

Abstract
Traditional high-throughput drug combination screening requires automatic pipetting of drugs into high density microtiter plates. Here, a drug-on-pillar platform is proposed for efficient combination drug screening. Using the proposed approach, combination drug screening can be carried out in a plug-and-play manner, allowing for high-throughput screening of large permutations of drug combinations at various concentrations, such that drug dispensing and cell-based screening can be temporally separated, and therefore can potentially be performed at distant laboratories. The dispensing is implemented using our recently developed microfluidic pneumatic printing platform, which features a low-cost disposable cartridge that minimizes cross contamination. Moreover, our previously developed drug nanoformulation method with amphiphilic telodendrimers has been utilized to maintain drug stability in a dry form, allowing for convenient drug storage, shipping, and subsequent rehydration. Combining the above features, we have implemented a 1260-spot drug combination array to study the effect of paired drugs against MDA-MB-231 triple negative human breast cancer cells. This study supports the feasibility of the drug-on-pillar platform for combination drug screening and has provided valuable insight in drug combination efficacy against breast cancer.
Graphical Abstract

INTRODUCTION
Combination chemotherapy has been used in the clinic for cancer care since the 1960s1. In the past two decades, target specific drugs such as tyrosine kinase inhibitors, proteasome inhibitors, and m-TOR inhibitors have been used in combination with standard cytotoxic chemotherapy2. It has become clear that some of the FDA-approved drugs for non-cancer indications may potentiate the anti-cancer activity of known chemotherapeutic drugs3. Thus, there is great interest in developing high-throughput approaches capable of testing large permutations of drug combinations in cultured cancer cell lines or patient-derived organoids, to effectively repurpose FDA-approved drugs. Dorshaw et al. recently reported on the therapeutic activity of over 5,000 pairs of FDA-approved cancer drugs against a panel of 60 well-characterized human tumor cell lines4. Synergistic drug combinations were found: clofarabine with bortezomib, and nilotinib with paclitaxel. Based on these encouraging results, clinical trials have been designed to test such drug combinations in human. With the advent of patient-derived tumor organoid culture using 3D-gel matrices or spheroid systems5, there is a need for rapid testing of patients’ cancer cells against drugs and drug combinations in a high-throughput and convenient manner5. We believe the novel plug-and-play, drug-on-pillar platform reported here will be able to fulfill the need for rapid discovery of novel synergistic drug combinations and selection of efficacious drug combinations for personalized cancer medicine.
Recent developments in microarray technologies have led to their successful application in cell microarrays and drug screening6. Arrays can be composed of single cells, cell monolayers, aggregates, spheroids, or even small living eukaryotes7. Microarray based screening involves two major steps: generation of cell microarrays followed by application of drug combinations or drug libraries to those arrays. The capability to create multiple spots on the microscale allows for high-throughput parallel cell-based assays8. Such microarray technologies are diverse and can be categorized into microwell type, surface patterned arrays, or microfluidics-based microarrays. Microwells, as the name suggests, describes arrays of wells where cells can be separately seeded and cultured. Many types of soft materials are being used to create the microwells. Mirjam et al. demonstrated the ability to grow single cells in 3D culture using polydimethylsiloxane (PDMS) microwells9. Hydrogels comprised of polyethylene glycol (PEG)10,11, chitosan12, or agarose13,14 have also been applied to create microchambers for cell growth. On the other hand, one can also pattern flat surfaces to generate separated spots for cell culture. Most commonly, alternating hydrophilic and hydrophobic coatings are patterned such that cell solution can only be spotted where the surface is hydrophilic. Other than hydrophilic/hydrophobic patterning, some moieties are used to provide adequate environments for cell growth, e.g. alginate15,16 and cellulose17, while cell-free surfaces are typically blocked using PEG18,19 or BSA20 to prevent non-specific absorption. With regards to scale, modern soft lithography methods can easily produce either wells or chemically- patterned surfaces with submicron precision21. However, this require an intermediate step of engineering custom photomasks for each specific pattern, which limits flexibility and increases the cost. Microfluidic approaches to generate cell compartments are an alternative technology. Cell arrays are enclosed within a fabricated microfluidic chip containing multiple branched channels leading to separated partitions22,23. The design of microfluidic chips provides high flexibility to implement different kinds of reactions. Pumps and valves are always used along with the chip for liquid manipulation. For example, Wang et al. reported a pneumatic valve controlled microfluidic chip for cell culturing and study23. Despite the high flexibility, one microfluidic chip is usually used for only one specific application. Given the design and fabrication complexity of such chips, the cost is very high and the applications limited. Another hurdle is their compatibility with existing biomedical approaches, including the well plate-based assays. Many protocols have been laboriously developed using traditional well plates, and may not easily transfer to microcell arrays.
A major area of promise for microscale array-based screening is high-throughput testing of drug combinations or libraries on compartmentalized cell arrays. The most straightforward approach is to dispense each drug onto the cell array using an automated dispenser or pipettor24,25, where biochemical reagents are loaded in a cartridge or pipette. Computer programs then coordinate the dispensing and stage movement, generating droplets on-demand at specific locations, thus generating any desired pattern in a high-throughput manner. However, such automated systems come with high equipment costs, thus are not widely available. Moreover, due to their high fabrication cost, dispensing heads are often recollected and washed for multiples use, and extra care must be taken to avoid cross contamination. Serial operation style of drug loading can also be very long for large scale arrays, and thus drug-cell reaction time is hard to control, introducing error. Some alternative technologies have been developed to dispense drugs to the cell array, including stamp26 and microfluidic22,27 methods,. In stamp method, drugs are pre-loaded onto a separate chip, and then “stamped” against the cell array for drug transference. Nevertheless, the pre-loading of drug libraries still requires sophisticated equipment, which is not accessible for most labs or institutes. Microfluidic methods utilize microscale networks to deliver multi-drug libraries into separate compartments. However, the design and fabrication of such networks is typically complicated. Unless mass-produced, the cost per analysis of microfluidic chip remains high, making it difficult for microfluidic methods to be widely applied.
