

Abstract
Genetic mutations in the phosphatase PTPN11 (SHP2) are associated with childhood leukemias. These mutations cause hyperactivation of SHP2 due to the disruption of the auto-inhibitory conformation. By targeting the activation-associated protein conformational change, we have identified an SHP2 inhibitor (E)-1-(1-(5-(3-(2,4-dichlorophenyl)acryloyl)-2-ethoxy-4-hydroxybenzyl)-1,2,5,6-tetrahydropyridin-3-yl)-1H-benzo[d]imidazol-2(3H)-one (LY6, 1) using computer-aided drug design database screening combined with cell-based assays. This compound inhibited SHP2 with an IC50 value of 9.8 µM, 7-fold more selective for SHP2 than the highly related SHP1. Fluorescence titration, thermal shift, and microscale thermophoresis quantitative binding assays confirmed its direct binding to SHP2. This compound was further verified to effectively inhibit SHP2-mediated cell signaling and proliferation. Furthermore, mouse and patient leukemia cells with PTPN11 activating mutations were more sensitive to this inhibitor than wild-type cells. This small molecule SHP2 inhibitor has a potential to serve as a lead compound for further optimization studies to develop novel anti-SHP2 therapeutic agents.
Graphical Abstract
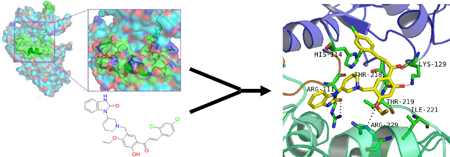
INTRODUCTION
SHP2 (encoded by PTPN11), a widely expressed non-receptor type protein tyrosine phosphatase (PTP), plays an essential role in several intracellular signal transduction processes, such as Ras-Erk, PI3K-Akt, Jak-Stat, NF-κB, and mTOR pathways1–3. It contains two Src homology 2 (SH2) domains at the N-terminus and a PTP domain at the C-terminus. The 2.0 Å X-ray crystal structure of SHP2 reveals that in the basal state, the catalytic site of SHP2 is autoinhibited by the N-terminal SH2 (N-SH2) domain. The N-SH2 domain of SHP2 functions as a conformational switch, which either binds to the PTP domain and directly blocks the active site, or binds to phospho-tyrosine (pY)-containing signaling partners and activates the enzyme by disrupting the intra-molecular inhibitory interaction4. The C-SH2 also plays an important role in recognition of bisphosphorylated ligands and contributes to binding energy and specificity, being an integral element of the switch5, 6. Although SHP2 dephosphorylates tyrosine phosphorylated signaling proteins, this phosphatase plays an overall positive role in transduction of the signals initiated from growth factors, cytokines and extracellular matrix proteins1–3, in sharp contrast to hematopoietic cell-specific SHP1 phosphatase, which shares a similar overall structure and a high homology with SHP2, but plays a negative role in cell signaling. The molecular mechanism for SHP2 function is not completely understood and is an area of intensive research. Both the SH2 domains and the PTP domains of SHP2 and SHP1 determine the signaling and thus functional specificities of these two highly related PTPs.
The critical role of SHP2 in cell signaling and other cellular activities is further underscored by its direct association with human diseases. Germline or somatic mutations in the PTPN11 gene that cause hyperactivation of SHP2 catalytic activity have been identified in 50% of patients with the developmental disorder Noonan Syndrome (NS) and various childhood leukemias, including juvenile myelomonocytic leukemia (JMML) (~35%), myelodysplastic syndrome (~10%), B cell acute lymphoblastic leukemia (~7%), and acute myeloid leukemia (~4%)7–11. In addition, activating mutations of PTPN11 have been found in sporadic solid tumors12. PTPN11 mutations, found in NS, leukemias, and solid tumors disrupt the intra-molecular interaction of SHP2, leading to a partially open conformation and thus activation of SHP2 due to greatly increased accessibility of the catalytic site11, 13. Several studies have demonstrated that a single PTPN11 activating mutation is sufficient to induce NS, JMML-like myeloproliferative neoplasm, and acute leukemias in mice14–16, suggesting that PTPN11 mutations play a causative role in these diseases.
The direct connection between activating mutations of PTPN11 and human diseases makes SHP2 an attractive target for developing mechanism-based therapeutic drugs. Identification of SHP2 inhibitors with high bio-activity will not only be helpful for drug discovery but also for understanding SHP2 function in cell signaling. However, it is extremely challenging to identify SHP2 selective inhibitors given the high homology in the catalytic site of SHP2 to those in other tyrosine phosphatases, especially the highly related SHP1 phosphatase. Over the last decade, several SHP2 inhibitors have been reported17–23. However, their cross reactivity for other PTPs, off-target effects, or biological functions in PTPN11 mutated tumor cells have not been well characterized. More recently, a potent and selective inhibitor of SHP2 was identified through high throughput screening24, 25. This inhibitor concurrently binds to the interface of the N-SH2, C-SH2, and PTP domains, inhibiting SHP2 activity through an allosteric mechanism. In the present study, we have chosen to target a different SHP2 surface pocket to identify small molecule compounds that suppress activation-associated conformational changes to functionally inhibit SHP2. Using computer-aided drug design (CADD) screening of a virtual database of compounds followed by experimental assays, we have identified a new low molecular weight compound that inhibits SHP2 activity. It is more than 7-fold selective for SHP2 over the closest homologue SHP1. Moreover, biological assays have verified that it is effective in blocking SHP2-mediated signaling and cellular functions in mouse and patient cells.
Results
Determination of a drug binding pocket in SHP2 for CADD database screening.
To identify small molecule inhibitors that are selective for SHP2 over SHP1, we performed thorough sequence and structural analyses for SHP2 and SHP1. The active site of SHP2 is almost identical to that of the SHP1 phosphatase. Compounds that target the catalytic core of SHP2 often inhibit SHP1. Therefore, our structure-based approach was aimed to target the allosteric change required for activation of SHP2, instead of the catalytic site. SiteMap was used to explore a potential drug-binding site in SHP2 for inhibitor discovery. Five possible binding sites in SHP2 were identified in this study (Figure 1A). The characters of these potential binding sites were assessed by calculating various properties, such as size, volume, amino acid exposure, enclosure, contact, hydrophobicity, hydrophilicity, and donor/acceptor ratio. Site 1 had the largest internal space and it was located at the interface between C-SH2 and PTP domains of SHP2. Site 4 was close to Site 1 and sat at the interface between N-SH2 and PTP domains. Site 3 was also located between N-SH2 and PTP domains. Both Site 2 and Site 5 were parts of the PTP domain. Site 3 and Site 5 were subsequently eliminated due to their low scores in the Sitemap algorithm. By analyzing chemical environments and structural conformations of Site 1 and the other two binding pockets we thought that compounds binding to Site 1 might tighten the connection of C-SH2 and PTP domains and stabilize the “closed” autoinhibited conformation of SHP2. More importantly, Site 1 of SHP2 showed specific features, as compared to the corresponding region of the homology SHP1 (Figure 1B). This pocket in SHP2 may confer biological selectivity to small molecule compounds that bind to and potentially stabilize the inhibitory conformation of SHP2. Therefore, Site 1 was selected as the docking site for database screening.
Figure 1. Identification of a drug docking site in SHP2 that has unique features in both amino acid composition and structure.
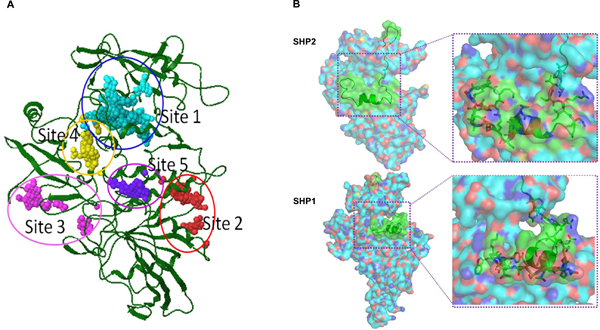
(A) SiteMap was used to explore potential binding sites in the SHP2 receptor for computational docking studies. Five possible binding sites were identified. The characters of potential binding sites were assessed by calculating various properties, such as size, volume, amino acid exposure, enclosure, contact, hydrophobicity, hydrophilicity, and donor/acceptor ratio. Site 1 in SHP2 was chosen for subsequent CADD in silico database screening. (B) Comparison between Site 1 in SHP2 and a corresponding site in SHP1. Different amino acid sequences between SHP2 and SHP1 are shown in green cartoons and key residues are shown in green carbon sticks.
CADD in silico screening and structural optimization.
