

Abstract
Leishmania donovani is a unicellular parasite that causes visceral leishmaniasis, a fatal disease in humans. In this study, a complete assembly of the genome of L. donovani is provided. Apart from being the first published genome of this strain (HU3), this constitutes the best assembly for an L. donovani genome attained to date. The use of a combination of sequencing platforms enabled to assemble, without any sequence gap, the 36 chromosomes for this species. Additionally, based on this assembly and using RNA-seq reads derived from poly-A + RNA, the transcriptome for this species, not yet available, was delineated. Alternative SL addition sites and heterogeneity in the poly-A addition sites were commonly observed for most of the genes. After a complete annotation of the transcriptome, 2,410 novel transcripts were defined. Additionally, the relative expression for all transcripts present in the promastigote stage was determined. Events of cis-splicing have been documented to occur during the maturation of the transcripts derived from genes LDHU3_07.0430 and LDHU3_29.3990. The complete genome assembly and the availability of the gene models (including annotation of untranslated regions) are important pieces to understand how differential gene expression occurs in this pathogen, and to decipher phenotypic peculiarities like tissue tropism, clinical disease, and drug susceptibility.
Introduction
Leishmaniasis is a group of diseases caused by protists of the genus Leishmania, which belong to the eukaryotic lineage Excavata and are classified within the order Trypanosomatida1. The list of pathogenic Leishmania species is large, even though the taxonomy of the genus is still under debate2. Nevertheless, some species are clearly linked to specific clinical syndromes3. Thus, Leishmania major most frequently causes cutaneous leishmaniasis (CL), which is characterized by the appearance of single to multiple skin ulcers, satellite lesions or nodular lymphangitis, but these affections more often resolve without treatment4. Leishmania braziliensis, which is exclusively distributed in South America, can cause mutilating mucocutaneous leishmaniasis (MCL). Leishmania infantum and Leishmania donovani are the causative species of the most severe form of disease, the visceral leishmaniasis (VL). According to recent estimates5, approximately 0.2 to 0.4 million VL cases and 0.7 to 1.2 million CL cases occur each year in the world. Unfortunately, there are no vaccines for human use and treatment of leishmaniasis relies on a small arsenal of drugs that, moreover, have important limiting factors in its use, like toxicity and emerging resistance6. Therefore, new developments, both vaccines and more effective drugs, are needed to control leishmaniasis. Better knowledge of the molecular biology of this parasite will help to achieve these objectives.
Most of the Leishmania species are digenetic, i.e. they need two hosts to complete their life cycle. Leishmania parasites are transmitted to vertebrate hosts by the bite of female phlebotomine sandflies; a remarkable association between sandflies and Leishmania species exists, being dictated by old relationships in evolutionary terms7. In the insect vector, the parasite survives and proliferates extracellularly in the alimentary tract; in contrast, in the vertebrate host, it adopts an obligatory intracellular form that thrives inside phagolysosomes8. The adaptation required to face these different host environments is achieved by modulating gene expression. However, Leishmania and related trypanosomatids are eukaryotes that possess unusual ways of controlling their gene expression9. Little, if any, regulation al the transcriptional level seems to exist; the genes, which are organized in large clusters with the same transcriptional orientation, but without any functional relationship, are constantly transcribed into long polycistronic precursor RNAs10. Following synthesis, the precursor transcripts are processed into mature mRNAs, each coding for an individual protein, by another unusual process known as trans-splicing11. The trans-splicing machinery, at specific positions, cuts the polycistronic precursor and a 39-nt long mini-exon (also named as spliced leader or SL) is added to the 5′-end of all mRNAs. In a coordinated manner, a poly-A tail is added to the 3′-end of mRNAs in such a way that polyadenylation of the upstream gene is directed by (and coupled to) the trans-splicing of the downstream gene12. The mini-exon contains a highly modified 5′-cap structure that, upon exportation of the mRNAs to the cytoplasm, is recognized by the translation machinery13. Although this polycistronic transcription might suggest that adjacent genes would have the same expression levels, it is known that this is not the case and strikingly different steady-state levels exist for collinear mRNAs14. Hence, mechanisms targeting post-transcriptional events, such as trans-splicing efficiency, mRNA nucleo-cytoplasmic transport, transcript degradation and translational efficacy are the relevant players controlling gene expression in Leishmania9,15. Ultimately, the fate of any given mRNA is determined by the ensemble of particular RNA-binding proteins (RBPs) that recognize specific sequences and/or structural motifs present in the mRNAs, mainly located at their untranslated regions (UTRs)16,17. In this context, physical delimitation of UTRs and definition of accurate gene models are paramount for deciphering the regulatory networks that control gene expression not only in Leishmania but in any eukaryotic cell18.
From a genomic approach, the basic step for establishing gene models is the determination of the complete genome sequence of an organism. The first sequenced genome for a Leishmania species was that of L. major (Friedlin strain); this was attained after a hard experimental labour, following meticulous strategies and involving large dedicated sequencing centres19. This genome assembly remains as one of the more robust Leishmania genomes determined to date, and, since then, only few and small modifications have been introduced20. During the last decade, the extraordinary progress in sequencing due to the development of the so-called next-generation sequencing (NGS) technologies together with a significant reduction of sequencing costs have enabled the determination of genome sequences for many Leishmania species and strains21. However, even though this information is really valuable, the quality of genome assemblies is lower than that achieved in 2005 for the L. major (Friedlin strain) genome. Nevertheless, in a recent work, by the combination of sequencing data derived from two NGS- platforms, the Pacific Biosciences (PacBio) technology, which produces long sequencing reads, and the Illumina technology, which yields shorter but more accurate sequences, a complete assembly for the L. infantum (JPCM5 strain) was obtained22.
On the other hand, the establishment of gene models requires not only the prediction of open reading frames (ORFs) but also the delimitation of UTRs, which are regions earmarked to govern the fate of the mRNA molecules inside the cell23–25. In fact, the Leishmania genome contains a remarkable number of genes sharing identical ORFs but differing substantially in their UTRs26–32. Moreover, several studies dealing with expression levels of particular genes in Leishmania have evidenced the relevance of the 5′- and/or 3′- UTRs in mediating differential transcript abundances and translation efficacies along the parasite life cycle33–37. For a long time, identifying the full set of transcripts present in a cell or organism, i.e. the transcriptome, was envisioned as impractical given that this task required the sequencing of large numbers of expressed sequence tag (EST) sequences, an approach that otherwise is biased for the abundance of the different RNA molecules. Again, in this research field, the application of NGS technologies for RNA sequencing (RNA-seq) has allowed to reveal the landscape and dynamics of particular transcriptomes with unprecedented level of depth and accuracy38. However, to date, there are few studies aimed to deciphering the transcriptomes in Leishmania39. L. major was the first species of the genus Leishmania in which a complete poly-A+ transcriptome was generated; a total of 10,285 transcripts were identified, 1,884 of which did not correspond to previously predicted genes and 410 miss-annotated ORFs were corrected14. L. mexicana was the second Leishmania species to have annotated its transcriptome, consisting of 9,169 transcripts matching with previously predicted ORFs and 936 novel transcripts40. Additionally, the L. major repertoire of snoRNAs was determined by affinity purification of the SNU13 and NHP2 RNPs and RNA-seq analysis; the study identified 81H/ACA and 80 C/D snoRNAs41.
Here, we describe the first complete assembly for the L. donovani genome, and this information, combined with RNA sequencing (RNA-seq), was used to generate a comprehensive transcriptome. Thus, our study is providing the gene models for this Leishmania species that will guide further investigations into the molecular mechanisms responsible for differential gene expression along its life cycle and in response to environmental stresses such as drug treatment.
Materials and Methods
Leishmania culture
Promastigotes of L. donovani (MHOM/ET/67/HU3) were grown at 26 °C in RPMI 1640-modified medium42. In detail, RPMI 1640 (Gibco, Ref 51800-043) medium was supplemented with 13.3 mM glutamine, 2.5 mM arginine, 0.3 mM cystine,1.7 mM glutamate, 62.1 mM proline, 0.6 mM ornithine, 3.8 mM glucose, 2.2 mM fructose, 5.1 mM malate, 2.8 mM α-ketoglutarate, 0.5 mM fumarate, 0.5 mM succinate, 25 mM Hepes, 50 µg/ml gentamicin, 2 × MEM vitamins (Gibco); after adjusting pH to 7.2, heat-inactivated foetal bovine serum (HIFBS, Gibco) was added to a final concentration of 20%. Alternatively, in the experiments dealing with the isolation of DNA for PacBio sequencing, promastigotes of the same strain were cultured at 26 °C in M199 medium (Sigma-Aldrich) supplemented with 10% HIFBS (Biowest), 40 mM Hepes (pH 7.4), 0.1 mM adenine, 10 µg/ml hemin, 1 µg/ml biotin, 2 ng/ml biopterin, 100 U/ml penicillin G and 0.1 mg/mL streptomycin sulphate.
