestinal Epithelial Cells. PLOS One 2013, 8, e62441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chandra LC; Traore D; French C; Marlow D; D’Offay J; Clarke SL; Smith BJ; Kuvibidila S White Button, Portabella, and Shiitake Mushroom Supplementation Up-Regulates Interleukin-23 Secretion in Acute Dextran Sodium Sulfate Colitis C57BL/6 Mice and Murine Macrophage J.744.1 Cell Line. Nutr. Res. 2013, 33, 388–396. [DOI] [PubMed] [Google Scholar]
- (26).Tanaka K.-i.; Tanaka Y; Suzuki T; Mizushima T Protective Effect of β-(1,3 → 1,6)-D-Glucan Against Irritant-Induced Gastric Lesions. Br. J. Nutr. 2011, 106, 475–485. [DOI] [PubMed] [Google Scholar]
- (27).Zhou M; Wang Z; Chen J; Zhan Y; Wang T; Xia L; Wang S; Hua Z; Zhang J Supplementation of the Diet with Salecan Attenuates the Symptoms of Colitis Induced by Dextran Sulphate Sodium in Mice Br. J. Nutr 2014, 111, 1822–1829. [DOI] [PubMed] [Google Scholar]
- (28).Liao G; Zhou Z; Burgula S; Liao J; Yuan C; Wu Q; Guo Z Synthesis and Immunological Studies of Linear Oligosaccharides of β‑Glucan As Antigens for Antifungal Vaccine Development. Bioconjug. J. 2015, 26, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jamois F; Ferrières V; Guégan J-P; Yvin J-C; Plusquellec D; Vetvicka V Glucan-Like Synthetic Oligosaccharides: Iterative Synthesis of Linear Oligo-β-(1,3)-Glucans and Immunostimulatory Effects. Glycobiology 2005, 15, 393–407. [DOI] [PubMed] [Google Scholar]
- (30).Vetvicka V; Saraswat-Ohri S; Vashishta A; Descroix K; Jamois F; Yvin J-C; Ferrières V New 4-Deoxy-(1,3)-β-D-Glucan-Based Oligosaccharides and Their Immunostimulating Potential. Carbohydr. Res. 2011, 346, 2213–2221. [DOI] [PubMed] [Google Scholar]
- (31).Liao G; Zhou Z; Liao J; Zu L; Wu Q; Guo Z 6‑O‑Branched Oligo-β-glucan-Based Antifungal Glycoconjugate Vaccines. ACS Infect. Dis. 2016, 2, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tanaka H; Kawai T; Adachi Y; Ohno N; Takahashi T β−(1,3) Branched Heptadeca- and Linear Hexadeca-Saccharides Possessing an Aminoalkyl Group as a Strong Ligand to Dectin-1. Chem. Commun. 2010, 8249–8251. [DOI] [PubMed] [Google Scholar]
- (33).Weishaupt MW; Matthies S; Seeberger PH Automated Solid-Phase Synthesis of a β-(1,3)-Glucan Dodecasaccharide. Chem. Eur. J. 2013, 19, 12497–12503. [DOI] [PubMed] [Google Scholar]
- (34).Ross GD; Vetvicka V; Yan J; Xia Y; Vetvickova J Therapeutic Intervention with Complement and β-Glucan in Cancer. Immunopharmacol. 1999, 42, 61–74. [DOI] [PubMed] [Google Scholar]
- (35).Xia Y; Vetvicka V; Yan J; Hanikyrova M; Mayadas T; Ross GD The β-Glucan-Binding Lectin Site of Mouse CR3 (CD11b/CD18) and its Function in Generating a Primed State of the Receptor that Mediates Cytotoxic Activation in Response to iC3b-Opsonized Target Cells. J. Immunology 1999, 162, 2281–2290. [PubMed] [Google Scholar]
- (36).Yan J; Vetvicka V; Xia Y; Coxon A; Carroll MC; Mayadas TN; Ross GD β-Glucan, a “Specific” Biologic Response Modifier that Uses Antibodies to Target Tumors for Cytotoxic Recognition by Leukocyte Complement Receptor Type 3 (CD11b/CD18). J. Immunology 1999, 163, 3045–3052. [PubMed] [Google Scholar]
- (37).Thornton BP; Vetvicka V; Pitman M; Goldman RC; Ross GD Analysis of the Sugar Specificity and Molecular Location of the β-Glucan-Binding Lectin Site of Complement Receptor Type 3 (CD11b/CD18). J. Immunology 1996, 156, 1235–1246. [PubMed] [Google Scholar]