To identify small molecular compounds that bind to and stabilize the auto-inhibited conformation to functionally inhibit SHP2, virtual chemical structure database screening and core-hopping procedures were performed. The drug-like structure database of the National Cancer Institute (NCI), USA, was screened by using Glide of Schrodinger 2010 based on the conformation of the Site 1 pocket in SHP2. NSC681129, ranked 21st in the positive hits from the screening, was considered as the most promising lead compound (Figure S1). NSC681129-based structural optimization was then performed to identify chemical structures that can potentially better stabilize the intramolecular interaction between SH2 and PTP domains in SHP2 (Figure S1). NSC681129 was divided into three parts - Fragment 1, Core, and Fragment 2. The Core structure and the two fragments were replaced with chemical segments from a fragment database to fill up Site 1 and keep SHP2 in a closed autoinhibited state. As a result, top 10 derivatives (LY1-LY10) with high docking scores were selected (Figure S2). These derivatives along with the top 100 hits from the initial database screening were subjected to PAINS (Pan Assay Interference Compounds) and aggregator examination26. Forty-seven structures, including NSC681129, were identified as potential PAINS-group containing structures (Supplementary PAINS Results). However, the 10 derivatives passed PAINS and aggregator tests and therefore were synthesized (Figure S3 and S9). These molecular candidates were re-docked into Site 1 to estimate their SHP2-binding affinities. The docking results showed that (E)-1-(1-(5-(3-(2,4-dichlorophenyl)acryloyl)-2-ethoxy-4-hydroxybenzyl)-1,2,5,6-tetrahydropyridin-3-yl)-1H-benzo[d]imidazol-2(3H)-one (LY6, 1) had the best docking score (Figure S2). The Glide scoring was conducted based on the sum of the Coulomb interaction energy, van der Waals energy, lipophilic and hydrogen-bonding terms, and penalty which included factors that hindered binding. Van der Waals interactions dominated all interactions between the ligands and the receptor. 1 was identified to have the strongest van der Waals interaction energy among all candidates, and it formed a strong H-bond with Glu250 and Thr218, indicating that 1 occupied the pocket better than other derivatives. The amino acids involved at the binding site in SHP2 included Arg111, Phe113, His114, Gly115, His116, Leu117, Leu125, Lys129, Leu136, Arg138, Thr218, Thr219, Arg220, Ile221, Ser228, Arg229, Glu232, Val243, Lys244, Gln245, Gly246, Glu249, Glu250 and Thr253.
Identification of 1 as a novel SHP2 inhibitor.
The ten candidate compounds were then subjected to SHP2 enzymatic assays with a tyrosine phosphorylated peptide as the substrate, which also served as an allosteric activator. These compounds inhibited SHP2 phosphatase activity to various extents (Figure S4). LY1 and LY7 were subsequently eliminated due to acute toxicity in cells. 1 was identified as the most active compound among the remaining compounds that inhibited SHP2 activity (Figure S4 and Figure 2A). As PTPN11 (SHP2) activating mutations are mainly associated with JMML, which is characterized by hypersensitity of myeloid progenitors to GM-CSF and IL-327, 28; it is critically important to determine the inhibitory activity of 1 against the homologous SHP1 phosphatase because SHP2 and SHP1 play opposing roles in these cytokine signaling pathways (due to the functional specificity determined by their SH2 domains). As shown in Figure 2B, 1 inhibited SHP2-catalyzed hydrolysis of the phospho-peptide substrate with an IC50 value of 9.8 μM, whereas the IC50 of 1 for SHP1 was 72.7 μM. Although LY3 and LY5 showed good effects on SHP2, they inhibited the SHP1 phosphatase to a similar extent (i.e., no selectivity between SHP2 and SHP1; data not shown). Therefore, LY3 and LY5 were eliminated.
Figure 2. Identification of 1 as a novel inhibitor of SHP2.
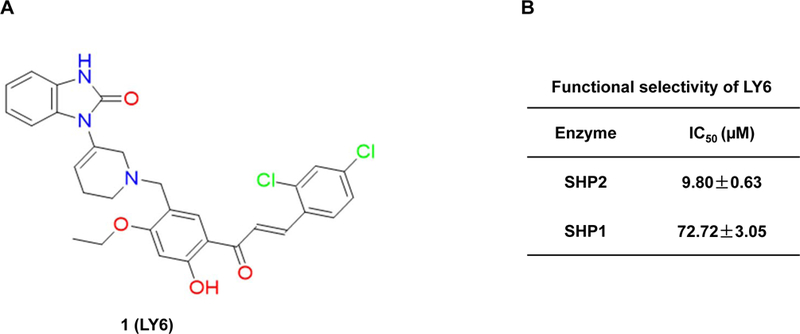
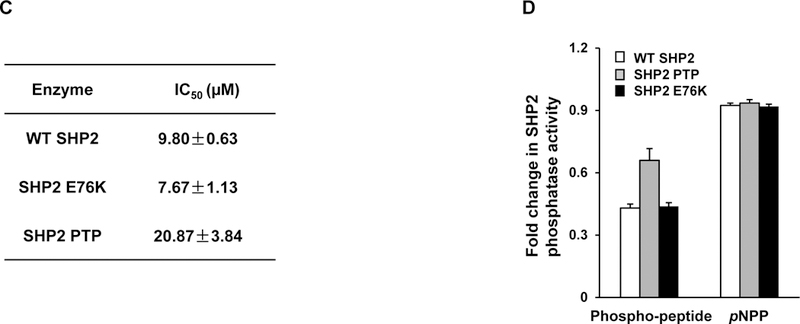
(A) Chemical structure of 1. (B) Phosphatase assays were carried out using full length SHP2 or SHP1 as the enzyme and a phospho-peptide as the substrate in the presence of various concentrations of 1, as described in Experimental Section. For IC50 determinations, six concentrations of 1 were tested. Results shown are the mean ± SD of three independent experiments. (C) Phosphatase activities of full length WT SHP2, full length SHP2 E76K, and the SHP2 PTP domain were assessed in the presence of 1 at various concentrations as above. IC50 values were then determined. Experiments were repeated independently three times, similar results were obtained in each experiment. Results shown are the mean ± SD of triplicates from one experiment. (D) Phosphatase activities of full length WT SHP2, full length SHP2 E76K, and the SHP2 PTP domain were determined in the presence of 1 (10 μM) using a phospho-peptide or pNPP as the substrate, as detailed in Experimental Section Experiments were repeated independently three times, similar results were obtained in each experiment. Results shown are the mean ± SD of triplicates from one experiment.
Docking simulation analyses demonstrated that 1 created three hydrogen bonds with Arg111, Lys129, and Arg229 (Figure S5A). Moreover, the dichloro-phenyl ring of 1 occupied the hydrophobic pocket made up of Leu117, Leu125, Leu136, and Leu216. 1 possessed an L-shaped structure and fitted well in the pocket of SHP2, connecting C-SH2 and PTP domains through hydrogen bonds and hydrophobic interactions. Binding energy calculation showed that 1 occupied Site 1 with high binding energy (Figure S6A), similar to the binding energy of the newly identified allosteric SHP-2 inhibitor SHP099 24 when docking into its binding site (http://www.RCSB.org; PDB ID: 5EHR). While 1 docked into Site 1 with docking score of −7.3, it could not dock into the SHP099 binding site due to limited space. Interestingly, 1 gained more binding energy when docking into Site 1 than did SHP099 when docking into its binding site (Figure S6B).
Molecular dynamics (MD) simulation of the 1/SHP2 binding interaction.
Many biological functions of proteins and their profound dynamic mechanisms, such as switching between active and inactive states29, cooperative effects30, allosteric transition31, and intercalations with drugs, can be revealed by studying their internal motions. In order to understand the allosteric mechanism of a ligand with its receptor, both the static structures and the dynamics information should be considered. Therefore, MD simulations were performed to further define the 1/SHP2 interaction. The root mean square deviation (RMSD) as compared with the initial conformation is a crucial criterion for evaluating the reliability and stability of drug/protein systems. The RMSD of four complexes (SHP2-1, SHP1-1, SHP2 alone, and SHP1 alone) versus the simulation times is illustrated in Figure S7A, in which the RMSD of the SHP2-1 interaction is lower than those of the other systems, indicating that the movement of SHP2 might be circumscribed when binding with 1, which would reduce the activity of SHP2 because activation of SHP2 requires protein conformational changes from the closed autoinhibited conformation to the open conformation. In contrast, the motion of SHP1 could hardly be suppressed by 1 during the simulation process.
In order to evaluate the motion of the key residues interacting with 1 at the binding site in SHP2 defined in Figure S5A, the root mean square fluctuations (RMSF) for all residues of the SHP2 protein were calculated. The most flexible region for SHP2 is made up of residues 111–206, corresponding to the C-SH2 domain. 1 was designed to concurrently bind to the C-SH2 domain and the PTP domain. The interaction between 1 and the receptor SHP2 made the whole system stable, decreasing the flexibility of the C-SH2 domain in the meantime. The fluctuating magnitude of SHP2 decreased when binding to 1 (Figure S7B), indicating that SHP2 is more stable when bond with 1. In contrast, 1 could hardly lower the movement of key residues in SHP1 (Figure S7A and S7B), further supporting that 1 is selective for SHP2 over SHP1. 10 ns MD simulation of the 1/SHP2 binding (Figure S5B) predicts that 1 creates three hydrogen bonds with Thr218, Arg220, and Arg229 of SHP2. The dichloro-phenyl ring of 1 is still in the hydrophobic region of the pocket consisted of Leu117, Leu125, Leu136, and Leu216. The hydrogen bond between Arg111 and 1 is broken under MD simulation, while Arg111 forms a salt bridge with Glu250. By comparing the binding modes of 1 after and before MD simulations, it appears that 1 stays tightly in the binding pocket of SHP2, indicating that the interaction between 1 and SHP2 is stable.
1 also inhibits leukemia-associated mutant SHP2.
The E76K mutation, localized in the N-SH2 domain, is the most common and active PTPN11 mutation found in leukemias. This mutation causes a partially open conformation in SHP2 due to the disruption of the intramolecular interaction. To further characterize the acting mechanisms of 1, we purified GST fusion proteins of wild-type (WT) full-length SHP2 and the full-length SHP2 E76K mutant, and compared sensitivities of these SHP2 proteins to 1. 1 inhibited the full length SHP2 E76K mutant in a dose-dependent manner, with an IC50 value of 7.67 μM, which is slightly lower than that for full length WT SHP2 (9.8 μM) (Figure 2C). The fact that the IC50 values of 1 for WT SHP2 and SHP2 E76K were in a similar range suggests that 1 was able to “close” WT SHP2 and SHP2 E76K, regardless of the status of their basal conformations. Another possibility is that the phospho-peptide substrate used in the phosphatase assay also functioned as an allosteric activator “opening up” protein conformations of WT SHP2 and SHP2 E76K, regardless of the basal status, and similarly opened conformations resulted in similar 1 concentrations required to achieve 50% inhibition of SHP2 enzymatic activity.