In this study, we propose a novel process for personalized drug screening, of which no expensive machines are required, and drug combination libraries can be generated and screened against tumor cells directly at clinical laboratories. In this approach, we have utilized our previously developed microfluidic pneumatic printing platform28–30 to generate drug combination libraries in a high-throughput manner on a pillar microarray. Advantages of our microfluidic printing approach are that the resulting drug loading cartridges are inexpensive and disposable, effectively eliminating cross contamination, and only small volumes (e.g. 100 μL) of drug stock solutions are needed.
A major technical barrier to produce stable, lyophilized drug combination chips is their hydrophobic nature, which is responsible for their poor water solubility. They can be easily dried out during manufacturing, but rehydration at the site of the experiment is difficult. To overcome this we leveraged our expertise in the use of cholic acid-based amphiphilic polymers (so-called telodendrimers) to nanoformulate potent hydrophobic anti-cancer drug such as paclitaxel into water soluble nanoparticles, nanotaxane31. Such technology permits long-term storage of drug as lyophilized powder, which months later can be subsequently rehydrated with water or saline prior to administration.
Here we applied nanoformulated drug combinations (using our established telodendrimer-based technology to confer better water solubity31) printed onto micropillar array chips, fabricated to fit by insertion into standard 1536 well plates, thus allowing for a direct plug-and-play screening process. In this way, drug combinatorial libraries can be designed, printed onto pillar arrays, stored dry, and shipped to clinical laboratories for direct screening with patients’ primary tumor cells or organoids, without the need for setting up expensive liquid dispensing equipment and handling of many drugs. The drug-on-pillar platform reported here allows the dispensing of cancer drugs and drug combinations at a central facility with robotics, and the shipment of the testing sets to any clinical facilities, where plug-and-play operations can occur. Here we establish and test a simple two-drug per pillar combinatorial library that is comprised of 7 different drugs, each at 3 different concentrations. The resulting 1260-spot pillar array was screened against MDA-MB-231 triple negative breast cancer cells. Notably, in addition to establishing solid technical proof-of-principle for this technology, several synergistic combinations emerged from this study, further indicating that this platform is practical and functional. When applied in conjunction with primary tumor organoid culture, the results may allow the medical oncologist to tailor specific drug combinations for specific patient.
EXPERIMENTAL SECTION
Micropillar array fabrication
4 mm-thick poly(methyl methacrylate) (PMMA) was laser patterned (Universal Laser Systems) into a 48-by-32 array of through-holes with a center-to-center distance of 2.25 mm (same as a Corning® 1536 well plate). The diameter of each hole was 1 mm. Four 2 mm-thick PMMA blocks were then bonded to the four corners of the through-hole array as a spacer. The table-like structure was then placed in a petri dish. PDMS prepolymer was prepared at a ratio of base:curing agent = 10:1, which was then slowly poured into the petri dish from the side. The petri dish was then placed in a leveled surface until PDMS liquid filled all the PMMA holes due to capillary force. The PDMS was then cured in 70 °C oven for 1 h, after which it was released from the PMMA mold, resulting in a complementary micropillar array.
Microfluidic pneumatic printing (MPP)
The MPP platform is composed of a printer head, a 2-axis traveling stage, the control software, and a custom circuit. The printer head is the key component of the platform and is formed as an assembly of two parts: 1) an electromagnetic valve array (The Lee Co.) that controls the compressed air and 2) a cartridge where chemical reagents are loaded and printed from. The valve array is mounted on a customized PMMA manifold with a common compressed air inlet. The cartridge is a two-layer structure fabricated by laser patterning (Universal Laser Systems) and plasma bonding of PDMS membranes, of which the two layers are comprised of nozzle layer and channel layer with inlet, respectively. The cartridge is assembled with the valve manifold via tubing connections. The outlet of each valve is either connected with compressed air, or atmosphere, depending on the voltage level of control signal provided by the circuit and software. In each printing cycle, a pulse voltage signal results in a compressed air pulse towards the cartridge, generating enough force for droplet ejection.
Before printing, several chemotherapeutic drugs were formulated into a micellar nanocarrier system with superior drug loading and narrow size distribution, that we have recently reported for in vivo drug delivery31. Nanoformulated drugs were first loaded into the cartridge through an inlet reservoir. PDMS micropillar arrays sterilized with 70% ethanol were placed on a 1536 well plate cover, which was mounted on the traveling stage. Calibration was performed so that the starting position was aligned with one corner of the micropillar array. The droplet pattern, including array size and number of droplets per spot, was designed in an Excel file, which was then imported into the control software, where other parameters including array dimensions, stage moving speed, and printing frequency were defined and used to coordinate the stage movement and dispensing of the printer head through a driver and custom circuit. Each cartridge integrates 8 channels, allowing for printing of 8 different drugs in one printing cycle. In this study, 7 different drugs are used, including doxorubicin, nilotinib, olaparib, capsaicin, tamoxifen, cisplatin, and tretinoin, while on each pillar either 1 or 2 different drugs are printed with the volume of each drug varying among 10 nL, 50 nL, or 100 nL. This resulted in a library size of . The same library was repeated 6 times for each micropillar array chip, resulting in a total array size of 210×6=1260, which could fit in a standard 1536 well plate. All these 7 drugs except for doxorubicin and cisplatin are almost insoluble in water. All these 7 drugs are soluble in methanol during the nanoformulation procedure. After the printing was complete, the drug-pillar chip was stored in a bio-safety cabinet so that the drugs could dry under sterile condition.
Cell culture in 1536-well plate
MDA-MB-231 cells were maintained in DMEM medium with 10% FBS, and 1% PS, in an incubator with 5% CO2. Prior to each experiment, fresh cell medium was added to cover the entire 1536 well plate, followed by plate centrifugation to remove air bubbles. Cells were spun down and resuspended before applied to the 1536 plate, resulting ~1000 cells per well after the cells sank to the bottom due to gravity. The cell seeded well plate was incubated for 24 h in a 5% CO2 incubator.
