

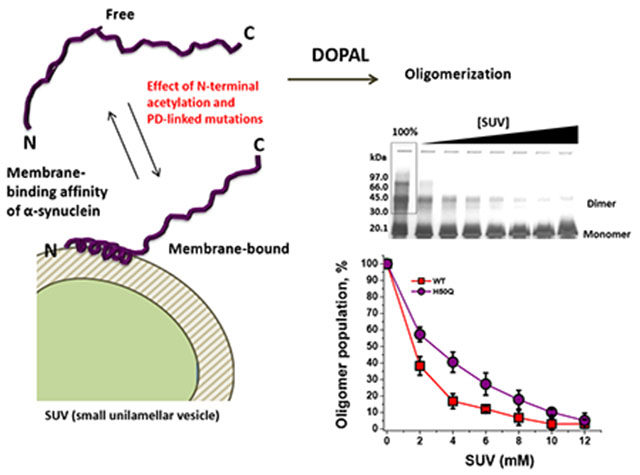
Abstract
Identifying the mechanisms by which the presynaptic protein α-synuclein (aSyn) is associated to neurodegeneration of dopamine neurons is a major priority in the Parkinson’s disease (PD) field. Studies indicate that DOPAL (3,4-dihydroxyphenylacetaldehyde), an aldehyde generated from the enzymatic oxidation of dopamine, may convert aSyn monomer into a neurotoxin via formation of covalently stabilized toxic oligomers. Herein we investigated the role of N-terminal acetylation and familial aSyn mutations (A30P, A53T, E46K, G51D and H50Q) on DOPAL-induced oligomerization of the protein. Our results indicate that wild-type (WT) N-terminally acetylated-aSyn (Ac-aSyn) is less prone to form oligomers upon incubation with DOPAL than the non-N-terminally acetylated protein. On the other hand, familial mutants from Ac-aSyn, particularly A53T, E46K and H50Q increased the formation of DOPAL-derived aSyn oligomers, especially large oligomers. Binding of aSyn to synaptic-like small unilamellar vesicles (SUVs) protected distinctively aSyn variants against the effects of DOPAL. While N-terminal acetylation increased the protective action of against DOPAL-induced aSyn oligomerization, A53T, A30P and H50Q mutations in Ac-aSyn had an opposite effect. It means that PD-linked mutations may not only perturb the affinity of aSyn for membranes but also influence the formation of DOPAL-mediated oligomers. Overall, our findings provide important evidences for the existence of a connection between familial mutations of aSyn, and their distinct affinity to lipid membranes, and the formation of potentially toxic oligomers of the protein mediated by DOPAL.
Keywords: alpha-synuclein, familial mutations, oligomers, DOPAL, lipid vesicles, N-terminal acetylation
Graphical Abstract
INTRODUCTION
Parkinson’s disease (PD) is a neurodegenerative disorder characterized histologically by the presence of intracellular deposits containing fibrillar aggregates of the protein α-synuclein (aSyn) (Lewy bodies and Lewy neurites).1,2 The central role of aSyn in PD pathogenesis emerged with the identification of rare missense mutations (e.g. A53T, A30P, E46K, and most recently H50Q, G51D and A53E) in autosomal dominant familial cases of the disease.3–8 These mutations are associated with different phenotypes and likely with different pathogenic mechanisms.9 In addition, multiplication of aSyn gene (SNCA) is also responsible for different phenotypes, whose severity seem to correlate with the overall number of gene copies.10,11
Structurally, aSyn monomer (14 kDa) can be divided into three distinct domains based on the amino acid composition: an amphipathic N-terminal domain; a central hydrophobic domain (denoted as Non-Amyloid-β Component: NAC); and an acidic C-terminal domain (Figure 1). Alpha-Syn behaves as an intrinsically disordered protein when free in solution12,13 that adopts an amphipathic helical structure at the N-terminal domain and NAC domain (1-100 residues) upon binding to detergent micelles or phospholipid vesicles.14 Recently, aSyn was reporte to be N-terminally acetylated in its physiological state15, probably by the enzyme acetyltransferase B (Nat-B).16 This post-translational modification (PTM) increased the transient helical propensity of the N-terminal residues in the free state,17,18 which causes an increase of the ability of the protein to bind to phospholipid liposomes, particularly to those most similar to synaptic vesicles, a known binding target of aSyn in vivo19 Important evidences suggest that aSyn plays an important role in the genesis, trafficking or fusion of synaptic vesicles at presynaptic nerve terminal, where the protein is proposed to promote SNARE-complex assembly as well as to inhibit SNARE-mediated vesicle fusion in a mechanism that dependent on both the binding of the protein to lipid membrane and the interaction with the protein synaptobrevin-2.20,21
Figure 1. Location of PD-linked mutations into aSyn sequence.
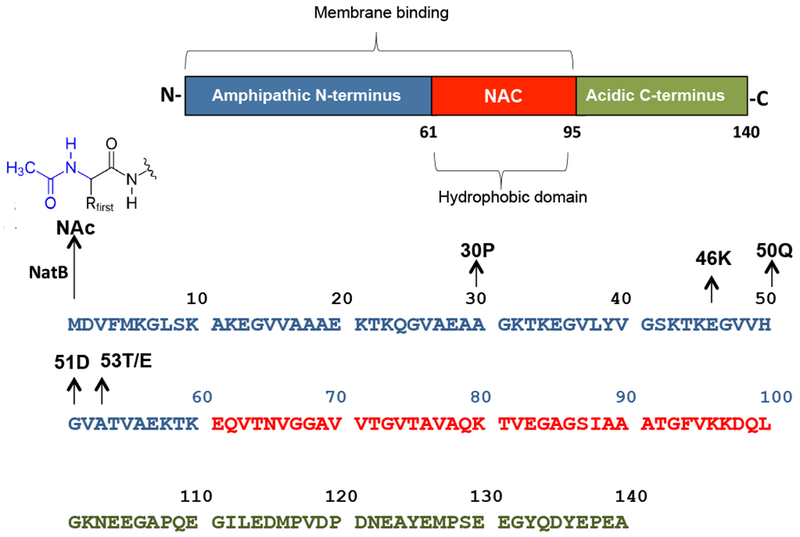
aSyn is a N-terminally acetylated protein with 140 residues that is divided into three distinct domains: an amphipathic N-terminal domain, a central hydrophobic domain (denoted as Non-Amyloid-β Component: NAC), and an acidic C-terminal domain. All familial aSyn mutations (A53T, A30P, E46K, H50Q, G5 ID and A53E) are located into N-terminal domain.
The precise contribution of aSyn to the pathogenesis in PD remains elusive. The most accepted hypothesis postulates that aSyn undergoes a conformational change from random-coil to a beta-sheet conformation leading to the formation of potentially toxic protein aggregates, in which oligomers instead of mature fibrils are the pathogenic agents.22 The selective vulnerability of dopaminergic neurons in PD suggests the existence of a toxic synergism between the aggregation of aSyn and dopamine (DA) metabolism.23–27 For instance, neurotoxicity associated with aSyn overexpression is remarkably reduced when DA synthesis is inhibited.28 Our rationale for this is the existence of modifier factors in DA neurons capable of altering the structural properties of aSyn toward either an impairment of its physiological function or the conversion of the protein into a neurotoxin upon aggregation.
One of the key molecules generated from DA metabolism that has gained particular attention is the highly cytotoxic aldehyde DOPAL (3,4-dihydroxyphenylacetaldehyde). The enzymatic oxidation of DA by monoamine oxidase (MAO-A and MAO-B) produces DOPAL29 whose levels were found enhanced in post mortem brains of PD patients.30 When injected into rat brains, DOPAL causes the loss of dopaminergic neurons accompanied by the accumulation of aSyn oligomers.31 Our group was pioneer in demonstrating that DOPAL interacts with aSyn via formation of Schiff-base and Michael-addition adducts with Lys residues, which leads to the formation of aSyn oligomers that are likely cross-linked by DOPAL.32 A recent study indicated that DOPAL-derived aSyn oligomers might induce DA leakage and cause damage of synaptic vesicles in neurons.33 Interestingly, the binding of aSyn to either lipid vesicles or detergent micelles is capable of inhibiting the oligomerization of aSyn induced by DOPAL.32 In this respect, an impairment of the affinity of aSyn for membranes could play a critical role in the protein dysfunction via either the conversion of the aSyn into a neurotoxic upon protein aggregation (gain-of-toxic function hypothesis) or the loss of its physiological function (loss-of-function hypothesis).
