

Abstract
The lipid phosphatase gene FIG4 is responsible for Yunis-Varón Syndrome and Charcot-Marie-Tooth Disease Type 4J, a peripheral neuropathy. We now describe four families with FIG4 variants and prominent abnormalities of CNS white matter (leukoencephalopathy), with onset in early childhood, ranging from severe hypomyelination to mild undermyelination, in addition to peripheral neuropathy. Affected individuals inherited biallelic FIG4 variants from heterozygous parents. Cultured fibroblasts exhibit enlarged vacuoles characteristic of FIG4 dysfunction. Two unrelated families segregate the same G>A variant in the +1 position of intron 21, in homozygous state in one family and compound heterozygous in the other. This mutation in the splice donor site of exon 21 results in read-through from exon 20 into intron 20 and truncation of the final 115 C-terminal amino acids of FIG4, with retention of partial function. The observed CNS white matter disorder in these families is consistent with myelination defects in the Fig4 null mouse (Chow et asl, 2007) and the known role of FIG4 in oligodendrocyte maturation (Mironova et al, 2016, 2018). The families described here expand the clinical spectrum of FIG4 deficiency to include leukoencephalopathy.
Keywords: VAC14, PIKFYVE, neurodegeneration, endolysosome, leukodystrophy, splice-site, CMT4J, oligodendrocyte, vacuolization, dysmyelination
INTRODUCTION
Neurological effects of FIG4 mutations were first described in a mouse mutant with a loss-of-function mutation that resulted in neuronal degeneration, dysmyelination in the CNS and PNS, and juvenile lethality (Chow et al 2007; Winters et al 2011; Mironova et al, 2016). The corresponding human disorder with homozygous loss-of-function of FIG4 is the Yunis-Varón Syndrome, a multi-system disorder with severe neurological and skeletal defects, CNS dysmyelination and juvenile lethality (Campeau et al, 2014). Partial loss-of-function of FIG4 can result in Charcot-Marie-Tooth disease (CMT) type 4J, an autosomal recessive peripheral neuropathy with myelin defects restricted to the PNS (Chow et al, 2007; Nicholson et al, 2011). In one consanguineous family, a homozygous missense variant of FIG4 results in polymicrogyria and psychiatric features, but no CNS demyelination (Baulac et al, 2014). Thus, to date, impaired CNS myelination has been associated with complete loss-of-function of FIG4 in Yunis-Varón Syndrome, but not with partial loss-of-function due to missense variants.
The FIG4 protein is a subunit of the PI(3,5)P2 biosynthetic complex that also contains PIKFYVE, a PI3P kinase, and VAC14, a scaffold protein. Recessive variants of VAC14 in human and mouse mimic the clinical and cellular defects of FIG4 deficiency (Jin et al, 2008; Lenk et al 2016,b; Stutterd et al, 2018). Variants that reduce VAC14 abundance also destabilize the FIG4 protein (Lenk et al, 2011; Zolov et al, 2012). The product of the biosynthetic complex, PI(3,5)P2, is a low abundance signaling lipid that activates ion channels in the lysosomal membrane, including TRPML1, TPC1 and TPC2 (Dong et al, 2010; Wang et al, 2012; She et al, 2018; Kirsch et al, 2018; Wilson et al, 2018). At the cellular level, low levels of PI(3,5)P2 lead to accumulation of large, acidic lysosome-derived vesicles (Chow 2007, Ferguson 2009, 2012). These enlarged vesicles appear to result from osmotic swelling of lysosomes secondary to deficient activation of ion channels in the lysosome membrane (Lenk and Meisler, 2014). FIG4 deficiency may thus be considered one of the lysosomal disorders affecting lysosomal membrane function rather than enzymatic degradation of macromolecules (McDonald and Krainc, 2017).
A direct role for FIG4 in CNS myelination is indicated by defective oligodendrocyte maturation and myelin biosynthesis in Fig4 null mice (Mironova et al, 2016). In addition, targeted inactivation of Fig4 in Schwann cells causes peripheral nerve demyelination, demonstrating a role in the PNS (Vaccari et al, 2015).
In this report we describe patients with rare variants of FIG4 in whom abnormalities of CNS white matter is a predominant feature. In view of these observations, we suggest that FIG4 should be considered a candidate gene in individuals presenting with primary defects in CNS white matter.
METHODS
DNA sequencing.
Families 1 and 2 were consented for clinical exome sequencing as part of their formal diagnostic work-up at Leeds Teaching Hospitals NHS Trust. Sequencing was performed using the Agilent SureSelectXT Focused Exome reagent (Agilent Technologies, Wokingham, UK) and sequenced on an Illumina HiSeq 2500 rapid mode flow cell (Illumina Inc., San Diego, CA, USA) with 2 × 101nt paired-end reads for family 1, and an Illumina NextSeq 500 with 2 × 151nt paired-end reads for family 2. A custom bioinformatics pipeline was applied, comprising Cutadapt v.1.9.1 for adaptor and quality trimming, BWA-mem for read alignment, GATK UnifiedGenotyper for variant calling, Alamut batch v.1.4.0 for variant annotation, and the GATK walkers DepthOfCoverage, CallableLoci and CountReads for generating coverage data. Analysis was restricted initially to a list of 98 genes known to be associated with white matter conditions, and then expanded to a manually-curated list of 2,060 genes associated with or candidates for human developmental and neuromuscular diseases. Identified FIG4 variants were also analyzed by bidirectional Sanger sequencing in probands and parents.
Family 3 consented to participate in a gene discovery project approved by the Royal Children’s Hospital Human Research Ethics Committee (HREC number 28097). Whole genome sequencing (WGS) was performed for both affected individuals and both parents using 2 × 150nt paired end reads on an Illumina X (Illumina Cambridge Ltd, Little Chesterford UK). Read alignment was performed using BWA-mem; variant calling of the nuclear genome was performed using GATK HaplotypeCaller v3.7, BCFtools was used to call mtDNA variants. (Li et al., 2009, McKenna et al., 2010) SnpEff v4.3m was used for variant annotation and a custom script was utilized for variant filtration and prioritization. (Cingolani et al., 2012). No additional relevant variants were observed in Family 3.
