

Abstract
Since the elucidation of the structure of anisatin in the late 1960’s, sesquiterpene lactones from various Illicium species of plants have captivated synthetic chemists worldwide resulting in a large body of synthetic work. In particular, Illicium sesquiterpenes containing the seco-prezizaane carbon framework have seen immense interest in recent years owing to both desirable structural and medicinal attributes. This synopsis will focus on recently developed synthetic strategies to access these compact, highly oxidized terpenoids.
Graphical Abstract
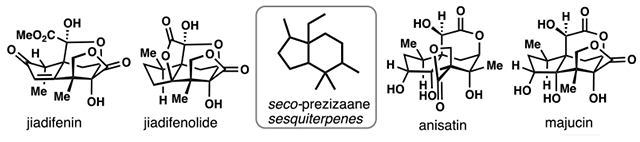
In 1968, Yamada and co-workers disclosed the X-ray crystal structure of the unusual β-lactone-containing toxin anisatin (1), thus ushering in an exciting new chapter in natural products chemistry.1,2 Over the next 50 years, over 100 natural products with apparent biosynthetic connections to 1 have been isolated from various Illicium species of plants, a family of short shrubs and trees indigenous to the Southeastern United States, Australasia, and certain regions of Asia.3 Among these natural products are a large proportion of sesquiterpenes which feature the 5,6-fused “seco-prezizaane” terpene skeleton shown and are endowed, in many cases, with exceedingly high levels of oxidation (Figure 1A). Majucin (2), jiadifenin (3), jiadifenolide (4), minwanenone (5), and pseudoanisatin (6), are all representative of this series and have served as attractive synthetic targets.
Figure 1.
(A) The seco-prezizaane skeleton and chemical structures of many highly-oxidized Illicium sesquiterpenes possessing it. (B) Other major Illicium sesquiterpene scaffolds, with representative example members.
The first reported synthetic work toward an Illicium family member was Woodward’s posthumous 1982 communication detailing a clever intramolecular glyoxylate ene reaction to construct an anisatin model system.4 This work was followed up several years later (1985) by Kende’s remarkably efficient synthesis of (±)-8-deoxyanisatin (10-deoxyanisatin, by modern numbering conventions).5 In 1990, the first seco-prezizaane natural product, namely anisatin itself, succumbed to total synthesis via a heroic synthetic effort by the group of Yamada.6 While the ensuing decade proved quiet, the 21st century witnessed a sharp increase in synthetic efforts toward this family of compounds and the majority of work has appeared after 2011. To date, almost twenty total syntheses of seco-prezizaane-containing Illicium sesquiterpenes have been reported as well as a swath of studies toward publications.7–10 In this synopsis, we will highlight recent synthetic efforts towards seco-prezizaane-containing Illicium sesquiterpenes aiming to complement Inoue’s review which covers up to 2009.11,12 Due to space constraints, syntheses of related allo-cedrane and anislactone-type sesquiterpenes, typified by 11-O-debenzoyltashironin (7) and merrilactone A (8) respectively, are not covered despite also being popular synthetic targets (Figure 1B).13–15
JIADIFENIN
In 2002, Fukuyama reported the isolation and neurotrophic activity of jiadifenin (3), a compact seco-prezizaane sesquiterpene from I. jiadifengpi.16 Unlike 1, 3 belongs to a subgroup of Illicium natural products which do not have reported convulsant properties but instead promote neurite outgrowth.16,17 The biochemical effects of Illicium sesquiterpenes have been the subject of considerable study as well and a major driving force behind their syntheses in general.17 The group of Danishefsky reported the first synthetic route to (±)-3 in 2004,7a an exercise which also provided some of the first pharmacophore mapping of the neurotrophic Illicium sesquiterpenes.7b
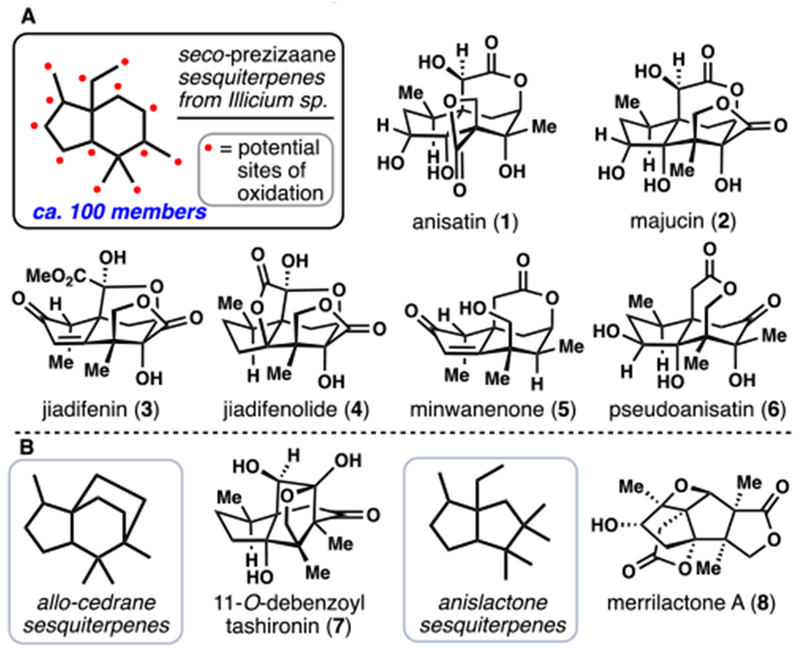
In 2011, Theodorakis reported the first asymmetric route to 3, a task facilitated by the availability of enantiopure Hajos-Parrish-like diketone building block 12, prepared in 3 straightforward steps from 1,3-cyclopentadione (9) (Scheme 1).7c Of note, the Robinson annulation, first utilized by Kende,5 serves as a recurring disconnection in Illicium sesquiterpene syntheses. From 12, cyclopentanone reduction and protection afforded an enone which was subjected to a Stiles-type α-carboxylation reaction with magnesium methyl carbonate (13). Following γ-deprotonation and alkylation with methyl iodide, bicycle 14 was generated. Global reduction of this material with lithium aluminum hydride, protection of the primary hydroxy group, oxidation of the secondary hydroxyl group and enolate triflation afforded 15 setting the stage for construction of the ubiquitous δ-valerolactone motif.
Scheme 1.
Theodorakis’s 2011 Total Synthesis of (–)-Jiadifenin (3)
Subjecting 15 to Pd-catalyzed methoxycarbonylation conditions (cat. Pd(PPh3)4), CO, MeOH) afforded the expected methyl ester 16 (60%) along with substantial amounts of cyclized product 17 (22%) which contains the requisite lactone. Moreover, 16 was easily converted to 17 upon exposure to trifluoroacetic acid. The electrophilic α,β-unsaturated lactone of 17 underwent clean basic epoxidation and following a similar oxidative cyclization to Danishefsky’s work,7a the crucial δ-valerolactone sector was installed. Following treatment with tetrabutylammonium fluoride, key tetracycle 18 was furnished; notably this material will also serve as a precursor to jiadifenolide (4) (vide infra). To advance 18 to (1R,10S)-2-oxo-3,4-dehydroxyneomajucin (ODNM) (22)–and ultimately 3–only five additional steps were required. First the secondary alcohol was eliminated with Martin sulfurane (19) and the resulting alkene hydrogenated to generate 20. Next Mn(III)-mediated allylic oxidation generated the required cyclopentenone. Finally, α-hydroxylation of the ester with 21 and α-methylation of the cyclopentenone gave 22. Utilizing Danishefsky’s protocol,7a 22 could also be converted into 3 in one step using the Jones reagent followed by treatment with methanol. Following up on Danishefsky’s work, a number of differentially oxidized congeners were evaluated for their neurite outgrowth thus greatly expanding the Illicium pharmacophore map.7e
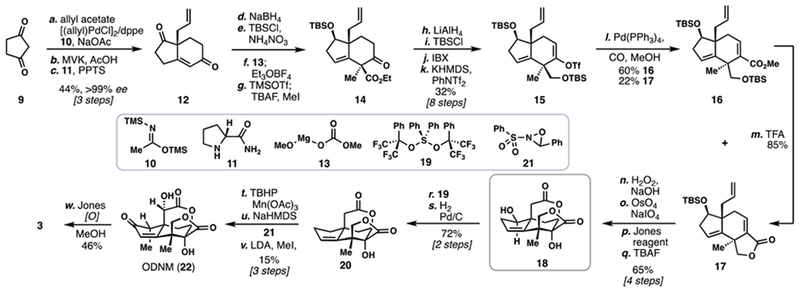
In 2012, a conceptually disparate strategy toward ODNM (22) and jiadifenin (3) was reported by the group of Zhai (Scheme 2).7d Notably this work introduced the Pauson-Khand disconnection into the seco-Prezizaane synthetic field–a strategy which will be revisited again. Allylic bromide 23 was first converted into protected, enantioenriched diol 25 via a three-step procedure consisting of coupling with a lithium acetylide generated from 24, Sharpless asymmetric dihydroxylation (in 93% ee), and dioxolane formation. From alkyne 25, the PMP group was removed under oxidative conditions and the resulting alcohol oxidized to an acid via Jones conditions. Following the coupling of this acid with (Z)-configured allylic alcohol 26, ester 27 was generated setting the stage for the first key C–C bond-forming step. Enolization of 27 and trapping with TMSCl generated the depicted silyl ketene acetal which upon warming underwent a diastereoselective Ireland-Claisen rearrangement, presumably via the chair-like transition state shown (see 28). Upon treating the reaction mixture with para-toluenesulfonic acid, lactone 29 was generated in good yield and with reasonable selectivity.