In this work, the effect of N-terminal acetylation and PD-linked mutations (A53T, A30P, E46K, G51D and H50Q) on the oligomerization of DOPAL-modified aSyn was investigated. N-terminal acetylation of wild-type (WT) aSyn significantly decreased the formation of DOPAL-induced oligomers of the protein. On the other hand, familial aSyn mutations, with exception of A30P and G51D, increased the oligomerization of DOPAL-derived N-terminally acetylated-aSyn (Ac-aSyn). N-terminal acetylation of aSyn-WT increased the protective role of lipid vesicles [small unilamellar vesicles (SUV)] against DOPAL-mediated oligomerization of the protein, which is correlated with a strengthening of the interaction protein-membrane caused by this PTM, whereas the mutations A53T, A30P and H50Q in Ac-aSyn decreased the protective role of SUVs, which favors the formation of oligomers in the presence of DOPAL plus membrane. On the other hand, E46K enhances the membrane-binding affinity of Ac-aSyn, which makes this variant less prone to form oligomers in the presence of DOPAL plus SUVs. Thereby, an impairment of the binding of aSyn to lipid membranes could play a pivotal role in the formation of toxic DOPAL-aSyn oligomers in neurons. Collectively, our findings provide new insights into how different events associated with PD such as the formation of toxic DA metabolites, PD-linked mutations, and membrane-binding affinity of aSyn could be interconnected to trigger degeneration of dopaminergic neurons.
RESULTS AND DISCUSSION
N-terminal acetylation decreases the oligomerization of aSyn induced by DOPAL.
Protein acetylation is one of the major PTMs found in eukaryotes proteins. In aSyn, N-terminal acetylation has gain particular attention particularly because it increases of the transient helical propensity of the N-terminal of the free protein and leads to an enhanced binding-affinity for synaptic vesicle-like membranes.17–19 However, most in vitro studies of aSyn have been done using the recombinantly expressed non-N-terminally acetylated protein, which in certain situations could provide misleading conclusions. Herein, recombinant Ac-aSyn was generated by co-expressing the protein with the enzyme Nat-B (Figure 1),34 and the effect of this PTM on DOPAL-induced oligomerization of the WT protein was addressed.
Figure 2 shows the effect of varying concentrations of DOPAL on the formation of oligomers of aSyn and Ac-aSyn, both WT, evaluated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (upper panels) and size-exclusion chromatography (SEC) (Figure 2A,B). In these experiments, the protein was incubated in the presence of DOPAL for 24 hours to assure that the steady state was reached. As previously reported, DOPAL forms covalent adducts with Lys residues of aSyn, which leads to the formation of SDS-resistant oligomers that can be easily visualized in SDS-PAGE. aSyn monomer (14 kDa) migrates in the gel with an apparent molecular mass of ~18 kDa, which is attributed to a poor binding of SDS molecules to the acidic C-terminal tail of the protein.35,36 We can also notice an increase in the molecular mass of aSyn monomer (N-terminally acetylated or not) in the gel with increasing concentrations of DOPAL, which may be attributed to the formation of DOPAL-Lys adducts. In addition, a change in the net charge of the protein due to the lost of positively charged Lys at N-terminal domain might affect the ability of aSyn to migrate in SDS-PAGE.
Figure 2. Effect of N-terminal acetylation on DOPAL-induced oligomerization of aSyn-WT.
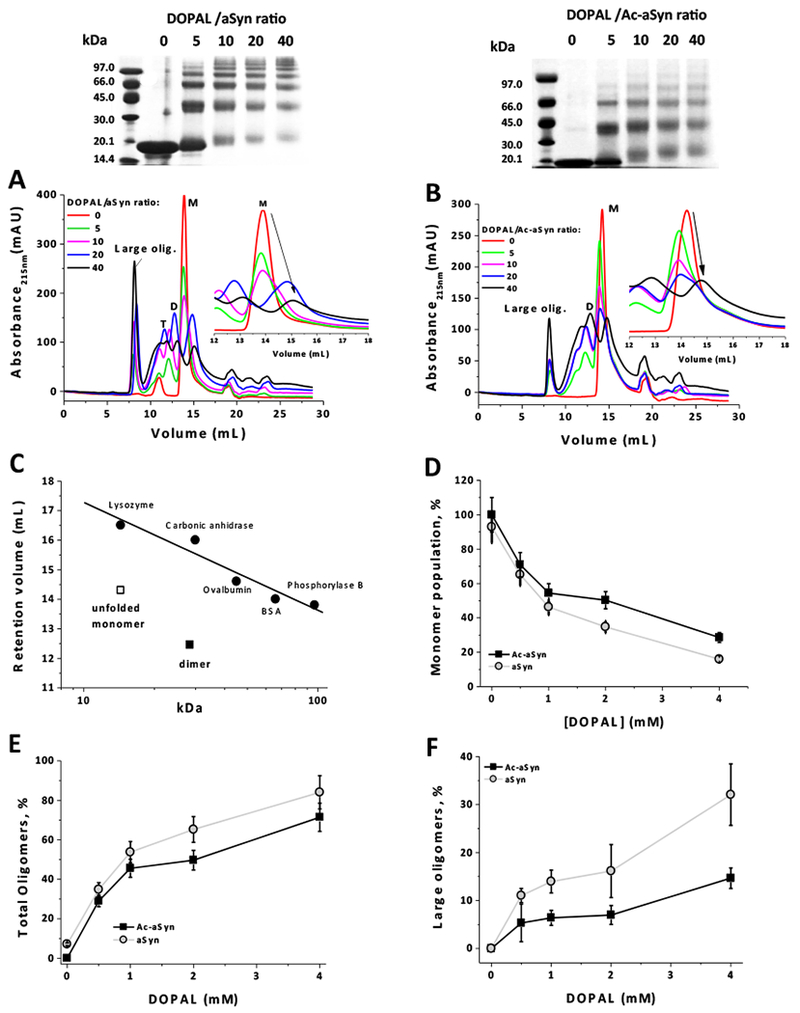
The formation of oligomers from aSyn and Ac-aSyn, both WT, (50 μM in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl) upon incubation for 24 hours at 25 °C in the presence of DOPAL (0.25, 0.5, 1 and 2 mM), was evaluated by SDS-PAGE and SEC-HPLC (A: aSyn; B: Ac-aSyn). (C) Retention volumes of aSyn unfolded monomer and dimer in a Superdex 200 10/300 GL column; a standard curve was generated by using the proteins lysozyme (14.4 kDa), carbonic anhidrase (30 kDa), ovalbumin (45 kDa), BSA (66 kDa) and phosphorylase B (97 kDa). The population of monomer (D), total oligomers (E) and large oligomers (F), after incubation with DOPAL, was quantified by the integration of the peak areas in SEC-HPLC. Results in D-F represents mean ± standard deviation of five independent experiments. M: monomer; D: dimer: T: trimer.
By inspecting aSyn oligomerization by SEC, we notice the formation of two distinct types of DOPAL-derived oligomers: large oligomers (eluting at ~ 8 mL) and small oligomers [dimer (D) and trimers (T)] (eluting at 10-13 mL); these latters can also be visualized by SDS-PAGE. Although Ac-aSyn is capable of forming both types of oligomers in the presence of DOPAL, their populations are reduced when compared with the non-acetylated protein. We can also notice that the elution volume of the peak corresponding to the aSyn monomer (M) (~ 14 mL in SEC) increases with the increase of the concentration of DOPAL, which suggests a strengthening of the intra-molecular contacts in the monomer (favoring a more compact conformation) mediated by DOPAL (Figure 2A,B - insets). Our previous study indicated that the formation of DOPAL-aSyn adducts increased the interaction of the N-terminus with the NAC domain,32 which corroborates to the hypothesis of a conformational change in aSyn promoted by DOPAL. Interestingly, this shift to higher elution volume of DOPAL-treated aSyn monomer is partially prevented by the N-terminal acetylation of the protein. It is worth mentioning that the intrinsically disorder nature of aSyn monomer is responsible for the premature elution of the protein in SEC (volume of elution corresponding to a 60-70 kDa protein) (Fig. 2C).