Family 4 underwent exome sequencing at the Center for Mendelian Genomics at the Broad Institute after written informed consent to participate in the Murdoch Children’s Research Institute Undiagnosed Diseases Project (RCH HREC 36291A).
Cell culture.
Patient cells were grown in RPMI 1640 medium supplemented with 15% FBS and 1% Antibiotic-Antimycotic solution (GIBCO) on vacuum gas plasma treated dishes (Corning) at 37°C with 5% CO2 supplementation. Vacuolization was assessed as previously described (Lenk et al, 2016). Briefly, cells were plated at 20,000 cells per cm2 for18 hours prior to assessment of vacuolation. Cells were imaged without selection and scored for vacuolation as described.
RESULTS
Clinical features of affected children in four families with FIG4 variants.
Family 1:
Patient 1 is currently four years of age. He presented at six months with developmental delay and hypotonia. His initially rapid head growth stabilized at the 75th centile by the age of two. He began sitting without support at 12 months and has made slow developmental progress, but at the age of four years is not walking without support. He has generalized hypotonia with absent deep tendon reflexes. His brain MRIs showed complete lack of cerebral myelination at 10 months and 27 months of age, as well as ventriculomegaly without CSF flow obstruction. (Figure 1A - D). His progressive peripheral neuropathy is similar to patients with CMT4J, but the CNS deficits are distinct.
Figure 1: Evidence of CNS myelin defects in affected individuals carrying FIG4 variants. MRI images.
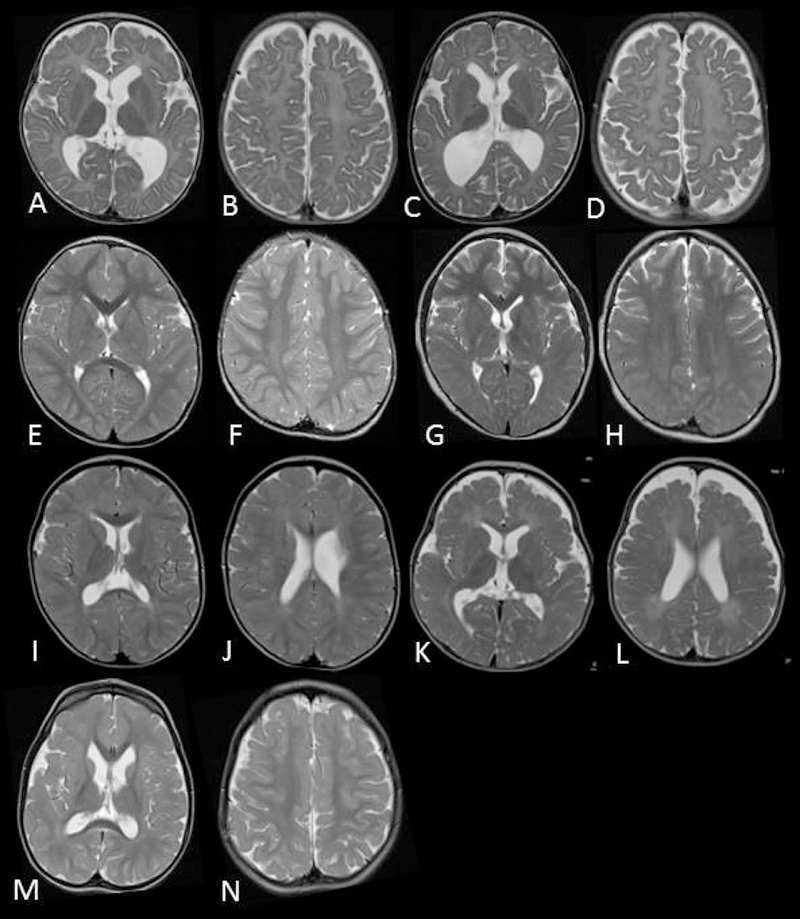
Axial T2 weighted MR images of patient 1(family 1) at 10 months (A,B) and 27 months (C,D) demonstrating complete absence of myelination in cerebral white matter and internal capsule with no improvement on follow up. Patient 2 (family 2) at 30 months (E,F) and 8 years (G,H) demonstrates high signal in the posterior limb of the internal capsule and diffuse high signal in the posterior periventricular and deep cerebral white matter. Family 3, sibling 1 at 25 months (I,J) demonstrates mild high signal in cerebral white matter with myelination present in deep and subcortical white matter. Family 3, sibling 2 at 18 months (K,L) demonstrates more striking hypomyelination with very little normal myelin visible. The MRI of patient 5 in family 4 at 3 years of age was largely normal with non-specific high signal in the periventricular and deep parietal white matter (M,N).
Family 2:
Patient 2 is the first child of consanguineous South Asian parents. He was born at term and had initial feeding difficulties and severe motor delay with hypotonia and weakness. At 11 years of age, he is nonambulant and has profound generalized weakness. He is nonverbal but able to indicate his needs and identify favorite objects. After a chest infection he required a tracheostomy and is fed by gastrostomy. Vision and hearing are normal. Nerve conduction studies demonstrate a severe motor and sensory demyelinating neuropathy. None of the skeletal features seen in Yunis-Varón Syndrome were observed. Brain MRIs demonstrate abnormal signal in the internal capsule and cerebral white matter which remained stable between 30 months and eight years of age. There is mild progressive volume loss of the cerebellar hemispheres (Figure 1E - H).
Family 3:
The two affected siblings in Family 3 are currently aged eleven years (Patient 3) and four years (Patient 4) and experienced onset of symptoms at 9 months and 12 months, respectively. Patient 3 presented with profound hypotonia, developmental delay and areflexia, with disease progression that includes scoliosis. Patient 4 presented with a mild degree of gross motor and developmental delay, depressed tendon reflexes and relative macrocephaly, and later developed strabismus. Both children have cognitive impairment; Patient 3 has low to borderline IQ and Patient 4 is nonverbal at four years of age. Nerve conduction studies in both siblings demonstrated lack of sensory responses and mild slowing of motor responses consistent with mild to moderate demyelinating peripheral neuropathy. Brain MRIs of both children demonstrate T2 hyperintensity in the cerebral white matter consistent with hypomyelination (Figure I - L). Patient 3 exhibited T2 hyperintensities without enhancement in the spinal cord and abnormal enhancement of cervical root and cauda equine suggestive of polyneuropathy (not shown). Patient 4 did not have spinal cord involvement.