Scheme 2.
Zhai’s 2012 Total Synthesis of (–)-Jiadifenin (3)
With enyne 29 in hand, work began on construction of the cyclopentenone ring. Following dioxolane removal and protection of the secondary alcohol as its TBS ether, a diastereoselective Pauson-Khand reaction ensued under the mediation of [Co2(CO)8] generating tricycle 30 in good yield (67%). To install the remaining carbon atoms of the target, the Zhai lab utilized a clever photochemical enone/allene [2+2] reaction to construct 31 in 72% yield–a small amount of diastereomer 32 was also generated in this reaction. Oxidative scission of the exocyclic alkene via ozonolysis followed by sodium methoxide-mediated cleavage of the resulting strained cyclobutanone led to ester 33. To complete the synthesis of ODNM (22), and therefore also 3, four additional transformations were required. A two-step Saegusa oxidation accomplished desaturation of the cyclopentanone ring and upon treatment of this compound with TBAF, the δ-valerolactone ring was generated. Finally, α-oxidation according to Theodorakis (NaHMDS then oxaziridine 21) furnished 22 and ultimately 3.
In 2015, a formal synthesis of 3 was reported by Y. Fukuyama and co-workers via Theodorakis’ advanced intermediate 20 (Scheme 3).7f Vinyl bromide 34, prepared in five steps, underwent a high-yielding Heck cyclization to furnish 35. After twelve additional manipulations, β-keto ester 36 was prepared and this material served as a substrate for an intramolecular palladium-mediated Tsuji-Trost allylation/lactonization cascade leading to tricycle 37 in an impressive 65% yield. Deprotection of this material and subjection to Grieco’s elimination sequence afforded 38 after hydroxylation of the β-ketoester with m-CPBA. Finally, diastereoselective ketone reduction and two-step oxidative cyclization (OsO4/NaIO4 then Ag(I)) arrived at 20, completing the formal synthesis.
Scheme 3.
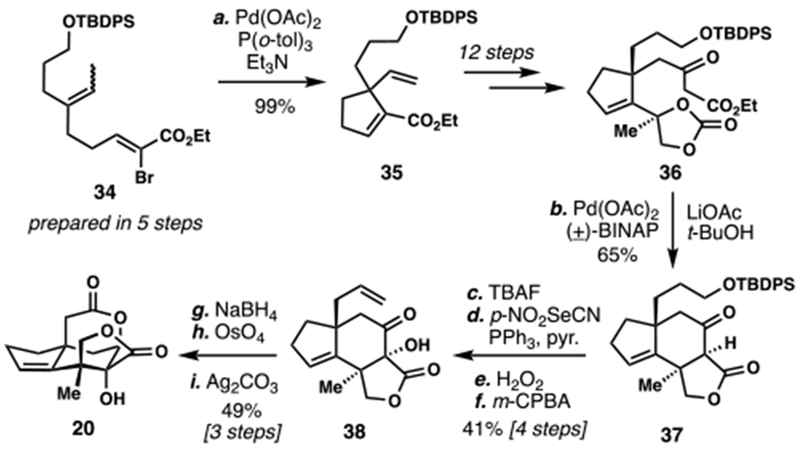
Y. Fukuyama’s 2015 Formal Synthesis of (±)-Jiadifenin (3)
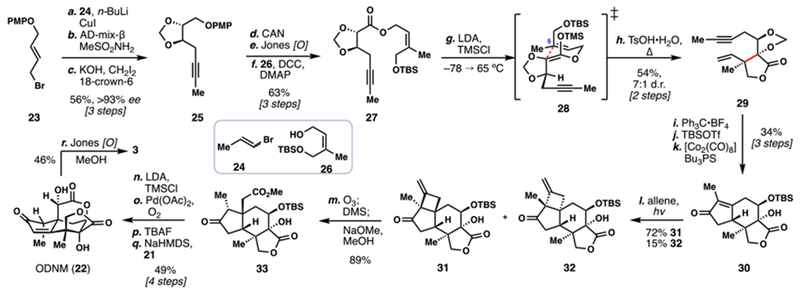
Recently, Micalizio and co-workers reported a distinct route to both 22 and 3 as well as the related Illicium member (2S)-hydroxy-3,4-dehydro-neomajucin (52) (Scheme 4).7g Key to the success of this route was a complex application of the hydroxyl-directed metallacycle-mediated formal [2+2+2] annulation of enynes with alkynes.18 The synthesis commenced with the preparation of key fragment 42. Beginning with enantiopure epoxide 39, copper-mediated oxirane opening with the Grignard reagent derived from vinylbromide 40 afforded alcohol 41. TBAF-mediated deprotection of this material, activation of the primary alcohol as its tosylate, and cyclization under basic conditions (NaH) formed a second epoxide which could be opened with propynyl lithium to give 42.
Scheme 4.
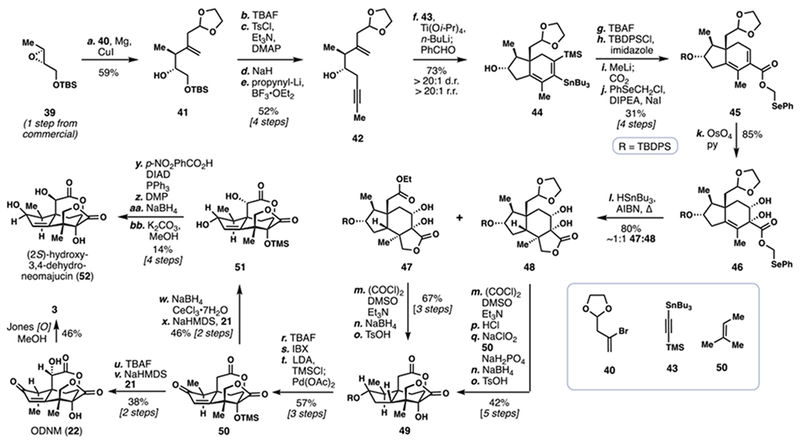
Micalizio’s 2016 Total Synthesis of (–)-Jiadifenin (3) and Related Majucinoids
In the first key step of the synthesis, the lithium alkoxide of enyne 42 was reacted with stannyl-substituted TMS-acetylene 43 and a reduced titanium complex prepared by mixing Ti(Oi-Pr)4 and n-BuLi, inducing a formal [2+2+2] annulation. Notably the product formed (i.e., diene 44) was generated as essentially a single regioisomer–and diastereomer–in excellent yield. Compound 44 was next treated with tetrabutylammonium fluoride, cleaving the carbon-silicon bond. The secondary hydroxyl group was protected as TBDPS ether. A tin-lithium exchange followed by lithiate quench with carbon dioxide appended a carboxylic acid which could be alkylated with chloromethyl phenylselenide to deliver 45. Following chemo- and diastereoselective osmium tetroxide-mediated dihydroxylation, selenophenyl methyl ester 46 was secured.