DNA and RNA isolation
RNA was prepared from around 4 × 108 promastigotes in the late logarithmic phase; after harvesting by centrifugation, the pellet was suspended in 1 ml of TRI Reagent (Sigma-Aldrich, product No. T9424). Manufacturer’s instructions were followed. Samples were kept at −70 °C for a week before proceeding with the phase separation. After thawing, 0.2 ml of chloroform was added, and the mixtures were shaken vigorously for 15 sec. After centrifugation, three phases were observed: a red organic phase (containing protein), an interphase (containing DNA), and a colorless upper aqueous phase (containing RNA). Both interphase and aqueous phases were processed separately for isolation of DNA and RNA, respectively. RNA samples were suspended in DEPC-treated water, and their concentrations were determined using the Nanodrop ND-1000 (Thermo Scientific); all samples showed A260/A280 ratios higher than 2.0. In addition, RNA integrity was checked in a bioanalyzer (Agilent 2100). The DNA samples were also quantified by absorbance at 260 nm using the Nanodrop, and the integrity analyzed by agarose gel electrophoresis. For PacBio sequencing, DNA was prepared following a classical phenol extraction method43.
Illumina sequencing of DNA and reads assembly
Library construction and paired-end library sequencing were performed at the Centro Nacional de Análisis Genómico (CNAG-CRG, Spain) using Illumina HiSeq. 2000 technology. A total of 16,980,871 paired-end, 101 bp sequence reads were generated. PrinseqQuality (http://prinseq.sourceforge.net/) was applied to quality filtering/trimming of reads (cut-off value, 20), and only reads with length ≥60-nt were used. Reads were assembled using the CLC Genomics Workbench version 5.0 (CLC Bio).
PacBio sequencing and de novo assembly
The single-molecule real-time (SMRT) sequencing technology developed by Pacific Biosciences (PacBio) was used for long-read sequencing. A total of 312,388 pre-filtered reads were generated on a PacBio RS II sequencing instrument. The sequencing service was provided by the Norwegian Sequencing Centre (www.sequencing.uio.no), a national technology platform hosted by the University of Oslo and supported by the “Functional Genomics” and “Infrastructure” programs of the Research Council of Norway and the South-Eastern Regional Health Authorities. Quality trimming of PacBio reads was done by default parameters as part of the HGAP pipeline (P_filter Module).
De novo genome assembly was carried out following a hierarchical genome-assembly process (HGAP44), using the HGAP v3 (PacBio, SMRT Analysis Software v2.3.0) and HGAP4 (PacBio, SMRT Link 4.0.0) protocols. Three different assemblies were performed with HGAP by varying the size of the expected genome (34 and 35 Mbp for HGAP3, and 35 Mbp for HGAP4).
Assembly refinements
The contigs, initially assembled by HGAP (varying the GenomeSize parameter) from the PacBio reads, were checked in order to discard those having a disproportionately low coverage (<40x) or short length (<15-Kb). To assign the correspondence between contigs and chromosomes, BLAST searches45 were performed between the assembled contigs and current L. major (Friedlin strain) genome19. Finally, a total of 41 contigs were determined as bona fide genomic sequences. Thirty-one of these contigs were found to correspond to complete chromosomes. To complete assemble the other five chromosomes, their corresponding contigs were joined using minimus2 pipeline46. Firstly, the gap size between contigs was calculated (lower than 5-kb in all cases) and, based on the Illumina reads, the gaps were closed by Gapfiller47, which takes into account the mean size of the paired-end reads.
On the other hand, the contigs generated from the Illumina sequencing reads (see above) were aligned to the PacBio reads-based assembly using LAST aligner (http://last.cbrc.jp/). This allowed the identification of Illumina contigs that aligned with the chromosomal ends and therefore to extend the chromosomes. Several tools were used to accurately extend the chromosomal ends. Thus, for chromosomes 7, 14, 31 and 36, the optimal extension was attained with MAFFT multiple-aligner software48. For chromosomes 10, 13, and 35, the best extension was obtained by BLAST alignment. Additionally, for the rest of chromosomes, the SSPACE-standard software49 was used.
Finally, sequence corrections were performed in the draft assembly using PacBio-utilities (indel-targets and indel-apply tools; https://github.com/douglasgscofield/PacBio-utilities). This tool uses paired end Illumina reads and it is designed for detecting single-base deletions introduced with low frequency in homopolymer strings by the PacBio platform. Sequence insertions/deletions (indels) were introduced when they were supported by more than 10 Illumina reads and the insertion/deletion was present in 80% (or above) of the reads mapping the concerned position. Furthermore, an in-house Python script, which uses the results provided by Pilon tool50, was designed to assign the position when ambiguous indels were detected. A total of 3,098 indels were corrected, of which 3,061 corresponded to sequence insertions and 37 to deletions.
Alignments and coverage maps
A coverage analysis on the newly assembled chromosomes was performed using both Illumina and PacBio reads. Illumina reads were aligned by Bowtie251 and PacBio bax.h5 reads were aligned by pbalign (which uses the BLASR method52). Coverage analysis was done from each alignment along the 36 chromosomes using the GenomeCoverageBed tool53. Coverage data were smoothed using an in-house Perl script that calculates the mean coverage over a one bp step sliding window with a size of 200 bp. The coverage plots were generated using GNUPLOT (http://www.gnuplot.info/).
SNP identification
Firstly, the BWA-MEM tool54 was used to align the Illumina reads to the L. donovani (HU3) genome. Afterwards, the picard tool (http://broadinstitute.github.io/picard/) was used to remove read duplicates. The selected reads were realigned to the genome by GATK (version 3.7; www.broadinstitute.org/gatk/). Finally, variant calling was done by two methods, the GATK HaplotypeCaller v. 3.755 and the Freebayes version 1.1.056. In both methods, the quality filters (depth above 9 and quality above 10) were applied.
Annotation of protein-coding genes and known non-coding RNAs
Bulk annotation of the assembled L. donovani HU3 genome was performed using Companion web server57 with the default settings, and selecting the L. major (Friedlin strain) annotation as a reference genome. OrthoMCL58 and BLAST searches were performed to establish orthology between L. donovani (HU3) and L. major (Friedlin) genes. All this information was combined into a GFF3 file using an in-house script written in Python. The automatic ID codes generated by Companion were accommodated to the transcript nomenclature (see below, transcriptome annotation section), and the annotated genes were named with the label LDHU3_XX.YYYY, where XX identifies the chromosome number and YYYY is the serial number assigned to the transcript in which the ORF is found. For structural RNAs, it is indicated the RNA type, the chromosomal location and a serial number of three digits (e.g. LDHU3_TRNA.02.001).
Transcriptome annotation
A total of 56,506,556 (2 × 76-nt) stranded RNA-seq reads, derived from three biological replicates, were generated using the Illumina HiSeq. 2000 technology (CNAG-CRG, Spain) as described elsewhere59. The L. donovani (HU3) transcriptome was generated following the pipeline described by Rastrojo et al.14. In brief, after going through the standard Illumina quality-filtered process, the reads were further analysed using FASTQC to assure adequate quality. RNA-seq reads were then mapped to the L. donovani de novo genome (generated in this work) using Bowtie2 aligner with default parameters. Lastly, mapped reads were assembled into transcripts using Cufflinks60 with default parameters. On the other hand, among the unaligned reads, a search was performed, using a Perl in-house script, looking for reads that contained eight or more nucleotides identical to the 3′-end of the SL sequence (AACTAACGCTATATAAGTATCAGTTTCTGTACTTTATTG). After removing the SL-derived nucleotides, the trimmed reads were mapped back to the L. donovani (HU3) to define the position of the corresponding SL-addition site (SAS). For the identification of poly-A addition sites (PAS), a similar procedure was performed. Here, the presence of an A-string longer than 5 nucleotides at the end of the reads was used to uncover potential PAS. A Perl script was developed to trim the transcripts generated by Cufflinks according to the positions of the mapped SAS and PAS. Finally, the transcripts were visualized and manually curated (if required) using the Integrative Genomics Viewer (IGV61). During this revision, some previously annotated ORFs had to be corrected because they started before the beginning of the transcript. For transcript nomenclature, a serial number (four-digits), according to its chromosomal location (from left to right) and increasing by 10, was added after the chromosome ID and labelled with the word ‘T’, e.g. LDHU3_01.T0010 (i.e. first transcript annotated on chromosome 1).
Determination of RNA levels from RNA-seq data
RNA-seq data were obtained from three different cultures of L. donovani HU3 promastigotes in order to determine relative levels of every transcript in the transcriptome. Transcript levels were expressed as FPKM (fragments per kilobase of transcript per million mapped reads). This parameter reflects the abundance of a transcript in the sample by normalizing for RNA length and the total read number62. In this study, Cufflinks tool60 was used to calculate the FPKM.