Drug treatment and cell viability determination
The drug combinatorial library was then applied by attaching the drug-pillar chip to the cover of 1536 well plate, followed by flipping over and covering on top of the well plate. The chip and plate cover were aligned so that drug-loaded pillars can be inserted into the wells and submerged in culture medium to ensure drug rehydration. Prior to the insertion, aspiration was applied to each well to prevent medium overflow due to pillar insertion. After the drug-pillar chip was applied, the whole assembly was incubated in a 5% CO2 incubator for 72 h.
After 72 h of treatment, the drug-pillar chip was removed from the top, and the wells emptied by decanting. Calcein AM solution (2 μM) was then added to the well plate and incubated for 15 min to stain live but not dead cells32. The calcein AM solution was then removed, and replaced with fresh medium. The plate was then scanned with a 1536 plate reader (Filtermax F5, Molecular Devices) with excitation and emission wavelengths of 495 and 515 nm, respectively. For single-drug experiment, the plate was also imaged by confocal microscopy (Zeiss LSM 800) to visualize cell morphology in the green fluorescent channel. Fluorescent intensities were quantified. All data were shown as the mean of at least three experiments independently accompanied by standard error of mean. The dose-response curves were plotted and IC50 values of single-drugs were evaluated by GraphPad Prism software (GraphPad Software, USA). For the combined drug response, the combination effects of two drugs (A and B) were analyzed in terms of combination index (CI) theorem of Chou-Talalay 33 by the following equation:
(Dx)A is the dose of drug A required to produce x% effect alone; (D)A is the dose of drug A required to produce the same x% effect in combination with (D)B. Similarly, (Dx)B is the dose of drug B required to produce x% effect alone; (D)B is the dose of drug B required to produce the same x% effect in combination with (D)A. The CI values were calculated by CompuSyn software based on IC50 values of each single drug and the dose-response data of drug combinations. The present experiment was carried out in non-constant ratio combinations and the CI values could be determined at their corresponding specified data points 34.
RESULTS AND DISCUSSION
Drug-on-pillar screening work flow
The entire drug printing and screening process is illustrated in Figure 1, which is comprised of three major steps. In Step 1, PDMS pillar array was fabricated by cast molding out of PMMA hole array (Figure 1A). The shape of pillar array was complementary to the PMMA mold. Thus, different sizes of pillar array could be achieved simply by laser patterning the PMMA chip with different sizes. Here, we implemented pillar array of 384 and 1536 scale (Figure S1), corresponding to standard multi-titer well plate footprints. However, denser pillar array can be easily fabricated with the same approach, if needed. Due to the wetting affinity of PDMS liquid to PMMA material, uncured PDMS could fill in the PMMA holes due to surface tension. In this way, the pillar height could be controlled by the PMMA thickness. In this study, we designed the pillar height to be 4 mm in order to make sure the pillar can be inserted and made in contact with medium in the multi-titer well plates, while avoiding overflow. Moreover, the resulted pillar would have a concave surface at the tip due to the affinity of PDMS material to PMMA (Figure 1D), which is preferable for drug loading.
Figure 1.
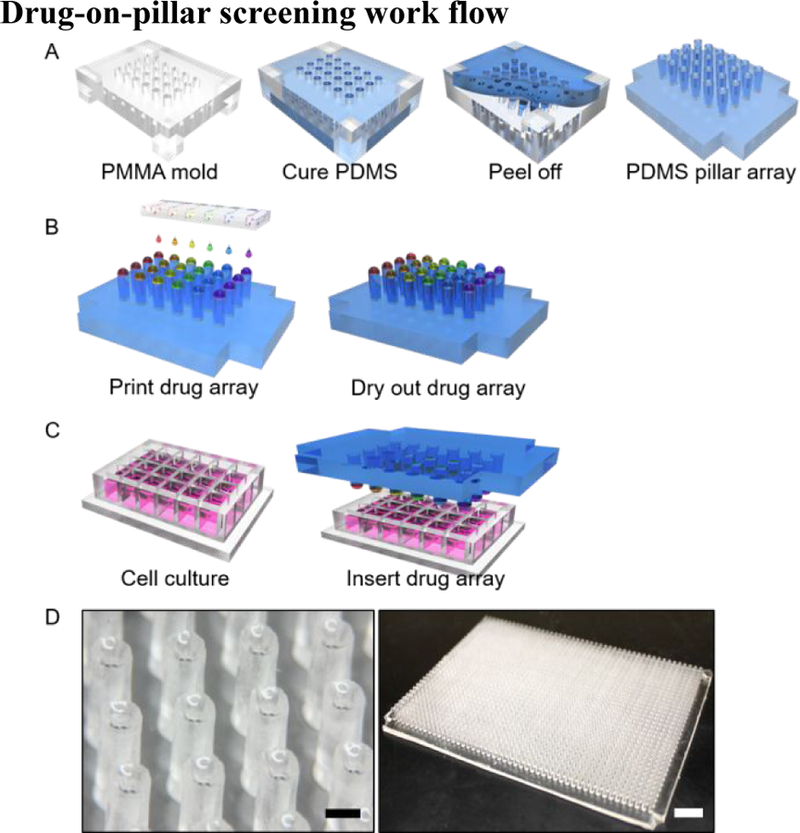
A-C, Plug-and-play style drug-on-pillar screening workflow: A) Step 1: cast molding of pillar array; B) Step 2: drug loading using microfluidic pneumatic printing; C) Step 3: plug in the drug array with multi-well plate for drug cell interaction. D) Microscopic picture of pillar array (left, scale bar: 1mm) and full view of a 1536 pillar array (right, scale bar: 1 cm)
In Step 2, drugs were automatically printed onto the top of each pillar, forming the drug-on-pillar combination library (Figure 1B). The previously reported microfluidic pneumatic printer28 was used to print the combinatorial drug pillar-arrays. As reported in our prior work, droplet size can be adjusted with a standard error of less than 5%, which indicates good accuracy of the printing platform. Taking advantages of soft lithography, modifications were made to improve the fabrication of the cartridge, resulting in a two-layer design instead of the four-layer one previously used (Figure S2). In this way, the cost and time consumption of cartridge fabrication could be further reduced, resulting in a disposable design, essential for biomedical applications where contamination is a serious concern. In traditional dispensing machines, due to the high cost of printer head and highly integrated design, cartridges are typically used multiple times, which could cause contamination if washing is not performed thoroughly. The newly fabricated microfluidic cartridge has 8 compartments, with one drug per compartment and each compartment controlled individually. If needed, additional compartments can be added to the cartridge design. The microfluidic printing platform is capable of implementing any customized drug pattern design in an automated fashion.