- (38).Vetvicka V; Thornton BP; Ross GD Soluble β-Glucan Polysaccharide Binding to the Lectin Site of Neutrophil or Natural Killer Cell Complement Receptor Type 3 (CD11b/CD18) Generates a Primed State of the Receptor Capable of Mediating Cytotoxicity of iC3b-Opsonized Target Cells. J. Clin. Invest. 1996, 98, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Czop JK; Austen KF A β-Glucan Inhibitable Receptor on Human Monocytes: Its Identity with the Phagocytic Receptor for Particulate Activators of the Alternative Complement Pathway. J. Immunology 1985, 134, 2588–2593. [PubMed] [Google Scholar]
- (40).Czop JK; Austen KF Properties of Glycans that Activate the Human Alternative Complement Pathway and Interact with the Human Monocyte β-Glucan Receptor. J. Immunology 1985, 135, 3388–3393. [PubMed] [Google Scholar]
- (41).Yan J; Allendorf DJ; Brandley B Yeast Whole Glucan Particle (WGP) β-Glucan in Conjunction with Antitumor Monoclonal Antibodies to Treat Cancer. Expert Opin. Biol. Ther 2005, 5, 691–702. [DOI] [PubMed] [Google Scholar]
- (42).Brown GD; Gordon S A New Receptor for β-Glucans. Nature 2001, 413, 36–37. [DOI] [PubMed] [Google Scholar]
- (43).Ariizumi K; Shen G-L; Shikano S; Xu S; Ritter R; Kumamoto T; Edelbaum D; Morita A; Bergstresser PR; Takashima A Identification of a Novel, Dendritic Cell-Associated Molecule, Dectin-1, by Subtractive cDNA Cloning. J. Biol. Chem. 2000, 275, 20157–20167. [DOI] [PubMed] [Google Scholar]
- (44).Goodridge HS; Reyes CN; Becker CA; Katsumoto TR; Ma J; Wolf AJ; Bose N; Chan ASH; Magee AS; Danielson ME; Weiss A; Vasilakos JP; Underhill DM Activation of the Innate Immune Receptor Dectin-1 Upon Formation of a ‘Phagocytic Synapse’. Nature 2011, 472, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Palma AS; Feizi T; Zhang Y; Stoll MS; Lawson AM; Díaz-Rodríguez E; Campanero-Rhodes MA; Costa J; Gordon S; Brown GD; Chai W Ligands for the β-Glucan Receptor, Dectin-1, Assigned Using “Designer” Microarrays of Oligosaccharide Probes (Neoglycolipids) Generated from Glucan Polysaccharides. J. Biol. Chem. 2006, 281, 5771–5779. [DOI] [PubMed] [Google Scholar]
- (46).Adams EL; Rice PJ; Graves B; Ensley HE; Yu H; Brown GD; Gordon S; Monteiro MA; Papp-Szabo E; Lowman DW; Power TD; Wempe MF; Williams DL Differential High-Affinity Interaction of Dectin-1 with Natural or Synthetic Glucans is Dependent Upon Primary Structure and is Influenced by Polymer Chain Length and Side-Chain Branching. J. Pharmacol. Expt. Ther. 2008, 325, 115–123. [DOI] [PubMed] [Google Scholar]
- (47).Descroix K; Vetvicka V; Laurent I; Jamois F; Yvin J-C; Ferrières V New Oligo-β-(1,3)-Glucan Derivatives as Immunostimulating Agents. Bioorg. Med. Chem. 2010, 18, 348–357. [DOI] [PubMed] [Google Scholar]
- (48).Brown J; O’Callaghan CA; Marshall ASJ; Gilbert RJC; Siebold C; Gordon S; Brown GD; Jones EY Structure of the Fungal β-Glucan-Binding Immune Receptor Dectin-1: Implications for Function. Protein Sci 2007, 16, 1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sylla B; Guégan J-P; Wieruszeski J-M; Nugier-Chauvin C; Legentil L; Daniellou R; Ferrières V Probing β-(1→3)-D-Glucans Interactions with Recombinant Human Receptors using High-Resolution NMR Studies. Carbohydr. Res. 2011, 346, 1490–1494. [DOI] [PubMed] [Google Scholar]