Results and Discussion
De novo assembly of the L. donovani (HU3 strain) genome
The L. donovani HU3 strain (MHOM/ET/67/HU3), also known as LV9 or L82, is one of the most widely used strains in studies regarding drug resistance and other molecular aspects63–65. It is a cloned line, as reported elsewhere66. Firstly, DNA from LdHU3 promastigotes was sequenced using Illumina technology, obtaining 16,980,871 (2 × 101 bp) reads with a median insert size of 295-bp. Taking into account the estimated size (32,4-Mb) for the L. donovani genome assembly reported by Downing et al.67, these reads would account for an average sequencing depth of 105x. Several assemblers were tried in order to get a de novo assembly, obtaining the best results with CLC Genomics Workbench software (CLC Bio; version 5.0). However, the attained assembly represented a fragmented genome composed by 2,545 contigs and 1,224 gaps that amounted for a total genome size of 30,221,623 bp, being the longest contig of 201,094-bp. Leishmania genomes are rich in repeated sequences (0.4–1 kb in length) that are scattered along the different chromosomes68,69, and this is a cause of conflict for assemblers that work with short reads as those generated by Illumina platforms70,71. In order to improve the genome assembly, we also obtained long-read sequences, generated by the PacBio single-molecule real-time (SMRT) sequencing technology72. A total of 312,388 reads, with an average length of 11,900-bp were obtained. According to the genome size of the L. donovani BPK strain (Table 1), these figures would account for an 89× mean coverage. Following the methodology detailed in the Materials and Methods section, a de novo assembly resulted in the generation of 85 contigs, even though 44 of them were discarded because of their small size and the fact that they were supported by a low number of reads (spurious contigs). Afterwards, using several assemblers and other bioinformatics tools, together with the contigs generated from Illumina-reads (see Assembly refinements in Methods section), it was possible to join some of the 41 contigs to a final number of 36 contigs, matching the number of chromosomes existing in this Leishmania species73. Chromosomes 10, 12, 16, 27 and 35 resulted from the joining of two PacBio contigs (Fig. 1), whereas the rest of chromosomes corresponded directly to individual PacBio contigs. As shown in Fig. 1, a continuous and homogenous distribution of reads was observed after the alignments of both types of sequences (Illumina and PacBio reads) against the assembled chromosomes; this finding supports that a correct assembly was attained.
Table 1.
Features of the L. donovani assembled genomes and sequencing strategies used.
| Features | Strain BPK282A1a | LD-974b | HU3c |
|---|---|---|---|
| Chromosomes (scaffolds) | 36 | 36 | 36 |
| Number of contigs | 2154 | 1100 | 36 |
| Annotated genes | 8195 | 8474 | 8595 |
| Annotated CDS | 8070 | 8336d | 8405 |
| Annotated pseudogenes | 13 | NAe | 47 |
| Annotated structural genes | 112 | 138 | 143 |
| Number of gaps | 2118 | 1064 | 0 |
| Number of Ns | 1192833 | NA | 0 |
| Haploide genome size (bp) | 31252135 | 27848322 | 33035865 |
| Illumina coverage median | 52 | ~110 | 105 |
| 454 GS FLX Titanium coverage median | 22 | — | — |
| PacBio coverage median | — | — | 89 |
Figure 1.

Read-depth analyses along the chromosomes generated by the fusion of two PacBio-assembled contigs (panels A–E). Coverage (log2 scale) was determined by sliding window analysis (bin 200 bp) with either Illumina (in blue) or PacBio (in red) reads, along chromosomes 10, 12, 16, 27 and 35. The size and position of the contigs used are shown by lines with arrow-heads. Panel F, relative somy of the L. donovani (HU3) chromosomes. The somy estimation was performed using a 2-loop method77. The median coverage of the genome is shown by a solid line, and it was assigned as 2, taking into account that diploid is considered the major ploidy status in Leishmania. The dotted lines indicated the estimated values for other somies. Graphs were generated from the median coverage values for each chromosome using the barplot function of R package (https://cran.r-project.org).
A singularity in the coverage was observed in the right end of chromosome 35 (Fig. 1E), the read depth of PacBio reads in the 3′ end (around 100-kb) was clearly higher than that observed in the rest of the chromosome. It was postulated the existence of an extrachromosomal amplicon covering this region. We analyzed whether this region may be related to the LD1 extrachromosomal amplicon, which has been observed very often in L. donovani and other Leishmania species63. BLAST analysis, using a partial sequence (7.1-kb) of the L. infantum LD1 amplicon determined by Myler et al.74, showed a 99% of sequence identity with the region 1,904,625-1,911,724 of the L. donovani chr35; interestingly, this region marks the point in which the PacBio coverage increases. Nevertheless, the Illumina coverage did not show a similar increase. A plausible explanation to this fact may be found in the different growth media used for culturing L. donovani promastigotes before DNA isolation for each one of the sequencing methodologies (see Material and Methods for further details). In brief, DNA for Illumina sequencing was isolated from promastigotes cultured in RPMI medium supplemented with 20% foetal bovine serum (FBS) and DNA isolation for PacBio sequencing was independently obtained from promastigotes of the same HU3 strain but cultured in M199 medium supplemented with 10% FBS. Our hypothesis is that the LD1 amplicon was generated during the culturing in M199 medium.
Table 1 summarizes the main features of the assembled genome for the strain HU3, generated in this work, and its comparison with the two other L. donovani assemblies published to date. This new assembly represents an improved genome regarding the current reference genome (BPK strain67), which is incomplete and presents some annotation deficiencies75. Thus, apart from eliminating the gaps, the genome size has been extended about 1.8 Mb in length and the total number of annotated genes has been increased by 400 genes, regarding the current reference L. donovani (BKP strain) genome. Additionally, after comparing the HU3 assembled genome and the BKP one, significant reorganizations in ten chromosomes were observed (see Figs S1 to S10 in Supplementary Information File). However, it is likely that these differences in chromosomal structure between both strains may be rather apparent than real, taking into account the fragmented assembly, currently available for the BKP strain. This question might be addressed when a full assembled genome for this strain is generated.
The assembly of the L. donovani HU3 genome cannot be considered as the final one, mainly considering the chromosomal extremities. Thus, telomeric TAGGGT repeats were found at the ends of many chromosomes, but not in all. They were found at the 5′ ends of the chromosomes 1, 5, 8, 14, 15, 16, 18, 22, 24, 27, 28, 32, 33 and 35, and at 3′ ends of the chromosomes 4, 7, 9, 12, 13, 15, 18, 20, 23, 24, 26, 27, 28, 29, 31, 32, 34 and 35. Chiurillo and co-workers76 have documented the existence in all L. donovani chromosomes of complex arrays of repeated and non-repeated sequences, adjacent to the telomeric repeats. It is likely that the complexity of these arrays and the intrinsic difficulty of sequencing the chromosomal ends have precluded the complete assembly of the extremities for all the L. donovani chromosomes.
Based on the Illumina read coverage, the somy for each chromosome was estimated by the 2-loop method77. The results indicated that most of the chromosomes in this strain are diploid (Fig. 1E). Chromosomes 5, 23, 26 and 33 would be trisomic and chromosome 31 appeared as pentasomic. The somy status is similar to that determined for other L. donovani strains67,78,79.
Additionally, we analyzed the degree of allelic heterozygosity by using two SNP-calling predictors, GATK HaplotypeCaller v. 3.7 and Freebayes version 1.1.0 (see Materials and Methods for further details). The results were very similar, GATK identified 4,622 SNPs, and 4,865 SNPs were annotated by Freebayes. Rogers et al.78, using also Illumina reads to call heterozygous SNPs in the reference genomes of L. major, L. infantum, L. braziliensis and L. mexicana, identified 297, 629, 44,588 and 12,531 SNPs, respectively. However, it is likely that the degree of heterozygosity may be a strain-specific trait rather than a species-specific one.
Table 2 shows the sizes and the number of annotated genes for each one of the 36 chromosomes that comprise the genome of this Leishmania species. The genome size and the number of annotated genes coding for proteins are very similar to those determined in other Leishmania species for which their genomes have been completely assembled. Thus, the L. major (Friedlin strain) genome has a size of 32,816,678 bp and 8,272 protein-coding genes were annotated19. The size of the L. infantum (JPCM5 strain) genome is 32,802,969 bp, and 8,645 protein-coding genes were annotated22. The size for the L. donovani (HU3 strain) genome assembled in this work was 33,035,865 bp, and the number of protein-coding genes was 8,405 (Table 1).
Table 2.
Chromosomal sizes and number of annotated genes in the L. donovani (HU3 strain) genome.
| Chromosome | Size (bp) | Annotated genes | Chromosome | Size (bp) | Annotated genes |
|---|---|---|---|---|---|
| 1 | 292351 | 85 | 19 | 718213 | 176 |
| 2 | 361533 | 73 | 20 | 741387 | 175 |
| 3 | 387516 | 99 | 21 | 764502 | 232 |
| 4 | 475442 | 128 | 22 | 740689 | 172 |
| 5 | 467653 | 154 | 23 | 785035 | 211 |
| 6 | 521439 | 138 | 24 | 860435 | 247 |
| 7 | 591743 | 134 | 25 | 897487 | 264 |
| 8 | 563529 | 135 | 26 | 1062302 | 277 |
| 9 | 574290 | 180 | 27 | 1162458 | 280 |
| 10 | 584800 | 153 | 28 | 1177572 | 327 |
| 11 | 601398 | 147 | 29 | 1263082 | 310 |
| 12 | 635222 | 130 | 30 | 1402142 | 389 |
| 13 | 649940 | 169 | 31 | 1543155 | 358 |
| 14 | 650104 | 160 | 32 | 1564662 | 422 |
| 15 | 658046 | 164 | 33 | 1553627 | 377 |
| 16 | 691230 | 178 | 34 | 1895418 | 485 |
| 17 | 701758 | 182 | 35 | 2008222 | 540 |
| 18 | 718943 | 172 | 36 | 2768540 | 772 |
| Genome | 33035865 | 8595 |
After sequencing the L. braziliensis and L. infantum genomes80, and their comparison with the L. major genome, it was surprising to realize the quite small number of species-specific genes existing in the three Leishmania species, taking into account both the large evolutionary distance that separates those species and the different pathologies they produce81. In particular, only five L. major–specific genes and 26 L. infantum–specific genes were identified80. Hence, we considered of interest to determine the number of species-specific genes existing in L. donovani by comparing them with the L. major annotated genes. L. donovani and L. infantum species are genetically almost indistinguishable82 and both cause VL in humans, whereas L. major causes cutaneous affections and diverged from the L. donovani/L. infantum complex parasites more than 10 million years ago7. Gene annotation of the assembled L. donovani HU3 genome was performed using the Companion web server57, selecting the L. major (Friedlin strain) annotation as a reference. An initial analysis indicated that 153 of the protein-coding genes annotated in the L. donovani genome did not have orthologues in the L. major database. However, an individual analysis of these predicted genes allowed us to determine that 97 out of the 153 protein-coding genes are present, but currently non-annotated, in the L. major genome. Another 41 L. donovani annotated genes showed remarkable sequence identity with L. major genomic regions, but the existence of stop codons suggested that these must be pseudogenes in L. major. Finally, for the remaining 15 L. donovani annotated genes (see Table 3), no significant sequence homology was found and, therefore, they can be considered as genes lacking orthologues in L. major.