In Step 3, the drug-pillar assembly and the multi-well plate containing cancer cells, in this case a standard 2D culture in 1536 microwell plate, were brought together in a pluggable fashion (Figure 1C). Placement of the drug pillar microarrays over the microwell plate allowed the drugs to rehydrate and diffuse into the liquid culture medium. Many small molecule or natural product macrocycle anti-cancer drugs are hydrophobic. Typically, most investigators dissolve these drugs in DMSO prior to dilution into the medium. In our drug-pillar platform, nanoformulated drugs are applied as a telodendrimer-based nanoplatform developed by us previously31. We utilized paclitaxel, a hydrophobic chemotherapeutic agent commonly used in the clinic and found that the nanoformulated paclitaxel in lyophilized dried powder form can be easily rehydrated with water or saline to form a clear solution of nanotaxane. In this study, we used a similar approach to nanoformulate our test set of hydrophobic drugs. To verify the stability and recovery of these nanoformulated drugs from the drug pillar-arrays, we compared the cytotoxic activity of freshly printed nanoformulated drugs to drug arrays printed 5 days earlier (Figure S3). Identical cytotoxic potency was observed. This result suggested that drugs or drug combination libraries can be nanoformulated prior to printing, and the resulting drug combination arrays can be stored in dry nanoformulated form and shipped to clinical institutes for screening. In this way, no expensive dispensing machine was required at the clinical test site, and the oncologists can design and outsource the drug combination library preparation to institutes specialized in drug dispensing, which can provide drug preparation services for a large number of clinical laboratories at the same time.
One may argue that there is no need for drug pillar-arrays, since nanoformulated drugs can be directly dispensed into multi-titer wells, dried, and then shipped. The problem with that approach is that most primary culture requires time (a few days to a week or two) to establish in enriched medium, therefore direct transfer of primary cells to drug-filled plate, such as patient-derived organoid cuture, is unlikely to work. Furthermore, the plug-and-play platform can be further miniaturized such that only a minimal number of cells will be needed from the patient. This would be difficult to achieve with standard multi-titer plates.
Single-drug experiment for comparability
The comparability of the present plug-and-play screening platform with conventional methods using 96-well plates was conducted with a single-drug assay. Figure 2B shows the printed drug array, in which 0, 10, 50, 100 nL of doxorubicin (1, 5 and 10 drops, respectively; stock conc. 1 mg/mL, 10 nL per drop) was printed in a separate 5 × 5 arrays, respectively, forming a 10 × 10 array. Drugs were dried out over night before culturing with cells to simulate the storing and transferring procedure. After a 48 h incubation with human breast cancer MDA-MB-231 cells, cell morphology was observed and captured every 24 h, as shown in Figure 2A. After doxorubicin treatment for 48 h, cell viability was detected with cell-permeant dye calcein AM, which was enzymatically converted to a green-fluorescent calcein by esterases in live cells but not dead cells. Fluorescence intensity, corresponding to the number of live cells, was determined by a confocal microscope in Figure 2C, and the cell viability variation after doxorubicin treatment was calculated by fluorescent intensity and represented as percentage of control. An inhibitory dose-response curve was fitted as shown in Figure 2D, and the IC50 value (50% of growth inhibition) of nanoformulated doxorubicin on MDA-MB-231 cells for 48 h exposure was calculated to be 3.27 μM. The bright circular field in Figure 2A is due to the presence of the pillar during the imaging of the bright field picture, which induces light reflection and transmission different that causes the bright circular field. However, the height of pillar is designed to not contact the bottom surface of the well plate. Moreover, final images (fluorescent pictures) are taken after removal of the pillar array, and results also shown no significant difference in the circular area, from which we conclude the pillar did not affect the result. Meanwhile, we performed a conventional method with 96-well plates for growth inhibition to determine IC50 values in parallel, and a comparable IC50 value of 2.97 μM was obtained. Hence, it can be concluded that this plug-and-play screening platform implemented by microfluidic printing was comparable with conventional methods in multi-well plates.
Figure 2.

Single-drug experiment with plug-and-play screening platform. a. Cell morphology alteration after 24-/48 h doxorubicin treatment (1, 5 and 10 drops, respectively; stock conc. 1 mg/ml, 10 nL per drop), magnification of 10X and 20X; b. The printed drug array after a dry overnight, in which 1, 5 and 10 drops of doxorubicin, respectively; stock conc. 1 mg/mL, 10 nL per drop; c. Fluorescence intensity alteration after a 48 h doxorubicin treatment, captured by confocal microscope; d. The inhibitory dose-response curves determined by the present plug-and-play screening platform and the conventional 96-well plate were fitted and the corresponding IC50 values were calculated.
Drug library design and high-throughput combinatorial drug screening
As a proof of concept, a drug library including 7 nanoformulated drugs with a series of concentrations (1, 5 and 10 drops, 10 nL per drop, stock conc. at 1 mg/mL) were combined randomly, composing 189 pairwise drug combinations, namely nilotinib (Nil), doxorubicin (Dox), olaparib (Ola), capsaicin (Cap), tamoxifen (Tam), cisplatin (Cis), and tretinoin (Tre), represented as A, B, C, D, E, F and G, as shown in Figure S4a. The numbers (1, 2, and 3) represented three concentrations for each drug. Along with 21 single-drug groups from 7 drugs with 3 concentrations, the total number of drug spots was 210, which constituted a 14 × 15 array, as shown in Figure S4B. Furthermore, a 1260-spot drug array was obtained with 6 repeats of 14 × 15 array in one 1536-well plate in Figure S5, and the vehicles were in the remaining empty wells of 1536-well plate. This drug pattern was demonstrated by printing food dyes onto a 1536-pillar array, as shown in Figure 3A. The total time it took to print such 1260 library was 15 min. After drug treatments in the plug-and-play screening platform for 48 h, cell viability of human breast cancer MDA-MB-231 cell was detected by calcein AM. 1260 spots with green-fluorescent densitometry alteration of calcein were analyzed by confocal microscopy and the corresponding growth inhibition was calculated and represented as percentage of control in a fluorescent heat map, as shown in Figure 3B.