- (50).Hanashima S; Ikeda A; Tanaka H; Adachi Y; Ohno N; Takahashi T; Yamaguchi Y NMR study of Short β-(1,3)-Glucans Provides Insights into the Structure and Interaction with Dectin-1. Glycoconj. J. 2014, 31, 199–207. [DOI] [PubMed] [Google Scholar]
- (51).Chabre YM; Roy R Design and Creativity in Synthesis of Multivalent Neoglycoconjugates. Adv. Carbohydr. Chem. Biochem. 2010, 63, 165–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pieters RJ Maximizing Multivalency Effects in Protein-Carbohydrate Interactions. Org. Biomol. Chem. 2009, 7, 2013–2025. [DOI] [PubMed] [Google Scholar]
- (53).Fasting C; Schalley CA; Weber M; Seitz O; Hecht S; Koksch B; Dernedde J; Graf C; Knapp E-W; Haag R Multivalency as a Chemical Organization and Action Principle. Angew. Chem. Int. Ed. 2012, 51, 10472–10498. [DOI] [PubMed] [Google Scholar]
- (54).Reynolds M; Pérez S Thermodynamics and Chemical Characterization of Protein-Carbohydrate Interactions: The Multivalency Issue. CR Chimie 2011, 14, 74–95. [Google Scholar]
- (55).Mo K-F; Li H; Mague JT; Ensley HE Synthesis of the β−1,3-Glucan, Laminarahexaose: NMR and Conformational Studies. Carbohydr. Res. 2009, 344, 439–447. [DOI] [PubMed] [Google Scholar]
- (56).Ferry A; Malik G; Guinchard X; Vetvicka V; Crich D Synthesis and Evaluation of Di- and Trimeric Hydroxylamine-Based β-(1→3)-Glucan Mimetics. J. Am. Chem. Soc. 2014, 136, 14852–14857. [DOI] [PubMed] [Google Scholar]
- (57).Sylla B; Legentil L; Saraswat-Ohri S; Vashishta A; Daniellou R; Wang H-W; Vetvicka V; Ferrières Oligo-β-(1→3)-glucans: Impact of Thio-Bridges on Immunostimulating Activities and the Development of Cancer Stem Cells. J. Med. Chem. 2014, 57, 8280–8292. [DOI] [PubMed] [Google Scholar]
- (58).Vetvicka V Synthetic Oligosaccharides - Clinical Application in Cancer Therapy. Anti-Cancer Agents Med. Chem. 2013, 13, 720–724. [DOI] [PubMed] [Google Scholar]
- (59).Ilardi EA; Vitaku E; Njardarson JT Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery. J. Med. Chem. 2013, 57, 2832–2842. [DOI] [PubMed] [Google Scholar]
- (60).Daeffler KN-M; Lester HA; Dougherty DA Functionally Important Aromatic−Aromatic and Sulfur−π Interactions in the D2 Dopamine Receptor. J. Am. Chem. Soc. 2012, 134, 14890–14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Motherwell WB; Moreno RB; Pavlakos I; Arendorf JRT; Arif T; Tizzard GJ; Coles SJ; Aliev AE Noncovalent Interactions of π-Systems with Sulfur: The Atomic Chameleon of Molecular Recognition. Angew. Chem. Int. Ed. 2018, 57, 1193–1198. [DOI] [PubMed] [Google Scholar]
- (62).Witczak ZJ; Poplawski T; Czubatka A; Sarnik J; Tokarz P; VanWert AL; Bielski R A Potential CARB-Pharmacophore for Antineoplastic Activity: Part 1. Bioorg. Med. Chem. Lett. 2014, 24, 1752–1757. [DOI] [PubMed] [Google Scholar]
- (63).Witczak ZJ; Sarnik J; Czubatka A; Forma E; Poplawski T Thio-Sugar Motif of Functional CARB-Pharmacophore for Antineoplastic Activity. Part 2. Bioorg. Med. Chem. Lett. 2014, 24, 5606–5611. [DOI] [PubMed] [Google Scholar]
- (64).Witczak ZJ; Kaplon P; Dey M Thio Sugars VII. Effect of 3-Deoxy-4-S-(β-D-gluco- and β-D-Galactopyranosyl)-4-thiodisaccharides and Their Sulfoxides and Sulfones on the Viability and Growth of Selected Murine and Human Tumor Cell Lines. Carbohydr. Res. 2003, 338, 11–18. [DOI] [PubMed] [Google Scholar]