Table 3.
L. donovani annotated genes without orthologues in the L. major genome.
| LDHU3.03.0460 | hypothetical protein |
| LDHU3.03.0900 | CDP-alcohol phosphatidyltransferase, putative |
| LDHU3.08.0620 | cyclopropane-fatty-acyl-phospholipid synthase |
| LDHU3.23.1360 | hypothetical protein |
| LDHU3.28.3370 | hypothetical protein |
| LDHU3.29.1980 | hypothetical protein |
| LDHU3.29.2050 | Amastin surface glycoprotein, putative |
| LDHU3.29.2080 | Amastin surface glycoprotein, putative |
| LDHU3.29.2100 | tuzin like protein, putative |
| LDHU3.31.2550 | hypothetical protein |
| LDHU3.32.3250 | hypothetical protein |
| LDHU3.33.4860 | Zn-finger in Ran binding protein, putative |
| LDHU3.34.4290 | hypothetical protein |
| LDHU3.36.0800 | sec. 14, cytosolic factor |
| LDHU3.36.5620 | Nucleotidyltransferase domain containing protein, putative |
The 15 LdHU3-specific CDSs (Table 3) are present in both the L. donovani BPK genome67 and the L. infantum genome22. Nevertheless, three of them are truncated in the BPK reference genome and seven were not annotated. In contrast, all of them are present in the L. infantum genome and 13 of them are currently annotated.
More pronounced differences between L. major and L. donovani genomes exist when pseudogenes are considered. Thus, among the L. donovani annotated genes, 47 were classified as pseudogenes based on either the presence of internal stop codons or incomplete ORF (Supplementary File 1, Table S1). Remarkably, except for one pseudogene that is also annotated as pseudogenic (LmjF.23.0910), the orthologues to the L. donovani pseudogenes seem to be functional genes in the L. major genome. Conversely, 38 out of the 39 pseudogenes currently annotated in the L. major genome19 seem to be functional genes in L. donovani. Therefore, it is likely that some phenotypical and virulence traits specific for each species can arise from differences in the degree of ‘pseudogenization’ of particular genes.
L. donovani (HU3 strain) genome contains many tandemly repeated genes
L. major (Friedlin strain) genome is considered the ‘gold standard’ among the Leishmania genome assemblies because it was the first to be sequenced19 and, surely, the best assembled until recently22. A synteny analysis comparing the L. major (Friedlin) and the L. donovani (HU3 strain) genomes indicated an extremely conservation of gene order, even though a few genomic reorganizations were observed (data not shown). However, significant differences regarding the number of copies in loci with tandemly repeated genes exist between both species (Fig. 2A). A total of 240 loci with tandemly repeated genes were identified, and the majority of the loci (168; listed in Supplementary File 1, Table S2) were found to contain identical number of copies in both species. In addition, 33 loci with tandemly repeated genes exist in both species, but the number of genes is different for each species (those loci are listed in Supplementary File 1, Table S3). An example is shown in Fig. 2B, the elongation factor1-alpha locus is composed by 23 tandemly linked genes in L. donovani (HU3 strain) whereas only seven copies were assembled in the L. major (Friedlin) genome. Finally, L. donovani (HU3) genome has 25 loci with tandemly repeated genes that are single copy in L. major (Friedlin), and the L. major genome contains 14 tandemly repeated loci that in L. donovani have only a gene (see Supplementary File 1, Tables S4 and S5, respectively, for lists of these loci). As an example, Fig. 2C illustrates the case of LDHU3_29.3240-3320 locus, coding for a hypothetical protein having a leucine-rich repeat domain, that is composed by seven genes in L. donovani but a sole gene copy (LmjF.29.2130) in the L. major genome. Several authors have proposed that differences in gene copy number may contribute to modulate gene expression levels, providing additional genetic contributions to species-specific differences in parasite tropism and disease outcome78,83.
Figure 2.

Comparison of tandemly repeated genes existing in the L. donovani (LdHU3) and the L. major (LmjF) genomes. Panel A, tandemly repeated genes in both species distributed according to the similarities/differences in the number of copies. Panel B, schematic representation of the loci coding for the elongation factor 1-alpha genes in the L. donovani and L. major genomes. The repeated copies are shown as green boxes in both species. The flanking ORFs are shown in red (ADP-ribosylation factor-like protein 1) and dark blue (receptor-type adenylate cyclase A). Panel C, genes encoding for a hypothetical protein (with a leucine-rich repeat domain) are shown as green boxes in both species. The flanking ORFs are shown in red (VIT family, putative) and dark blue (C-8 sterol isomerase-like protein).
Transcriptome of L. donovani HU3 strain
After the assembly of the genome for this L. donovani strain, the definition of the poly-A+ transcriptome was undertaken (see Methods section for procedure details). Finally, 10,893 transcripts/gene models were defined (see Supplementary file 2 for the complete list), of which 8,452 corresponded to genes with annotated protein-coding sequences (CDS), whereas the remaining 2,441 lacked annotated CDS. In the latter group, SL addition sites (SAS) were observed in most of them (2,301, 94%), even many of them presented two or more alternative SAS (1,781, 73%). Regarding those annotated as protein-coding transcripts, SAS were identified for nearly all the transcripts (8430, 99.7%) and two or more alternative SAS were defined for 7,792 (92.2%) of them. Moreover, 1,565 transcripts contained alternative SAS within the predicted ORF. In fact, for 249 genes, the ORF predicted by bioinformatics tools had to be corrected during the annotation of the L. donovani HU3 genome (above) due to the finding that transcripts were shorter than the predicted ORF, and no alternative SAS were found outside the ORF. In those cases, the ORF predicted by bioinformatics means had to be re-annotated taking into account the first, in-phase ATG initiation codon, located within the delineated transcript. An example of such events is shown in Fig. 3. Thus, the transcript LDHU3_05.T0940, delimited by SL and polyadenylation addition sites and supported by the RNA-seq coverage (panel A–C), was found to start downstream of the automatic CDS annotation, done by bioinformatics tools (panel D). Therefore, a new ORF had to be defined within the transcript sequence (panel E). Finally, after combining all this information, a plausible gene model (i.e. gene LDHU3_05.0940) was generated (panel F).
Figure 3.

Illustration of the process followed for correcting the automatic annotation of CDS based on the transcript definition. Panel A, mapping of RNA-seq reads in the genomic region expanding the LDHU3_05.0940 gene. Panel B, mapping of SL-containing RNA-seq reads (two SL-addition sites (SASs) were mapped, the main site was covered by 72 reads and the secondary –small arrow- by only 2). Panel C, LDHU3_05.T0940 transcript annotation based on the position of the main SAS and the polyadenylation site (not shown). Panel D, automatic CDS annotation generated by Companion. Panel E, manually corrected CDS annotation, after delimiting the transcript. Panel F, proposed gene model for the LDHU3_05.0940 gene.
Among the 2,441 transcripts lacking CDS annotation, 31 contained structural RNAs and the rest were categorized as novel transcripts. CDS annotation was done using Companion server, and this tool was set to predict new putative polypeptides from ORF larger than 200 bp. However, an ever growing number of short CDS-encoded peptides are being characterized and found to play physiological functions in metazoans, plants and in unicellular organisms84,85. Therefore, we must be cautious in considering that most of these novel transcripts are non-coding RNAs (ncRNAs). Transcripts lacking gene-annotation were also identified during the transcriptome annotation of two other Leishmania species, L. major14 and L. mexicana40. In order to know the degree of conservation of these novel transcripts among the three species, a BLASTn analysis was done using these three groups of novel transcripts and the results are summarized in Table 4. Thus, 1,513 out of the 2,410 novel transcripts annotated in this work for L. donovani can be categorized as homologues to novel transcripts annotated in L. major. The number of L. donovani novel transcripts with homologues in the L. mexicana was 899, but it may bear in mind that only 936 novel transcripts were annotated in the L. mexicana transcriptome40. It is worth noting that more restricted criteria were used to define a “novel transcript” in the L. mexicana transcriptome analysis than in the other two analyses (L. major and L. donovani); in the former, only transcripts having an ORF longer than >75 nucleotides were categorized as novel transcripts40. Nevertheless, this analysis showed the existence of a significant number of species-specific transcripts (Table 4), whose functional roles need to be addressed in future works.