Figure 3.

Drug printing patterns and fluorescent microscopic readouts through the drug-on-pillar microfluidic high-throughput combinatorial drug screening platform. A) A 14 × 15 array containing 189 pairwise drug combinations for 7 drugs with 3 concentrations and 21 single-drug groups, eventually a 1260 drug spots with six repeats in one 1536-well plate was formed. This drug pattern was demonstrated by printing food dye on 1536-pillar array; B) The screening result from the plug and play high-throughput combinatorial drug screening platform, represented by green-fluorescent densitometry alteration of calcein AM.
For single-drug groups, the corresponding inhibitory dose-response curve was fitted in Figure 4. The corresponding IC50 value against MDA-MB-231 cells for 48 h exposure was calculated to be 1.35μM (Tre), 2.18μM (Ola), 3.27μM (Dox), 10.84μM (Nil), 15.87μM (Cap), 15.89μM (Cis), and 26.01μM (Tam). The combination drug response was analyzed by combination index (CI) theorem of Chou-Talalay to quantitatively determine synergism, additive effect, or antagonism33,34.
Figure 4.

The inhibitory dose-response curve of single drugs. The inhibitory dose-response curves determined by the present plug-and-play screening platform, represented by cell viability of human breast cancer MDA-MB-231 cell using calcein AM staining and the corresponding IC50 values were calculated.
The combinatorial drug response of 189 pairwise drug combinations were calculated by CompuSyn software 33,34 based on IC50 values of single drugs and the dose-response data for drug combinations, as shown in Table 1. The present experiment was carried out in non-constant ratio combinations and the CI values could be determined at their corresponding specified data points 34. The values of CI<1 (*, CI<1 in Table 1) demonstrate the pairwise combinations with synergistic effect, while the values of CI=1 or >1 represent the pairwise combinations with an additive or antagonistic effect, respectively. Among 189 pairwise drug combinations in our present study, only a limited number of combinations showed significant synergistic effects. For example, combinations of olaparib and tamoxifen, as well as olaparib and tretinoin, showed the most potent synergistic effect with the values of CI<1 (*, CI<1), as shown in Table 1. In the case of olaparib (PARP inhibitor) and tamoxifen (ER antagonist), we speculate that the observed synergism is the result of PARP regulation of cellular response to estrogen. Namely, PARP1 has been shown to physically bind to and poly(ADP-ribosyl)ate ERα, resulting in increased binding to target genes 35. In the MCF7 breast cancer cell line, PARP1 is required for ERα transcriptional activity to occur in response to 17β-estradiol 36, and is potentially a co-activator of ERβ 37. Simultaneous targeting of both PARP1 and ER may therefore be leading to non-additive downregulation of ER target genes. Further experiments using a finer series of dosages will be needed to more systematically evaluate their synergy, as well as verify their mechanism of action. Synergistic effects were also observed in combinations of tamoxifen with either tretinoin or cisplatin, especially at lower concentrations of tretinoin and cisplatin (1 drop and 5 drops). Synergism was also detected in combination tamoxifen and capsaicin, which was significant at lower concentrations of capsaicin (1 drop and 5 drops). A similar observation was found in combinations of Nil-Tre and Nil-Tam; at the lower concentrations of nilotinib (1 drop and 5 drops). Other pairwise drug combinations with potential synergistic effect or nearly additive effect (0.9<CI<1.1), and antagonistic effect (CI>1) can be found in Table 1. The majority of the remaining pairwise drug combinations showed additive or antagonistic effect indicated by CI values >1. For example, doxorubicin shows a strong antagonistic effect with cisplatin; the CI values was determined to be 2~10, at the higher concentrations of doxorubicin. In addition to cisplatin, doxorubicin also shows strong antagonistic effect with capsaicin or nilotinib, most of the CI values were found to be greater than 4.
Table 1.
The CI values of combinatorial drug.