We also tested the inhibitory effect of 1 on GST-SHP2 PTP domain without SH2 domains. It inhibited the phosphatase activity of SHP2 PTP, although SHP2 PTP had a completely open conformation. This result indicates that the binding of 1 with SHP2 PTP somehow interfered the peptide substrate-enzyme interaction. However, the fact that the IC50 value (20.87 μM) of 1 for the SHP2 PTP domain was >2-fold higher than that for full length WT SHP2 (Figure 2C) supports that 1 functions by stabilizing the inhibitory closed conformation of full length SHP2. To further characterize the acting mechanisms of 1, we also tested its effect on SHP2 catalytic activity using para-Nitrophenylphosphate (pNPP), a general small compound substrate for all PTPs. pNPP is much smaller than the phospho-peptide substrate and can easily get access to SHP2 catalytic core. Dephosphorylation of this small interrogate substrate does not require a substantial conformational change in SHP2. Indeed, 1 did not affect the capability of SHP2 to dephosphorylate pNPP (Figure 2D), regardless of what form of SHP2 protein was used as the enzyme, suggesting that 1 binding does not interfere the access of this small substrate to SHP2 catalytic core.
Furthermore, we performed enzymatic kinetic studies (Lineweaver-Burk plot) in the absence or presence of 1. As shown in Figure 3A, Vmax decreased while Km increased when the 1/SHP2 binding increased with increasing concentrations of 1, suggesting that 1 is a mixed type inhibitor against SHP2 (not a competitive or non-competitive inhibitor). We also tested the inhibitory effect of 1 on SHP2 by extensively washing the enzyme-inhibitor complex. The results indicated that the 1/SHP2 binding was stable (Figure S8A). In addition, to exclude the possibility that the inhibition of SHP2 by 1 was a promiscuous effect, we tested the activity of 1 in the presence of the detergent Triton X-100 and found that the 1-SHP2 complex was stable and that the inhibitory function of 1 was not decreased by 0.001% Triton X-100 in the assay buffer (Figure S8B). However, the inhibition of SHP2 by 1 was diminished by 0.01% Triton X-100, suggesting that the 1/SHP2 binding affinity is low. To further determine whether 1 directly bond to the SHP2 protein, we performed the SHP2 fluorescence quenching assay, taking advantage of the seven tryptophans in the protein. As demonstrated in Figure 3B, this drug exhibited strong quenching of SHP2 fluorescence in a dose-dependent manner. Thermal shift assays also verified the binding of 1 and SHP2, as evidenced by dose-dependent right shifts in melting temperature (Tm) (Figure 3C). Furthermore, quantitative MicroScale Thermophoresis (MST) binding affinity assays demonstrated specific binding between 1 and His-tagged SHP2 and that their interaction was not irreversible. The equilibrium dissociation constant (Kd) of the 1/SHP2 complex was 69 ± 3 µM (Figure 3D). The Kd value of 1 derived from the MST assay was much lower than its IC50 value from the phosphatase assay, likely because the buffer conditions, e.g., chemical components and their concentrations, pH, etc. were significantly different in the two assays (different solution conditions could effectively modify the strength of any intermolecular interactions holding a particular ligand-protein complex together).
Figure 3. 1 directly binds to the SHP2 protein and functions as an inhibitor.
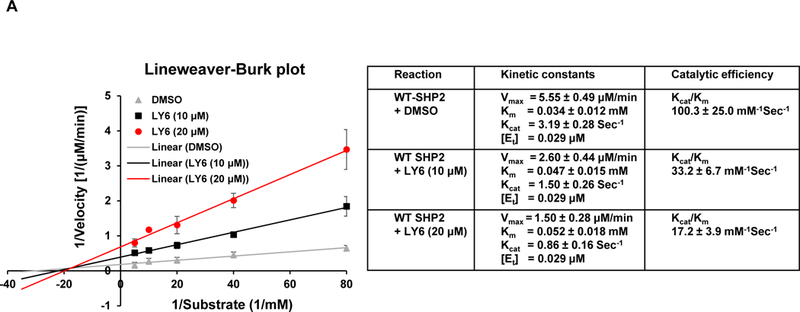
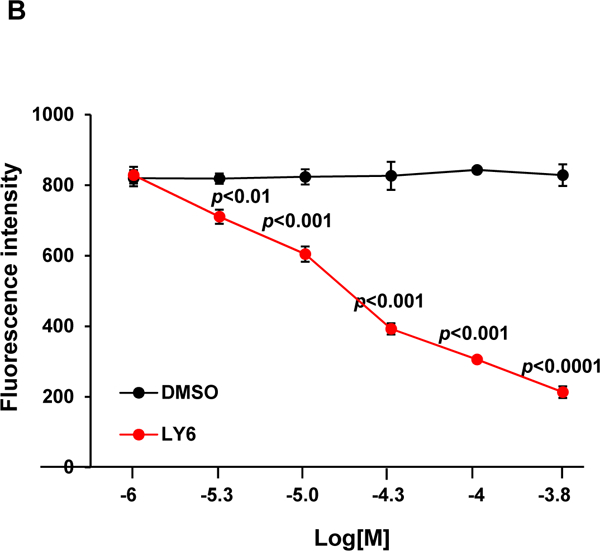
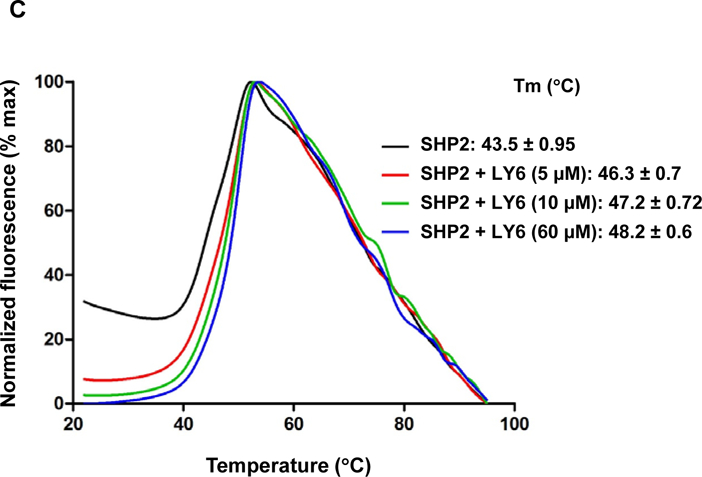
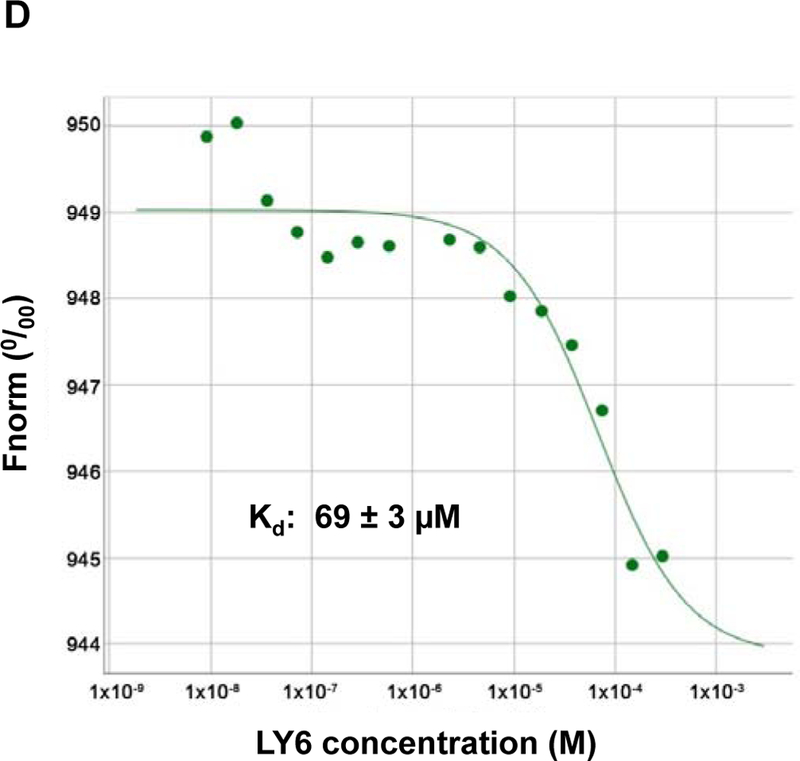
(A) Enzymatic kinetic analyses in the presence or absence of 1 were performed using full length WT SHP2 (0.1 µg) as the enzyme. 1 concentrations used in this experiment were 0, 10, and 20 μM. Lineweaver-Burk plot was generated to show 1-mediated SHP2 inhibition. Experiments were repeated independently three times. Results shown are the mean ± SD of three independent experiments. (B) Fluorescence titration of SHP2 was performed by increasing the concentrations of 1 while maintaining the WT SHP2 protein concentration at 3.0 µM. The fluorescence intensity is plotted against the log concentration in mol/L (Log[M]). Experiments were repeated independently three times, similar results were obtained in each experiment. Data shown are the mean ± SD of triplicates from one representative experiment. (C) Thermal shift assays were performed as described in Experimental Section. Representative dose response melting curves of His-tagged SHP2 in the presence of 1 are shown. Tm data shown are the mean ± SD of three independent experiments. (D) MST binding affinity assays were performed as described in Experimental Section. One representative dose-response curve of 1 binding to His-tagged SHP2 is shown. Fnorm, normalized fluorescence. Experiments were repeated independently three times. Reported Kd is the mean ± SD of three independent experiments.