The quantification of the population of DOPAL-derived aSyn oligomers was done by the integration of the peak areas in SEC in which the sum of areas of the peaks corresponding to each oligomeric state (monomer eluting at ~14 mL and oligomers eluting from 8 to ~12 mL) was taken as 100%. A baseline correction was performed for all chromatograms taking into account any drift/shift of the chromatogram in the analysis of the peak areas. Peaks eluting at volumes higher than 15 mL (EDTA and minor protein contaminants), whose areas were not affected upon incubation with DOPAL, were not considered. Figure 2D,E confirmed that N-terminal acetylation significantly diminishes the formation of aSyn-DOPAL oligomers and increases the population of the monomer. Additionally, N-terminal acetylation seems to cause a significant reduction of the population of large oligomers (elution at ~ 8 mL), at least for the WT protein (Figure 2F).
PD mutations affect the oligomerization Ac-aSyn mediated by DOPAL.
Multiple studies have been carried out to address the impact of PD familial mutations on the aggregation propensity and toxic properties of aSyn. Evidences suggest that A53T, E46K and H50Q increase the formation of mature fibril formation,37–41 while A30P favors the formation of oligomers instead of fibrils.40–42 G51D, on the other hand, seems to retard the aggregation process.39 In this context, we set out to investigate the role of the familial mutations A53T, A30P, E46K, H50Q and G51D (Figure 1) on the propensity of Ac-aSyn to form DOPAL-mediated oligomers. In this work, we did not evaluate the role of N-terminal acetylation on DOPAL-induced oligomerization of familial aSyn mutants. It means the all experiments described here for aSyn mutants were done using N-terminally acetylated protein, and the results compared with the Ac-aSyn WT.
Figure 3A-E reveals that all five mutations have a remarkable impact on both the content and the size of DOPAL-derived oligomers produced from Ac-aSyn, as observed by SEC or SDS-PAGE (upper panels). Quantification of the population of total oligomers indicates that the mutations A53T, E46K and H50Q increased the oligomerization of Ac-aSyn upon incubation with DOPAL, while A30P and G51D have no effect in comparison with the WT protein (Figure 3F). In contrast with A30P and G51D variants, A53T and H50Q exhibited an increased population of large oligomers in the presence of DOPAL (Figure 3G). The wide-range of sizes for the oligomers produced from E46K makes any estimation of the population of large oligomers from this variant difficult. The distinct propensity of aSyn variants to form large oligomers in the presence of DOPAL might have important implications for the toxicity of these species, mainly taking into account a recent study that demonstrated that the stabilization of large rather than small oligomers seems to be associated with an enhancement of the toxicity of aSyn-DOPAL oligomers.43 A key outcome of our study is the observation that familial aSyn mutations are capable of modulating the formation of oligomers of the protein in the presence of DOPAL. Interestingly, oligomers produced from A30P and A53T, when grown in the presence of DA, exhibited a remarkable gain in stability, in comparison with the WT protein, which suggests that these mutations may affect not only the formation of oligomers stabilized by DOPAL but also by DA.27
Figure 3. Effect of PD-linked mutations on DOPAL-induced oligomerization of Ac-aSyn.
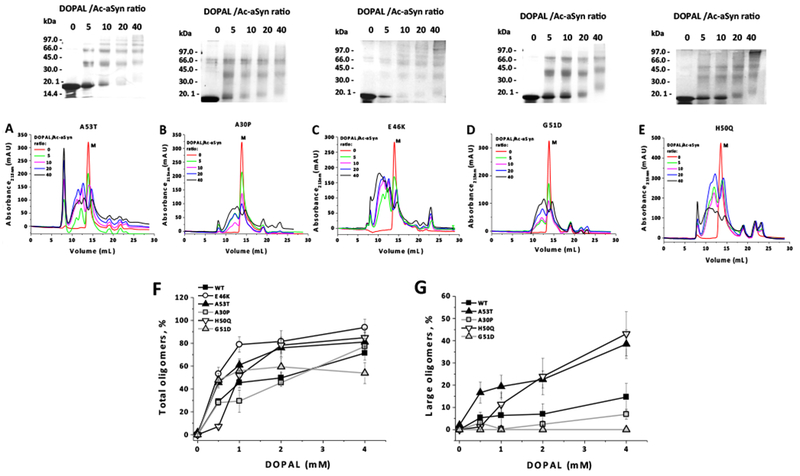
Oligomerization of Ac-aSyn mutants (A53T, A30P, E46K, G5 ID and H50Q) (50 μM in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl) upon incubation for 24 hours at 25 °C in the presence of DOPAL (0.25, 0.5, 1 and 2 mM), evaluated by SEC (A-E). The population of total oligomers (F) and large oligomers (G) from WT and PD mutants of Ac-aSyn, in the presence of DOPAL, was quantified by the integration of the peak areas in SEC. In F and G, for each DOPAL/Ac-aSyn ratio, the experiment was performed in quintuplicate, and the average value and standard deviation were calculated based on three to five measures of the population of oligomers.
It is worth noting that, with exception of E46K, the other mutations A30P, A53T, G51D and H50Q do not affect any residue target by DOPAL, namely Lys and Met residues.32,33 DOPAL-treated E46K variant was capable of forming oligomers with a wide-range of sizes, while the other variants exhibited two distinct populations of DOPAL-derived oligomers (small and large oligomers). Although we did not characterize the chemical structure of Lys-DOPAL adducts formed by the incubation of aSyn variants with DOPAL, one possible explanation for E46K is the presence of an additional Lys residue, which could react with DOPAL. Our previous work had associated the formation of low molecular weight oligomers from aSyn-DOPAL (mostly dimers and trimers) to steric hindrance associated to the formation of intra/inter-polypeptide cross-links.32 In this respect, an additional reactive Lys-DOPAL adduct in E46K could increase the probability of cross-links between adjacent protein chains.
Another plausible hypothesis for the findings outlined above is based on the fact that the familial mutations may perturb the conformation of the free state of aSyn in solution, which could impact the reactivity of the protein to DOPAL. Although aSyn behaves as an intrinsically unfolded protein under physiological conditions, the protein seems to adopt an ensemble of conformations that are maintained by intramolecular long-range contacts between N- and C-terminal, and NAC and C-terminal residues. A perturbation of these contacts in both the site of the mutation and C-terminal domain of PD familial mutants was demonstrated,44,45 resulting in the formation of fibrillation-prone intermediates in case of A53T, E46K and H50Q, but not for G51D and A30P variants. For G51D, this perturbation in long-range contacts is suggested to lead to an increased rigidity of the monomer, which has been attributed, among other factors, to the presence of additional contacts involving Lys residues (K58 and K60)44 Lys58 ad 60 was reported to form covalent adducts with DOPAL.33 Curiously, G51D exhibited a reduced propensity to form oligomers upon incubation with DOPAL, mainly large oligomers. This auto-inhibitory electrostatic interaction in G51D could also be the reason for the slower aggregation of this mutant in the absence of DOPAL.39,44 In general, an increase of the intramolecular contacts in aSyn monomer seems to have an inhibitory action on the formation of DOPAL-derived aSyn oligomers. For instance, oxidation of Met residues to Met-sulfoxide increases the long-range contacts in WT-aSyn monomer, which is accompanied by a diminished ability of the protein to form oligomers in the presence of DOPAL.43
Binding of aSyn to phospholipid vesicles inhibits the formation of DOPAL-mediated oligomers of the protein.
The binding of aSyn to phospholipid vesicles has been well studied by several groups and is believed to be necessary for the physiological function of the protein in regulating synaptic vesicle trafficking.20,46 In this respect, structural perturbations that disrupt interactions between the protein (particularly the NAC domain) and the phospholipid membrane are associated with the formation of potentially toxic oligomers of the protein at the membrane surface.47 Recently, our group demonstrated that the incubation of aSyn-WT in the presence of either SDS micelles or small unilamellar vesicles (SUVs) reduces the formation of oligomers induced by DOPAL.32 From these results, we hypothesize that an impaired membrane interaction of aSyn with lipid membranes could play a pivotal role in the formation of oligomers of the protein mediated by DOPAL.