| Ola-Tam | Ola-Tre | Tam-Tre | Tam-Cis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tam (Drops) | Combination Intex (CI) |
Tre (Drops) | Combination Intex (CI) |
Tre (Drops) | Combination Intex (CI) |
Cis (Drops) | Combination Intex (CI) |
||||||||
| Ola (Drops) | Ola (Drops) | Tam (Drops) | Tam (Drops) | ||||||||||||
| 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | ||||
| 1 | 0.65310* | 1.47217 | 0.82157* | 1 | 0.27756* | 0.79402* | 0.66028* | 1 | 0.12875* | 0.25304* | 0.40744* | 1 | 0.21194* | 0.47944* | 0.46650* |
| 5 | 0.50163* | 0.48219* | 0.66615* | 5 | 0.64385* | 0.93317* | 1.44454 | 5 | 0.32363* | 0.83603* | 0.62365* | 5 | 068378* | 0.93755* | 0.92834* |
| 10 | 0.45848 | 0.61190* | 0.71237* | 10 | 0.60102* | 0.58760* | 0.78370* | 10 | 0.78332* | 1.45322 | 1.25311 | 10 | 1.21246 | 1.42042 | 1.32280 |
|
Cap-Tam |
Cis-Tre |
Nil-Tre |
Nil-Tam |
||||||||||||
| Tam (Drops) | Combination Intex (CI) |
Tre (Drops) | Combination Intex (CI) |
Tre (Drops) | Combination Intex (CI) |
Tam (Drops) | Combination Intex (CI) |
||||||||
| Cap (Drops) | Cis (Drops) | Nil (Drops) | Nil (Drops) | ||||||||||||
| 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | ||||
| 1 | 0.20820* | 0.71509* | 1.35507 | 1 | 1.04152 | 0.49349* | 0.98342* | 1 | 0.85842* | 0.76347* | 1.05067 | 1 | 0.32548* | 1.10506 | 3.42433 |
| 5 | 0.44361* | 0.92425* | 1.49784 | 5 | 0.24496* | 0.58262* | 1.67863 | 5 | 0.65562* | 0.77043* | 1.40077 | 5 | 0.39691* | 0.78806* | 1.36773 |
| 10 | 0.50786* | 0.98764* | 1.57417 | 10 | 0.50006* | 1.16152 | 1.93432 | 10 | 0.87929* | 1.43968 | 1.61391 | 10 | 0.51410* | 0.90337* | 1.38978 |
|
Cap-Tre |
Nill-Cis |
Dox-Tre |
Cap-Cis |
||||||||||||
| Tre (Drops) | Combination Intex (CI) |
Cis (Drops) | Combination Intex (CI) |
Tre (Drops) | Combination Intex (CI) |
Cis (Drops) | Combination Intex (CI) |
||||||||
| Cap (Drops) | Nil (Drops) | Dox-(Drops) | Cap (Drops) | ||||||||||||
| 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | ||||
| 1 | 2.49504 | 0.75550* | 1.26651 | 1 | 0.29623* | 0.90987* | 1.61823 | 1 | 0.45571* | 0.86613* | 1.31753 | 1 | 0.33489* | 1.03897 | 1.77594 |
| 5 | 0.58158* | 0.84714* | 1.32983 | 5 | 0.81604* | 1.30334 | 1.78392 | 5 | 1.18081 | 1.53084 | 1.26625 | 5 | 0.91034* | 1.64054 | 2.27665 |
| 10 | 0.83450* | 1.52140 | 1.86865 | 10 | 1.33447 | 1.80258 | 2.39999 | 10 | 1.74578 | 2.06148 | 2.79286 | 10 | 1.54962 | 2.16294 | 2.80159 |
|
Dox-Tam |
Ola-Cis |
Ola-Cap |
Dox-Cap |
||||||||||||
| Tam (Drops) | Combination Intex (CI) |
Cis (Drops) | Combination Intex (CI) |
Tre (Drops) | Combination Intex (CI) |
Cap (Drops) | Combination Intex (CI) |
||||||||
| Dox (Drops) | Nil (Drops) | Ola-(Drops) | Dox (Drops) | ||||||||||||
| 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | ||||
| 1 | 7.79926 | 9.41277 | 10.8397 | 1 | 0.84093* | 1.96886 | 1.64439 | 1 | 0.47762* | 1.55271 | 2.56721 | 1 | 0.84026* | 4.76273 | 5.51714 |
| 5 | 2.91221 | 1.95826 | 1.24397 | 5 | 1.53134 | 2.37974 | 2.91431 | 5 | 1.7266 | 3.61031 | 5.90278 | 5 | 2.37791 | 6.10412 | 8.38702 |
| 10 | 0.54474* | 1.08101 | 1.45915 | 10 | 1.76827 | 3.68168 | 4.45954 | 10 | 2.90287 | 5.56389 | 7.45382 | 10 | 4.06555 | 7.61526 | 8.03917 |
|
Nil-Ola |
Nill-Cap |
Nill-Dox |
Ola-Dox |
||||||||||||
| Ola (Drops) | Combination Intex (CI) |
Cap (Drops) | Combination Intex (CI) |
Dox (Drops) | Combination Intex (CI) |
Dox (Drops) | Combination Intex (CI) |
||||||||
| Nil (Drops) | Nil (Drops) | Nil (Drops) | Ola (Drops) | ||||||||||||
| 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | 1 | 5 | 10 | ||||
| 1 | 0.58030* | 1.16794 | 1.95791 | 1 | 0.35381* | 1.02499 | 2.18424 | 1 | 0.79058* | 1.22547 | 1.22547 | 1 | 2.25306 | 0.93978* | 1.71674 |
| 5 | 1.35551 | 2.69480 | 3.75598 | 5 | 1.04574 | 1.84586 | 2.57392 | 5 | 4.37652 | 4.22207 | 5.63754 | 5 | 1.14035 | 1.59425 | 2.49591 |
| 10 | 2.31817 | 4.80135 | 6.66358 | 10 | 2.02962 | 2.59638 | 3.20377 | 10 | 6.06199 | 6.83303 | 5.83905 | 10 | 2.14189 | 3.25897 | 4.08724 |
|
Dox-Cis |
|||||||||||||||
| Cis (Drops) | Combination Intex (CI) |
||||||||||||||
| Dox (Drops) | |||||||||||||||
| 1 | 5 | 10 | |||||||||||||
| 1 | 1.78383 | 7.95590 | 9.90485 | ||||||||||||
| 5 | 1.12480 | 2.59548 | 4.53774 | ||||||||||||
| 10 | 1.48288 | 2.73447 | 2.87856 | ||||||||||||
Note: The IC50 values of different single-drugs shown in Figure 4 are calculated for the subsequent determination of CI value in pairwise drug combinations. The combinatorial drug response of 189 pairwise drug combinations was analyzed by combination index (CI) theorem of Chou-Talalay. The CI values were calculated by CompuSyn software based on IC50 values of single drugs and the dose-response data of drug combinations, which were to demonstrate the pairwise combinations with synergistic effects (CI value <1, *), additive effects (CI value =1) or antagonistic effects (CI value >1), respectively.