In a second key C–C bond-forming step in the synthesis, a 5-exo radical cyclization was envisioned to generate the butyrolactone ring, and thus the entire seco-prezizaane skeleton.19 Subjecting 46 to classic tin-mediated reductive radical cyclization conditions led to the expected product 48 in a diastereoselective fashion. Interestingly, however, this material was accompanied by an almost equimolar quantity of ethyl ester 47, which was believe to be formed by a process involving intramolecular radical C–H abstraction of the weak dioxolane C–H bond followed by fragmentation. Product formation could be biased toward 47 by replacing Bu3SnH with TMS3SiH. This finding was welcome as the ester oxidation state found in 47 was more ideal for conversion into 49–only a Swern oxidation, stereoselective reduction, and acid-mediated lactonization were required. In contrast, five steps were needed to advance 48 into 49. Tricycle 49 could be converted into enone 50 via a sequence consisting of fluoride-mediated desilylation, IBX oxidation, and desaturation via Saegusa’s protocol. Advanced intermediate 50 served as an entry point into several seco-prezizaane natural products. First, ODNM (22) could be prepared by deprotection (TBAF) and α-oxidation of the lactone under basic conditions, which also correctly epimerized the methyl-bearing stereocenter. As demonstrated before, 22 could be converted into 3. The authors also utilized 50 in the construction of (2S)-hydroxy-3,4-dehydro-neomajucin (52), a neurotrophic family member which had not been previously synthesized. Diastereoselective ketone reduction of 50 followed by lactone α-hydroxylation fashioned 51. Finally, a four-step double hydroxyl inversion sequence generated 52. This work attests to the power of both modern metal-mediated and classic radical-based cascades to forge challenging carbon-carbon bonds in the context of complex molecule synthesis.
JIADIFENOLIDE
Fukuyama’s 2009 disclosure of jiadifenolide (4), a compact seco-prezizaane with a propellane core and very high neurotrophic activity set off a frenzy within the synthetic community.20 4 was demonstrated to induce neurite outgrowth of rat cortical neurons at concentrations as low as 10 nM, and more recently, a similar effect has been seen in human induced pluripotent stem cells.21 While isolated less than a decade ago, 4 has seen a multitude of total and formal syntheses which will be discussed below.
In addition to their previously reviewed route to ODNM and jiadifenin, the group of Theodorakis reported the first total synthesis of 4 in 2011 (Scheme 5).8a Utilizing previously disclosed bislactone 18, a clever sequence was described to construct the hallmark propellane system. First, the trisubstituted olefin was epoxidized with m-CPBA. Then, oxidation of the secondary alcohol triggered an epoxide elimination/trans-lactonization cascade, smoothly affording propellane 55. Hydrogenation of the enone followed by silyl protection of the secondary alcohol prepared 56, a suitable substrate for late-stage installation of the final methyl group. By opting for a palladium-catalyzed cross-coupling sequence from a vinyl triflate intermediate, 57 was rapidly assembled. Hydrogenation and sequential lactone oxidations of 57 then completed the first synthesis of 4.
Scheme 5.
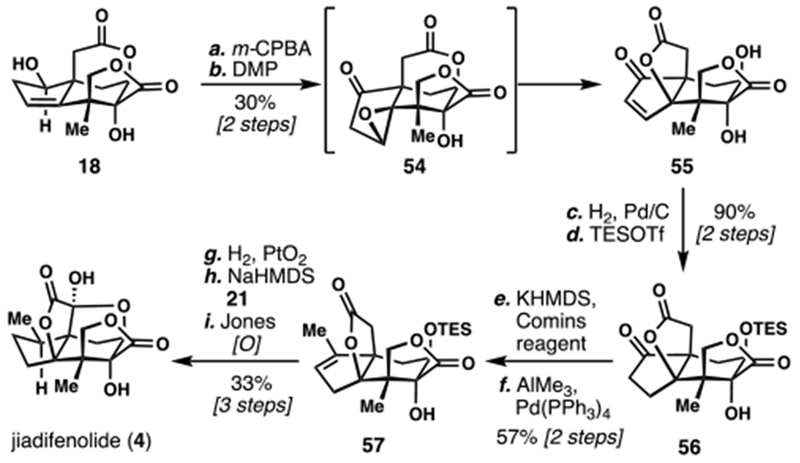
Theodorakis’s 2011 Total Synthesis of (–)-Jiadifenolide (4)
Following that seminal work, Sorensen disclosed his group’s efforts towards the same target, featuring the first example of a C(sp3)–H bond activation in the context of Illicium terpene synthesis (Scheme 6).8b Beginning from pulegone-derived building block 58, the venerable Robinson annulation was once again called on to construct the 5,6-fused ring system seen in 59. Straightforward functionalization of this intermediate, including double methylation, dioxolane protection, ester reduction, and oxidation afforded 61. An intriguing use of TosMIC (62), a reagent typically reserved for heterocycle synthesis,22 led to aldehyde chain extension and nitrile formation. Acidic methanolysis both instigated propellane formation by nitrile hydrolysis and cleavage of dioxolane protecting group, unveiling 63, the key entry point for an ambitious C–H oxidation.
Scheme 6.
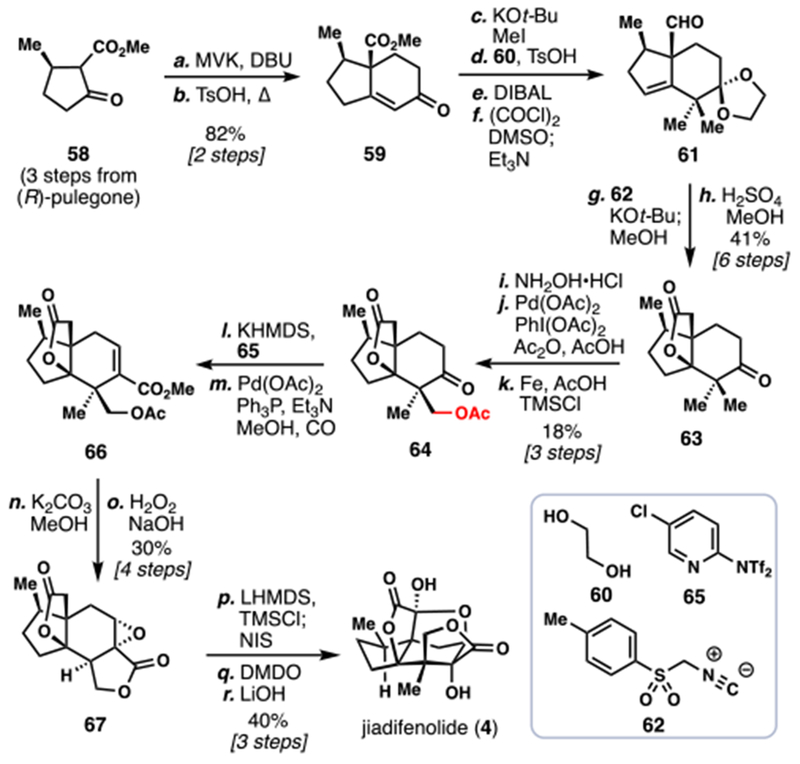
Sorensen’s 2014 Synthesis of (–)-Jiadifenolide (4).
Taking inspiration from Sanford’s methodology,23,24 ketone 63 was converted to an intermediate oxime, which served as a directing group for a palladium-catalyzed C–H acetoxylation reaction. Remarkably, synthetically useful amounts of the desired acetoxylated product 64 could be isolated after oxime cleavage. Unfortunately, differentiation of the gem-dimethyl group was not perfect – oxidation of the other methyl group was observed, along with a small amount of doubly-oxidized product – speaking to the challenges associated with planning late stage C–H activation reactions in synthesis. Palladium-catalyzed carbonylation of the vinyl triflate of 64 installed the final carbon atom for the synthesis, giving 66 which was well-suited for a lactonization/epoxidation sequence leading to 67. Similar to Theodorakis’s endgame, double oxidation of the propellane lactone was now required. A creative iodination/iodoso-Pummerer rearrangement sequence installed that requisite oxidation, which completed the synthesis of 4 after basic hydrolysis.