Firstly, we chose to focus on the effect of N-terminal acetylation on the oligomerization of aSyn-WT in the presence of DOPAL plus SUV. For that, aSyn and Ac-aSyn monomer, both WT, in the presence of varying concentrations of SUV (0, 2, 4, 6, 8, 10, or 12 mM) was incubated with DOPAL (10-fold molar excess over protein) for 24 hours at 25 °C, and then the samples were analyzed by SDS-PAGE (Figure 4A,B). These assays were done using vesicles with a lower negative charge content (DOPE: DOPS: DOPC = 5:3:2 w/w/w) and high curvature, which are presumably similar to that of synaptic vesicles, a binding target of the protein in vivo. The relative population of oligomers was quantified from SDS-PAGE by densitometry in which the intensity of oligomer bands in the absence of SUV was considered as 100%. In this experiment, only oligomers that appear in SDS-PAGE were taken into account. Figure 4C indicates that an increase of the concentration of SUV reduced the relative population of oligomers, being the protective effect of SUV against DOPAL-induced oligomerization higher for Ac-aSyn compared with the non-acetylated protein. We also observed an increase of the intensity of the band of the monomer upon the incubation of the protein with DOPAL plus SUV (6 mM) for both proteins (Fig. 4D). Unfortunately, we were not able to measure the population of monomer in function of SUV concentration because of the distortion of the monomer band in the gel caused by SUV at concentrations higher than 6 mM. Taken together, these data indicate that N-terminal acetylation strengths the protective role of SUV against DOPAL-induced the oligomerization of aSyn-WT. Interestingly, a previous study demonstrated that the enhanced helicalfolding propensity of Ac-aSyn has an inhibitory effect on the aggregation of the protein incubated in the presence of GM1, a lipid enriched in presynaptic membranes.48
Figure 4. Effect of N-terminal acetylation on the protective role of SUVs on DOPAL-induced oligomerization of aSyn-WT.

aSyn (A) and Ac-aSyn (B) monomer (50 μM in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl), in the presence of varying concentrations of SUV (0, 2, 4, 6, 8, 10, or 12 mM), was incubated with 500 μM DOPAL for 24 hours at 25 °C, and then the samples analyzed by 15% SDS-PAGE. (C) Population of SDS-resistant oligomers determined by quantitative densitometry of proteins in SDS-PAGE stained with Coomassie Blue, using ImageJ software.59 (D) Population of monomer and oligomers from aSyn and Ac-aSyn upon the incubation with DOPAL in the absence or the presence of 6 mM SUV. Results in C and C represent mean ± standard deviation of three to five independent experiments.
Our next objective was to determine how PD mutations affect the protective role of SUV against the oligomerization of Ac-aSyn induced by DOPAL. Although the content of DOPAL-derived oligomers decreased for all mutants with the increase of the concentration of SUV, the extension of this inhibition seems to be affected distinctively by the mutations (Figure 5A-E). The variants A53T, A30P and H50Q of Ac-aSyn were found to be less protected by SUV than the WT protein. On the other hand, E46K exhibited a very slight increased protection at 2 mM SUV, whereas G51D was similar to WT. For all aSyn variants, the relative population of monomer increased upon the incubation with DOPAL plus SUV (6 mM) (Figure 5, panels on the right). However, in some cases, we did not observe an increase of the monomer content proportionally to the decrease of oligomer population, which might be attributed to the fact that only oligomers that are visualized in the gel were quantified. In all our studies, the formation of insoluble aggregates upon incubation of aSyn with DOPAL was not observed, irrespective the concentration of DOPAL or the presence of SUV.
Figure 5. Protective role of SUVs on DOPAL-induced oligomerization of Ac-aSyn mutants.
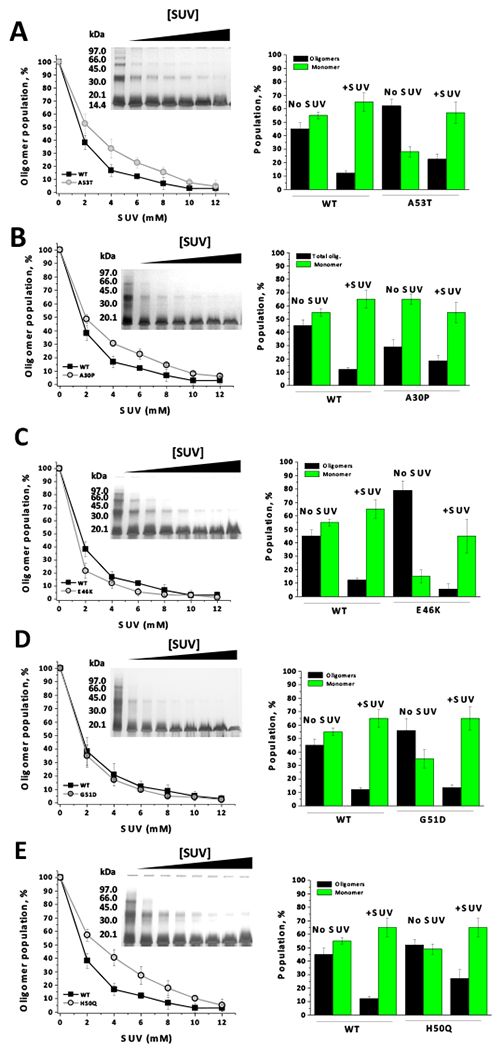
Ac-aSyn mutants (A53T, A30P, E46K, G51D and H50Q) (50 μM in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl), in the presence of varying concentrations of SUV (0, 2, 4, 6, 8, 10, or 12 mM), were incubated with 500 μM DOPAL for 24 hours at 25 °C, and then the samples analyzed by 15% SDS-PAGE (A-E). On the right, population of monomer and oligomers from Ac-aSyn (WT or mutant) upon the incubation with DOPAL in the absence or the presence of 6 mM SUV. Results represents mean ± standard deviation of three to five independent experiments.
Ruzafa et al. suggested a protective role of N-terminal acetylation of aSyn against SDS-induced aggregation.49 In this study, N-acetylation seems to favor a conformational transition in which non-aggregating oligomers become stabilized. Interestingly, the influence of N-acetylation in promoting this transition becomes impaired by the presence of the mutations A30P, E46K and A53T. In our study with SUV and DOPAL, A30P and A53T were capable of reducing the protective role of N-terminal acetylation against DOPAL-induced aSyn oligomerization in the presence of SUV.
The results outlined above indicate that PD-linked mutations may affect DOPAL-induced oligomerization of Ac-aSyn via distinct mechanisms, depending on whether the process takes place in either the presence or the absence of lipid membrane. For instance, A30P formed oligomers (total oligomers) in the presence of DOPAL similarly to WT. However, in the presence of SUV, A30P is less protected than WT against DOPAL-induced oligomerization of Ac-aSyn, which leads to the formation of higher content of oligomers from A30P when in the presence of SUV plus DOPAL. For A53T and H50Q, the formation of DOPAL-derived oligomers was higher than for WT, irrespective the presence of SUV. On the other hand, E46K formed less oligomers in the presence of SUV than WT, even though the former seems to lead to an increased content of oligomers in the absence of SUV. Collectively, these findings suggest that interaction of aSyn with lipid membranes inhibits the formation of DOPAL-induced oligomers and, more importantly, this inhibitory effect depends on the familial mutations.
The presence of either SDS or lipid vesicles has the ability of inhibiting in a dose-dependent manner the formation of DA-mediated oligomerization of aSyn,50 similarly to that observed for DOPAL. In the presence of DA and its oxidative intermediates, aSyn forms SDS-resistant soluble oligomers that are off-pathway to fibril formation.23–27 Therefore, the binding affinity of aSyn to lipid membranes seems to play a central role in controlling the formation of oligomers from the protein induced by either DOPAL or DA.
Disruption of contacts between aSyn and phospholipid vesicles favors the formation of DOPAL-induced aSyn oligomers.