1 inhibits SHP2-mediated cell signaling and cellular function.
PTPN11 (SHP2) activating mutation-associated JMML exhibits hypersensitivity to GM-CSF and IL-327, 28, and previous studies have demonstrated that SHP2 promotes IL-3-stimulated cell proliferation by enhancing activation of Erk, Akt, and Jak232, in sharp contrast to the SHP1 phosphatase, which negatively regulates these cytokines signaling. It is critically important to test the effects of any potential SHP2 inhibitors in these cytokines induced signaling and cellular responses. Ba/F3, an IL-3 dependent murine pro-B lymphoma cell line, was utilized to determine the effects of 1 on SHP2-mediated cell signaling and cellular functions. This compound significantly suppressed cell growth (Figure 4A), in agreement with the overall positive role of SHP2 catalytic activity in cellular response to IL-3. To determine whether 1 functioned by inhibiting SHP2-mediated cell signaling, we examined the effects of 1 on IL-3-induced signaling processes. Following IL-3 stimulation, activation of Erk, Akt, Jak2, and Stat5, determined by their phosphorylation levels, was inhibited by 1 (Figure 4B and 4C), and this effect was clearly 1 dose-dependent, consistent with the previous finding that SHP2 catalytic activity was required for optimal activation of IL-3-induced signaling pathways32. These results suggest that 1 suppresses IL-3 signaling and cellular responses through a mechanism involving inhibition of SHP2 catalytic activity.
Figure 4. 1 inhibits IL-3-induced signaling and cell proliferation.
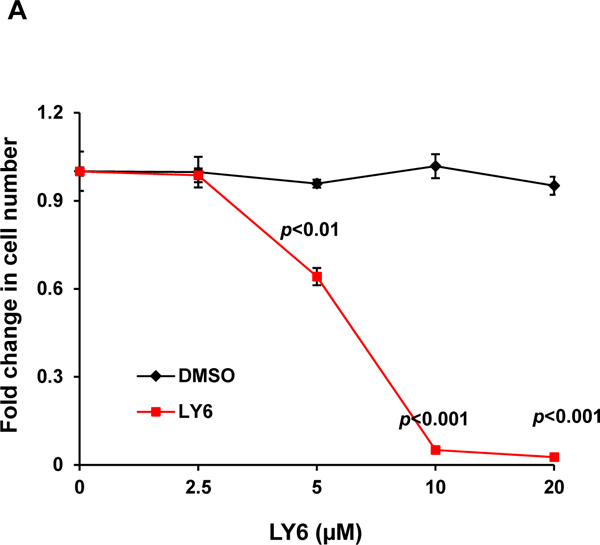
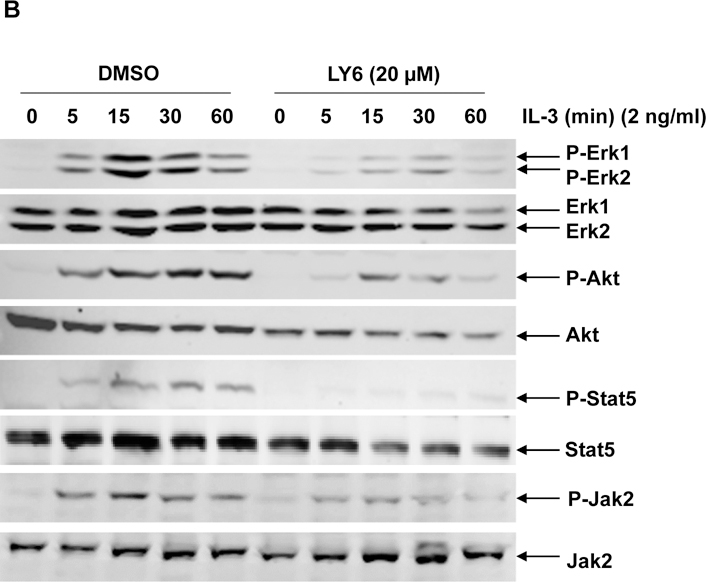
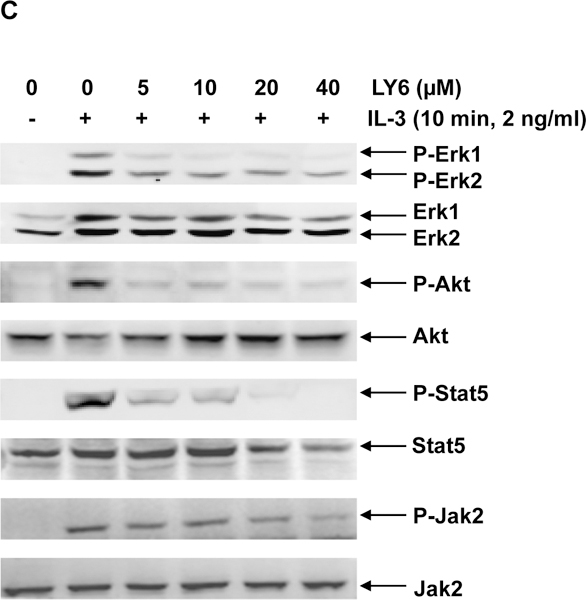
(A) Ba/F3 cells were cultured in IL-3 (1.0 ng/mL) containing medium supplemented with 1 at the indicated concentrations or DMSO. Cell numbers were determined 48 hours later using a One Solution Cell Proliferation Assay kit. Experiments were performed three times with similar results obtained in each. Data shown are the mean ± SD of triplicates from one representative experiment. (B) Ba/F3 cells were deprived of IL-3 overnight. Cells were treated with 1 (20 µM) for 4 hours and then stimulated with IL-3 (2.0 ng/mL) for the indicated times. (C) Ba/F3 cells were deprived of IL-3 overnight. Cells were treated with 1 at the indicated concentrations for 4 hours and then stimulated with IL-3 (2.0 ng/mL) for 10 min. Whole cell lysates were prepared. Levels of p-Erk, p-Akt, p-Jak2, and p-Stat5 were determined by immunoblotting analyses. Blots were striped and reprobed with anti-Erk, anti-Akt, anti-Jak2, and anti-Stat5 antibodies to check for protein loading. Experiments were repeated independently three times. Representative results from one experiment are shown.
1 has minimal off-target effects.
To confirm that the cellular effects of 1 were mediated by acting on SHP2, PTPN11 (SHP2) knockout cells were treated with 1. PTPN11 conditional knockout (PTPN11fl/fl/Ade-Cre+) and WT control (PTPN11+/+/Ade-Cre+) mouse embryonic fibroblast (MEFs) were treated with 1 or vehicle. Proliferation of WT cells was markedly decreased by this inhibitor, consistent with the positive role that SHP2 plays in growth factor signaling1–3. In contrast, 1 did not show a significant effect on PTPN11-depleted MEFs (Figure 5A). The observation that cells lacking SHP2 were insensitive to 1 suggests that SHP2 is the main target of this compound.
Figure 5. Effects of 1 are diminished in SHP2 knockout cells while cells harboring hyperactivated SHP2 are more sensitive to 1.
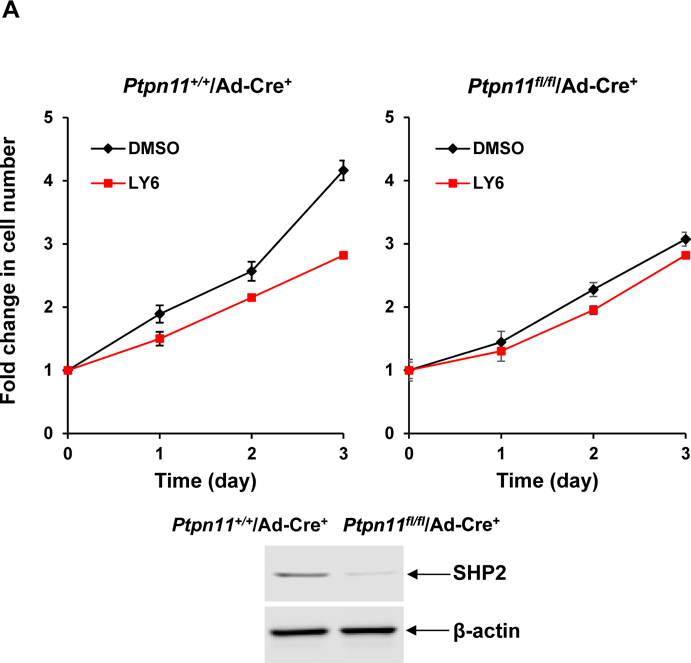
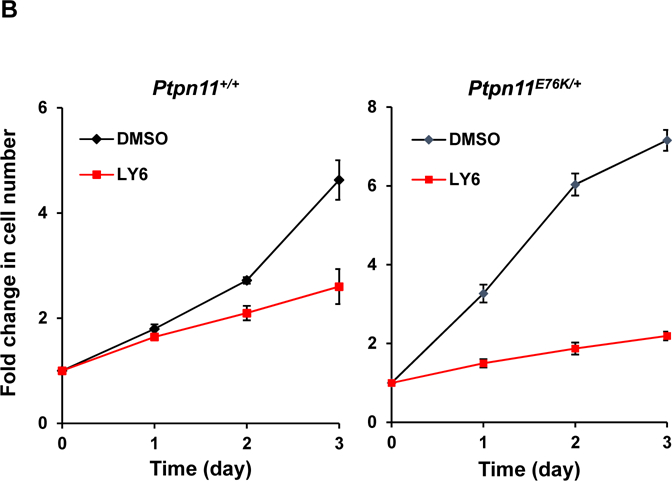
(A) Ptpn11 conditional knockout (Ptpn11fl/fl/Ade-Cre+) mouse embryonic fibroblasts (MEFs) and WT (Ptpn11+/+/Ade-Cre+) control cells were infected with adeno-Cre viruses and then treated with 1 at 6 μM or DMSO. Cell growth rates were determined at 24, 48 and 72 hours using a One Solution Cell Proliferation Assay kit. SHP2 levels in knockout and control cells were analyzed by immunoblotting analyses with anti-SHP2 antibody. Blots were stripped and reprobed with anti-β-actin antibody to monitor protein loading. Experiments were performed three times. Similar results were obtained in each. Data shown are the mean ± SD of triplicates from one representative experiment. (B) Ptpn11E76K/+ and Ptpn11+/+ control MEFs were treated with 1 (6 μM). Cell numbers were determined using a One Solution Cell Proliferation Assay kit. Experiments were performed three times. Similar results were obtained in each. Data shown are the mean ± SD of triplicates from one representative experiment.