Shortly after Sorensen’s work, Paterson reported a conceptually distinct approach to constructing the polycyclic ring system of 4 (Scheme 7).8c Rather than planning a Robinson annulation, a key samarium(II) iodide-mediated reductive cyclization was envisioned.
Scheme 7.
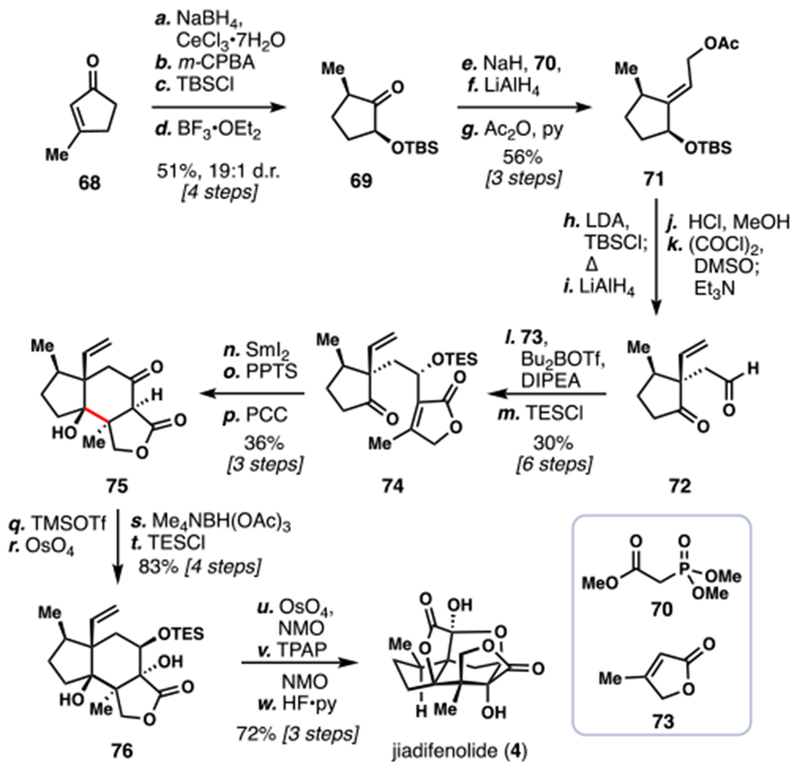
Paterson’s 2014 Total Synthesis of (±)-Jiadifenolide (4)
A four-step sequence from simple 3-methyl-cyclopentenone (68) quickly prepared building block 69, which underwent facile intermolecular Horner-Wadsworth-Emmons (HWE) olefination with phosphonate 70. Reduction of the intermediate ester (LiAlH4) and acetate protection provided ample amounts of 71. Silyl ketene acetal synthesis followed by heating initiated a highly diastereoselective Ireland-Claisen rearrangement, cleanly setting one of the challenging quaternary centers. Reduction of the resulting material, silyl-group cleavage, and oxidation produced aldehyde 72. Fittingly, a boron enolate of butenolide 73 was prepared and reacted with 72 to furnish 74 after silyl protection of the secondary alcohol. In the key step of the synthesis, samarium(II) iodide reduced the ketone in 74 with concomitant ketyl radical addition into the pendant butenolide moiety, generating 75 as a single diastereomer after straightforward functional group manipulations. This impressive transformation set adjacent stereocenters with high fidelity and completed the 5,6-fused ring system in a single operation. Two-step Rubottom-type oxidation of 75 installed a tertiary alcohol, which was then used to direct reduction of the adjacent ketone, delivering 76 after silyl protection. Finally, dihydroxylation of the monosubstituted alkene and oxidative lactonization gave an intermediate keto-lactone from which 4 was prepared after desilylation.
Possibly inspired by Paterson’s key samarium(II) iodide-mediated cyclization, Zhang and Gademann both emulated that disconnection in their own total and formal syntheses of 4 (Scheme 8). Zhang’s total synthesis began from pulegone-derivative 77, which was allylated and oxidatively cleaved to yield aldehyde 78, a compound parallel to Paterson’s 72.8e As such, it was also treated with the boron enolate of 73, giving rise to 79, which was advanced to cyclization precursor 80 after straightforward functional group manipulations. Once again, samarium(II) iodide was employed to construct the strategic bond, and key aldehyde 81 was isolated after subsequent oxidation. 81 was then poised to undergo a novel [4+1] annulation reaction with TMS-diazomethane. In this step, the anion of TMS-diazomethane is thought to add into the aldehyde, triggering a Brook rearrangement and subsequent O–H insertion into the adjacent tertiary alcohol. The authors also provided a substrate scope for this transformation to demonstrate its utility for tetrahydrofuran synthesis. A sequence of three consecutive oxidations and basic hydrolysis then completed the synthesis of 4.
Scheme 8.
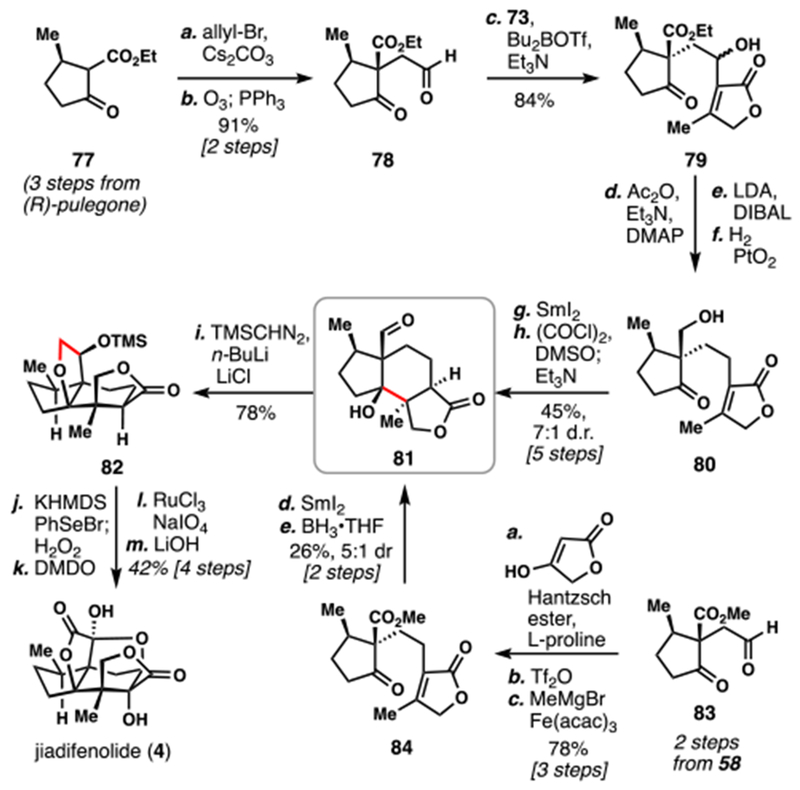
Zhang’s 2015 Total Synthesis and Gademann’s 2016 Formal Synthesis of (–)-Jiadifenolide (4)
Gademann’s formal synthesis of 4 was disclosed as part of a larger collection of seco-prezizaane synthetic studies and targeted aldehyde 81 as the formal synthesis endpoint.8f Beginning from aldehyde 83, prepared in the same way as Zhang’s 78, a proline-catalyzed aldol/dehydration reaction with tetronic acid afforded a highly electrophilic enone which was reduced in the same operation with a Hantzsch ester. Triflation followed by iron-catalyzed cross-coupling reaction cleanly appended an additional methyl group onto the structure to provide 84, the key cyclization precursor. Though somewhat less diastereoselective than the other substrates with stronger biasing elements, cyclization of 84 with samarium(II) iodide nevertheless proceeded efficiently and the intermediate ester could be reduced to aldehyde 81, completing the formal synthesis of 4.