Table 4.
Analysis of the conservation of the novel transcripts identified in the L. donovani (LdHU3), L. major (LmjF) and L. mexicana (LmxM) transcriptomes.
| Transcripts | LdHU3a | LmjFb | LmxMc |
|---|---|---|---|
| Total novel transcripts | 2410 | 2143 | 936 |
| Homologs in the LdHU3 transcriptome | — | 1349 | 590 |
| Homologs in the LmjF transcriptome | 1513 | — | 598 |
| Homologs in the LmxM transcriptome | 893 | 762 | — |
| Conserved in the three species | 770 | 617 | 444 |
| Species specific transcripts | 773 | 647 | 192 |
aThis work.
bLmjF_cbm_v1_MT_v1.2.gtf transcriptome available in Leish-ESP Web server (http://leish-esp.cbm.uam.es/L_major_downloads.html).
cSee ref.40.
The transcript coding for poly(A) polymerase is also processed by cis-splicing in L. donovani
During gene annotation, LDHU3_29.3990 gene, coding for a putative poly(A) polymerase (PAP), was initially annotated as pseudogene due to the presence of premature stop codons. Nevertheless, the homologous genes in T. brucei and T. cruzi are known to represent an exception to the general rule that genes in trypanosomatids are intron-less. Mair and co-workers86, by comparison of PAP genes in T. brucei and T. cruzi, suggested that an intronic sequence of 653 and 302 nt, respectively, would be present. Afterwards, these authors demonstrated experimentally that this intron is removed by cis-splicing in T. brucei, yielding a translatable mRNA expressing the complete PAP protein86. In both organisms, the intron occurs at identical positions within the CDS and obey the GT/AG rule of cis-splicing introns. In order to determine whether the orthologous gene in L. donovani (i.e., LDHU3_29.3990), after transcription, is also processed by cis-splicing, we firstly analysed the distribution of RNA-seq reads on the genomic region expanding the gene (Fig. 4A). The presence of a sudden drop in coverage around the genomic coordinate 1,173,206 caught our attention (this position is covered only by 24 reads, whereas the rest of the transcript is covered by around 400 reads). This valley was not observed when DNA-seq reads were mapped, indicating that it is not a region with special complexity for sequencing. Thus, looking for a possible cis-splicing processing of the LDHU3_29.3990 gene, the genomic sequence was analysed to define the putative exonic sequences, based on the amino acids encoded in the three direct reading frames. Interestingly, two putative exons accounting for the complete protein sequence were depicted. Based on this picture, a pair of oligonucleotides were designed, each one in a different exonic sequence, and used for PCR amplification from oligo d(T)-primed cDNA. An amplification product of around 1,100-nt was observed when the cDNA was used as template, whereas the expected PCR product of 1,949-nt in length was observed when genomic DNA was used as template (Fig. 4B). After cloning the 1,100-nt product, three independent clones were sequenced. In all three, the cloned sequence was identical and its length was 1,108-nt. The analysis of the sequence allowed us to determine that the LDHU3_29.3990 gene had an intron of 841-nt in length that would be removed post-transcriptionally by cis-splicing. Figure 4C shows a schematic representation of the intronic region of the LDHU3_29.3990 gene. It is remarkable the high sequence conservation existing between T. brucei (and T. cruzi too) and L. donovani intron sequences at the 5′-end. The first ten nucleotides are identical, whereas at the 3′-end only four nucleotides are conserved (Fig. 4C). In T. brucei, it was demonstrated that punctual substitutions of any of the 10 conserved nucleotides at 5′-end of the intron led to a total inhibition of the intron removal86. The authors suggested that this sequence conservation is being dictated by the sequence complementarity existing between the exon1-intron boundary and the U1 snRNA, putatively involved in the splicing process.
Figure 4.
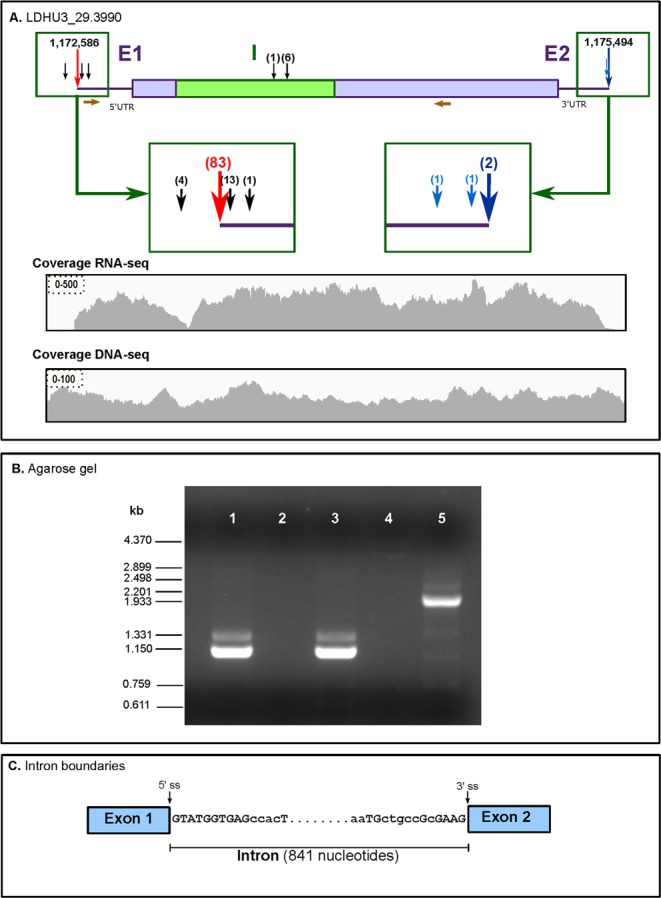
Processing by cis-splicing of the transcript encoding for the poly-A polymerase in L. donovani. Panel A, gene model for LDHU3_29.3990; E1 and E2, exons; I, intron. The red arrow indicates the position of the main SAS and the alternative SASs are indicated by black arrows (the number of RNA-seq mapped to each SAS is shown in parentheses). Blue arrows point to the poly-A addition sites. SASs mapped in the intron sequence are indicated by arrows above it. The position of the primers used for the PCR amplification are shown (maroon arrows; forward: 5′-GCGAGTTTCT GAAGTGCTGC-3′; reverse: 5′-TTCAGCACTG GGAACAGGTC-3′). The distribution (coverage) of Illumina reads along the region in study obtained after mapping of either RNA-derived reads (coverage RNA-seq) or DNA-derived reads (coverage DNA-seq) are also shown. Panel B, electrophoresis of PCR products on a 1% agarose gel; lanes 1 and 3, PCR amplification using cDNA derived from L. donovani total RNA and using for retrotranscription either SuperScript III (lane 1) or ThermoScript (lane 3) retrotranscriptases; lanes 2 and 4, PCR amplification from L. donovani total RNA (without previous retrotranscription step); lane 5, PCR amplification from L. donovani total DNA. Relative migration and size of molecular weight markers (Φ29 DNA digested with HindIII) are shown on the left. Uncropped gel shown in Supplementary Information Fig. S12. Panel C, schematic representation of the exon-intron junctions as determined after sequencing of the RT-PCR amplicon. Conserved nucleotides (upper case) in the equivalent intron existing in the gene coding for poly-A polymerase in T. brucei86. The positions of 5′ and 3′ splice sites (5′ ss and 3′ ss, respectively) are indicated.
The coverage of RNA-seq reads over the LDHU3_29.3990 genomic locus did not show a clear decline along the intronic region, suggesting a non-efficient cis-splicing and intron removal, at least in the promastigote stage. The existence of a low read depth in position close to the 5′ splice site (Fig. 4A) would be an indication that pre-mRNA is being cut at that position, but intron removal from exon 2 would be occurring inefficiently. Also, it should be noted the existence of two SAS in the intronic sequence, just upstream from the second exon. Mair et al.86 also found that some RNA molecules derived from the orthologue gene in T. brucei are processed by trans-splicing at a cryptic site located within the intron. It would be interesting to know whether this shortened mRNA is translated and, if so, whether a functional protein is generated.
There is another documented case of cis-splicing in trypanosomatids. The Trypanosoma brucei gene Tb927.8.1510, encoding an ATP-dependent RNA helicase (DBP2B), has been found to be processed by cis-splicing87. Similarly, the orthologous gene in L. major (LmjF.07.0340) was annotated as cis-spliced gene by Peter Myler at TriTrypDB. To address the possibility that the orthologous gene in L. donovani (LDHU3_07.0430) is also processed by cis-splicing, a search for splice junctions among the RNA-seq reads was performed using the Tophat2 tool aligner88. The results were clear, a significant number of RNA-seq reads covering the exon-junction site were identified; this information allowed us to precisely map the position of the two exons in the genomic sequence and to determine that the gene is split by a 4,470-bp long intron (see Fig. S11 in Supplementary Information File 1).