X-rays demonstrate severe scoliosis and thin shafts of the tubular bones with over tubulation (Patient 3), delayed bone age (Patient 4) and thin metacarpals, coxa valga and dolicocephaly in both siblings (Supp. Figure S1).
Family 4:
Three affected siblings born to unrelated parents in Family 4 (Patients 5, 6 and 7) have a novel phenotype of severe global developmental delay, autistic features and maculopathy. The proband (Patient 5) was first noted to have plagiocephaly, delayed milestones, abnormal tone and choreoathetoid movements in infancy. The involuntary movements improved spontaneously by age 22 months. He also experienced gastroesophageal reflux with frequent vomiting that improved by 15 months of age with medical therapy and thickened feeds. At 11 years, he has limited communication and is incontinent. He has had no developmental regression. His brain imaging at three years of age demonstrated mild T2 hyperintensity of the periventricular white matter and mild ventricular dilatation (Figure 1 M,N). Follow up imaging demonstrated persistently delayed myelination, mild-moderate ventricular dilatation, reduction in white matter bulk, and mildly increased T2 and FLAIR signal within the deep white matter around the posterior bodies of the lateral ventricles. His male and female siblings exhibited a similar clinical course with delayed milestones and autistic features but without abnormal movements.
The ocular findings in each affected child in Family 4 were initially described as bull’s eye maculopathy (Supp. Figure S2A,B). Preferential Cardiff acuity testing suggested vision of 6/12 equivalent. There was normal ocular movement, healthy ocular media and no strabismus. The more severely affected sib (Patient 6) also has optic atrophy, retinal atrophy and vascular attenuation (Supp. Figure S2C,D). An optical coherence tomogram under anesthetic identified outer retinal atrophy, loss of foveal architecture and generalized retinal thinning including loss of the retinal nerve fibre layer with temporal optic disc pallor (Supp. Figure 2E,F).
The father was clinically unaffected, but the mother was diagnosed with multiple sclerosis at the age of 39, based on clinical presentation and MRI findings. She presented with progressive dizziness and ataxia of several weeks’ duration. Review of her brain MRI demonstrated classical features of multiple sclerosis including widespread supra- and infratentorial demyelination, meeting the diagnostic criteria (Polman et al, 2011). Spinal cord imaging was normal. One episode of left arm numbness lasting for weeks occurred seven years prior to presentation, and there was fatigue during the last few years. Examination demonstrated truncal and right limb ataxia and subtle pyramidal deficit but no cognitive deficits. Ataxia improved after treatment with intravenous methylpredisolone. She was commenced on disease modulating therapy with fingolimod soon after the diagnosis of multiple sclerosis and remains clinically stable.
Identification and inheritance of FIG4 mutations.
Family 1:
The affected individual is a compound heterozygote for the nonsense variant p.Trp246Ter (NM_014845.5: c.737G>C) (allele 1) and a variant in the consensus +1 G nucleotide of the donor splice site of exon 21 (c. 2459+1G>A) (allele 2) (Figure 2A). Genotyping of the parents demonstrated that both variants were inherited (Figure 2A). In the gnomAD database (Lek et al, 2016), allele 1 (rs776005417) is present in 10 heterozygotes and no homozygotes (allele frequency = 0.00005) and allele 2, the splice site mutation (rs747768373), is present in 4 heterozygotes and no homozygotes (allele frequency = 0.000014).
Figure 2: Recessive inheritance of FIG4 variants in four families.
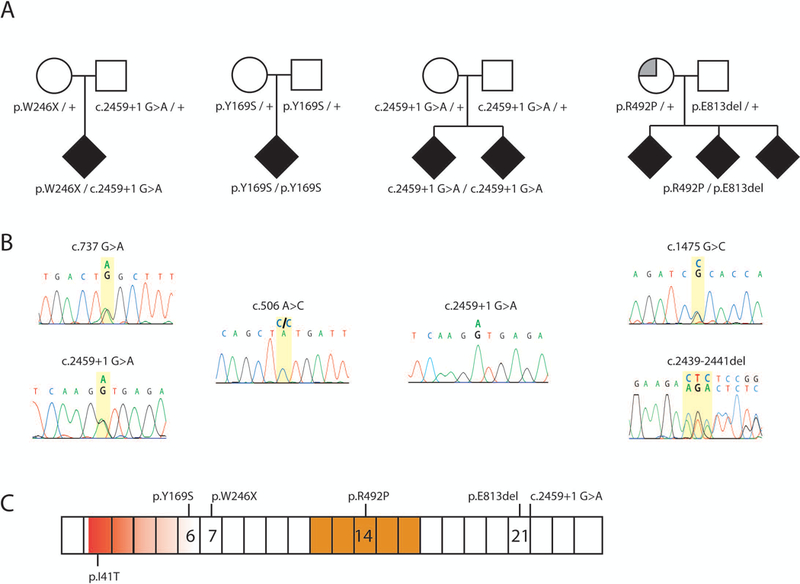
(A) Affected individuals (solid symbols) are compound heterozygotes for inherited variants of FIG4. Carrier parents were unaffected (open symbols) except for the mother in Family 4 who was diagnosed with multiple sclerosis (partially filled symbol). (B) Genomic DNA sequences from affected individuals demonstrate two inherited mutant alleles. (C) Locations of variants on the FIG4 protein. Exon borders are marked. Red, VAC14 protein interaction domain; gold, phosphatase catalytic active site. FIG4 cDNA, NM_014845.5
Family 2:
The affected individual is homozygous for the missense variant p.Tyr169Ser (c.506A>C). He is the first child of consanguineous South Asian parents. This ultra-rare missense variant is located in an evolutionarily conserved region of the protein and within 6 residues of the Yunis-Varón null mutation p.Leu175Pro (Campeau et al, 2014; Figure 2). This variant is present in three heterozygotes of South Asian origin in the gnomAD database and no homozygotes (allele frequency of 0.00001). Another substitution of this amino acid residue, p.Tyr169Cys, is found in in gnomAD in seven heterozyogtes and no homozygotes.