Tumor cells carrying SHP2 activating mutations are more sensitive to 1 than control cells with WT SHP2.
We next assessed the effect of 1 on mutant cells with a tumor-associated PTPN11 activating mutation (PTPN11E76K). Treatment of both WT and PTPN11E76K/+ MEFs with 1 significantly decreased cell growth (Figure 5B). Interestingly, PTPN11E76K/+ mutant cells were much more sensitive than WT cells to SHP2 inhibition although the IC50 value of 1 for the SHP2 E76K mutant was only slightly lower than that for WT SHP2. Additionally, human lung cancer cells H661 carrying the activating mutation (N58S) in PTPN11 and human lung cancer cells H596 with WT PTPN11 were treated with 1. This compound inhibited proliferation of both two cell lines in a dose-dependent manner. Importantly, H661 cells were more sensitive to 1 than H596 cells (Figure 6A). Moreover, 1-treated H661 cells showed pronounced G2/M arrest (Figure 6B), consistent with the positive role of SHP2 in cell cycle progression33.
Figure 6. Lung cancer cells harboring hyperactivated SHP2 are more sensitive to 1 than lung cancer cells expressing WT SHP2.
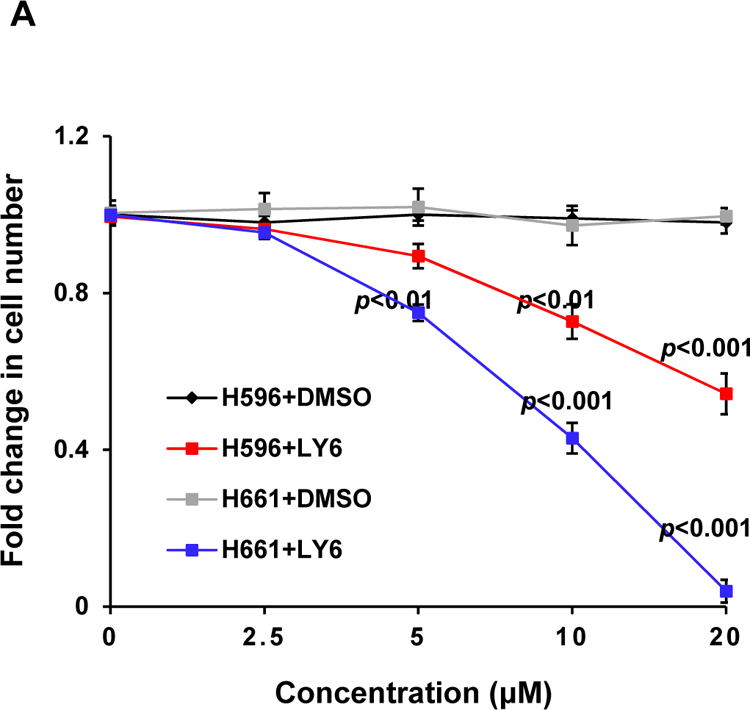
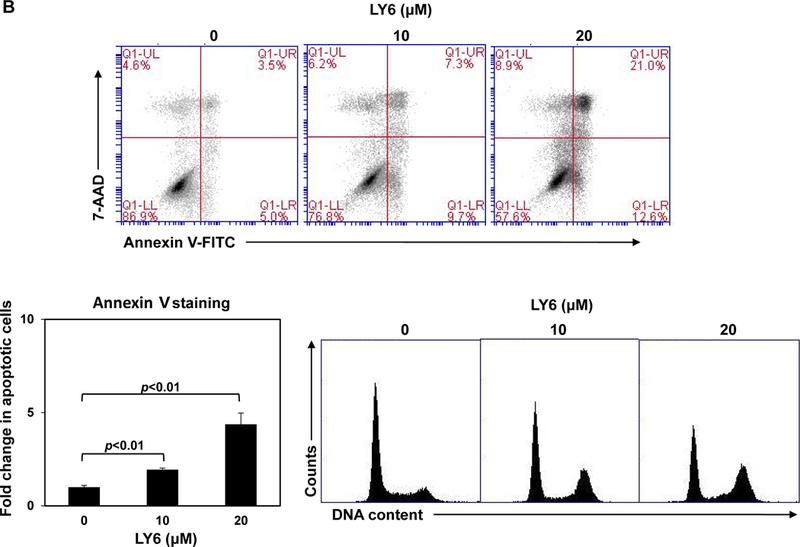
(A) Human lung cancer cell line H661 carrying the PTPN11N58S/+ mutation and H596 with WT PTPN11 were treated with 1 at the indicated concentrations for 48 hours. DMSO-treated cells were included as negative controls. Cell numbers were determined using a One Solution Cell Proliferation Assay kit. Experiments were repeated three times, similar results were obtained in each experiment. Data are presented as the mean ± SD of triplicates from one representative experiment. (B) H661 cells were treated with 1 at 0, 10, and 20 μM. Apoptotic cells were quantified 24 hours later by FACS using an Annexin V staining kit. Cell cycle changes were determined by propidium iodide staining followed by FACS analyses. Experiments were performed three times. Apoptosis data presented are the mean ± SD of three experiments. Cell cycle profiles are representatives of three independent experiments.
We next determined whether 1 could suppress colony formation and proliferation of mouse and human leukemic cells with PTPN11 activating mutations. Myeloid progenitors from PTPN11E76K/+ mice showed greatly increased responses to GM-CSF as compared to WT counterparts. These SHP2 E76K-expressing progenitors were much more sensitive than control cells to 1 inhibition of colony-forming capabilities over a range of concentrations (Figure 7A). Consistent with this data, colony formation of myeloid blasts from JMML patients with the PTPN11E76K/+ mutation was exquisitely sensitive to 1 (Figure 7B). Overall, these results suggest that 1 is able to overcome the dominant effects of SHP2 activating mutations in primary mouse and patient cells, and that SHP2 is a useful therapeutic target for PTPN11 mutation-associated hematological malignancies.
Figure 7. Mouse and JMML patient leukemic cells are more sensitive to 1 than normal control cells.
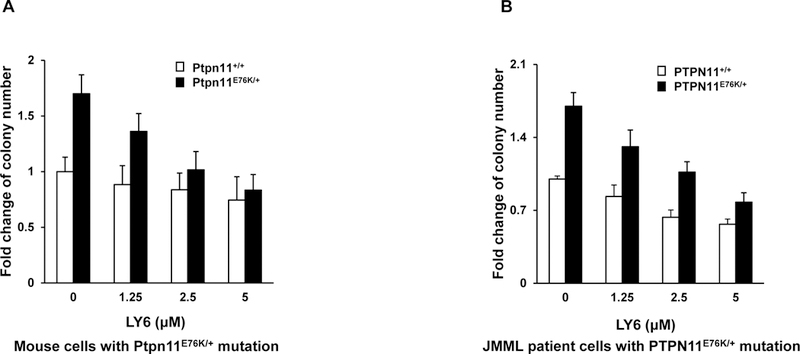
Bone marrow cells harvested from Ptpn11E76K/+/Mx1-Cre+ and Ptpn11+/+/Mx1-Cre+ mice were plated in methylcellulose medium containing GM-CSF (1.0 ng/mL) and 1 at the indicated concentrations or DMSO. Hematopoietic colonies that were generated were enumerated 7 days later and normalized against the number of colonies derived from WT control cells treated with DMSO. Data are presented as the mean ± SD of three independent experiments. (B) Splenocytes/marrow cells/blood cells from JMML patients with the PTPN11E76K/+ mutation and apheresis samples from healthy donors were plated in methylcellulose medium containing GM-CSF (1.0 ng/mL) and 1 at the indicated concentrations or control DMSO. Leukemic colonies that were produced were enumerated 14 days later and normalized against the number of colonies derived from the cells treated with DMSO. Three patient samples and three apheresis samples were tested in three independent experiments. Similar results were obtained in each. Data are presented as the mean ± SD of triplicates from one patient sample.