Determination of RNA levels from RNA-seq data
RNA-seq data produce digital counts of transcript abundance, allowing to quantify transcript levels in a straightforward manner. Nevertheless, basic for an accurate determination of expression levels is the availability of appropriate gene models, as determined in this work for L. donovani. To measure relative expression levels of the transcripts, we used RNA-seq reads derived from three biological replicates of L. donovani promastigotes to determine FPKM values for the 10,893 gene models established in this study. Table 5 lists the 50 most abundant transcripts in logarithmic phase promastigotes. It is not surprising that the list is headed by two histone transcripts and that another seven histone genes were also present among the 50 most abundant transcripts. Similarly, the high cellular requirements of tubulin agree with the high expression levels determined for seven different alpha-tubulin genes. Also, transcripts coding for 13 ribosomal proteins were found among the top 50 transcripts. In agreement with previous studies in other Leishmania species14,89, HSP70 (type-II gene) and KMP11 transcripts were also found among the most abundant transcripts. Many of the most expressed genes in L. donovani were also identified among the most highly expressed genes in the L. tropica promastigote stage83. Additionally, among the highly expressed genes, four lacking of CDS annotation (LDHU3.10.T1390, LDHU3.27.T1760, LDHU3.30.T1180, and LDHU3.36.T5090) were present. Given their abundance, further analysis should be performed in order to determine whether they have protein-coding capacity or definitively represent non-coding RNAs (ncRNAs).
Table 5.
The 50 most abundant transcripts in L. donovani (HU3 strain) promastigotes.
| Transcript | Protein product | FPKM (± SD) |
|---|---|---|
| LDHU3_09.T1660 | histone H2B | 3677.33 (± 38.61) |
| LDHU3_06.T0020 | histone H4 | 3642.62 (± 45.66) |
| LDHU3_27.T1700 | hypothetical protein | 3388.42 (± 41.35) |
| LDHU3_29.T2670 | histone H2A | 2428.46 (± 38.17) |
| LDHU3_31.T1470 | hypothetical protein, conserved | 2317.52 (± 21.57) |
| LDHU3_13.T0480 | Alba | 2160.44 (± 29.93) |
| LDHU3_27.T1760 | Non annotated CDS | 2045.59 (± 36.76) |
| LDHU3_15.T0010 | histone H4 | 1902.71 (± 37.44) |
| LDHU3_17.T1420 | META domain containing protein | 1710.65 (± 17.22) |
| LDHU3_13.T0630 | 40 S ribosomal protein S12 putative | 1668.54 (± 21.42) |
| LDHU3_27.T0170 | WW/Zinc finger domain containing protein | 1611.85 (± 21.22) |
| LDHU3_10.T1400 | histone H3 | 1605.27 (± 26.92) |
| LDHU3_28.T3970 | HSP70 (gene HSP70-II) | 1565.85 (± 12.38) |
| LDHU3_15.T1550 | nucleoside transporter 1 | 1503.34 (± 11.69) |
| LDHU3_30.T0880 | 40 S ribosomal protein S30 | 1473.70 (± 64.82) |
| LDHU3_19.T0030 | histone H2B | 1470.15 (± 23.58) |
| LDHU3_23.T2590 | Nucleoside 2-deoxyribosyltransferase | 1450.04 (± 24.23) |
| LDHU3_36.T5090 | Non annotated CDS | 1450.04 (± 28.18) |
| LDHU3_36.T5100 | EF-hand domain containing protein | 1447.41 (± 26.13) |
| LDHU3_26.T2920 | ribosomal protein L38 | 1275.35 (± 28.52) |
| LDHU3_13.T0400 | alpha tubulin | 1258.35 (± 15.09) |
| LDHU3_13.T0370 | alpha tubulin | 1256.55 (± 15.07) |
| LDHU3_13.T0360 | alpha tubulin | 1238.15 (± 14.97) |
| LDHU3_30.T1180 | No annotated CDS | 1223.66 (± 16.36) |
| LDHU3_19.T0040 | histone H2B | 1207.16 (± 28.86) |
| LDHU3_13.T0350 | alpha tubulin | 1206.51 (± 14.76) |
| LDHU3_36.T1660 | fructose-16-bisphosphate aldolase | 1154.03 (± 15.42) |
| LDHU3_33.T4780 | 40S ribosomal protein S13 | 1122.95 (± 28.51) |
| LDHU3_33.T1080 | 60S ribosomal protein L6 | 1111.59 (± 23.27) |
| LDHU3_15.T1290 | 40S ribosomal protein S3 | 1107.49 (± 18.96) |
| LDHU3_24.T2850 | 60S ribosomal protein L12 | 1082.74 (± 24.63) |
| LDHU3_21.T1370 | histone H2A | 1074.84 (± 24.22) |
| LDHU3_10.T1390 | No annotated CDS | 1063.97 (± 24.84) |
| LDHU3_20.T1990 | 40S ribosomal protein S11 | 1056.10 (± 21.5) |
| LDHU3_35.T0570 | 40S ribosomal protein S3A | 1035.42 (± 18.85) |
| LDHU3_35.T2890 | kinetoplastid membrane protein-11 | 1033.21 (± 15.14) |
| LDHU3_24.T2590 | 60S ribosomal protein L26 | 1030.16 (± 21.2) |
| LDHU3_13.T0340 | alpha tubulin | 1010.16 (± 12.49) |
| LDHU3_19.T0050 | histone H2B | 1007.14 (± 26.18) |
| LDHU3_21.T2650 | Tubulin C domain containing protein | 1004.59 (± 11.1) |
| LDHU3_08.T0720 | Amastin surface glycoprotein | 1002.89 (± 9.58) |
| LDHU3_26.T3080 | 60S ribosomal protein L35 | 995.32 (± 18.85) |
| LDHU3_36.T2660 | inosine-guanosine transporter | 995.13 (± 10.33) |
| LDHU3_32.T3530 | L-Lysine transport protein | 991.13 (± 8.017) |
| LDHU3_32.T0990 | RNA binding protein | 986.09 (± 14.73) |
| LDHU3_33.T1490 | hypothetical protein conserved | 970.46 (± 19.07) |
| LDHU3_13.T0390 | alpha tubulin | 963.41 (± 11.52) |
| LDHU3_16.T1490 | 60S ribosomal protein L39 | 951.77 (± 21.3) |
| LDHU3_35.T0740 | 60S ribosomal protein L18a | 949.84 (± 16.3) |
| LDHU3_13.T0380 | alpha tubulin | 943.59 (± 11.4) |
In a previous work from our group, we determined the 50 most abundant transcripts in L. major promastigotes14. A comparison between both studies evidenced, apart from the structural abundant proteins indicated above, a similar high expression of the following transcripts: LDHU3_31.T1470 (coding for a conserved hypothetical protein), LDHU3_13.T0480 (Alba protein), LDHU3_15.T1550 (nucleoside transporter 1), LDHU3_36.T5100 (EF-hand domain containing protein), LDHU3_08.T0720 (amastin), and LDHU3_36.T2660 (inosine-guanosine transporter).
Conclusions
As proved recently with the L. infantum genome assembly22, combination of the Illumina sequencing accuracy and the long sequence reads obtained by the PacBio platform has resulted adequate to obtain an almost complete assembly of the L. donovani genome.
The availability of a well-assembled genome is pivotal for undertaking global genomics, transcriptomics and proteomics studies with confidence. Here, based on the assembled genome, we have delineated the poly-A+ transcriptome, consisting in 10,893 transcripts (see Supplementary File 2 for the complete list). Remarkably, 2,410 are transcripts that do not contain annotated ORFs. Furthermore, transcript location served to correct several hundreds of miss-annotated ORFs, which are usually predicted by bioinformatics tools (Companion, in this study). As reported for L. major14 and for L. mexicana40, the transcriptome of L. donovani also shows a remarkable heterogeneity in the sites used for the mini-exon (SL) and polyadenylation addition. Thus, alternative trans-splicing and polyadenylation may represent an additional and relevant points for controlling gene expression in Leishmania.
Analysis of gene expression, undertaken either individually or genome wide, requires accurate gene models in which, in addition to the ORF, the UTRs must be precisely delimitated. In fact, most of the regulatory elements involved in post-transcriptional mechanisms of gene expression accumulate in UTRs90, and regulation of gene expression in Leishmania and other trypanosomatids is essentially post-transcriptional9,15. Additionally, the availability of a complete L. donovani transcriptome will allow to carry out transcriptome profiling associated with drug resistance in this species without the necessity of using data derived from other Leishmania species59,91.
As shown previously for other Leishmania species80, the L. donovani genome is not very different to the genome of other species regarding the number of different proteins that are encoded. Thus, only fifteen out of the 8,405 genes annotated as protein-coding lacked orthologues in the L. major genome. This number of genes may seem too low to explain the different pathologies that these species produce in humans: L. donovani causes fatal viscerocutaneous affectations, whereas L. major infection generates self-curing cutaneous lesions. However, large differences were found regarding the number of distinct pseudogenes and the gene copy number variations existing between both species. Thus, in agreement with other comparative genomics studies78,83, we suggest that pseudogene formation and variations in gene copy numbers may be greatly contributing to the genetic basis for disease tropism.