Family 3.
Both affected individuals in Family 3 are homozygous for the exon 21+1 splice site mutation c.2459+1G>A (rs747768373) described above in Family 1. Both parents in Family 3 are of European non-Finnish origin. The four individuals in the gnomAD database who carry this variant are from European (non-Finnish) and African populations.
Family 4.
The three affected siblings in Family 4 are compound heterozygotes for a maternally inherited in-frame deletion (c.2439_2441del; p.Glu813del) and a paternally inherited missense variant (c.1475G>C; p.Arg492Pro). The deletion of glutamate residue 813 changes the surrounding protein sequence from SerGluGluAsp to SerGluAsp. This deletion was observed in three out of 246,074 alleles in gnomAD but not in homozygous state (allele frequency 0.000012 (Lek et al., 2016). The paternal allele is the novel missense variant p.Arg492Pro. Arginine 492 is an invariant residue located within the C(X5)RT motif of the phosphatase active site. Substitution of this residue results in loss of phosphatase activity (Guan and Dixon 1991; Liu and Bankaitis, 2010) and p.Arg492Pro is absent from the gnomAD database. Another substitution of this residue, p.Arg492Cys, is present in gnomAD in heterozygous state in two out of 246,238 alleles but not in homozygous state.
Functional analysis of FIG4 variants in patient fibroblasts and transfected cells.
Cultured fibroblasts were obtained from affected individuals carrying the FIG4 variants. Fibroblast RNA was analyzed to determine the effects of splice site and nonsense variants on transcript processing. Live cell microscopy of fibroblast cultures was carried out to detect enlarged vacuoles characteristic of fibroblasts with deleterious variants of FIG4. These acidic vesicles are of lysosomal origin and LAMP1 and LAMP2 localize to their membranes (Lenk and Meisler, 2014).
Family 1:
The transcript of allele 1 with a stop codon in exon 7 is a predicted substrate for nonsense-mediated decay. To evaluate the stability of the allele 1 transcript, we compared the sequence of exon 7 in genomic DNA and fibroblast RNA. Heterozygosity for the stop codon was detected by Sanger sequencing of the genomic PCR product, but only the wildtype sequence was present in the RT-PCR product (Figure 3). This result indicates that the transcript is subject to nonsense-mediated decay. The truncated protein that terminates at residue 246 is unlikely to be produced at significant levels, making this a null allele.
Figure 3: Nonsense-mediated decay of the allele 1 transcript in Family 1.
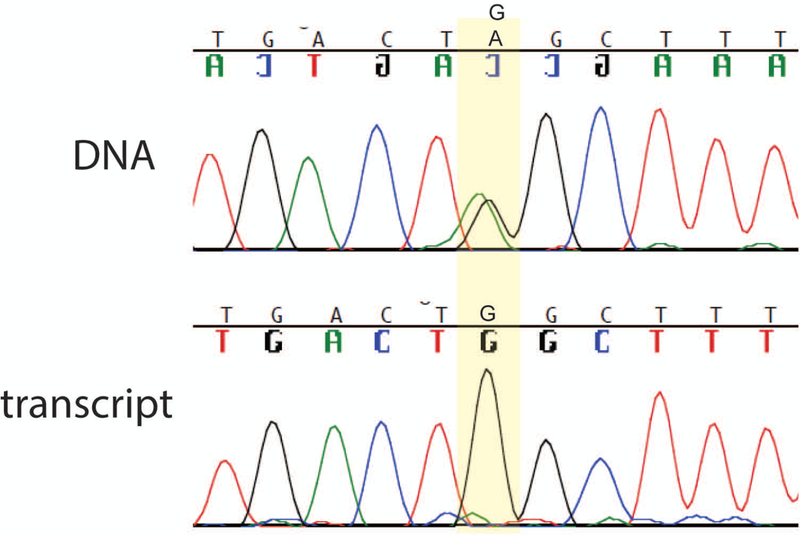
The affected individual is heterozygous for variant W246X in exon 7. The sequence of the wildtype tryptophan codon TGG and the stop codon TAG are both evident in the PCR product from genomic DNA (top). In contrast, the product amplified by RT-PCR of fibroblast RNA contains the tryptophan codon TGG, but the stop codon is not detectable. The data demonstrates the instability of the stop-codon containing transcript, which is a predicted substrate for nonsense-mediated decay.
Allele 2 in Family 1 contains a G>A substitution at the +1 position of intron 21 that is predicted to inactivate the splice donor site for exon 21 (Figure 4A). Loss of the +1 G nucleotide can lead either to skipping of the affected exon or to other alternative splicing. RT-PCR with a forward primer in exon 20 and a reverse primer in exon 22 did not detect transcripts that skip exon 21. To identify the aberrant transcript, we therefore carried out 3’ RACE with a reverse oligo dT primer and a forward primer in exon 20. Two products were obtained from heterozygous RNA, the predicted wildtype product containing exons 20, 21, 22 and 23, and a shorter unique product of 325 bp (Figure 4B, arrow). Sequencing the gel-purified fragments demonstrated correct splicing from exon 21 to 22 in the wildtype product, but read-through from exon 20 into intron 20 in the mutant 3’ RACE product (Figure 4C). The mutant product terminates at a cryptic polyadenylation signal beginning at position +176 in intron 20 (Figure 4 D). The mutant transcript encodes a predicted protein of 816 residues that terminates with 24 amino acids encoded by intron 20 and lacks the 115 C-terminal residues of the wildtype protein.
Figure 4. Retention of intron 20 in the allele 2 transcript in Family 1.