To shed light on the mechanisms lying behind the protective action of phospholipid membrane against the oligomerization of aSyn mediated by DOPAL, we investigated the ability of the protein to bind to SUV. For this purpose, we utilized far-UV CD, a technique that reports on the increase in aSyn α-helicity upon interaction of the protein with SUV. CD spectra for aSyn and Ac-aSyn monomer, both WT, in the presence of varying concentrations of SUVs are shown in Figure 6A and B, respectively. A higher helicity is observed for Ac-aSyn compared with the non-Ac protein at equivalent SUV concentrations, which is evidenced by measuring the mean residue molar ellipticity at 222 nm ([θ]MR,222) as function of lipid concentration (Figure 6C). The titration curve for aSyn is shifted significantly to the right compared with Ac-aSyn, indicating the N-terminal acetylation increased the affinity to SUV. Fits of the titration data using Equation 2 yielded Kd values of 2.8 ± 0.6 and 9.2 ±3.8 μM for Ac-aSyn and aSyn, respectively. This result is consistent with other studies that showed that the N-terminal acetylation increases the transient helical propensity of the N-terminal ~10 residues in the free state, which results in an increase the binding of WT monomer to vesicles of lower negative charge content and higher curvature.17–19 From these results, we infer that, in the presence of SUV plus DOPAL, naturally occurring Ac-aSyn would be less prone to oligomerize than the non-Ac protein, likely due to its increased affinity for synaptic-like lipid vesicles caused by the N-terminal acetylation.
Figure 6. Effect of N-terminal acetylation on the binding-affinity of aSyn-WT monomer to SUVs analyzed by far-UV CD.
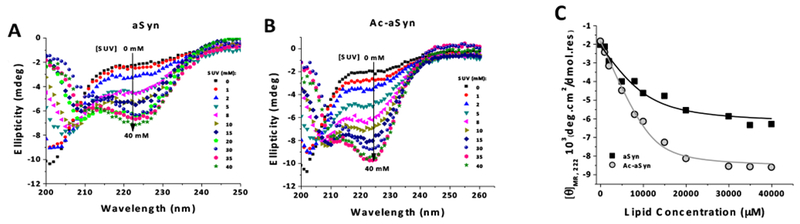
CD spectra of aSyn and Ac-aSyn monomer (40 μM in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl) incubated at 25 °C with increasing concentrations of SUVs (0 to 40 mM) are shown in (A) and (B), respectively. (C) Mean residue molar ellipticity at 222 nm ([θ]MR,222) as function of lipid concentration for aSyn and Ac-aSyn. The data were fit to Equation 2 to calculate the values for Kd (Table 1). SUV composition: 60% DOPC, 25% DOPE, and 15% DOPS.
Familial aSyn mutations have been associated with different effects on membrane binding affinity: while A30P has a reduced affinity for phospholipid membranes,46 A53T and H50Q do not seem to alter the binding affinity,51,52 and E46K shows enhanced binding.51,53 On the other hand, G5 ID appears to decrease the membrane affinity.54 However, most of these studies were done with the non-Ac form of the protein. Herein, we evaluated the familial mutants of Ac-aSyn monomer in terms of both the propensities to acquire an α-helical conformation upon binding to phospholipid membranes (far-UV CD) and the site-specific perturbations of the different domains of membrane-bound protein (2D NMR). CD spectra for the lipid titrations assays for familial variants of Ac-aSyn monomer are shown in Figure 7A-E. The determination of the values of dissociation constants (Kd) from the curves of [θ]MR,222 versus lipid concentrations (Figure 7F and Table 1) indicate that, relative to the WT protein (Kd = 2.8 ± 0.6 μM), E46K increased the affinity to SUV (Kd = 1.1± 0.8 μM), while A30P reduced it (5.1 ± 1.2 μM), similarly to that previously reported for the non-Ac forms of these mutants. Conversely, A53T and H50Q, which were reported not to affect the affinity of aSyn to membranes, exhibited a significant reduction of the affinity of the protein for SUV [Kd values: 4.3 ± 0.8 μM (A53T) and 5.4 ± 1.1 μM (H50Q)]. G51D exhibit a value of Kd (3.7 ±1.2 μM) nearly close to that found for the WT. Previous studies on G51D variant indicated that this mutation would interfere negatively with the interaction between aSyn and lipid membranes by placing a negative charge on the non-polar face of the amphipathic α11/3 helix.47 Our results indicate that N-terminal acetylation might counterbalance the impaired membrane binding caused by G51D mutation.
Figure 7. Effect of PD-linked mutations on the membrane binding-affinity of Ac-aSyn monomer analyzed by far-UV CD.
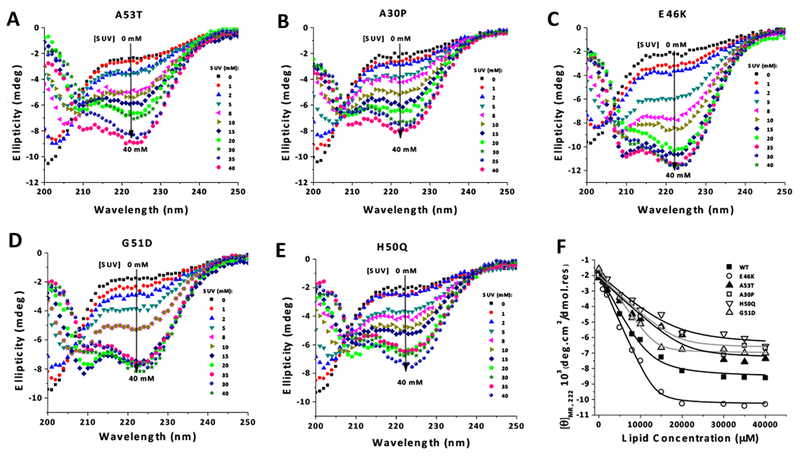
CD spectra of familial variants of Ac-aSyn monomer (A53T, A30P, E46K, G51D and H50Q) (40 μM in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl), incubated at 25 °C with increasing concentrations of SUVs (0 to 40 mM) are shown in (A-E). (F) Data from [θ]MR,222 as function of lipid concentration were fit to Equation 2 to calculate the values for Kd (Table 1). SUV composition: 60% DOPC, 25% DOPE, and 15% DOPS.
Table 1.
Binding affinity of Ac-aSyn (WT and PD mutants) to SUV evaluated by both the fractional populations of membrane-associated aSyn conformers determined by NMR (10 °C) and the dissociation constant (Kd) measured by far-UV CD (25 °C).
| Ac-aSyn | Total bound 1 − {I/I0}mean,3-25 |
Hidden 1 − {I/I0}mean,66-80 |
Exposed* |
Kd (μM) (from far-UV CD) |
|---|---|---|---|---|
| WT | 0.73 | 0.63 | 0.10 | 2.8 ± 0.6 |
| A30P | 0.62 | 0.40 | 0.22 | 5.1 ± 1.2 |
| A53T | 0.66 | 0.56 | 0.10 | 4.3 ±0.8 |
| E46K | 0.76 | 0.66 | 0.10 | 1.1 ± 0.8 |
| G51D | 0.71 | 0.39 | 0.32 | 3.7 ± 1.2 |
| H50Q | 0.72 | 0.54 | 0.18 | 5.4 ±1.1 |
Calculated as the difference between the total bound population and the hidden population.
The examination of data above suggests that the membrane-binding affinity of Ac-aSyn variants may represent an important factor for formation of oligomers in the presence of DOPAL. For instance, E46K exhibited an increased affinity for SUV and this mutant has a lower propensity to form oligomers upon incubation with SUV plus DOPAL. On the other hand, the variants A53T, A30P and H50Q were less protected by SUV against DOPAL effects than WT, and those mutations were also associated to a diminished affinity to SUV. G51D behaves similarly to WT in term of propensity to form oligomers in the presence of SUV plus DOPAL, and both have similar affinities for SUV accordingly to the Kd values determined by far-UV CD.