In 2015, Shenvi reported an impressive gram-scale synthesis of 4 (Scheme 9).8d Like many previous works, this synthesis had planned a key annulation reaction to construct the 5,6-fused ring system; specifically, a butenolide heterodimerization between 87 and 88 was envisioned. 87 could be prepared in only three steps from (+)-citronellal by dehydration (BTPP, NfF), oxidative alkene cleavage (O3; DMS), and a hetero-Pauson Khand reaction (Mo(CO)6) of enyne 85. A four-step route traversing intermediate dibromide 86 was also developed to ensure greater material throughput. Likewise, 88 was synthesized rapidly – in only two steps, diketene acetone adduct (89) was condensed with hydroxyacetone to afford butenolide 88 in good yield on multi-gram scales. In the key step, the lithium enolate of 87 was joined with 88 with exquisite diastereoselectivity to forge 92 via 90, presumably due to the intermediacy of a chelated intermediate, like 91. With 92 in hand, completion of the synthesis of 4 was straightforward: enol hydroxylation and directed reduction converted 92 to trans-diol 93. A further two-step oxidation sequence, involving sequential bromination and hydroxylation events, advanced 93 on to 4, completing the synthesis in just eight steps. In a subsequent publication, 92 was also shown to go on to 7, speaking to the power of this route in also accessing allo-cedrane members.
Scheme 9.
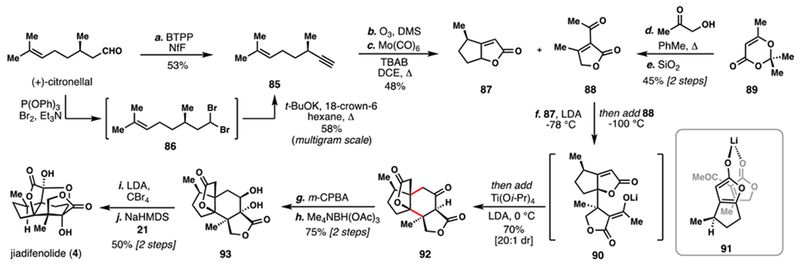
Shenvi’s 2015 Gram-Scale Total Synthesis of (–)-Jiadifenolide (4)
ANISATINOIDS & PSEUDOANISATINOIDS
In contrast to the prolific work seen toward majucinoid synthesis, studies on anisatinoids and pseudoanisatinoids have been somewhat less frequent in the 21st century. Since Yamada’s groundbreaking work, only a single additional synthesis of 1 has been completed and will be presented in this section. Additionally, Mehta’s 2007 synthesis of ent-minwanenone (ent-5) stood as the only synthesized member of the latter category for nearly a decade.9a,b
T. Fukuyama’s synthesis of 1 relied on a very non-obvious dearomatization sequence for key bond formations (Scheme 10).9c Beginning with compound 94 (prepared in four steps from commercial materials), propargylation of the primary alcohol followed by functional group manipulations afforded alkyne 95. Dearomatization of the electron-rich aromatic core (PhI(OAc)2, MeOH) of 95 generated an intermediate protected ortho-quinone which then underwent smooth intramolecular Diels-Alder reaction with the pendant alkyne upon heating. HWE olefination of the remaining ketone with phosphonate 96 followed by reduction cleanly led to primary alcohol 97. This compound was alkylated with Bu3SnCH2I, a cuing element for a [2,3]-sigmatropic rearrangement. Indeed, lithium-tin exchange initiated that transformation, setting the second seco-prezizaane quaternary center with high fidelity. Benzylation and strained olefin cleavage afforded intermediate 98 in short order.
Scheme 10.
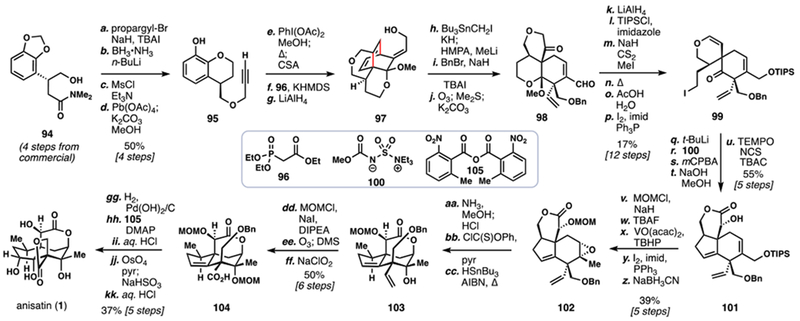
T. Fukuyama’s 2012 Total Synthesis of (–)-Anisatin
Unfortunately, while this opening sequence offered rapid access to a highly-substituted cyclohexane ring a further 11 steps were required to close the five-membered ring and complete the seco-prezizaane ring system. First, primary iodide 99 was prepared in six steps from 98; then, lithium-halogen exchange created an organometallic nucleophile that added into the pendant ketone. Further functional group manipulations advanced that intermediate on to 101. Although the seco-prezizaane skeleton was now in place, extraneous oxidation at C12 and C15 were also present and required an additional 8 steps to address, proceeding through 102. From 103, the terminal alkene was exhaustively oxidized to carboxylic acid 104, and the signature spiro-β-lactone of 1 was formed by employing anhydride 105 as an activating reagent. Protecting group cleavages and olefin dihydroxylation finally arrived at 1 completing a synthesis that truly exemplifies the diversity of insight possible for creating any given structure.
Our own group’s work in both the pseudoanisatinoid and majucinoid areas represents a significant departure from the previously discussed syntheses. We focused on utilizing a semisynthetic approach, predicated on the ability to convert a pre-existing 15-carbon sesquiterpene into various Illiciums via sequential C–C bond reorganization and site-selective C(sp3)–H bond activation (Scheme 11).9d,e
Scheme 11.
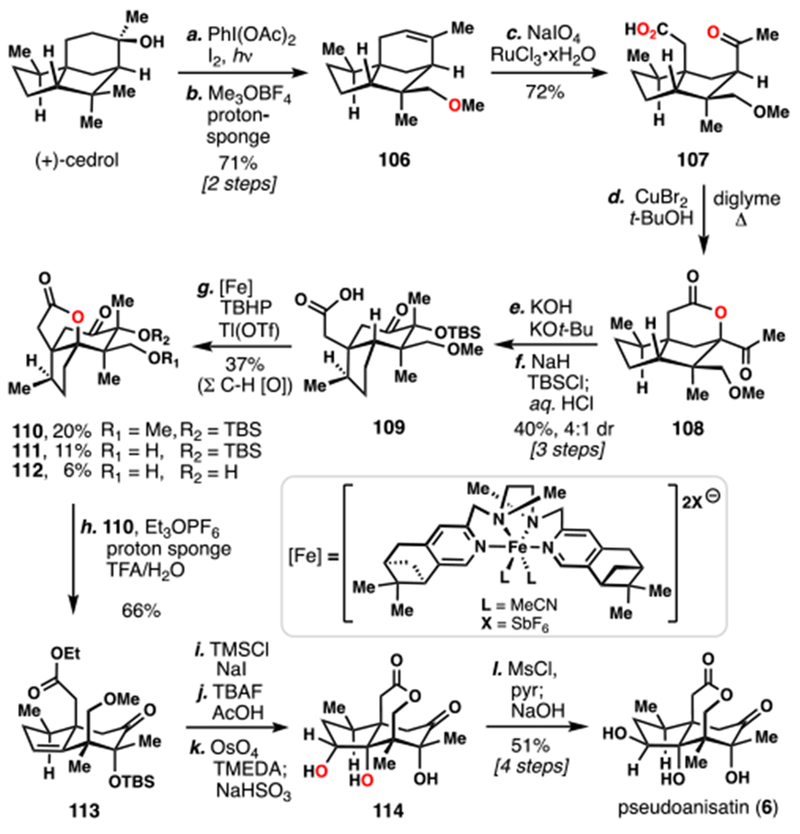
Maimone’s 2016 Synthesis of (+)–Pesudoanisatin (6) from (+)–Cedrol
Owing to both availability and biosynthetic considerations, (+)-cedrol was chosen as starting material and converted into a cyclic ether via Suarez’s hypoiodite-mediated C–H functionalization (Scheme 11).25 The strained tetrahydrofuran ring formed could then be smoothly eliminated under the alkylative action of methyl Meerwein’s salt to provide alkene 106. Further oxidations were introduced by cleavage of the trisubstituted olefin with in situ generated RuO4, giving keto-acid 107 in good yield. A challenging intramolecular oxidative lactonization was found to proceed under the action of CuBr2 giving lactone 108. At this point, the 5,5-fused cedrane system needed to be converted to the 5,6-fused seco-prezizaane one, and an abiotic α-ketol rearrangement was chosen for this task.26 Thus, treating 108 with strong base (KOH, KOt-Bu) effected lactone hydrolysis with concomitant ring shift, giving seco-prezizaane core 109 after silylation. In a key step of this synthesis, the exceptionally hindered C4 methine position was successfully oxidized by carboxylic acid-directed non-heme iron(oxo)-catalysis.27,28 Although this step was not perfect – a designer pinene-derived ligand architecture was required for sufficient catalyst activity and multiple products of excessive C–H oxidation were isolated (see 111 and 112) – it nevertheless provided lactone 110 in serviceable enough yield for the completion of the synthesis. The lactone of 110 could be eliminated by the action of ethyl Meerwein’s salt, giving rise to ethyl ester 113. Two-step deprotection of the methyl ether (in situ TMSI) and silyl ether (TBAF, AcOH) led to an intermediate ε-lactone, which could undergo directed, diastereoselective dihydroxylation using conditions developed by Donohoe.29 Following inversion of the secondary alcohol by activation of it as the mesylate and subsequent displacement–by a presumed intramolecular carboxylate nucleophile–the first synthesis of pseudoanisatin (6) was completed.