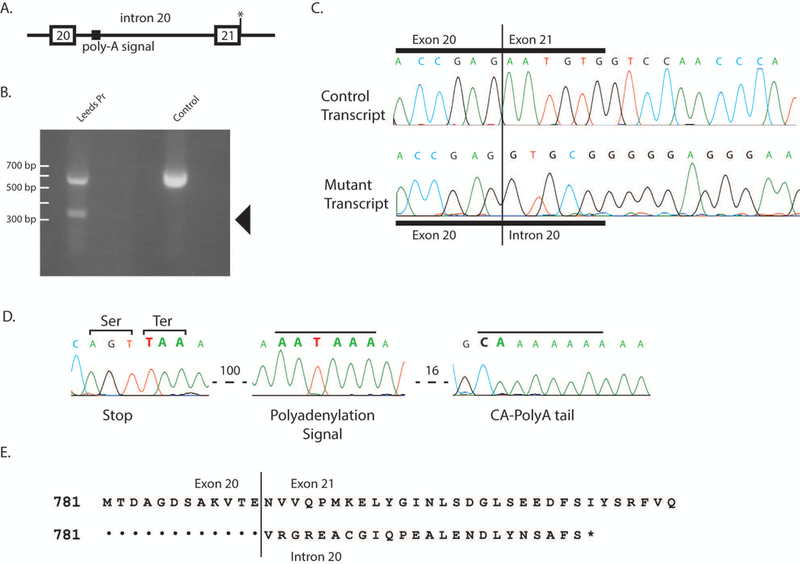
(A) location of splice site mutation at exon 21+1. (B) Aberrant, short 3’ RACE product obtained from fibroblast RNA isolated from the affected individual in Family 1 (arrow). (C) Sanger sequence of purified 3’ RACE products demonstrates correct splicing from exon 20 to exon 21 in one transcript and readthrough from exon 20 into intron 20 in the other transcript. (D) Additional sequence of the aberrant, read-through 3’ RACE product demonstrates the polyadenylation signal in intron 20, 100 bp downstream from exon 20. (E) Amino acid sequence of the truncated protein encoded by the read-through transcript.
Eighty percent of cultured patient fibroblasts contained enlarged vacuoles (Figure 5A, B), demonstrating that the truncated protein encoded by allele 2 is not fully functional. The clinical features of the patient are much less severe than patients with Yunis-Varón Syndrome, suggesting that the mutant protein does retain partial function. Patient 1 is thus heterozygous for one loss-of-function allele and one partial-loss-of-function allele.
Figure 5. Patient fibroblasts from Family 1 and Family 2 exhibit the characteristic vacuolization caused by deleterious variants of FIG4.
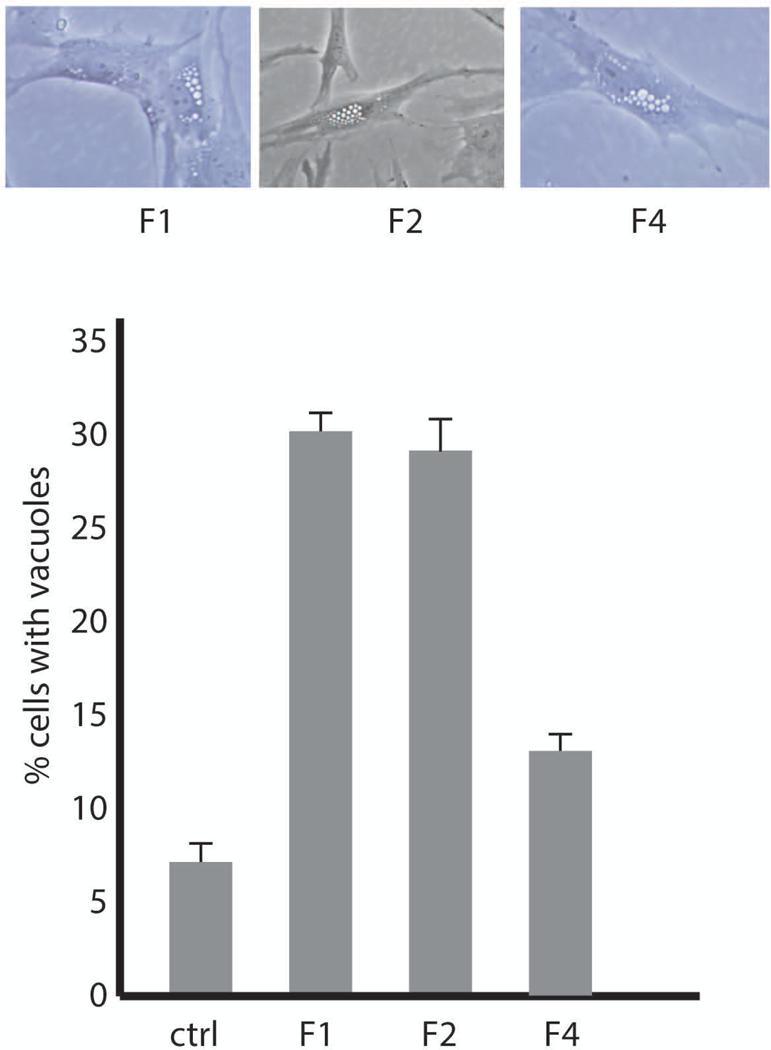
A) Live cell microscopy of cultured fibroblasts from affected individuals. B) Quantitation of the extent of vacuolization. Values represent means ± SD.
Family 2:
The affected individual is homozygous for the missense mutation p.Tyr169Ser. Tyr169 is located in a conserved region of the protein whose specific function is not known. Examination of cultured fibroblasts revealed the characteristic enlarged vacuoles described above for patient 1, with a similar frequency of vacuolated cells (Figure 5A, B). p.Tyr169Ser thus appears to be a partial-loss-of-function allele.
Family 3.
The affected siblings in Family 3 are homozygous for the exon 21 +1 mutation characterized in Family 1 (Figure 3). The relatively mild phenotype of these children, compared to FIG4 null individuals with Yunis-Varón Syndrome is consistent with the evidence above that the C-terminal truncated protein retains partial activity.
Family 4.