Our next objective was to understand how site-specific perturbations of the different domains of membrane-bound aSyn contribute to the inhibitory action of SUV on DOPAL-induced oligomers of the protein. To compare the familial aSyn mutants in terms of SUV binding affinity, 200 μM of 15N-labeled Ac-aSyn monomer (WT and mutants) were incubated with or without 8 mM SUV (lipid:protein ratio, 40:1 mol/mol), and 1H-15N-HSQC spectra were recorded. All NMR experiments were done at 10 °C. Binding of the monomer to SUVs occurs in the slow-exchange limit on the NMR frequency scale, which results in a resonance linewidth too broad for detection due to the slow rotational motion of the membrane-bound protein. Thus NMR experiments allow us to correlate the normalized signal attenuation of resonances from specific residues, obtained in the presence of liposomes, to the binding of these residues to the membrane. Selected regions of the 1H-15N-HSQC spectrum of WT and familial mutants of Ac-aSyn in the absence (green) or presence (magenta) of SUVs are shown in Figure 8A-F. The intensity of cross-peaks in 1H-15N HSQC spectra in the presence of SUV (I) was normalized by the signal in the absence of SUV (I0) (Figure 8G-L). In this case, the ratio I/I0 corresponds to the proportion of molecules in the mixture aSyn plus SUV in which the residue remains mobile and in solution, and the signal attenuation (1 − I/I0) is the proportion of molecules in which the residue is tightly bound to the lipid vesicle.51
Figure 8. Membrane-binding affinity of familial mutants of Ac-aSyn probed by 1H-15N-HSQC.

Selected region of the 1H-15N-HSQC spectra of PD mutants or WT Ac-aSyn, in the absence (green) or the presence (magenta) of 8 mM SUV (A-F). The intensity of cross-peaks in the 1H-15N HSQC spectra of Ac-aSyn in the presence of SUV (I) was normalized by the signal in the absence of SUV (I0) (G-L). NMR data were acquired at 10 °C using 200 μM of 15N-labeled Ac-aSyn monomer in 20 mM sodium phosphate buffer, pH 7.5, 100 mM NaCl, 10 % D2O.
Considering that interactions involving an N-terminal segment spanning residues 1–25 are critical for membrane binding and for the adoption of an α-helical structure,55 fractional population of molecules bound to the membrane was determined from the mean attenuation of residues 3-25 (‘total bound population’). Table 1 shows that 73% of WT molecules were bound to the membrane, which is reduced to 62 and 66% for A30P and A53T variants, respectively. A slight increase of the population bound was observed for E46K (76%), while G51D and H50Q exhibited values of 71 and 72 %, respectively. The weaker affinity for SUV observed for A30P and A53T can explain why these mutants are less protected by the presence of SUV against the oligomerization induced by DOPAL. Corroborating this hypothesis, E46K exhibited an increased total bound population as well as a higher inhibitory affect of SUV against the formation of oligomers upon incubation with DOPAL, compared with the WT protein. However, these data do not seem to be sufficient to explain the effect of SUV on the oligomerization of H50Q and G51D. Although these mutants exhibited nearly the same total membrane-bound populations, SUV was able to inhibit more significantly the formation of oligomers from G51D than H50Q in the presence of DOPAL.
To gain additional information on the interaction of aSyn mutants with SUV, we examine the conformation of the hydrophobic domain NAC at the membrane, which is described to have an important role on the oligomerization of aSyn at membrane surface.47 The fractional population of protein in either dissociated state (referred to here as ‘exposed’) or associated state (‘hidden’) we estimated the mean attenuation of residues 66-80. The population in the exposed state was determined by subtracting the hidden population from the total bound population (residues 3-25). For WT, only ~10% was in the exposed membrane-bound conformation, compared with ~63% in the hidden state. For A30P and H50Q the hidden population decreases to 40 and 54%, respectively. On the other hand, for the total E46K protein, ~66% is the hidden state. Although H50Q and WT have similar total bound populations, H50Q has a higher population in the exposed state than WT (18% for H50Q), which could explain why H50Q is less protected by SUV than the WT protein against the formation of oligomers in the presence of DOPAL. Although A53T has an exposed population similar to the WT protein, A53T has a lower total bound population. Besides A30P, G51D mutant has the higher exposed population (32%) among the familial aSyn mutants analyzed. It means that A30P or G51D mutation led to a significant reduction in lipid membrane interactions involving the central hydrophobic domain NAC. A key outcome of our studies is the observation of the existence of a correlation between the population in the exposed state and the formation of DOPAL-derived aSyn oligomers. An increase of the population of the protein in a completely membrane-dissociated state or with the hydrophobic domain in an exposed state may influence the protective role of SUV against the formation of DOPAL-mediated aSyn oligomers.
Intriguingly, H50Q and G51D exhibited similar populations in both the exposed and total-bound states but very distinct propensity to form oligomers upon incubation with SUV and DOPAL. However, Kd value for G51D obtained by using far-UV CD indicates a membrane binding affinity similar to that found for the WT protein, which is consistent with a similar protection against DOPAL promoted by SUV. The apparent inconsistence between CD and NMR data in case of G51D might be attributed to the difference of temperatures between these experiments: the determination of the binding affinity of aSyn to liposomes by NMR experiments was done at 10°C, while CD signal measurements were at 25°C. G51D substitution is proposed to have a disruptive effect on membrane binding because of the high-energy barrier caused by inserting a charged aspartate residue in the hydrophobic region of the bilayer.47 In this case, an increase of temperature of 15 °C (far-UV CD versus NMR) could be responsible to overcome this energy barrier and hence favor the binding of G51D to lipid membrane at 25 °C, accordingly to that observed by using far-UV CD.
In addition to the effect of the familial mutations on the ability of Ac-aSyn to bind phospholipid vesicles outlined above, the formation of aSyn-DOPAL adducts was demonstrated to reduce both the affinity of the protein for SUVs and the formation of helical conformations upon membrane binding.32 Importantly, DOPAL-induced impairment of aSyn binding to SUV does not require the oligomerization of the protein. Although the physiological function of aSyn is not completely understood, multiple lines of evidence suggest that the protein has some role in the genesis, trafficking, or fusion of synaptic vesicles at presynaptic nerve terminals.20,21 All these functions are directly dependent of the binding of aSyn to lipid membrane via its N-terminal domain, which adopts an amphipathic helical conformation upon binding to membranes. Importantly, 11 of 15 Lys residues found in aSyn are located in the N-terminal domain, which is the primary target of DOPAL.32,56 Taken these findings into account, we suggest that both familial aSyn mutations (at least for A30P, A53T and H50Q) and the formation of adducts with DOPAL may favor the releasing the protein from membrane surfaces and, hence, enhance the susceptibility of the protein to undergo DOPAL-induced oligomers.
CONCLUSIONS
In this work, we demonstrate that both N-terminal acetylation and familial mutations of aSyn are capable of interfering with the formation of oligomers of the protein induced by DOPAL. In light of a previous report showing that the interaction of aSyn with lipid membranes inhibits the formation of DOPAL-derived oligomers of the protein, our data pointed out that an impaired membrane binding of the monomer caused by PD-linked mutation favors the formation of oligomers induced by DOPAL. Therefore, PD-linked mutations could affect DOPAL-induced oligomerization of aSyn distinctively depending on whether the process takes place in either the presence or the absence of lipid membrane. Collectively, our findings provide a significant contribution to understand how familial mutations, N-terminal acetylation and membrane-binding affinity of aSyn affect the propensity of the protein to form potentially toxic aggregates in dopaminergic neurons.
METHODS
Expression and purification of aSyn.
Alpha-Syn (WT) and Ac-aSyn (WT and mutants) were produced as described.16,57 Firstly, we prepared competent E. coli BL21 (DE3) cells previously transformed with N-acetyltransferase complex B (Nat-B) plasmid expressing chloramphenicol resistance,34 gently provided by Dr. Dan Mulvihill, University of Kent, UK. Next, E. coli BL21 (DE3) cells (transformed or not with Nat-B), were transformed with a plasmid encoding aSyn (expressing ampicillin resistance) and then were grown in rich media containing chloramphenicol/ampicillin at 37 °C to an optical density (600 nm) of 0.6. Protein expression was induced with 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) for 4 hours at 37 °C. Purification of aSyn consisted of acid precipitation and ammonium sulfate cuts.57 For the purification of Ac-aSyn, a saturation of ammonium sulfate of 40% was applied (instead of 50% for the non-Ac protein). Recombinant Ac-aSyn uniformly labeled with 15N for NMR experiments was produced by the media swap method.58 The purified protein was lyophilized and stored at −20 °C. N-terminally acetylation of both WT and variants of aSyn was confirmed by using mass spectrometry.
Preparation of SUVs.