- (65).Witczak ZJ Thio Sugars: Biological Relevance as Potential New Therapeutics. Curr. Med. Chem. 1999, 6, 165–178. [PubMed] [Google Scholar]
- (66).Robina I; Vogel P; Witczak ZJ Synthesis and Biological Properties of Monothiosaccharides. Curr. Org. Chem. 2001, 5, 1177–1214. [Google Scholar]
- (67).Driguez H In Topics in Current Chemistry: Glycoscience; Driguez H, Thiem J, Eds.; Springer: Berlin, 1997; Vol. 187, p 85–116. [Google Scholar]
- (68).Whistler RL; Rowell RM Derivatives of α-D-Glucothiopyranose. J. Org. Chem. 1966, 31, 1514–1516. [Google Scholar]
- (69).Tsui H-C; Paquette LA Reversible Charge-Accelerated Oxy-cope Rearrangements. J. Org. Chem. 1998, 63, 9968–9977. [Google Scholar]
- (70).Yuasa H; Tamura J.-i.; Hashimoto H Synthesis of Per-O-alkylated 5-Thio-D-glucono-1,5-lactones and Transannular Participation of the Ring Sulphur Atom of 5-Thio-D-glucose Derivatives on Solvolysis under Acidic Conditions. J. Chem. Soc, Perkin Trans 1 1990, 2763–2769. [Google Scholar]
- (71).Tsuda Y; Sato Y; Kanemitsu K; Hosoi S; Shibayama K; Nakao K; Ishikawa Y Thio-sugars 1. Radical-Promoted Thione-Thiol Rearrangement of Cyclic Thionocarbonates: Synthesis of 5-Thioglucose. Chem. Pharm. Bull. 1996, 44, 1465–1475. [Google Scholar]
- (72).Korytnyk W; Valentekovic-Horvath S; Dodson-Simmons O Synthesis and Properties of 2,3,4,6-Tetra-O-acetyl-5-thio-α- and -β-D-Glucopyranosyl Bromide. Carbohydr. Res. 1982, 108, 293–297. [Google Scholar]
- (73).Jamois F; Le Goffic F; Yvin JC; Plusquellec D; Ferrières V How to Improve Chemical Synthesis of Laminaribiose on a Large Scale. Open Glycosci 2008, 1, 19–24. [Google Scholar]
- (74).Hummel G; Hindsgaul O Solid-Phase Synthesis of Thio-oligosaccharides. Angew. Chem. Int. Ed. 1999, 38, 1782–1784. [DOI] [PubMed] [Google Scholar]
- (75).Al-Masoudi NAL; Hughes NA Synthesis of 5-Thio-D-Allose and the Methyl 5-Thio-α- and -β-D-Allopyranosides. Carbohydr. Res. 1986, 148, 25–37. [Google Scholar]
- (76).Bennett S; von Itzstein M; Kiefel MJ A Simple Method for the Preparation of Thioglycosides of N-Acetylneuraminic Acid Carbohydr. Res. 1994, 259, 293–299. [DOI] [PubMed] [Google Scholar]
- (77).Ratajczak F; Greffe L; Cottaz S; Driguez H Convergent Synthesis of 4-Thiomaltooligosaccahrides. Synlett 2003, 1253–1254. [Google Scholar]
- (78).Gais H-J Cyclic Dithiohemiacetals - Synthesis and Properties. Angew. Chem. Int. Ed. 1977, 16, 196–197. [Google Scholar]
- (79).Vetvicka V; Yvin J-C Effects of Marine β−1,3 Glucan on Immune Reactions. Int. Immunopharmacol. 2004, 4, 721–730. [DOI] [PubMed] [Google Scholar]
- (80).Vetvicka V; Vetvickova J Glucans and Cancer: Comparison of Commercially Available beta-Glucans - Part IV. Anticancer Res. 2018, 38, 1327–1333. [DOI] [PubMed] [Google Scholar]
- (81).Plytycz B; Rozanowska M; Seljelid R Quantification of Neutral Red Pinocytosis by Small Numbers of Adherent Cells: Comparative Studies. Folia Biologica 1992, 40, 3–9. [PubMed] [Google Scholar]
- (82).Vetvicka V; Fornusek l.; Kopecek J; Kaminkova J; Kasparek L; Vranova M Phagocytosis of Human Blodd Leukocytes: A Simple Micromethod. Immunol. Lett. 1982, 5, 97–100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.