DISCUSSION AND CONCLUSIONS
In this study, by using CADD in silico database screening in combination with experimental assays, we have identified a new SHP2 inhibitor. SHP2, encoded by PTPN11, is an important signaling component downstream of growth factor/cytokine receptors and cell adhesion molecules. Activating mutations of PTPN11 that cause enhanced catalytic activity are associated with human diseases, such as Noonan syndrome and childhood leukemias7, 10–12. Importantly, these mutations play a causative role in the pathogenesis of these diseases14–16. Therefore, identification of highly selective SHP2 inhibitors is greatly needed and would facilitate the development of therapeutic drugs for these diseases. However, it has been challenging to develop SHP2 selective inhibitors that directly target the active site due to a high homology in these sites among tyrosine phosphatases. To circumvent this problem, a few allosteric inhibitors of SHP2 that inhibit the conformational change required for activation of this enzyme have recently been successfully developed23–25. Our structure-based approach in this study was also targeting the activation-associated conformational change, but a different surface pocket formed between C-SH2 and PTP domains. We have identified a novel SHP2 inhibitor (1) through CADD database screening, structural optimization, and cell-based assays.
Structure-activity relationship analyses of optimized derivatives suggest that three parts of 1 function cooperatively in keeping the “closed” auto-inhibited conformation of SHP2. (1) The Core part of 1 is responsible for an interaction with the PTP domain. The hydroxyl group of the Core part (beta-methyloxygenly phenol group) forms firm connections with key residues of the PTP domain via H-bond interactions. The methoxyl group and benzene ring interact with Lys129 and Tyr515 via intermolecular forces. (2) R1 of Fragment 1 acts as a linker of the SH2 domain to stabilize its position by hydrophobic effects and VDW interactions. (3) R2 and R3 of Fragment 2 provide benzene ring effects and possibly other molecular interactions with the SH2 domain. In addition, PTP and SH2 domains are firmly connected via two Cl atoms of Fragment 2. 1 inhibits SHP2 activity toward a pY-peptide substrate, but not the small compound substrate pNPP (Figure 2D). This is because pY-peptide substrates rely more on SHP2 protein conformational changes than pNPP in order to get access to the catalytic core to be dephosphorylated. It is likely that 1 functions by inhibiting the activation-associated allosteric change of SHP2, as designed, not targeting the active site. Computational modeling shows that the structure of 1 complements very well the binding pocket in SHP2 in docking simulation. In addition, MD simulation demonstrates that movement of the SHP2 protein is suppressed upon binding with 1, further supporting that the auto-inhibitory protein conformation of SHP2 is stabilized by 1.
PTPN11 (SHP2) activating mutations are associated with JMML, which display a characteristic hypersensitive growth pattern in response to GM-CSF and IL-327, 28. 1 inhibited IL-3-stimulated cell proliferation and suppressed IL-3-induced Ras-Erk, PI3K-Akt, and Jak2-Stat5 pathways. Given the positive roles that the SHP2 phosphatase plays in these signaling pathways, the function of 1 in this context is likely mediated by inhibition of the catalytic activity of SHP2. Interestingly, mouse and human leukemia cells carrying the activating mutation SHP2 E76K were much more sensitive than control cells with WT SHP2 to the inhibition by 1 (Figure 7), although the SHP2 E76K protein was only slightly more sensitive to 1 than WT SHP2 (Figure 2C). Part of the reason for this might be the addiction of leukemic cells to this disease causing oncoprotein. The differential sensitivity of PTPN11 activated leukemia cells and normal control cells to SHP2 inhibition provides proof of concept that SHP2 is a “druggable” target for the treatment of PTPN11-associated malignancies.
1 appears to function as an allosteric inhibitor of SHP2, despite a low SHP2-binding affinity. This inhibitor and the allosteric SHP-2 inhibitor SHP099 recently identified 24 have different binding sites and activities. 1 was designed to bind to the surface pocket of SHP2 formed between C-SH2 and PTP domains. This drug docking site (Site 1) is different from the binding site of SHP099 24, which concomitantly binds to N-SH2, C-SH2 and PTP domains (http://www.RCSB.org; PDB ID: 5EHR). Given that the structures and the SHP2-binding sites of these two inhibitors are completely different, they have distinct biochemical activities in inhibiting the function of WT SHP2 and SHP2 variants. This is especially the case when different phospho-peptides or interrogate compounds were used as the substrate in phosphatase assays. More biochemical/biological studies with tumor-associated mutant SHP2 and SHP2-mutated tumor cells will be necessary to further compare the functions of these two inhibitors. It is important to point out that while 1 is a drug-like small molecule that is bioavailable in cells, 1/SHP2 binding affinity is low. Further structure-activity relationship analysis for this compound is needed to establish independent intellectual property positions that will allow pharmacological validation of its effect on SHP2 inhibition for the development of analogues for further preclinical and clinical studies. In addition, cocrystallization of 1 and SHP2 is required to reveal the structural bases of their binding interaction, which will be essential for structural optimization of 1. The structural bases of the 1/SHP2 binding can also address the mechanisms of action of this inhibitor and other related questions regarding the biochemical activities of ths inhibitor.
EXPERIMENTAL SECTION
Reagents and Chemicals.
The CellTiter 96® AQueous One Solution Cell Proliferation Assay kit was purchased from Promega (Madison, WI, USA). Antibodies specific for p-Erk (E-4), Erk (C-16), Jak2 (C-20), and SHP2 (C-18) were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA). Antibodies against p-Akt (S473), Akt, and p-Stat5 (Y694) were obtained from Cell Signaling Technology (Beverly, MA, USA). Stat5 antibody was purchased from BD Biosciences (San Jose, CA, USA). Antibodies specific for p-Jak2 (Y1007/1008) and phosphor-tyrosine (pY) were purchased from Millipore Corporation (Temecula, CA, USA). Horseradish peroxidase conjugated goat-anti-rabbit and anti-mouse IgG were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). Dimethyl sulfoxide and other chemicals used for buffer solutions were provided by Fisher Scientific (Pittsburgh, PA, USA). LY series compounds, including 1, were obtained from Tianjin Institute of Pharmaceutical Research. (Tianjin, China), and dissolved in DMSO to prepare a stock solution (20 mM) for subsequent experiments. The purity of all test compounds was ≥ 98% by HPLC.
Compound Synthesis and Characterization.
1 was synthesized using 1-(2,4-dihydroxyphenyl) ethan-1-one and diethyl sulfate as starting materials with 4-step reactions (Figure S3). Synthesis of other derivatives is described in Figure S9.
1-(4-ethoxy-2-hydroxyphenyl) ethan-1-one.
A mixture of 1-(2,4-dihydroxyphenyl) ethan-1-one (19.8g, 130 mmol) and diethyl sulfate (40.5g, 520 mmol) was stirred in 300 mL acetone at room temperature for 24 hours. The solution was heated to reflux for 1 hour and then filtered. Desolventized mixture was washed by 50 mL water 3 times and extracted by 100 mL Ethyl Acetoacetate 3 times. A crude product [(Building block (2)] was obtained. The product was used in the next step without any further purification.
1-(5-(chloromethyl)-4-ethoxy-2-hydroxyphenyl) ethan-1-one.
Building block (2) (15.0g, 83 mmol) was dissolved in chloromethyl methyl ether (14.0g, 122 mmol). The solution was diluted in 100 mL acetic acid and stirred at ambient temperature for 24 hours. After removing the solvent under reduced pressure, the product [(Building block (3)] (7.7g, 33.6 mmol) was obtained with yield of 27.5%.
1-(1-(5-acetyl-2-ethoxy-4-hydroxybenzyl)-1,2,5,6-tetrahydropyridin-3-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one.
K2CO3 (1.74g, 12.6 mmol) and 1-(1,2,5,6- tetrahydropyridin-3-yl)-1H-benzo[d]imidazol-2(3H)-one (11g, 50.4 mmol) were added into the solution of Building block (3) (7.7g, 33.6 mmol) in 30 mL Ethyl Acetoacetate at room temperature and stirred for 24 hours. The solvent was removed under reduced pressure, and then washed by brine and 1 M hydrochloric acid 3 times, respectively. The solution was extracted by Ethyl Acetoacetate after adjusting pH to 8–9 by 1 M K2CO3. After the volatile solvent of the reaction was evaporated, 15.3g yellow pale oily raw product [(Building block (4)] was obtained.
(E)-1-(1-(5-(3-(2,4-dichlorophenyl)acryloyl)-2-ethoxy-4-hydroxybenzyl)-1,2,5,6-tetrahydropyridin-3-yl)-1H-benzo[d]imidazol-2(3H)-one.
Building block (4) (1.12g, 2.5 mmol), 2,4-dichlorobenzaldehyde (0.88g, 5 mmol), and 0.7g KOH (0.7g, 12 mmol) were mixed and stirred in 9 mL ethanol and 2 mL water at room temperature for 24 hours. The product was recrystallized in 15 mL ethanol after discarding the volatile solvent. 0.3g compound (22.4%) was obtained as the final product (LY6, 1). The purity and the structure of this final product were determined by High-performance liquid chromatography (HPLC) and Nuclear magnetic resonance (NMR), respectively (Supplementary HPLC and NMR results).
Virtual Screening and Core-Hopping Procedure in Silico.
The database of small structures from NCI, USA was screened by the Glide docking package of the Schrodinger suite 201034 based on the 3D structure of residues in the designated binding pocket in SHP2. Protein and small compound structures were geared up by Protein Preparation Wizard and LigPrep modules embedded in Schrodinger suite 2010 (www.schrodinger.com)35, respectively. All investigated structures in the NCI database were docked into the defined pocket using the rigid docking model with the stand-precision scoring function34, 36 to estimate the binding affinities. In order to enhance binding affinity, the program Core-Hopping36 in Schrodinger suite 2010 was utilized in this study, which had dual functions (including fragment-based replacing and molecular docking). OPLS2005 force field37, 38 was recruited in all assays performed in silico.