Finally, we have demonstrated that the transcripts coding for the poly(A) polymerase (PAP) and for an ATP-dependent RNA helicase in L. donovani are processed by cis-splicing, following a similar pattern of processing to that described for the orthologous genes in other trypanosomatids. These results suggest an ancient acquisition of these particular events of cis-splicing in the evolutionary line of these protists, where cis-splicing is considered essentially absent. Surely, a relevant physiological function could be behind the conservation of this, apparently fossilized, molecular mechanism.
Supplementary information
Acknowledgements
We are extremely grateful to all the support, advice and help received by our colleagues from the Genomics and Next Generation Sequencing Core Facility at the CBMSO. This work was supported by grants (to B.A. and J.M.R.) from Proyecto del Ministerio de Economía, Industria y Competitividad SAF2017-86965-R (co-funded with FEDER funds), and by the Network of Tropical Diseases Research RICET (RD16/0027/0008) and FEDER. Institutional grants from the Fundación Ramón Areces and Banco de Santander to the CBMSO are also acknowledged.
Author Contributions
Conceived and designed the experiments: F.C.R., J.M.R. and B.A. Parasites cultures and preparation of DNA and RNA samples: J.C.S., F.G. and J.M.R. Genome assembly: S.G.-d.l.F. and R.P.P. Genome annotation: S.G.-d.l.F. and E.C. Transcriptome annotation: E.C. and A.R. RT-PCR experiments: E.C. and L.T. Drawing of figures: E.C., S.G.-d.l.F. and J.M.R. E.C. drafted the manuscript and all authors contributed to the writing. J.M.R. and B.A. edited the final manuscript. All authors have read and approved the final manuscript.
Data Availability
Both genomic and transcriptomic raw data have been deposited in The European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/). The assembled genome and transcriptome sequences together with annotations files were uploaded under the Study accession number PRJEB23341 and Study unique name: ena-STUDY-CBMSO-06-11-2017-13:36:27:181–100. Additionally, Fasta files with the genome sequence and transcriptome are downloadable at the Leish-ESP web site (http://leish-esp.cbm.uam.es/).
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Esther Camacho and Sandra González-de la Fuente contributed equally.
Contributor Information
Jose M. Requena, Email: jmrequena@cbm.csic.es
Begoña Aguado, Email: baguado@cbm.csic.es.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-42511-4.
References
- 1.Moreira D, Lopez-Garcia P, Vickerman K. An updated view of kinetoplastid phylogeny using environmental sequences and a closer outgroup: proposal for a new classification of the class Kinetoplastea. Int J Syst Evol Microbiol. 2004;54:1861–1875. doi: 10.1099/ijs.0.63081-0. [DOI] [PubMed] [Google Scholar]
- 2.Van der Auwera G, Dujardin JC. Species typing in dermal leishmaniasis. Clin Microbiol Rev. 2015;28:265–294. doi: 10.1128/CMR.00104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGwire BS, Satoskar AR. Leishmaniasis: clinical syndromes and treatment. QJM. 2014;107:7–14. doi: 10.1093/qjmed/hct116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Vries HJ, Reedijk SH, Schallig HD. Cutaneous leishmaniasis: recent developments in diagnosis and management. Am J Clin Dermatol. 2015;16:99–109. doi: 10.1007/s40257-015-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvar J, et al. Leishmaniasis worldwide and global estimates of its incidence. Plos One. 2012;7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hefnawy A, Berg M, Dujardin JC, De Muylder G. Exploiting Knowledge on Leishmania Drug Resistance to Support the Quest for New Drugs. Trends Parasitol. 2017;33:162–174. doi: 10.1016/j.pt.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Akhoundi M, et al. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS neglected tropical diseases. 2016;10:e0004349. doi: 10.1371/journal.pntd.0004349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Requena, J. M. The Stressful Life of pathogenic Leishmania species. In: Stress Response in Microbiology (Ed. Requena, J.M.), 323–346 (Caister Academic Press, 2012).
- 9.Clayton CE. Gene expression in Kinetoplastids. Curr Opin Microbiol. 2016;32:46–51. doi: 10.1016/j.mib.2016.04.018. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Calvillo S, et al. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol. Cell. 2003;11:1291–1299. doi: 10.1016/S1097-2765(03)00143-6. [DOI] [PubMed] [Google Scholar]
- 11.Liang XH, Haritan A, Uliel S, Michaeli S. trans and cis splicing in trypanosomatids: mechanism, factors, and regulation. Eukaryot Cell. 2003;2:830–840. doi: 10.1128/EC.2.5.830-840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeBowitz JH, Smith HQ, Rusche L, Beverley SM. Coupling of poly(A) site selection and trans-splicing in Leishmania. Genes Dev. 1993;7:996–1007. doi: 10.1101/gad.7.6.996. [DOI] [PubMed] [Google Scholar]
- 13.Freire ER, Sturm NR, Campbell DA, de Melo Neto OP. The Role of Cytoplasmic mRNA Cap-Binding Protein Complexes in Trypanosoma brucei and Other Trypanosomatids. Pathogens. 2017;6:55. doi: 10.3390/pathogens6040055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rastrojo A, et al. The transcriptome of Leishmania major in the axenic promastigote stage: transcript annotation and relative expression levels by RNA-seq. BMC Genomics. 2013;14:223. doi: 10.1186/1471-2164-14-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Requena JM. Lights and shadows on gene organization and regulation of gene expression in Leishmania. Front Biosci. 2011;17:2069–2085. doi: 10.2741/3840. [DOI] [PubMed] [Google Scholar]
- 16.Kramer S, Carrington M. Trans-acting proteins regulating mRNA maturation, stability and translation in trypanosomatids. Trends Parasitol. 2011;27:23–30. doi: 10.1016/j.pt.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romaniuk MA, Cervini G, Cassola A. Regulation of RNA binding proteins in trypanosomatid protozoan parasites. World J Biol Chem. 2016;7:146–157. doi: 10.4331/wjbc.v7.i1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gehring NH, Wahle E, Fischer U. Deciphering the mRNP Code: RNA-Bound Determinants of Post-Transcriptional Gene Regulation. Trends Biochem Sci. 2017;42:369–382. doi: 10.1016/j.tibs.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Ivens AC, et al. The Genome of the Kinetoplastid Parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alonso G, Rastrojo A, Lopez-Perez S, Requena JM, Aguado B. Resequencing and assembly of seven complex loci to improve the Leishmania major (Friedlin strain) reference genome. Parasites & vectors. 2016;9:74. doi: 10.1186/s13071-016-1329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aslett M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38:D457–462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez-de la Fuente S, et al. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci Rep. 2017;7:18050. doi: 10.1038/s41598-017-18374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuersten S, Goodwin EB. The power of the 3′ UTR: translational control and development. Nat Rev Genet. 2003;4:626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- 24.Mazumder B, Seshadri V, Fox PL. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem Sci. 2003;28:91–98. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 25.Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell. 2009;136:719–730. doi: 10.1016/j.cell.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soto M, Requena JM, Alonso C. Isolation, characterization and analysis of the expression of the Leishmania ribosomal PO protein genes. Mol Biochem Parasitol. 1993;61:265–274. doi: 10.1016/0166-6851(93)90072-6. [DOI] [PubMed] [Google Scholar]
- 27.Soto M, et al. Genomic organization and expression of two independent gene arrays coding for two antigenic acidic ribosomal proteins of Leishmania. J Biol Chem. 1993;268:21835–21843. [PubMed] [Google Scholar]
- 28.Requena JM, Soto M, Quijada L, Alonso C. Genes and chromosomes of Leishmania infantum. Mem Inst Oswaldo Cruz. 1997;92:853–858. doi: 10.1590/S0074-02761997000600022. [DOI] [PubMed] [Google Scholar]
- 29.Zilka A, Garlapati S, Dahan E, Yaolsky V, Shapira M. Developmental Regulation of Heat Shock Protein 83 in Leishmania. 3′ Processing and mRNA stability control transcript abundance, and translation is directed by a determinant in the 3′-untranslated region. J Biol Chem. 2001;276:47922–47929. doi: 10.1074/jbc.M108271200. [DOI] [PubMed] [Google Scholar]
- 30.Soto M, et al. Leishmania infantum possesses a complex family of histone H2A genes: structural characterization and analysis of expression. Parasitology. 2003;127:95–105. doi: 10.1017/S0031182003003445. [DOI] [PubMed] [Google Scholar]
- 31.Larreta R, et al. The expression of HSP83 genes in Leishmania infantum is affected by temperature and by stage-differentiation and is regulated at the levels of mRNA stability and translation. BMC Mol Biol. 2004;5:3. doi: 10.1186/1471-2199-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Folgueira C, et al. The translational efficiencies of the two Leishmania infantum HSP70 mRNAs, differing in their 3′-untranslated regions, are affected by shifts in the temperature of growth through different mechanisms. J Biol Chem. 2005;280:35172–35183. doi: 10.1074/jbc.M505559200. [DOI] [PubMed] [Google Scholar]
- 33.Boucher N, et al. A common mechanism of stage-regulated gene expression in Leishmania mediated by a conserved 3′-untranslated region element. J Biol Chem. 2002;277:19511–19520. doi: 10.1074/jbc.M200500200. [DOI] [PubMed] [Google Scholar]
- 34.Mishra KK, Holzer TR, Moore LL, LeBowitz JH. A negative regulatory element controls mRNA abundance of the Leishmania mexicana Paraflagellar rod gene PFR2. Eukaryot Cell. 2003;2:1009–1017. doi: 10.1128/EC.2.5.1009-1017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray A, Fu C, Habibi G, McMaster WR. Regions in the 3′ untranslated region confer stage-specific expression to the Leishmania mexicana a600-4 gene. Mol Biochem Parasitol. 2007;153:125–132. doi: 10.1016/j.molbiopara.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 36.David M, et al. Preferential translation of Hsp83 in Leishmania requires a thermosensitive polypyrimidine-rich element in the 3′ UTR and involves scanning of the 5′ UTR. Rna. 2010;16:364–374. doi: 10.1261/rna.1874710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quijada L, Soto M, Alonso C, Requena JM. Identification of a putative regulatory element in the 3′-untranslated region that controls expression of HSP70 in Leishmania infantum. Mol Biochem Parasitol. 2000;110:79–91. doi: 10.1016/S0166-6851(00)00258-9. [DOI] [PubMed] [Google Scholar]
- 38.Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 39.Patino LH, Ramirez JD. RNA-seq in kinetoplastids: A powerful tool for the understanding of the biology and host-pathogen interactions. Infect Genet Evol. 2017;49:273–282. doi: 10.1016/j.meegid.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Fiebig M, Kelly S, Gluenz E. Comparative life cycle transcriptomics revises Leishmania mexicana genome annotation and links a chromosome duplication with parasitism of vertebrates. PLoS Pathog. 2015;11:e1005186. doi: 10.1371/journal.ppat.1005186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eliaz D, et al. Genome-wide analysis of small nucleolar RNAs of Leishmania major reveals a rich repertoire of RNAs involved in modification and processing of rRNA. RNA biology. 2015;12:1222–1255. doi: 10.1080/15476286.2015.1038019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson PR, et al. Restriction endonuclease analysis of Leishmania kinetoplast DNA characterizes parasites responsible for visceral and cutaneous disease. Am J Trop Med Hyg. 1984;33:808–819. doi: 10.4269/ajtmh.1984.33.808. [DOI] [PubMed] [Google Scholar]
- 43.Requena JM, Lopez MC, Jimenez-Ruiz A, de la Torre JC, Alonso C. A head-to-tail tandem organization of hsp70 genes in Trypanosoma cruzi. Nucleic Acids Res. 1988;16:1393–1406. doi: 10.1093/nar/16.4.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chin CS, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 45.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nadalin F, Vezzi F, Policriti A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics. 2012;13(Suppl 14):S8. doi: 10.1186/1471-2105-13-S14-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 50.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012;13:238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:111011–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv, 1207.3907v1202 (2012).