To gain synthetic entry into majucinoids even further oxidation was required (Scheme 12). In order to circumvent previous challenges encountered in acid-directed methine C–H oxidation, substrate 116 was prepared in three straightforward steps from (+)-cedrol, by way of alkene 115. Subjecting 116 to hypoiodite photolysis conditions smoothly oxidized the same methine position, and 117 could be isolated in excellent yield. Inspired by Waegell’s work on simpler substrates,30 we treated 117 with in situ generated RuO4 in order to perform a double C–H activation/C–C oxidation cascade and arrive at keto-lactone 118 in a single step. Additional oxidations required for the majucinoids were installed by treating 118 with selenium(IV) oxide. Remarkably, all C–H bonds next to the ketone in 119 were oxidized. Reduction and an acetate cleavage/isomerization cascade took this material (i.e. 119) on to enol lactone 120. Once again, an α-ketol rearrangement was desired to isomerize the 5,5-fused ring system to the 5,6-fused one. This time, while oxidation to the α-hydroxy ketone was more facile (DMDO), that intermediate was far more labile and would only undergo ring shift under thermal conditions, providing diol 93 after directed reduction. As diol 93 was prepared in both Shenvi’s and Theodorakis’s routes to 4, isolation of that intermediate constituted a formal synthesis. As we were interested in also accessing compounds that had not previously been synthesized, we advanced 93 to alkene 121 by an intramolecular translactonization/dehydration sequence. 121 was reported by Theodorakis to go on to 3 and 22. Finally, a three-step sequence of hydroxylation, ruthenium-catalyzed epimerization, and dihydroxylation completed the synthesis of 2, conclusively demonstrating the viability of an oxidative route to accessing multiple family members including highly oxidized majucinoids.
Scheme 12.
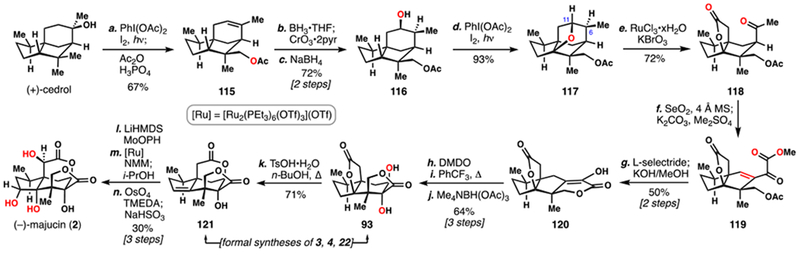
Maimone’s 2017 Synthesis of (–)-Majucin (2) and Related Majucinoids
In summary, a host of different strategies have been explored for seco-prezizaane Illicium sesquiterpene synthesis. From strategic annulations and radical cyclization cascades, to elaborate dearomatizations, transition metal couplings, and creative use of the chiral pool, Illicium syntheses truly showcase a wide variety of synthetic strategies and tactics, making their study inherently valuable for the synthetic practitioner. It is reasonable to speculate at this point where the future of Illicium sesquiterpene chemistry lies – what questions are there left to answer? We note from a synthetic chemistry perspective that routes to anisatinoids and pseudoanisatinoids are still comparably underdeveloped relative to majucinoid synthesis and perhaps future success will be seen in that area. Nevertheless, we are confident that future syntheses will doubtlessly provide fertile ground for new, inspiring chemistries. From a medicinal and biological perspective many questions still remain with regard to the (poly)pharmacology of these compounds, atomic level descriptions of their binding, and ultimately whether they, or designed derivatives, are effective agents to treat human neurological disease. Many of these questions can be greatly aided by the advances in synthetic chemistry described herein.
ACKNOWLEDGMENT
Financial support is provided by the NIH (GM116952). T.J.M. is an Arthur C. Cope Scholar and Research Corporation Cottrell Scholar and acknowledges unrestricted support from Novartis, Bristol-Myers Squibb, Amgen, and Eli Lilly. M.L.C. acknowledges UC-Berkeley and the NSF for a Berkeley graduate fellowship and NSF pre-doctoral fellowship (DGE-1106400) respectively. L. F. T. N. thanks the São Paulo Research Foundation (FAPESP) for a graduate fellowship (2017/18029-6).
Biographies
BIOGRAPHIES

Matthew Condakes received his A.B. and A.M. degrees in chemistry summa cum laude from Harvard University in 2014. While there, he conducted research on the synthesis of novel macrolide antibiotic analogues under the guidance of Prof. Andrew Myers. His passion for complex molecule synthesis then led him to pursue graduate work with Prof. Tom Maimone at the University of California, Berkeley, as an NSF predoctoral fellow. He earned his Ph.D. in 2018 for work on developing new synthetic strategies and methodologies, particularly those seen in the context of Illicium sesquiterpene syntheses.

Luiz Novaes received his B.S. (2013) and M.S. (2015) degrees in Chemistry from University of Campinas under the supervision of Prof. Ronaldo Pilli. In 2015, he joined the group of Prof. Julio Pastre (University of Campinas) for his Ph.D. studies on the total synthesis of natural terpenes. In March of 2018, Luiz moved to UC Berkeley as a visiting Ph.D. student in the Maimone research group.

Tom Maimone was born and raised in Warsaw, NY. He completed his B.S. degree in chemistry at UC-Berkeley in 2004 and his Ph.D. at The Scripps Research Institute in 2009. During these periods, he conducted total synthesis research in the labs of Prof. Dirk Trauner and Phil Baran respectively. From 2009-2012 he was an NIH post-doctoral fellow at MIT under the tutelage of Prof. Stephen Buchwald. In 2012 Tom joined the faculty at UC-Berkeley where his group works in the area of natural products chemistry.