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE):1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS):1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) (5:3:2 w/w/w) were purchased from Avanti Polar Lipids (Alabaster, AL). Phospholipid mixtures containing 60% DOPC, 25% DOPE, 15% DOPS (molar concentrations) were prepared by drying a mixture of the different lipids dissolved in chloroform under nitrogen gas and resuspending the lipid film in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl at 25 °C. SUVs were prepared by pulse-sonicating the phospholipid suspensions in a bath sonicator for 10 min in 2-min increments. The size of the resulting SUVs (hydrodynamic radii of 30-50 nm) was determined by dynamic light scattering using a Zetasizer Nano ZS instrument (Malvern Instruments, Worcestershire, UK).
Oligomerization of aSyn induced by DOPAL.
A solution of 100 μM of recombinant aSyn monomer in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl, was incubated for 24 hours at 25 °C, no agitation, in the presence of 0, 0.5, 1, 2 or 4 mM of DOPAL (Cayman Chemical, Ann Arbor, USA). Aliquots were withdrawn at different times and analyzed by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or size-exclusion chromatography (SEC) using a Superdex 200 10/300 GL column (void volume: 7.8 mL) (GE Healthcare, Little Chalfont, UK) using an AKTA Purifier 10 equipment (GE Healthcare) (flow-rate 0.7 mL/min). The percentage of oligomers formed was determined by the relative areas in SEC-HPLC. To investigate the protective effect of SUV on DOPAL-induced aSyn oligomerization, 50 μM recombinant aSyn monomer in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl was mixed with varying concentrations of SUV (0, 2, 4, 6, 8, 10, or 12 mM), following by the addition of 500 μM of DOPAL. Samples were incubated for 24 hours at 25 °C, no agitation, and then analyzed by 15% SDS-PAGE. In these experiments, the population of the different oligomeric states of aSyn was evaluated by using quantitative densitometry of proteins, in SDS-PAGE stained with Coomassie Blue, using ImageJ software.59 Protein samples (in the absence or the presence of SUV) were centrifuged at 10,000 rpm before the analysis by SEC-HPLC or SDS-PAGE. No insoluble aggregates were observed.
NMR spectroscopy.
NMR experiments were performed using a 600 MHz (Weill Cornell Medical College, Cornell University, New York City, USA) Bruker Avance spectrometer equipped with cryogenic probes. Phase-sensitive two-dimensional 1H-15N Heteronuclear Single Quantum Coherence (1H-15N-HSQC) spectra were recorded using Echo-anti Echo gradient selection. TopSpin 3.2 was used for data acquisition. All spectra were processed with NMRPipe60 and analyzed with CCPN software. Amide resonance assignments were performed according to previously reported chemical shift assignments for intrinsically unfolded aSyn.61–63 Binding of aSyn monomer to SUV was determined via 1H-15N-HSQC experiments in which 200 μM of 15N-labeled Ac-aSyn monomer in 20 mM sodium phosphate buffer, pH 7.5, 100 mM NaCl, 10% D2O was incubated in the absence or the presence of 8 mM of SUV. All NMR experiments were conducted at 10 °C.
Far-UV circular dichroism (CD).
The gain of content of α-helix structure of aSyn monomer, which occurs upon the binding to SUV, was evaluated by far-UV circular dichroism (CD) in a Chirascan Spectropolarimeter (Applied Photophysics, UK). Solutions of 40 μM of aSyn monomer in 20 mM sodium phosphate, pH 7.5, 100 mM NaCl, in the absence or presence of varying concentration of SUVs were analyzed in a 0.2 mm quartz cuvette at 25 °C. The ellipticity at 222 nm was measured and the background associated with buffer or SUV solutions was subtracted. The mean residue molar ellipticity at 222 nm (θMR,222) was determined using the equation (1):
where θ222 is the measured ellipticity at 222 (millidegrees), C is the protein concentration (Molar), n = 140 (number of amino acid residues), and l is the path length of the cuvette in cm (0.02 cm). Lipid titration curves generated by plotting [θ]MR,222 versus the lipid concentration were analyzed as previously described47,64 by fitting to the equation (2):
where R is the measured [θ]MR,222 at a given lipid concentration, R0 is the [θ]MR,222 in the absence of lipid, Rf is the [θ]MR,222 in the presence of saturating lipid, L is the total lipid concentration, C is the total protein concentration, Kd is the apparent macroscopic dissociation equilibrium constant, and N is the binding stoichiometry (lipids/protein).
ACKNOWLEDGMENT
C.F. is grateful to Dr. Dan Mulvihill, University of Kent, UK, for providing Nat-B plasmid. We gratefully acknowledge Trudy Fiona Ramlall for help with protein production and Dr. Shifeng Xiao, Dr. Guohua Lv, and Dr. Clay Bracken for help with NMR experiments.
Funding
This work was supported by the National Counsel of Technological and Scientific Development (CNPq).
Footnotes
Authors declare there is no conflict of interest regarding the publication of this articl
REFERENCES
- (1).Spillantini MG, Crowther RA, Jakes R, Hasegawa M, and Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U. S. A 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, and Goedert M (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- (3).Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, and Nussbaum RL (1997) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- (4).Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, and Riess O (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet 18, 106–108. [DOI] [PubMed] [Google Scholar]
- (5).Zarranz JJ, Alegre J, Esteban JCG, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Tortosa EG, del Ser T, Muñoz DG, and Yebenes JG (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol 55, 164–173. [DOI] [PubMed] [Google Scholar]
- (6).Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, Verny C, and Brice A (2013) French Parkinson’s Disease Genetics Study Group. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol 73, 459–471. [DOI] [PubMed] [Google Scholar]
- (7).Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, and Schapira AH (2013) A novel α-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, and Paetau A (2014) A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35, 2180 e1–5 . [DOI] [PubMed] [Google Scholar]
- (9).Petrucci S, Ginevrino M, and Valente EM, (2016) Phenotypic spectrum of α-synuclein mutations: New insights from patients and cellular models. Parkinsonism Relat. Disord 22, 16–20. [DOI] [PubMed] [Google Scholar]
- (10).Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, and Hardy KG (2003) Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- (11).Harlin CMC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, and Destée A (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 [DOI] [PubMed] [Google Scholar]
- (12).Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf OM, Masliah E, and Lashuel HA (2012) α-Synuclein in the central nervous system and from erythrocytes, mammalian cells and E. coli exists predominantly as a disordered monomer. J. Biol. Chem 287, 15345–15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Weinreb PH, Zhen W, Poon AW, Conway KA, and Lansbury PT Jr. (1996) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. [DOI] [PubMed] [Google Scholar]
- (14).Eliezer D, Kutluay E, Bussell R Jr., and Browne G (2001) Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- (15).Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, and Chilcote TJ (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem 281, 29739–29752. [DOI] [PubMed] [Google Scholar]
- (16).Fauvet B, Fares MB, Samuel F, Dikiy I, Tandon A, Eliezer D, and Lashuel HA (2012) Characterization of semisynthetic and naturally Nα-acetylated α-synuclein in vitro and in intact cells: implications for aggregation and cellular properties of α-synuclein. J. Biol. Chem 287, 28243–28262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Maltsev AS, Ying J, and Bax A (2012) Impact of N-terminal acetylation of α-synuclein on its random coil and lipid binding properties. Biochemistry 51, 5004–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kang L, Moriarty GM, Woods LA, Ashcroft AE, Radford SE, and Baum J (2012) Nterminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci 21, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dikiy I, and Eliezer D (2014) N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound α-synuclein and increases its affinity for physiological membranes. J. Biol. Chem 289, 3652–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC, (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Diao J, Burré J, Vivona S, Cipriano DJ, Sharma M, Kyoung M, Südhof TC, and Brunger AT, (2013) Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2:e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, and Riek R, (2011) In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U. S. A 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bisaglia M, Tosatto L, Munari F, Tessari I, de Laureto PP, Mammi S, and Bubacco L (2010) Dopamine quinones interact with α-synuclein to form unstructured adducts. Biochem. Biophys. Res. Commun 394, 424–428. [DOI] [PubMed] [Google Scholar]