The above result indicates that drug concentrations can greatly influence the CI values. Therefore, for drug combination testing and for repurposing of non-cancer drug for cancer applications, one will need to test not just two or three combinations at a single drug dose level, but drug combinations at multiple dose levels. Most drug combination screening studies reported in the literature do not consider pharmacodynamic separation of drug combinations, and the drugs are usually added to the cells at the same time38. It is now known that some drug combinations work better if they are administered in sequence. For example, Erlotinib, an EGFR tyrosine kinase inhibitor, when given concomitantly with cytotoxic drugs, are antagonistic. To be effective, cytotoxic drug needs to be given before Erlotinib on different days. The plug-and-play platform is particularly suitable for testing such drug combination sequence as two different drug pillar-arrays can be “plugged” in sequentially on two different days into the same multi-well plate. Collectively, the present plug-and-play platform provides an approach to screen the potential synergistic pairwise drug combinations of large number of drugs against patients’ cancer cells in a fully customized manner, rapidly and conveniently. As the technology for patient-derived organoid culture has become a routine, on-site testing of patients’ primary cancer cells with drug pillar-arrays could guide the oncologists to select the most appropriate drug combinations for a specific patient. Thus, we expect this technology will have a significant impact on precision cancer medicine in the future.
CONCLUSION
In this study, we have introduced a new and highly efficient approach for drug combination screening based on drug-printed micropillar arrays. The proposed drug-on-pillar array allows for convenient in vitro drug screening in a plug-and-play manner, which greatly reduces operational cost and time. The current pillar array has been designed to be compatible with standard 1536 multi-well plates, thus can be easily adaptable to traditional cell-based assays, such as live and dead determination using dye and commercially available plate reader. If needed, the pillar array footprint can be further miniaturized to afford higher density testing than the standard 1536 wells. The fabricated pillar arrays have a concave top surface, providing better capability of drug loading. Fabrication methods for pillar array is easy and inexpensive, and can be readily mass produced. The drug-on-pillar design has been combined with our previously developed microfluidic pneumatic printing platform, which is used for drug library loading in this study. The microfluidic printing platform allows for fully customizable drug combination library implementation, thus providing a potential tool for personalized medicine. At the same time, the microfluidic printing platform still demonstrates several advantages including inexpensive and disposable cartridge, capability of multi-channel integration, as well as non-contamination features. Moreover, the use of telodendrimers to nanoformulate drugs or drug combinations allows printed drugs to be stored in a dry form, shipped, and rehydrated during cell-based or organoid cytotoxic or functional assays. To sum up, the combination of the drug-on-pillar design, the microfluidic printing platform, and the drug nanoformulation provide a whole new approach for combination drug screening, of which drug loading and cell response assay can be carried out efficiently and conveniently. Future work includes expansion of the multi-channel integration capability from 12 channels to 24 channels, as well as enclosing the printing platform inside a sterile enclosure. We will also optimize the drug dosing range coverage (e.g. 0, 1, 2, 4, and 8 drops, instead of 0, 1, 5 and 10 drops) and use lower concentration of drug in the stock solution (0.2mg/mL instead of 1mg/mL) for CI value determination. We envision that in future clinical application, synergistic drug combinations against primary cell or organoid culture derived from patients’ biopsy specimens can be rapidly identified by this high-throughput platform. Such combinations can then be evaluated in patient-derived xenograft (PDX) models39–41, which is very expensive and time-consuming, prior to clinical implementation.
We have demonstrated the feasibility of the proposed drug-on-pillar approach through a single-drug study of doxorubicin against human breast cancer MDA-MB-231 cells. The resulting IC50 values of doxorubicin were comparable to that obtained using standard multi-well plate based IC50 assay. To further study the synergistic effects of different drug combinations, we have designed and implemented a combinatorial drug-on-pillar library with 1260 spots, and screened them against MDA-MB-231 cells cultured in a standard 1536 well plate. Moreover, as a result of the combination drug library screening, we have identified lead single drugs against MDA-MB-231 cells, including tretinoin, olaparib, and doxorubicin (consistent with existing reports), as well as drug combinations with synergistic effects, including olaparib and tamoxifen, olaparib and tretinoin, as well as tamoxifen and tretinoin. The results have not only demonstrated the feasibility of proposed drug-on-pillar approach, and its compatibility with standard 1536 multi-well plate based assays, but have also provided data for further study of combination drug therapy against human breast cancer.
Supplementary Material
ACKNOWLEDGMENT
This research work has been supported in part by the National Science Foundation Awards ECCS-0846502 and DBI-1256193, National Institute of Health Awards R21CA173243, NIEHS-P42ES004699, R01CA115483, R01EB012569, and also by the Fundamental Research Funds for the Central Universities lzujbky-2018–135. JL acknowledges the assistance from Kuo-Hao Tseng for fabrication and Qi Gao for statistical analysis.