- 57.Steinbiss S, et al. Companion: a web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016;44:W29–34. doi: 10.1093/nar/gkw292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li L, Stoeckert CJ, Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rastrojo A, et al. Genomic and transcriptomic alterations in Leishmania donovani lines experimentally resistant to antileishmanial drugs. Int J Parasitol Drugs Drug Resist. 2018;8:246–264. doi: 10.1016/j.ijpddr.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 63.Tripp CA, Myler PJ, Stuart K. A DNA sequence (LD1) which occurs in several genomic organizations in Leishmania. Mol Biochem Parasitol. 1991;47:151–156. doi: 10.1016/0166-6851(91)90174-5. [DOI] [PubMed] [Google Scholar]
- 64.Seifert K, et al. Characterisation of Leishmania donovani promastigotes resistant to hexadecylphosphocholine (miltefosine) International journal of antimicrobial agents. 2003;22:380–387. doi: 10.1016/S0924-8579(03)00125-0. [DOI] [PubMed] [Google Scholar]
- 65.Garcia-Hernandez R, Manzano JI, Castanys S, Gamarro F. Leishmania donovani develops resistance to drug combinations. PLoS neglected tropical diseases. 2012;6:e1974. doi: 10.1371/journal.pntd.0001974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tovar J, Cunningham ML, Smith AC, Croft SL, Fairlamb AH. Down-regulation of Leishmania donovani trypanothione reductase by heterologous expression of a trans-dominant mutant homologue: effect on parasite intracellular survival. Proc Natl Acad Sci USA. 1998;95:5311–5316. doi: 10.1073/pnas.95.9.5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Downing T, et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011;21:2143–2156. doi: 10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ubeda JM, et al. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol. 2014;12:e1001868. doi: 10.1371/journal.pbio.1001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Requena JM, et al. Genomic cartography and proposal of nomenclature for the repeated, interspersed elements of the Leishmania major SIDER2 family and identification of SIDER2-containing transcripts. Mol Biochem Parasitol. 2017;212:9–15. doi: 10.1016/j.molbiopara.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 70.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 73.Wincker P, et al. The Leishmania genome comprises 36 chromosomes conserved across widely divergent human pathogenic species. Nucleic Acids Res. 1996;24:1688–1694. doi: 10.1093/nar/24.9.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Myler PJ, Venkataraman GM, Lodes MJ, Stuart KD. A frequently amplified region in Leishmania contains a gene conserved in prokaryotes and eukaryotes. Gene. 1994;148:187–193. doi: 10.1016/0378-1119(94)90688-2. [DOI] [PubMed] [Google Scholar]
- 75.Pawar H, Kulkarni A, Dixit T, Chaphekar D, Patole MS. A bioinformatics approach to reanalyze the genome annotation of kinetoplastid protozoan parasite Leishmania donovani. Genomics. 2014;104:554–561. doi: 10.1016/j.ygeno.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 76.Chiurillo MA, et al. Cloning and characterization of Leishmania donovani telomeres. Exp Parasitol. 2000;94:248–258. doi: 10.1006/expr.2000.4499. [DOI] [PubMed] [Google Scholar]
- 77.Mondelaers A, et al. Genomic and Molecular Characterization of Miltefosine Resistance in Leishmania infantum Strains with Either Natural or Acquired Resistance through Experimental Selection of Intracellular Amastigotes. PLoS One. 2016;11:e0154101. doi: 10.1371/journal.pone.0154101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rogers MB, et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21:2129–2142. doi: 10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Imamura H, et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. Elife. 2016;5:e12613. doi: 10.7554/eLife.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peacock CS, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lynn MA, McMaster WR. Leishmania: conserved evolution–diverse diseases. Trends Parasitol. 2008;24:103–105. doi: 10.1016/j.pt.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 82.Fraga J, et al. Evolution and species discrimination according to the Leishmania heat-shock protein 20 gene. Infect Genet Evol. 2013;18:229–237. doi: 10.1016/j.meegid.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 83.Iantorno, S. A. et al. Gene Expression in Leishmania Is Regulated Predominantly by Gene Dosage. mBio8, 10.1128/mBio.01393-17 (2017). [DOI] [PMC free article] [PubMed]
- 84.Couso JP, Patraquim P. Classification and function of small open reading frames. Nat Rev Mol Cell Biol. 2017;18:575–589. doi: 10.1038/nrm.2017.58. [DOI] [PubMed] [Google Scholar]
- 85.Delcourt V, Staskevicius A, Salzet M, Fournier I, Roucou X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics. 2018;18:e1700058. doi: 10.1002/pmic.201700058. [DOI] [PubMed] [Google Scholar]
- 86.Mair G, et al. A new twist in trypanosome RNA metabolism: cis-splicing of pre-mRNA. Rna. 2000;6:163–169. doi: 10.1017/S135583820099229X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jae N, et al. Essential role of a trypanosome U4-specific Sm core protein in small nuclear ribonucleoprotein assembly and splicing. Eukaryot Cell. 2010;9:379–386. doi: 10.1128/EC.00353-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Quijada L, Soto M, Alonso C, Requena JM. Analysis of post-transcriptional regulation operating on transcription products of the tandemly linked Leishmania infantum hsp70 genes. J Biol Chem. 1997;272:4493–4499. doi: 10.1074/jbc.272.7.4493. [DOI] [PubMed] [Google Scholar]
- 90.Mayr C. Regulation by 3′-Untranslated Regions. Annu Rev Genet. 2017;51:171–194. doi: 10.1146/annurev-genet-120116-024704. [DOI] [PubMed] [Google Scholar]
- 91.Verma A, et al. Transcriptome profiling identifies genes/pathways associated with experimental resistance to paromomycin in Leishmania donovani. Int J Parasitol Drugs Drug Resist. 2017;7:370–377. doi: 10.1016/j.ijpddr.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gupta AK, Srivastava S, Singh A, Singh S. De Novo Whole-Genome Sequence and Annotation of a Leishmania Strain Isolated from a Case of Post-Kala-Azar Dermal Leishmaniasis. Genome Announc. 2015;3:e00809. doi: 10.1128/genomeA.00809-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Both genomic and transcriptomic raw data have been deposited in The European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/). The assembled genome and transcriptome sequences together with annotations files were uploaded under the Study accession number PRJEB23341 and Study unique name: ena-STUDY-CBMSO-06-11-2017-13:36:27:181–100. Additionally, Fasta files with the genome sequence and transcriptome are downloadable at the Leish-ESP web site (http://leish-esp.cbm.uam.es/).