The three affected siblings in this family are compound heterozygotes for the amino acid deletion p.Glu813del (allele 1) and the missense mutation p.Arg492Pro at the CX5RT motif of the phosphatase active site. This invariant arginine residue stabilizes the dephosphorylation transition state and mutation of this residue results in loss of enzymatic activity in related enzymes (Guan and Dixon, 1991; Liu and Bankaitis, 2010). To determine whether allele 1 is deleterious, we examined cultured fibroblasts by light microscopy. The presence of enlarged vacuoles was evident (Figure 5). The fact that the affected individuals do not have Yunis-Varón syndrome together with the vacuolar phenotype suggests that allele 1 is a partial loss-of-function allele (Figure 5).
DISCUSSION
The leukodystrophies are genetically determined disorders primarily affecting central nervous system white matter, irrespective of the structural white matter component involved, the molecular process affected, or the disease course (Kevelam et al 2016). A classification of leukodystrophies grouping disorders according to cellular pathology has been proposed (van der Knaap & Bugiani 2017). In this classification FIG4 leukodystrophy would be considered a myelin disorder, since FIG4 has a direct role in oligodendrocyte maturation and Schwann cell myelination, in addition to its functions in neurons and other cells (Mironova et al 2016, Vaccari et al, 2015).
The clinical consequences of partial loss-of-function variants of FIG4 are highly heterogeneous, ranging from peripheral neuropathy in CMT4J to polymicrogyria with psychiatric components (Nicholson et al, 2011; Baulac et al, 2014). Comparison of the effects of partial loss-of-function variants on enzymatic activity might help to clarify the distinction between the variants described in the present study and the variants seen in CMT4J, such as p.Ile41Thr (Nicholson et al, 2011). CNS hypomyelination is not a recognized feature of CMT4J.
The identification of four unrelated families with white matter defects and biallelic deleterious variants of Fig4 confirms the important role of FIG4 in myelinating glia. Our findings underline the importance of considering FIG4 as a candidate gene in genomic studies of patients with myelination defects. Because of the variable phenotypes presented (skeletal features in one of the four families, macular features in one family, and variable degrees of CNS myelination across the cohort), phenotypic prioritization of FIG4 in specific cases may be difficult, although peripheral neuropathy appears to be a common feature.
The exon 21 splice site variant was identified in heterozygous state opposite a null allele in Family 1 and in homozygous state in Family 3. The read-through transcript encodes a truncated protein containing 792 residues of the full length protein (907 amino acids) plus 24 residues encoded by intron 20. The truncated protein retains the protein interaction domain (Manford et al, 2010), the catalytic active site (Guan and Dixon, 1991; Liu and Bankaitis, 2010) and has partial function. The missing 115 residues are poorly conserved through evolution and are absent from yeast Fig4p. In addition to the two families described here, this variant was identified in a patient with Charcot-Marie-Tooth disease; the other allele in this patient was not described (DiVincenzo et al, 2014).
The active-site mutation p.Arg492Pro in Family 4 was inherited in trans with a single amino acid deletion. The three affected siblings have a severe but non-lethal developmental disorder, indicating that one or both alleles retain partial function. We previously studied the active site mutation p.Cys486Ser and found that expression of this mutant as a transgene extended the survival of Fig4 null mice (Lenk et al, 2016). These two active-site mutants indicate that the FIG4 protein also has a non-enzymatic function such as stabilization of the PI(3,5)P2 biosynthetic complex in vivo. The presence of maculopathy in all three siblings suggests that this is part of their syndrome, but further work will be required to exclude another cause. There has been one report of a macular phenotype in Yunis-Varon Syndrome (Corona-Rivera et al, 2011), suggesting that children with FIG4-associated CNS myelination defects should undergo ocular examinations.
The affected individuals in Family 3 have skeletal abnormalities that overlap with Yunis-Varón syndrome. Patient 3 has progressive scoliosis requiring surgical management. Systemic x-rays demonstrate thin shafts in the tubular bones and over tubulation, a feature also seen in Yunis-Varón syndrome. His younger brother had no clinical evidence of skeletal disease but his x-rays demonstrate delayed bone age. Both siblings also exhibit thin metacarpals, coxa valga and dolicocephaly. These subtle skeletal abnormalities support the view that hypomyelination and Yunis-Varón syndrome are representative of a spectrum of FIG4 deficiency disease.
The role of FIG4 in oligodendrocyte maturation was previously studied in Fig4 null mice. Primary oligodendrocytes in culture develop enlarged LAMP1-positive vesicles that accumulate myelin-associated glycoprotein (MAG) and fail to migrate to the nascent myelin sheet, demonstrating dependence of myelin biosynthesis on FIG4 function (Mironova et al, 2016). In addition, induced knockout of Fig4 in the adult mouse prevents repair of a chemically-induced white matter lesion (Mironova et al, 2017). These findings provide a cellular mechanism to explain the effects of pathogenic variants of Fig4 on CNS white matter. The observed heterozygosity for a deleterious allele in an individual with multiple sclerosis reported here may be an incidental finding, but the role of FIG4 in myelination suggests that follow-up studies would be worthwhile.
In summary, we identified four families with novel FIG4 genotypes and CNS white matter disease varying from severe hypomyelination to mild undermyelination, in addition to peripheral neuropathy. Impaired FIG4 function was revealed by the presence of large vacuoles in cultured patient fibroblasts. Abnormal mRNA splicing and nonsense-mediated decay were characterized in two families. Inheritance of unique combinations of variants with partial loss-of-function of FIG4 results in disease severity that lies between the lethal Yunis-Varón syndrome and the milder CMT4J. The clinical management of patients with pathogenic variants in FIG4 should include assessment of peripheral neuropathy, CNS hypomyelination and skeletal disease. The prominent CNS white matter defects in three of these four families indicate that FIG4 is should be considered a candidate gene in individuals presenting with leukoencephalopathy.
Supplementary Material
Table 1.