REFERENCES
- (1).(a) Lane JF; Koch WT; Leeds NS; Gorin G On the Toxin of Illicium Anisatum. I. The Isolation and Characterization of a Convulsant Principle: Anisatin. J. Am. Chem. Soc 1952, 74, 3211 [Google Scholar]; (b) Yamada K; Takada S; Nakamura S; Hirata Y The structure of anisatin Tetrahedron Lett. 1965, 52, 4797. [Google Scholar]
- (2).Yamada K; Takada S; Nakamura S; Hirata Y The structures of anisatin and neoanisatin: Toxic sesquiterpenes from Illicium Anisatum L. Tetrahedron 1968, 24, 199. [Google Scholar]
- (3).Fukuyama Y; Huang J-M Chemistry and neurotrophic activity of seco-prezizaane- and anislactone-type sesquiterpenes from Illicium species in Studies in Natural Products Chemistry (Atta-ur-Rahman, Ed.) 2005, 32, 395. [Google Scholar]
- (4).Lidner DL; Doherty JB; Shoham G; Woodward RB Intramolecular ene reaction of glyoxylate esters: an anisatin model study. Tetrahedron Lett. 1982, 23, 5111. [Google Scholar]
- (5).Kende AS; Chen J Stereoselective total synthesis of (±)-8-deoxyanisatin J. Am. Chem. Soc 1985, 107, 7184. [Google Scholar]
- (6).Niwa H; Nisiwaki M; Tsukada I; Ishigaki T; Ito S; Wakamatsu K; Mori T; Ikagawa M; Yamada K Stereocontrolled total synthesis of (–)-anisatin: a neurotoxic sesquiterpenoid possessing a novel spiro β-lactone J. Am. Chem. Soc 1990, 112, 9001. [Google Scholar]
- (7).(a) For total and formal syntheses of jiadifenin (3) and (1R,10S)-2-oxo-3,4-dehydroxyneomajucin (ODNM), see: Cho YS; Carcache DA; Tian Y; Li Y-M; Danishefsky SJ Total Synthesis of (±)-Jiadifenin, a Non-peptidyl Neurotrophic Modulator. J. Am. Chem. Soc 2004, 126, 14358. [DOI] [PubMed] [Google Scholar]; (b) Carcache DA; Cho YS; Hua Z; Tian Y; Li Y-M; Danishefsky SJ Total Synthesis of (±)–Jiadifenin and Studies Directed to Understanding its SAR: Probing Mechanistic and Stereochemical Issues in Palladium-Mediated Allylation of Enolate-Like Structures. J. Am. Chem. Soc 2006, 128, 1016. [DOI] [PubMed] [Google Scholar]; (c) Trzoss L; Xu J; Lacoske MH; Mobley WC; Theodorakis EA Enantioselective Synthesis of (–)-Jiadifenin, a Potent Neurotrophic Modulator Org. Lett 2011, 13, 4554. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang Y; Fu X; Chen J; Zhai H Total Synthesis of (–)-Jiadifnin Angew. Chem., Int. Ed 2012, 51, 9825. [DOI] [PubMed] [Google Scholar]; (e) Trzoss L; Xu J; Lacoske MH; Mobley WC; Theodorakis EA Illicium Sesquiterpenes: Divergent Synthetic Strategy and Neurotrophic Activity Studies. Chem. - Eur. J 2013, 19, 6398. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Harada K; Imai A; Uto K; Carter RG; Kubo M; Hioki H; Synthesis of jiadifenin using Mizoroki-Heck and Tsuji-Trost reactions. Tetrahedron 2015, 71, 2199. [Google Scholar]; (g) Cheng X; Micalizio GC Synthesis of Neurotrophic Seco-prezizaane Sesquiterpenes (1R,10S)-2-Oxo-3,4-dehydroneo-majucin, (2S)-Hydroxy-3,4-dehydroneomajucin, and (−)-Jiadifenin. J. Am. Chem. Soc 2016, 138, 1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) For total and formal syntheses of jiadifenolide (4), see: Xu J; Trzoss L; Chang WK; Theodorakis EA Enantioselective Total Synthesis of (–)-Jiadifenolide. Angew. Chem. Int. Ed 2011, 50, 3672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Siler DA; Mighion JD; Sorensen EJ An Enantiospecific Synthesis of Jiadifenolide. Angew. Chem. Int. Ed 2014, 53, 5332. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Paterson I; Xuan M; Dalby SM Total Synthesis of Jiadifenolide. Angew. Chem. Int. Ed 2014, 53, 7286. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu HH; Martinez MD; Shenvi RA An eight-step gram-scale synthesis of (–)-jiadifenolide. Nat. Chem 2015, 7, 604. [DOI] [PubMed] [Google Scholar]; (e) Shen Y; Li L; Pan Z; Wang Y; Li J; Wang K; Wang X; Zhang Y; Hu T; Zhang Y Protecting-Group-Free Total Synthesis of (–)-Jiadifenolide: Development of a [4+1] Annulation toward Multisubstituted Tetrahydrofurans. Org. Lett 2015, 17, 5480. [DOI] [PubMed] [Google Scholar]; (f) Gomes J; Daeppen C; Liffert R; Roesslein J; Kaufmann E; Heikinheimo A; Neuburger M; Gademann K Formal Total Synthesis of (–)-Jiadifenolide and Synthetic Studies toward seco-Prezizaane-Type Sesquiterpenes. J. Org. Chem 2016, 81, 11017. [DOI] [PubMed] [Google Scholar]
- (9).(a) For total syntheses of other seco-prezizaane family members, see: Mehta G; Shinde HM Total synthesis of the novel seco-prezizaane sesquiterpenoid (+)-1S-minwanenone. Tetrahedron Lett. 2007, 48, 8297 [Google Scholar]; (b) Mehta G; Shinde HM An Approach to seco-Prezizaane Sesquiterpenoids: Enantioselective Total Synthesis of (+)-1S-Minwanenone. J. Org. Chem 2012, 77, 8056. [DOI] [PubMed] [Google Scholar]; (c) Ogura A; Yamada K; Yokoshima S; Fukuyama T Total Synthesis of (–)-Anisatin. Org. Lett 2012, 14, 1632. [DOI] [PubMed] [Google Scholar]; (d) Hung K; Condakes ML; Morikawa T; Maimone TJ Oxidative Entry into the Illicium Sesquiterpenes: Enantiospecific Synthesis of (+)-Pseudoanisatin J. Am. Chem. Soc 2016, 138, 16616. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Condakes ML; Hung K; Harwood SJ; Maimone TJ Total Syntheses of (–)-Majucin and (–)-Jiadifenoxolane A, Complex Majucin-type Illicium Sesquiterpenes. J. Am. Chem. Soc 2017, 139, 17783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) For select studies toward seco-prezizaane sesquiterpenes, see: Niwa H; Mori T; Hasegawa T; Yamada K Stereocontrolled Synthesis of Polyfunctionalized trans-Hydrindan Systems: A Model Study toward Anisatin. J. Org. Chem 1986, 51, 1015. [Google Scholar]; (b) Loh T-P; Hu Q-Y Synthetic Studies toward Anisatin: A Formal Synthesis of (±)-8-Deoxyanisatin. Org. Lett 2001, 3, 279. [DOI] [PubMed] [Google Scholar]; (c) Mehta G; Shinde HM; Kumaran RS Model synthetic studies toward jiadifenin and majucin type seco-prezizaane natural products via a stereo- and enantioselective approach. Tetrahedron Lett. 2012, 53, 4320. [Google Scholar]
- (11).Urabe D; Inoue M Total syntheses of sesquiterpenes from Illicium species. Tetrahedron 2009, 65, 6271. [Google Scholar]
- (12).(a) For additional reviews discussing Illicium syntheses, see: Xu J; Lacoske MH, Theodorakis EA Neurotrophic Natural Products: Chemistry and Biology. Angew. Chem. Int. Ed 2014, 53, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li L; Shen Y; Zhang Y Synthetic Progress of Jiadifenolide. Chin. J. Org. Chem 2016, 36, 439. [Google Scholar]; (c) Brill ZG; Condakes ML; Ting CP; Maimone TJ Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev 2017, 117, 11753. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schwan J; Christmann M Enabling strategies for step efficient syntheses. Chem. Soc. Rev 2018, 47, 7985. [DOI] [PubMed] [Google Scholar]
- (13).(a) For total and formal synthesis of 11-O-debenzoyltashironin (7), see: Cook SP; Polara A; Danishefsky SJ The Total Synthesis of (±)-11-O-Debenzoyltashironin J. Am. Chem. Soc 2006, 128, 16440. [DOI] [PubMed] [Google Scholar]; (b) Polara A; Cook SP; Danishefsky SJ Multiple chirality transfers in the enantioselective synthesis of 11-O-debenzoyltashironin. Chiroptical analysis of the key cascade. Tetrahedron Lett. 2008, 49, 5906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mehta G; Maity P A total synthesis of 11-O-methyldebenzoyltashironin. Tetrahedron Lett. 2011, 52, 1749; [Google Scholar]; (d) Mehta G; Maity P Explorations en route to synthesis of tashironins: singlet oxygen cycloaddition to 1,3-cyclopentadienes embedded within allo-cedrane framework Tetrahedron Lett. 2011, 52, 5161. [Google Scholar]; (e) Ohtawa M; Krambis MJ; Cerne R; Schkeryantz JM; Witkin JM; Shenvi RA Synthesis of (–)-11-O-Debenzoyltashironin: Neurotrophic Sesquiterpenes Cause Hyperexcitation. J. Am. Chem. Soc 2017, 139, 9637. [DOI] [PubMed] [Google Scholar]