Molecular Dynamics (MD) Simulation.
To examine whether the inhibitor remains bound in the presence of explicit solvent from a dynamic point of view, the MD simulation was performed with GROMACS 96–53a6 force fields39 with the periodic boundary conditions by using the GROMACS 4.5 package for Linux. The topology files and charges for the ligand atoms were generated by the Dundee PRODRG2.5 Server40. After minimization of the potential energy of the whole system, 10 ns MD simulations were carried out with a time interval of 2 fs; corresponding coordinates were stored every 10 fs. The PME algorithm was used to calculate the electrostatic interactions. All simulations were run under the periodic boundary condition with NVT ensemble by using the V-rescale coupling algorithm to keep the temperature at 310 K and pressure at 1atm. All bonds were constrained by using the LINCS algorithm. The GROMACS 4.5 package was utilized to analyze the results. The initial complex structures of 1-SHP1 and 1-SHP2 for MD simulation were from docking calculations (Supplementary PDB File 1 and 2).
Protein-Ligand Binding Energy Calculation.
The binding energy was calculated as follows. The coulomb interaction energies between protein and ligand were computed by equation (1) where qi and qj were the formal charge associated with atomic centers i and j, which were the residues in the C-SH2 and PTP domain, respectively. Rij was the distance between them. F = 138.935 and εr was the dielectric constant.
| (1) |
The van der Waals interaction energies between protein and ligand equals:
| (2) |
| (3) |
| (4) |
As the parameters Cij(6) and Cij(12) depend on pairs of atom types, they were taken from a matrix of Lennard-Jones parameters. Rij was the distance between them.
In Vitro Phosphatase Activity Assay.
Purified SHP2 and SHP1 phosphatases purchased from Enzo Life Sciences (Farmingdale, NY, USA) were used as enzymes and a phospho-peptide (H-Glu-Phe-pTyr-Ala-Glu-Val-Gly-Arg-Ser-Pro-Pro-Asp-Pro-Ala-Lys) (BML-P157, Enzo Life Sciences Inc., NY, USA) was used as the substrate, which also functions as an activator of SHP2 by binding to the N-SH2 domain. The assay determines free phosphate generated by dephosphorylation of the substrate by using the Malachite Green reagent (Sigma, St. Louis, MO, USA). In brief, 0.1 μg of SHP2 or SHP1 was incubated in 40 μL of the assay buffer (25 mM Tris-HCl, pH 7.4, 50 mM NaCl, 5 mM DTT, and 2.5 mM EDTA) with test compounds at various concentrations at room temperature for 30 min. The substrate was then added to a final concentration of 50 µM. The mixture was incubated at 30 °C for 30 min. Finally, 50 μL of Malachite Green solution was added and OD620 was measured after 10 min. Under these conditions, the absorbance readings of the phosphatase assays with SHP2 E76K or SHP2 PTP as the enzyme were still within the linear calibration range. For the phosphatase assay with pNPP as the substrate, 0.1 μg of SHP2 or its mutants was incubated in 60 µL of the assay buffer as above with 1 at various concentrations at 37 °C for 15 min, followed by addition of 90 µL of pNPP to the final concentration of 0.9 mg/ml. The mixture was incubated at 37 °C for 15 min and OD405 was then measured.
Fluorescence Titrations.
Purified full length WT SHP2 GST-fusion protein was diluted into 20 mM Tris-HCl, pH 7.5. Fluorescence spectra were recorded with a Luminescence Spectrometer LS50 (Perkin-Elmer, Boston, MA, USA). Titrations were performed by increasing the concentration of the compound while maintaining the SHP2 protein concentration at 3.0 μM. Contributions from background fluorescence of the inhibitor were accounted for by subtracting the fluorescence of the inhibitor alone from the protein-inhibitor solution. The excitation wavelength was 295 nm and fluorescence was monitored from 360 to 500 nm. All reported fluorescence intensities were relative values and were not corrected for wavelength variations in detector response.
Thermal Shift Assay.
Thermal shift assays were performed as previously described 41, 42. In brief, in a 20 µl final reaction system, purified His-tagged SHP2 protein (1 µg) in the buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 50 mM imidazole) was mixed with 10 µl 5x Sypro Orange Protein Gel Stain (Life Technologies) (diluted in 100 mM HEPES, pH 7.5). 0.2 µl compound 1 dissolved in DMSO was added to final concentrations of 5, 10, and 60 µM. 1% (V/V) DMSO was used as the control. Samples were heated in a QuantStudio 3 Real-Time PCR System (ThermoFisher Scientific) from 22°C to 95°C in steps of 1°C every 50 sec. Melting curves were analyzed with Protein Thermal Shift Software Version 1.3 (ThermoFisher Scientific) to determine melting temperature (Tm).
MicroScale Thermophoresis (MST) Assay.
Purified His-tagged SHP2 protein (200 nM) in 100 µL buffer (50 mM Tris-HCl, pH 8.0, 10 mM NaCl, and 0.005% Tween 20) was labeled with red-tris-NTA dye using a Monolith NT™ His-tag labeling kit (NanoTemper Technologies) following the protocol provided by the manufacturer. 1 (600 μM) was diluted with PBST (PBS buffer containing 0.005% Tween 20) in 15 2-fold serial dilutions. 10 μL labeled protein was mixed with 10 µL diluted 1 at various concentrations, and the mixtures were loaded to standard capillaries. Binding assays were run with a red filter on a Monolith NT.115 (NanoTemper Technologies). Automated MST measurement was performed with an MST power of 50%. The resulting dose-response curve was fitted to a one-site binding model to extract a Kd value.
Western Blot Analysis.
Ba/F3 cells were starved overnight in serum-free and cytokine-free RPMI1640. Cells were then treated with 1 for 4 hours before stimulation with IL-3 (2 ng/mL). Stimulated cells were harvested and lysed on ice with RIPA buffer containing 50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF and protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Equivalent amounts of protein (50 μg) were resolved on 10% SDS-PAGE and transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). Membranes were blocked with 2% BSA in TBS (20 mM Tris-HCl, pH 7.4 and 150 mM NaCl) for 1 hour at room temperature and probed with primary antibodies overnight at 4°C. Blots were washed with TBST and exposed to HRP conjugated goat-anti-mouse or goat-anti-rabbit secondary antibodies for 1 hour at room temperature. Immunoreactive bands were detected by using ECL Plus Reagents (GE Healthcare, Piscataway, NJ, USA).
Colony-Forming Unit Assay.
Mouse bone marrow cells (2×104 cells/mL) or patient splenocytes or blood cells (5×104 cells/mL) were assayed for colony forming units (CFUs) in 0.9% methylcellulose IMDM containing 30% FBS, glutamine (10−4 M), β-mercaptoethanol (3.3×10−5 M), GM-CSF (1.0 ng/mL), and varying concentrations of 1 or the equivalent amount of DMSO. After 7 days (mouse bone marrow cells) or 14 days (patient cells) of culture at 37°C in a humidified 5% CO2 incubator, myeloid colonies (CFU-GM and CFU-M) were counted under an inverted microscope.
Statistical Analysis.
Data are presented as mean ± SD. Statistical significance was determined using unpaired two-tailed Student’s t test. p<0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health grant HD087760 and a Hyundai Hope on Wheels Scholar grant (to C.K.Q.).
Abbreviations used:
- SHP-2
Src homology 2 domain-containing phosphatase 2
- PTP
Protein tyrosine phosphatase
- SH2
Src homology 2
- pY
Phosphotyrosine
- pNPP
para-Nitrophenylphosphate
- NS
Noonan Syndrome
- JMML
Juvenile myelomonocytic leukemia
- WT
Wildtype
- CFU
Colony forming unit
- CADD
Computer-aided drug design
- MST
MicroScale Thermophoresis
- NMR
Nuclear magnetic resonance
- HPLC
High-performance liquid chromatography
Footnotes
CONFLICT-OF-INTEREST DISCLOSURE
The authors declare no competing financial interests.
ANCILLARY INFORMATION
Supporting Information: Supplementary Figure S1 to Figure S9; Top hits from the initial virtual database screening; PAINS results of the top 100 hits from the initial database screening and the ten derivatives of NSC681129; The complex structure of 1 and SHP1 in the PDB format (PDB file 1); The complex structure of 1 and SHP2 in the PDB format (PDB file 2); Molecular formula strings of 1 (CSV file); Proton NMR data of 1; Proton NMR spectra data of 1; NMR results of other derivatives synthesized; HPLC data of all derivatives synthesized.