Clinical description of affected individuals.
| Family ID |
Family 1 | Family 2 | Family 3 | Family 4 | |||
|---|---|---|---|---|---|---|---|
| Patient ID |
Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 |
| Referrer | Vadlamani/Livingston | Vadlamani/Livingston | Stutterd | Tan | |||
| Sex | male | male | male | male | male | male | female |
| Current age | 4 years | 11 years | 11 years | 4 years | 11 years | 8 years | 6 years |
| Family history/ consanguinity/ ethnicity | No FH/ non consang/ white british | Anglo-celtic | Anglo-celtic | ||||
| Pregnancy complication | No | No | MRI evidence of prenatal ischaemic cerebral injury | No | No | No | No |
| Birth complication | No | No | birth asphyxia | No | Previous first trimester miscarriages | No | No |
| Age of disease onset | 5 months | Birth | < 9 months | 12 months | Neonatal period | Infancy | Infancy |
| Presenting symptom | delayed motor development | Feeding difficulties and evelopmental delay | Hypotonia, developmental delay, areflexia | developmental delay | Feeding difficulties, global developmental delay, choreoathetoid movements and bullseye maculopathy | Global developmental delay and bullseye maculopathy | Global developmental delay and bullseye maculopathy |
| Progressive disease | No | Yes | Yes | No | No | No | No |
| GMFCS/motor development at last review | GMFCS III | GMFCS V | GMFCS III | GMFCS II | GMFCS II | GMFCS II | GMFCS II |
| Movement disorder | No | No | cerebellar ataxia | mild tremor | Choreoathetoid movements | No | No |
| Other neurological complication eg. Seizure | No | No | No | No | No | No | No |
| Cognitive impairment | Few words, impaired understanding | Yes, moderate cognitive impairment | low-borderline IQ | No speech at 4y | Limited communication | Limited communication | Limited communication |
| Vision impairment | No | No | Myopia, strabismus | Myopia, strabismus | No, but maculopathy | No, but maculopathy | No, but maculopathy |
| Other health problem or congenital disorder | No | tracheostomy and gastrostomy | No | Feeding issues and vomiting in infancy | No | Explosive bowel movements streaked with blood in infancy; no pathology identified; self-resolved by 10 months | |
| Nerve conduction study results | Not done | prolonged latencies, very slow condusction velocity, low CMAP | Mild-moderate demyelinating neuropathy | Mild-moderate demyelinating neuropathy; median n:22.8 m/s, tibial n:24.3 m/s | Not done | Not done | Not done |
| Age of MRIs (brain +/− spine) | 10 months and 27 months | 30 months and 8 years. | 12 months and 4 years | 15 months | 13 months, then 3 years | N/A | N/A |
| Pattern of white matter abnormality on MRI | diffuse hypomyelination | T2 hyperintensity in deep cerebral white matter and posterior limb of internal capsule. Mild cerebellar atrophy | Periventricular T2 hypomyelination. Nerve roots enhancement suggestive of polyneuropathy. | Periventricular hypomyelination. | Ventricular dilatation detected at 13 months; reduction in white matter myelination at 3 years | N/A | N/A |
| Skeletal abnormality (clinically or radiologically) | No | No | Radiological finding: thin shafts of the tubular bones with over tubulation, thin metacarpals, coxa valga, dolicocephaly | Radiological findings: delayed bone age, thin metacarpals, coxa valga, dolicocephaly | No skeletal anomaly clinically | No skeletal anomaly clinically | No skeletal anomaly clinically |
| Chr. position (Allele 1) | Chr6(GRCh37):g.110059618G>A | Chr6(GRCh37):g.110056361A>C | Chr6(GRCh37):g.110113868G>A | Chr6(GRCh37):g.110113868G>A | Chr6(GRCh37):g.110086256G>C | Chr6(GRCh37):g.110086256G>C | Chr6(GRCh37):g.110086256G>C |
| Chr. position (Allele 2) | Chr6(GRCh37):g.110113868G>A | (homozygous) | (homozygous) | (homozygous) | Chr6(GRCh37):g.110113847_110113849del | Chr6(GRCh37):g.110113847_110113849del | Chr6(GRCh37):g.110113847_110113849del |
| cDNA variant (Allele 1) | NM_014845.5: c.737G>A | NM_014845.5(FIG4):c.506A>C p.(Tyr169Ser) | 2459+1G>A homozygous | 2459+1G>A homozygous | NM_014845.5:c.1475G>C | NM_014845.5:c.1475G>C | NM_014845.5:c.1475G>C |
| cDNA variant (Allele 2) | NM_014845.5:c.2459+1G>A | (homozygous) | (homozygous) | (homozygous) | NM_014845.5:c.2439_2441del | NM_014845.5:c.2439_2441del | NM_014845.5:c.2439_2441del |
| Protein change (Allele 1) | p.Trp246Ter | p.Tyr169Ser | N/A | N/A | p.Arg492Pro | p.Arg492Pro | p.Arg492Pro |
| Protein change (Allele 2) | N/A | (homozygous) | (homozygous) | (homozygous) | p.Glu813del | p.Glu813del | p.Glu813del |
Acknowledgements:
We thank the patients and their families for participating in this study. We thank James Tellez, Northern Genetics Service, Institute of Genetic Medicine, Centre for Life, Newcastle upon Tyne, for RNA analysis of Family 1 that was not included in this paper. The authors declare that they have no conflict of interests in regard to this study.
Funding to MHM and GML was provided by NIGMS R01 GM24872. Sequencing of Family 4 was carried out by the Center for Mendelian Genomics at the Broad Institute of MIT and Harvard and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm. CAS was supported by NHMRC Postgraduate Scholarship (GNT1133266) and the Royal Children’s Hospital/Murdoch Childrens Research Institute Flora Suttie Neurogenetics Fellowship made possible by the Thyne-Reid Foundation and the Macquarie Foundation. Additional funding was provided by the NHMRC Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Program.