Footnotes
ASSOCIATED CONTENT
Supporting Information
(i) Pictures of pillar arrays with different sizes, (ii) Microfluidic printing cartridge and its fabrication process, (iii) Nanoformulated drug stability verification, (iv) Drug library design, and (v) drug library layout are included the electronic supplementary material (pdf). The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Li MC; Whitmore WF Jr.; Golbey R; Grabstald H JAMA 1960, 174, 1291–1299. [DOI] [PubMed] [Google Scholar]
- (2).Yardley DA Breast Cancer (Auckl) 2013, 7, 7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ben Sahra I; Le Marchand-Brustel Y; Tanti JF; Bost F Mol Cancer Ther 2010, 9, 1092–1099. [DOI] [PubMed] [Google Scholar]
- (4).Holbeck SL; Camalier R; Crowell JA; Govindharajulu JP; Hollingshead M; Anderson LW; Polley E; Rubinstein L; Srivastava A; Wilsker D; Collins JM; Doroshow JH Cancer Res 2017, 77, 3564–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fatehullah A; Tan SH; Barker N Nat Cell Biol 2016, 18, 246–254. [DOI] [PubMed] [Google Scholar]
- (6).Du G; Fang Q; den Toonder JM Anal Chim Acta 2016, 903, 36–50. [DOI] [PubMed] [Google Scholar]
- (7).Barata D; van Blitterswijk C; Habibovic P Acta Biomater 2016, 34, 1–20. [DOI] [PubMed] [Google Scholar]
- (8).Xu F; Wu J; Wang S; Durmus NG; Gurkan UA; Demirci U Biofabrication 2011, 3, 034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ochsner M; Dusseiller MR; Grandin HM; Luna-Morris S; Textor M; Vogel V; Smith ML Lab Chip 2007, 7, 1074–1077. [DOI] [PubMed] [Google Scholar]
- (10).Gobaa S; Hoehnel S; Roccio M; Negro A; Kobel S; Lutolf MP Nat Methods 2011, 8, 949–955. [DOI] [PubMed] [Google Scholar]
- (11).Yoshimoto K; Ichino M; Nagasaki Y Lab Chip 2009, 9, 1286–1289. [DOI] [PubMed] [Google Scholar]
- (12).Fukuda J; Khademhosseini A; Yeo Y; Yang X; Yeh J; Eng G; Blumling J; Wang CF; Kohane DS; Langer R Biomaterials 2006, 27, 5259–5267. [DOI] [PubMed] [Google Scholar]
- (13).Moskaluk CA; Stoler MH Diagn Mol Pathol 2002, 11, 234–238. [DOI] [PubMed] [Google Scholar]
- (14).Kang G; Lee JH; Lee CS; Nam Y Lab Chip 2009, 9, 3236–3242. [DOI] [PubMed] [Google Scholar]
- (15).Rowley JA; Madlambayan G; Mooney DJ Biomaterials 1999, 20, 45–53. [DOI] [PubMed] [Google Scholar]
- (16).Alsberg E; Anderson KW; Albeiruti A; Franceschi RT; Mooney DJ J Dent Res 2001, 80, 2025–2029. [DOI] [PubMed] [Google Scholar]
- (17).Petersen N; Gatenholm P Appl Microbiol Biotechnol 2011, 91, 1277–1286. [DOI] [PubMed] [Google Scholar]
- (18).Lesaicherre ML; Uttamchandani M; Chen GY; Yao SQ Bioorg Med Chem Lett 2002, 12, 2079–2083. [DOI] [PubMed] [Google Scholar]
- (19).Charles PT; Stubbs VR; Soto CM; Martin BD; White BJ; Taitt CR Sensors (Basel) 2009, 9, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Riquelme MV; Zhao H; Srinivasaraghavan V; Pruden A; Vikesland P; Agah M Sens Biosensing Res 2016, 47–54.
- (21).Xia Y; Whitesides GM Angew Chem Int Ed Engl 1998, 37, 550–575. [DOI] [PubMed] [Google Scholar]
- (22).Hung PJ; Lee PJ; Sabounchi P; Lin R; Lee LP Biotechnol Bioeng 2005, 89, 1–8. [DOI] [PubMed] [Google Scholar]
- (23).Kim J; Taylor D; Agrawal N; Wang H; Kim H; Han A; Rege K; Jayaraman A Lab Chip 2012, 12, 1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhu X; Zheng Q; Yang H; Cai J; Huang L; Duan Y; Xu Z; Cen P Expert Opin Drug Discov 2012, 7, 761–770. [DOI] [PubMed] [Google Scholar]
- (25).Meyvantsson I; Warrick JW; Hayes S; Skoien A; Beebe DJ Lab Chip 2008, 8, 717–724. [DOI] [PubMed] [Google Scholar]
- (26).Wu J; Wheeldon I; Guo Y; Lu T; Du Y; Wang B; He J; Hu Y; Khademhosseini A Biomaterials 2011, 32, 841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Upadhyaya S; Selvaganapathy PR Lab Chip 2010, 10, 341–348. [DOI] [PubMed] [Google Scholar]
- (28).Li J; Carney RP; Liu R; Fan J; Zhao S; Chen Y; Lam KS; Pan T Anal Chem 2018, 90, 5833–5840. [DOI] [PubMed] [Google Scholar]
- (29).Ding Y; Li J; Xiao W; Xiao K; Lee J; Bhardwaj U; Zhu Z; Digiglio P; Yang G; Lam KS; Pan T Anal Chem 2015, 87, 10166–10171. [DOI] [PubMed] [Google Scholar]
- (30).Li J; Carney RP; Liu R; Fan J; Zhao S; Lam KS; Pan T 2017. 19th International Conference on Solid-State Sensors, Actuators and Microsystems (Transducers) 2017, 2195–2198. [Google Scholar]
- (31).Li Y; Xiao K; Luo J; Xiao W; Lee JS; Gonik AM; Kato J; Dong TA; Lam KS Biomaterials 2011, 32, 6633–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Burroughs SL; Duncan RS; Rayudu P; Kandula P; Payne AJ; Clark JL; Koulen P; Kaja S J Neurosci Methods 2012, 203, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chou TC Pharmacol Rev 2006, 58, 621–681. [DOI] [PubMed] [Google Scholar]
- (34).Chou TC Cancer Res 2010, 70, 440–446. [DOI] [PubMed] [Google Scholar]
- (35).Zhang F; Wang Y; Wang L; Luo X; Huang K; Wang C; Du M; Liu F; Luo T; Huang D; Huang K J Biol Chem 2013, 288, 11348–11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ju BG; Lunyak VV; Perissi V; Garcia-Bassets I; Rose DW; Glass CK; Rosenfeld MG Science 2006, 312, 1798–1802. [DOI] [PubMed] [Google Scholar]
- (37).Schiewer MJ; Knudsen KE Mol Cancer Res 2014, 12, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Davies AM; Ho C; Lara PN Jr.; Mack P; Gumerlock PH; Gandara DR Clin Lung Cancer 2006, 7, 385–388. [DOI] [PubMed] [Google Scholar]
- (39).Hidalgo M; Amant F; Biankin AV; Budinska E; Byrne AT; Caldas C; Clarke RB; de Jong S; Jonkers J; Maelandsmo GM; Roman-Roman S; Seoane J; Trusolino L; Villanueva A Cancer Discov 2014, 4, 998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gao H; Korn JM; Ferretti S; Monahan JE; Wang Y; Singh M; Zhang C; Schnell C; Yang G; Zhang Y; Balbin OA; Barbe S; Cai H; Casey F; Chatterjee S; Chiang DY; Chuai S; Cogan SM; Collins SD; Dammassa E, et al. Nat Med 2015, 21, 1318–1325. [DOI] [PubMed] [Google Scholar]
- (41).Aparicio S; Hidalgo M; Kung AL Nat Rev Cancer 2015, 15, 311–316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.