REFERENCES
- 1.Neel BG; Gu H; Pao L The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends in biochemical sciences 2003, 28, 284–93. [DOI] [PubMed] [Google Scholar]
- 2.Tonks NK Protein tyrosine phosphatases: from genes, to function, to disease. Nature reviews. Molecular cell biology 2006, 7, 833–46. [DOI] [PubMed] [Google Scholar]
- 3.Xu D; Qu CK Protein tyrosine phosphatases in the JAK/STAT pathway. Frontiers in bioscience : a journal and virtual library 2008, 13, 4925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hof P; Pluskey S; Dhe-Paganon S; Eck MJ; Shoelson SE Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–50. [DOI] [PubMed] [Google Scholar]
- 5.Eck MJ; Pluskey S; Trub T; Harrison SC; Shoelson SE Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature 1996, 379, 277–80. [DOI] [PubMed] [Google Scholar]
- 6.Barford D; Neel BG Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure 1998, 6, 249–54. [DOI] [PubMed] [Google Scholar]
- 7.Loh ML; Vattikuti S; Schubbert S; Reynolds MG; Carlson E; Lieuw KH; Cheng JW; Lee CM; Stokoe D; Bonifas JM; Curtiss NP; Gotlib J; Meshinchi S; Le Beau MM; Emanuel PD; Shannon KM Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–31. [DOI] [PubMed] [Google Scholar]
- 8.Tartaglia M; Martinelli S; Cazzaniga G; Cordeddu V; Iavarone I; Spinelli M; Palmi C; Carta C; Pession A; Arico M; Masera G; Basso G; Sorcini M; Gelb BD; Biondi A Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood 2004, 104, 307–13. [DOI] [PubMed] [Google Scholar]
- 9.Loh ML; Reynolds MG; Vattikuti S; Gerbing RB; Alonzo TA; Carlson E; Cheng JW; Lee CM; Lange BJ; Meshinchi S PTPN11 mutations in pediatric patients with acute myeloid leukemia: results from the Children’s Cancer Group. Leukemia 2004, 18, 1831–4. [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia M; Niemeyer CM; Fragale A; Song X; Buechner J; Jung A; Hahlen K; Hasle H; Licht JD; Gelb BD Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nature genetics 2003, 34, 148–50. [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia M; Mehler EL; Goldberg R; Zampino G; Brunner HG; Kremer H; van der Burgt I; Crosby AH; Ion A; Jeffery S; Kalidas K; Patton MA; Kucherlapati RS; Gelb BD Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nature genetics 2001, 29, 465–8. [DOI] [PubMed] [Google Scholar]
- 12.Bentires-Alj M; Paez JG; David FS; Keilhack H; Halmos B; Naoki K; Maris JM; Richardson A; Bardelli A; Sugarbaker DJ; Richards WG; Du J; Girard L; Minna JD; Loh ML; Fisher DE; Velculescu VE; Vogelstein B; Meyerson M; Sellers WR; Neel BG Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer research 2004, 64, 8816–20. [DOI] [PubMed] [Google Scholar]
- 13.Keilhack H; David FS; McGregor M; Cantley LC; Neel BG Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. The Journal of biological chemistry 2005, 280, 30984–93. [DOI] [PubMed] [Google Scholar]
- 14.Chan G; Kalaitzidis D; Usenko T; Kutok JL; Yang W; Mohi MG; Neel BG Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood 2009, 113, 4414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu D; Liu X; Yu WM; Meyerson HJ; Guo C; Gerson SL; Qu CK Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. The Journal of experimental medicine 2011, 208, 1977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Araki T; Mohi MG; Ismat FA; Bronson RT; Williams IR; Kutok JL; Yang W; Pao LI; Gilliland DG; Epstein JA; Neel BG Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nature medicine 2004, 10, 849–57. [DOI] [PubMed] [Google Scholar]
- 17.Chen L; Sung SS; Yip ML; Lawrence HR; Ren Y; Guida WC; Sebti SM; Lawrence NJ; Wu J Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Molecular pharmacology 2006, 70, 562–70. [DOI] [PubMed] [Google Scholar]
- 18.Geronikaki A; Eleftheriou P; Vicini P; Alam I; Dixit A; Saxena AK 2-Thiazolylimino/heteroarylimino-5-arylidene-4-thiazolidinones as new agents with SHP-2 inhibitory action. Journal of medicinal chemistry 2008, 51, 5221–8. [DOI] [PubMed] [Google Scholar]
- 19.Hellmuth K; Grosskopf S; Lum CT; Wurtele M; Roder N; von Kries JP; Rosario M; Rademann J; Birchmeier W Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 7275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawrence HR; Pireddu R; Chen L; Luo Y; Sung SS; Szymanski AM; Yip ML; Guida WC; Sebti SM; Wu J; Lawrence NJ Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. Journal of medicinal chemistry 2008, 51, 4948–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X; He Y; Liu S; Yu Z; Jiang ZX; Yang Z; Dong Y; Nabinger SC; Wu L; Gunawan AM; Wang L; Chan RJ; Zhang ZY Salicylic acid based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2). Journal of medicinal chemistry 2010, 53, 2482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu W; Yu B; Xu G; Xu WR; Loh ML; Tang LD; Qu CK Identification of cryptotanshinone as an inhibitor of oncogenic protein tyrosine phosphatase SHP2 (PTPN11). Journal of medicinal chemistry 2013, 56, 7212–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chio CM; Lim CS; Bishop AC Targeting a cryptic allosteric site for selective inhibition of the oncogenic protein tyrosine phosphatase Shp2. Biochemistry 2015, 54, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen YN; LaMarche MJ; Chan HM; Fekkes P; Garcia-Fortanet J; Acker MG; Antonakos B; Chen CH; Chen Z; Cooke VG; Dobson JR; Deng Z; Fei F; Firestone B; Fodor M; Fridrich C; Gao H; Grunenfelder D; Hao HX; Jacob J; Ho S; Hsiao K; Kang ZB; Karki R; Kato M; Larrow J; La Bonte LR; Lenoir F; Liu G; Liu S; Majumdar D; Meyer MJ; Palermo M; Perez L; Pu M; Price E; Quinn C; Shakya S; Shultz MD; Slisz J; Venkatesan K; Wang P; Warmuth M; Williams S; Yang G; Yuan J; Zhang JH; Zhu P; Ramsey T; Keen NJ; Sellers WR; Stams T; Fortin PD Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–52. [DOI] [PubMed] [Google Scholar]
- 25.Garcia Fortanet J; Chen CH; Chen YN; Chen Z; Deng Z; Firestone B; Fekkes P; Fodor M; Fortin PD; Fridrich C; Grunenfelder D; Ho S; Kang ZB; Karki R; Kato M; Keen N; LaBonte LR; Larrow J; Lenoir F; Liu G; Liu S; Lombardo F; Majumdar D; Meyer MJ; Palermo M; Perez L; Pu M; Ramsey T; Sellers WR; Shultz MD; Stams T; Towler C; Wang P; Williams SL; Zhang JH; LaMarche MJ Allosteric Inhibition of SHP2: Identification of a potent, selective, and orally efficacious phosphatase inhibitor. Journal of medicinal chemistry 2016, 59, 7773–82. [DOI] [PubMed] [Google Scholar]
- 26.Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. Journal of medicinal chemistry 2010, 53, 2719–40. [DOI] [PubMed] [Google Scholar]
- 27.Emanuel PD; Bates LJ; Castleberry RP; Gualtieri RJ; Zuckerman KS Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood 1991, 77, 925–9. [PubMed] [Google Scholar]
- 28.Birnbaum RA; O’Marcaigh A; Wardak Z; Zhang YY; Dranoff G; Jacks T; Clapp DW; Shannon KM Nf1 and Gmcsf interact in myeloid leukemogenesis. Molecular cell 2000, 5, 189–95. [DOI] [PubMed] [Google Scholar]
- 29.Sun SX; Li XB; Liu WB; Ma Y; Wang RL; Cheng XC; Wang SQ; Liu W Design, synthesis, biological activity and molecular dynamics studies of specific protein tyrosine phosphatase 1B inhibitors over SHP-2. International journal of molecular sciences 2013, 14, 12661–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou KC Low-frequency resonance and cooperativity of hemoglobin. Trends in biochemical sciences 1989, 14, 212–3. [DOI] [PubMed] [Google Scholar]
- 31.Chou KC The biological functions of low-frequency vibrations (phonons). 4. Resonance effects and allosteric transition. Biophysical chemistry 1984, 20, 61–71. [DOI] [PubMed] [Google Scholar]
- 32.Yu WM; Hawley TS; Hawley RG; Qu CK Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene 2003, 22, 5995–6004. [DOI] [PubMed] [Google Scholar]
- 33.Bennett AM; Hausdorff SF; O’Reilly AM; Freeman RM; Neel BG Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Molecular and cellular biology 1996, 16, 1189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of medicinal chemistry 2006, 49, 6177–96. [DOI] [PubMed] [Google Scholar]
- 35.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of computer-aided molecular design 2013, 27, 221–34. [DOI] [PubMed] [Google Scholar]
- 36.Li XB; Wang SQ; Xu WR; Wang RL; Chou KC Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PloS one 2011, 6, e28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shivakumar D; Harder E; Damm W; Friesner RA; Sherman W Improving the prediction of absolute solvation free energies using the next generation OPLS force field. Journal of chemical theory and computation 2012, 8, 2553–8. [DOI] [PubMed] [Google Scholar]
- 38.Shivakumar D; Williams J; Wu Y; Damm W; Shelley J; Sherman W Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. Journal of chemical theory and computation 2010, 6, 1509–19. [DOI] [PubMed] [Google Scholar]
- 39.Oostenbrink C; Soares TA; van der Vegt NF; van Gunsteren WF Validation of the 53A6 GROMOS force field. European biophysics journal : EBJ 2005, 34, 273–84. [DOI] [PubMed] [Google Scholar]
- 40.Schuttelkopf AW; van Aalten DM PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta crystallographica. Section D, Biological crystallography 2004, 60, 1355–63. [DOI] [PubMed] [Google Scholar]
- 41.Pantoliano MW; Petrella EC; Kwasnoski JD; Lobanov VS; Myslik J; Graf E; Carver T; Asel E; Springer BA; Lane P; Salemme FR High-density miniaturized thermal shift assays as a general strategy for drug discovery. Journal of biomolecular screening 2001, 6, 429–40. [DOI] [PubMed] [Google Scholar]
- 42.Lo MC; Aulabaugh A; Jin G; Cowling R; Bard J; Malamas M; Ellestad G Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Analytical biochemistry 2004, 332, 153–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.