REFERENCES
- Baulac S, Lenk GM, Dufresnois B, Ouled Amar Bencheikh B, Couarch P, Renard J, Larson PA, Ferguson CJ, Noe E, Poirier K, Hubans C, Ferreira S, Guerrini R, Ouazzani R, El Hachimi KH, Meisler MH, & Leguern E, (2014). Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology, 82, 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau PM, Lenk GM, Lu JT, Bae Y, Burrage L, Turnpenny P, Roman Corona-Rivera J, Morandi L, Mora M, Reutter H, Vulto-van Silfhout AT, Faivre L, Haan E, Gibbs RA, Meisler MH, & Lee BH, (2013). Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. American journal of human genetics, 92, 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, & Meisler MH (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature, 448, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona-Rivera JR, Romo-Huerta CO, López-Marure E, Ramos FJ, Estrada-Padilla SA, Zepeda-Romero LC. (2011) New ocular findings in two sisters with Yunis-Varón syndrome and literature review. Eur J Med Genet. 54:76–81. [DOI] [PubMed] [Google Scholar]
- DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, Braastad CD, Bishop CM, Jaremko M, Wang Z, Liaquat K, Hoffman CA, York MD, Batish SD, Lupski JR, Higgins JJ (2014). The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. 2:522–529, Suppl Table 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, and Xu H(2010). controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun, 13, 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson CJ, Lenk GM, & Meisler MH, (2009). Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Human molecular genetics, 18, 4868–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson CJ, Lenk GM, Jones JM, et al. (2012). Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Human molecular genetics, 21, 3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anals of Biochemistry. 1991 [DOI] [PubMed] [Google Scholar]
- Jin N, Chow CY, Liu L, Zolov SN, Bronson R, Davisson M, Petersen JL, Zhang Y, Park S, Duex JE, Goldowitz D, Meisler MH, & Weisman LS, (2008). VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. The EMBO journal, 27, 3221–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevelam SH, Steenweg ME, Srivastava S, Helman G, Naidu S, Schiffmann R, Blaser S, Vanderver A, Wolf NI, van der Knaap MS. (2016). Update on Leukodystrophies: A Historical Perspective and Adapted Definition. Neuropediatrics, 47(6), 349–354 [DOI] [PubMed] [Google Scholar]
- Kirsch SA, Kugemann A, Carpaneto A, Böckmann RA, Dietrich P. (2018) Phosphatidylinositol-3,5-bisphosphate lipid-binding-induced activation of the human two-pore channel 2. Cell Mol Life Sci. 2018 April 28. doi: 10.1007/s00018-018-2829-5. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek et al, 2016 gnomADLek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB., et al. (2016) Analysis of protein-coding genetic variation in 60, 706 humans. Nature 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk GM, Ferguson CJ, Chow CY, Jin N, Jones JM, Grant AE, Ueno T, Koike M, Uchiyama Y, Kominami E, & Tanaka K, (2011). Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet, 7, e1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk GM, & Meisler MH, (2014). Mouse models of PI(3,5)P2 deficiency with impaired lysosome function. Methods in Enzymology, 534, 245–260. http://10.1016/B978-0-12-397926-1.00014-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk GM, Frei CM, Miller AC, Wallen RC, Mironova YA, Giger RJ, Meisler MH (2016a) Rescue of neurodegeneration in the Fig4 null mouse by a catalytically inactive FIG4 transgene.Hum Mol Genet 25:340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk GM, Szymanska K, Debska-Vielhaber G, et al. (2016b). Biallelic Mutations of VAC14 in Pediatric-Onset Neurological Disease. American Journal of Human Genetics, 99, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironova YA, Lenk GM, Lin JP, Lee SJ, Twiss JL, Vaccari I, Bolino A, Havton LA, Min SH, Abrams CS, Shrager P, Meisler MH, & Giger RJ, (2016). PI(3,5)P2 biosynthesis regulates oligodendrocyte differentiation by intrinsic and extrinsic mechanisms. eLife, 5, e13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironova YA, Lin J-P, Kalinski A, Huffman L, Lenk GM, Havton LA, Meisler MH and Giger RJ (2018) Protective Role of the Lipid Phosphatase Fig4 in the Adult Nervous System. Human Molec. Genet. 27:2443–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson G, Lenk GM, Reddel SW, Grant AE, Towne CF, Ferguson CJ, Simpson E, Scheuerle A, Yasick M, Hoffman S, Blouin R, Brandt C, Coppola G, Biesecker LG, Batish SD, Meisler MH, (2011). Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG4. Brain 134, 1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O’Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS. (2011) Diagnostic criteria for multiple sclerosis. Ann.Neurol.2011, 69, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She J, Guo J, Chen Q, Zeng W, Jiang Y, Bai XC (2018) Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 556:130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutterd C, Diakumis P, Bahlo M, Fanjul Fernandez M, Leventer RJ, Delatycki M, Amor D, Chow CW, Stephenson S, Meisler MH, Mclean C, & Lockhart PJ, (2017). Neuropathology of childhood-onset basal ganglia degeneration caused by mutation of VAC14. Annals of Clinical and Translational Neurology, 4(12), 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccari I, Carbone A, Previtali SC, Mironova YA, Alberizzi V, Noseda R, Rivellini C, Bianchi F, Del Carro U, D’Antonio M, Lenk GM, Wrabetz L, Giger RJ, Meisler MH, & Bolino A, (2015). Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Human molecular genetics, 24, 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Knaap MS, Bugiani M (2017). Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol, 134 (3), 35−−382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, and Xu H (2012). TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell, 12, 372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ZN, Scott AL, Dowell RD, Odorizzi G. (2018) PI(3,5)P(2) controls vacuole potassium transport to support cellular osmoregulation. Mol Biol Cell. 29:1718–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters JJ, Ferguson CJ, Lenk GM, Giger-Mateeva VI, Shrager P, Meisler MH, & Giger RJ, (2011). Congenital CNS hypomyelination in the Fig4 null mouse is rescued by neuronal expression of the PI(3,5)P(2) phosphatase Fig4. The Journal of neuroscience, 31, 17736–17751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolov SN, Bridges D, Zhang Y, Lee WW, Riehle E, Verma R, Lenk GM, Converso-Baran K, Weide T, Albin RL, Saltiel A,R, Meisler MH, Russell MW, & Weisman LS, (2012). In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A, 23, 17472–17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.