- (14).For total and formal syntheses of merrilactone A (8), see: (a) ref. 11 and references cited therein.; (b) Shi L; Meyer K; Greaney MF Synthesis of (±)-Merrilactone A and (±)-Anislactone A. Angew. Chem., Int. Ed 2010, 49, 9250. [DOI] [PubMed] [Google Scholar]; (c) Chen J; Gao P; Yu F; Yang Y; Zhu S; Zhai H Total Synthesis of (±)-Merrilactone A. Angew. Chem. Int. Ed 2012, 51, 5897. [DOI] [PubMed] [Google Scholar]; (d) Nazef N; Davies RDM; Greaney MF Formal Synthesis of Merrilactone A Using a Domino Cyanide 1,4-Addition–Aldol Cyclization. Org. Lett 2012, 14, 3720. [DOI] [PubMed] [Google Scholar]; (e) Liu W; Wang B Synthesis of (±)-Merrilactone A by Desymmetrization Strategy. Chem. - Eur. J 2018, 24, 16511. [DOI] [PubMed] [Google Scholar]
- (15).For total and formal syntheses of anislactone A, along with synthetic studies, see: Hong B-C; Shr Y-J; Wu J-L; Gupta AK; Lin K-J Novel [6 + 2] Cycloaddition of Fulvenes with Alkenes: A Facile Synthesis of the Anislactone and Hirsutane Framework. Org. Lett 2002, 4, 2249. [DOI] [PubMed] [Google Scholar]; (b) see ref. 14b
- (16).Yokoyama R; Huang J-M; Yang C-S; Fukuyama Y New seco-Prezizaane-Type Sesquiterpenes, Jiadifenin with Neurotrophic Activity and 1,2-Dehydroneomajucin from Illicium jiadifengpi. J. Nat. Prod 2002, 65, 527. [DOI] [PubMed] [Google Scholar]
- (17).(a) Kakemoto E; Okuyama E; Nagata K; Ozoe Y Interaction of anisatin with rat brain γ-aminobutyric acida receptors: allosteric modulation by competitive antagonists. Biochem. Pharmacol 1999, 58, 617. [DOI] [PubMed] [Google Scholar]; (b) Schmidt TJ Toxic Activities of Sesquiterpene Lactones: Structural and Biochemical Aspects. Curr. Org. Chem 1999, 3, 577. [Google Scholar]; (c) Wilson RM; Danishefsky S Applications of Total Synthesis to Problems in Neurodegeneration: Fascinating Chemistry along the Way. J. Acc. Chem. Res 2006, 39, 539. [DOI] [PubMed] [Google Scholar]; (d) Richers J; Pöthig A; Herdtweck E; Sippel C; Hausch F; Tiefenbacher K Synthesis and Neurotrophic Activity Studies of Illicium Sesquiterpene Natural Product Analogues. Chem. Eur. J 2017, 23, 3178. [DOI] [PubMed] [Google Scholar]; (e) see ref. 13e.; (f) Witkin JM; Shenvi RA; Li X; Gleason SD; Weiss J; Morrow D; Catow JT; Wakulchik M; Ohtawa M; Lu H-H; Martinez MD; Schkeryantz JM; Carpenter TS; Lightstone FC; Cerne R Pharmacological characterization of the neurotrophic sesquiterpene jiadifenolide reveals a non-convulsant signature and potential for progression in neurodegenerative disease studies. Biochem. Pharmacol 2018, 155, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Greszler S; Reichard HA; Micalizio GC Asymmetric Synthesis of Dihydroindanes by Convergent Alkoxide-Directed Metallacycle-Mediated Bond Formation. J. Am. Chem. Soc 2012, 134, 2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hung K; Hu X; Maimone TJ Total Synthesis of Complex Terpenoids Employing Radical Cascade Processes. Nat. Prod. Rep 2018, 35, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kubo M; Okada C; Huang J-M; Harada K; Hioki H; Fukuyama Y Novel Pentacyclic seco-Prezizaane-Type Sesquiterpenoids with Neurotrophic Properties from Illicium jiadifengpi. Org. Lett 2009, 11, 5190. [DOI] [PubMed] [Google Scholar]
- (21).Shoji M; Nishioka M; Minato H; Harada K; Kubo M; Fukuyama Y; Kuzuhara T Neurotrophic activity of jiadifenolide on neuronal precursor cells derived from human induced pluripotent stem cells Biochem. Biophys. Res. Commun 2016, 470, 798. [DOI] [PubMed] [Google Scholar]
- (22).Kaur T; Wadhwa P; Sharma A Arylsulfonylmethyl isocyanides: a novel paradigm in organic synthesis RSC Adv 2015, 5, 52769. [Google Scholar]
- (23).Desai LV; Hull KL; Sanford MS Palladium-Catalyzed Oxygenation of Unactivated sp3 C–H Bonds. J. Am. Chem. Soc 2004, 126, 9542. [DOI] [PubMed] [Google Scholar]
- (24).(a) For recent examples in total synthesis, see: Sharpe RJ; Johnson JS A Global and Local Desymmetrization Approach to the Synthesis of Steroidal Alkaloids: Stereocontrolled Total Synthesis of Paspaline. J. Am. Chem. Soc 2015, 137, 4968. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meng Z; Yu H; Tao W; Chen H; Wan M; Yang P; Edmonds DJ; Zhong J; Li A Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families. Nature Commun. 2015, 6, 6096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trotta AH Total Synthesis of Oridamycins A and B. Org. Lett 2015, 17, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhou S; Guo R; Yang P; Li A Total Synthesis of Septedine and 7-Deoxyseptedine J. Am. Chem. Soc 2018, 140, 9025. [DOI] [PubMed] [Google Scholar]
- (25).(a) Brun P; Waegell B Heterocyclisation intramoleculaire d’alcools possedant un squelette cedranique. Oxydation par le tetraacetate de plomb et par l’oxyde de mercure et le brome. Tetrahedron 1976, 32, 1137. [Google Scholar]; (b) Dorta RL; Francisco CG; Freire R; Suárez E Intramolecular hydrogen abstraction. The use of organoselenium reagents for the generation of alkoxy radicals. Tetrahedron Lett. 1988, 29, 5429. [Google Scholar]
- (26).For a review of the α-ketol rearrangement, see: Paquette LA; Hofferberth JE The α-Hydroxy Ketone (α-Ketol) and Related Rearrangements. Organic Reactions 2003, 62, 477. [Google Scholar]
- (27).(a) Chen M; White MC A predictably selective aliphatic C-H oxidation reaction for complex molecule synthesis. Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]; (b) Bigi MA; Reed SA; White MC Directed Metal (Oxo) Aliphatic C–H Hydroxylations: Overriding Substrate Bias. J. Am. Chem. Soc 2012, 134, 9721. [DOI] [PubMed] [Google Scholar]
- (28).For a recent example of successful iron-catalyzed, acid-directed C–H oxidation in synthesis, see: Ye Q; Qu P; Snyder SA Total Syntheses of Scaparvins B, C, and D Enabled by a Key C–H Functionalization. J. Am. Chem. Soc 2017, 139, 18428. [DOI] [PubMed] [Google Scholar]
- (29).(a) Donohoe TJ; Moore PR; Waring MJ; Newcombe NJ The directed dihydroxylation of allylic alcohols. Tetrahedron Lett. 1997, 38, 5027. [Google Scholar]; (b) Donohoe TJ; Mitchell L; Waring MJ; Helliwell M; Bell A; Newcombe NJ Homoallylic alcohols and trichloroacetamides as hydrogen bond donors for directed dihydroxylation. Tetrahedron Lett. 2001, 42, 8951. [Google Scholar]; (c) Donohoe TJ Development of the directed dihydroxylation reaction. Synlett 2002, 8, 1223. [Google Scholar]
- (30).(a) Tenaglia A; Terranova E; Waegell B Ruthenium-catalyzed C–H bond activation oxidation of bridged bicyclic and tricyclic alkanes. Tetrahedron Lett. 1989, 30, 5271 [Google Scholar]; (b) Tenaglia A; Terranova E; Waegell B Ruthenium-catalyzed carbon-hydrogen bond activation. Oxyfunctionalization of nonactivated carbon-hydrogen bonds in the cedrane series with ruthenium tetraoxide generated in situ. J. Org. Chem 1992, 57, 5523. [Google Scholar]