- (24).Leong SL, Pham CL, Galatis D, Tavoletti MTF, Perez K, Hill AF, Masters CL, Ali FE, Barnham KL, and Cappai R (2009) Formation of dopamine-mediated α-synuclein-soluble oligomers requires methionine oxidation. Free Radic. Biol. Med 46, 1328–1337. [DOI] [PubMed] [Google Scholar]
- (25).Li J, Zhu M, Bog ABM, Monte DAD, and Fink AL (2004) Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson’s and Alzheimer’s disease. FASEB J 18, 962–964. [DOI] [PubMed] [Google Scholar]
- (26).Conway KA, Rochet JC, Bieganski RM, and Lansbury PT (2001) Kinetic stabilization of the α-synuclein protofibrils by dopamine-α-synuclein adduct. Science 294, 1346–1349. [DOI] [PubMed] [Google Scholar]
- (27).Follmer C, Romão L, Einsiedler CM, Porto TC, Lara FA, Moncores M, Weissmuller G, Lashuel HA, Lansbury P, Neto VM, Silva JL, and Foguel D (2007) Dopamine affects the stability, hydration and packing of protofibrils and fibrils of wild-type and variants of α-synuclein. Biochemistry 46, 472–482. [DOI] [PubMed] [Google Scholar]
- (28).Xu J, Kao SY, Lee FJ, Song W, Jin LW, and Yankner BA (2002) Dopamine-dependent neurotoxicity of α-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat. Med 8, 600–606. [DOI] [PubMed] [Google Scholar]
- (29).Burke WJ, Li SW, Williams EA, Nonneman R, and Zahm DS (2003) 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson’s disease pathogenesis. Brain Res 989, 205–213. [DOI] [PubMed] [Google Scholar]
- (30).Goldstein DS, Sullivan P, Holmes C, Kopin IJ, Basile MJ, and Mash DC, (2011) Catechols in post-mortem brain of patients with Parkinson disease. Eur. J. Neurol 18, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O’Dell M, Li SW, Pan Y, Chung HD, and Galvin JE (2008) Aggregation of α-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol 115, 193–203. [DOI] [PubMed] [Google Scholar]
- (32).Follmer C, Cerqueira EC, Franco DYY, Araujo GD, Pinheiro AS, Domont GB, and Eliezer D (2015) Oligomerization and membrane-binding properties of covalent adducts formed by the interaction of α-synuclein with the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL). J. Biol. Chem 290, 27660–27679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Plotegher N, Berti G, Ferrari E, Tessari I, Zanetti M, Lunelli L, Greggio E, Bisaglia M, Veronesi M, Girotto S, Serra MD, Perego C, Casella L, and Bubacco L (2017) DOPAL derived α-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep 7, 40699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Johnson M, Coulton AT, Geeves MA, and Mulvihill DP (2010) Targeted amino-terminal acetylation of recombinant proteins in E. coli. Plos One 5:e15801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Huang C, Ren G, Zhou H, and Wang CC (2005) A new method for purification of recombinant human α-synuclein in Escherichia coli. Protein Expr. Purif 42, 173–177. [DOI] [PubMed] [Google Scholar]
- (36).Giehm L, Lorenzen N, and Otzen DE (2011) Assays for α-synuclein aggregation. Methods 53, 295–305. [DOI] [PubMed] [Google Scholar]
- (37).Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mellier MAL, Ruggeri FS, Mbefo MK, Vercruysse F, Dietler G, Lee SJ, Eliezer D, and Lashuel HA (2014) The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity. J. Biol. Chem 289, 21856–21876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, Ghosh S, Jha NN, Kumar A, and Maji SK (2013) The Parkinson’s disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 52, 6925–6927. [DOI] [PubMed] [Google Scholar]
- (39).Rutherford NJ, Moore BD, Golde TE, Giasson BI (2014) Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α-synuclein. J. Neurochem 131, 859–867. [DOI] [PubMed] [Google Scholar]
- (40).Giasson BI, Uryu K, Trojanowski JQ, and Lee VM (1999) Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem 274, 7619–7622. [DOI] [PubMed] [Google Scholar]
- (41).Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, and Citron M (1999) Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J. Biol. Chem 274, 9843–9846. [DOI] [PubMed] [Google Scholar]
- (42).Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, and Lansbury PT Jr (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U. S. A 97, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Phelippe do Carmo-Gonçalves P, Lucas Alex do Nascimento LA, Cortines JR, Eliezer D, Romão L, Follmer C (2018) Exploring the role of methionine residues on the oligomerization and neurotoxic properties of DOPAL-modified α-synuclein. Biochem. Biophys. Res. Comm 505, 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ranjan P, and Kumar A (2017) Perturbation in long-range contacts modulates the kinetics of amyloid formation in α-synuclein familial mutants. ACS Chem. Neurosci 8, 2235–2246. [DOI] [PubMed] [Google Scholar]
- (45).Dedmon MM, Larsen LK, Christodoulou J, Vendruscolo M, and Dobson CM (2005) Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc 127, 476–477. [DOI] [PubMed] [Google Scholar]
- (46).Jensen PH, Nielsen MS, Jakes R, Dotti CG, and Goedert M (1998) Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem 273, 26292–26294. [DOI] [PubMed] [Google Scholar]
- (47).Ysselstein D, Joshi M, Mishra V, Griggs AM, Asiago JM, McCabe GP, Stanciu LA, Post CB, and Rochet JC (2015) Effects of impaired membrane interactions on α-synuclein aggregation and neurotoxicity. Neurobiol. Dis 79, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bartels T, Kim NC, Luth ES, and Selkoe DJ (2014) N-α-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS One 9, e103727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ruzafa D, Hernandez-Gomez YS, Bisello G, Broersen K, Morel B, and Conejero-Lara F (2017) The influence of N-terminal acetylation on micelle-induced conformational changes and aggregation of α-synuclein. PLoS One 12, e0178576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Pham CL, and Cappai R (2013) The interplay between lipids and dopamine on α-synuclein oligomerization and membrane binding. Biosci. Rep 33, e00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bodner CR, Maltsev AS, Dobson CM, and Bax A (2010) Differential phospholipid binding of α-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 49, 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bussell R Jr., and Eliezer D (2004) Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated α-synuclein. Biochemistry 43, 4810–4818. [DOI] [PubMed] [Google Scholar]
- (53).Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, and Goedert M (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett 576, 363–368. [DOI] [PubMed] [Google Scholar]
- (54).Fares MB, Bouziad NA, Dikiy I, Mbefo MK, Jovičić A, Kiely A, Holton JL, Lee SJ, Gitler AD, Eliezer D, and Lashuel HA (2014) The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet 23, 4491–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bartels T, Ahlstrom LS, Leftin A, Kamp F, Haass C, Brown MF, and Beyer K (2010) The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J 99, 2116–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Allen JWW, DuMond JF, Levine RL, and Bax A (2016) Toxic dopamine metabolite DOPAL forms an unexpected dicatechol pyrrole adduct with lysines of α-synuclein. Angew. Chem. Int. Ed. Engl 20, 7374–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Cerqueira EC, Carmo-Gonçalves P, Pinheiro AS, Cortines J, and Follmer C (2013) α-Synuclein as an intrinsically disordered monomer – fact or artefact? FEBS J 280, 4915–4927. [DOI] [PubMed] [Google Scholar]
- (58).Marley J, Lu M, and Bracken C (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75. [DOI] [PubMed] [Google Scholar]
- (59).Abramoff MD, Magalhaes PJ, and Ram SJ (2004) Image processing with ImageJ. Biophot. Int 11, 36–42. [Google Scholar]
- (60).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (61).Bermel W, Bertini I, Felli IC, Lee YM, Luchinat C, and Pierattelli R (2006) Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J. Am. Chem. Soc 128, 3918–391. [DOI] [PubMed] [Google Scholar]
- (62).Chandra S, Chen X, Rizo J, Jahn R, and Südhof T (2003) A broken α-helix in folded α-synuclein. J. Biol. Chem 278, 15313–15318. [DOI] [PubMed] [Google Scholar]
- (63).Eliezer D, Kutluay E, Bussell R Jr., and Browne G (2001) Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- (64).Shvadchak VV, Lockhart LJF, Yushchenko DA, and Jovin TM (2011) Specificity and kinetics of α-synuclein binding to model membranes determined with fluorescent excited state intramolecular proton transfer (ESIPT) probe. J. Biol. Chem 286, 13023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]