

Summary
Systemic lupus erythematosus (SLE) is a complex autoimmune disorder whose pathology involves multiple immune cell types, including B and T lymphocytes as well as myeloid cells. While it is clear that autoantibody‐producing B cells, as well as CD4+ T cell help, are key contributors to disease, little is known regarding the role of innate lymphoid cells such as natural killer (NK) cells in the pathogenesis of SLE. We have characterized the phenotype of NK cells by multi‐color flow cytometry in a large cohort of SLE patients. While the overall percentage of NK cells was similar or slightly decreased compared to healthy controls, a subset of patients displayed a high frequency of NK cells expressing the proliferation marker, Ki67, which was not found in healthy donors. Although expression of Ki67 on NK cells correlated with Ki67 on other immune cell subsets, the frequency of Ki67 on NK cells was considerably higher. Increased frequencies of Ki67+ NK cells correlated strongly with clinical severity and active nephritis and was also related to low NK cell numbers, but not overall leukopenia. Proteomic and functional data indicate that the cytokine interleukin‐15 promotes the induction of Ki67 on NK cells. These results suggest a role for NK cells in regulating the immune‐mediated pathology of SLE as well as reveal a possible target for therapeutic intervention.
Introduction
Systemic lupus erythematosus (SLE) is characterized by the loss of immune tolerance to self‐antigens, resulting in the production of pathogenic autoantibodies which ultimately leads to inflammation and organ damage 1. Forty to 70% of patients develop lupus nephritis (LN), a major cause of morbidity and mortality in SLE. Obtaining optimal responses to therapy in lupus nephritis is clinically challenging. At present, the gold standard for LN diagnosis is a renal biopsy, but this invasive procedure can result in complications, and histological results are not always consistent with clinical symptoms 2, 3. Moreover, repeated biopsies to determine treatment success or flare status are impractical. Non‐invasive biomarkers such as proteinuria, anti‐dsDNA antibodies and C3 and C4 complement levels provide a low specificity and sensitivity for LN 4. There is a need for improved biomarkers for the diagnosis of LN as well as the response to therapy to this and other serious end‐organ manifestations.
SLE disease pathogenesis has been largely attributed to B and T cells, components of the adaptive immune system, which are able to recognize specific self‐antigens, resulting in the generation of autoantibodies. Despite their important and emerging roles in host defense, cytotoxicity and secretion of potentially pathogenic cytokines, the role of innate lymphoid cells, including natural killer (NK) cells, in promoting or protecting from pathology in SLE remains largely unexplored. NK cells, as part of the innate immune compartment, are largely known for their ability to recognize and kill target cells without prior antigen sensitization 5. NK cells also play a role in controlling adaptive immune responses through elimination of activated T cells, which has been shown to be inhibited by type I interferons (IFNs), a prominent cytokine present in SLE disease 6, 7, 8, 9.
NK cell function and homeostasis are sensitive to changes in inflammatory environments such as those in virus infections, cytokine therapy and autoimmune disease 10, 11, 12, 13, 14, 15. Several studies have demonstrated that NK cell number and cytotoxic function are diminished in SLE 16, 17, 18, 19. Moreover, the expression of NK cell receptors, surface molecules which can regulate the function of NK cells, are altered during SLE, suggesting other functional abnormalities of these cells 20, 21, 22, 23. One of the most potent factors regulating NK cells is interleukin (IL)‐15, a cytokine required for NK cell development and homeostasis, and can also drive NK cell proliferation as well as cytokine production 24. IL‐15 production by dendritic cells and other myeloid cells can be driven by type I IFNs and, interestingly, serum concentrations of IL‐15 are significantly increased in patients with SLE 25, 26. This notwithstanding, associations between the IFN/IL‐15 axis and NK cell function in SLE and SLE disease activity have yet to be investigated.
Here we have characterized peripheral blood NK cells in a large well‐phenotyped cohort of patients with SLE and found that NK cell expression of Ki67, a marker of proliferation, is significantly correlated with disease severity and nephritis. Additionally, Ki67+ NK cells correlated with serum levels of IL‐15 and type‐I IFN‐induced genes. In‐vitro experiments demonstrated that IL‐15, but not type I IFN, was able to up‐regulate NK cell expression of Ki67. These results suggest that NK cell expression of Ki67 is an indicator of SLE severity, with IL‐15 as a possible driver.
Experimental procedures
Peripheral blood collection
Lupus blood samples were obtained from the NIH Clinical Center Blood Bank (Bethesda, MD, USA), as approved by the National Institute of Arthritis and Musculoskeletal and Skin Diseases/National Institutes of Health and isolated as described above. The demographics and clinical characteristics of these donors are shown in Supporting information, Table S1. Healthy donor blood was either obtained from the NIH blood bank or from MedImmune or AstraZeneca employees who were anonymously enrolled in the MedImmune Research Specimen Collection Program. Donors with HIV infection, hepatitis B or C virus, human T lymphotropic virus or syphilis were excluded. Written consent for blood draws was obtained from the donor. Peripheral blood mononuclear cells (PBMCs) were isolated from CPT tubes (BD Biosciences, San Jose, CA, USA) following centrifugation.
Study approval
For healthy donors of MedImmune employees, all protocols and informed consent forms were approved by Chesapeake Institutional Review Board (Protocol 2010‐001, version 4.0). For lupus donors, the studies were approved by the Institutional Review Board of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (protocol 94‐AR‐0066).
Clinical outcomes
Active nephritis was defined as either one of the following at the time of visit: (1) active urinary sediment: red blood cells (RBC), white blood cells (WBC) or mixed cellular casts; (2) more than 10 RBCs or more than five WBC per high‐power field on urine microscopy; (3) new‐onset proteinuria with 3 months of sample collection or an increase by more than 500 mg protein in urine in 24 h; and (4) renal biopsy showing active inflammation. The Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) was used to determine disease activity. Lupus nephritis classification was determined using the World Health Organization (WHO) classification system.
Flow cytometry and antibodies
For multi‐color flow cytometry, PBMC were stained using the following antibodies (clone names in parentheses): CD45 (HI30), CD19 (HIB19), Ki67 (B56), CD4 (RPA‐T4), CD56 (HCD56 and NCAM16.2), CD8a (RPA‐T8), NKG2A (REA110), NKp30 (p30‐15), NKG2C (REA205), NKG2D (1D11), NKp46 (9E2), CD16 (3G8), CD57 (NK‐1), CD3 (SP34‐2), CD11c (B‐ly6), CD38 (HB7), CD95 (DX2) and immunoglobulin (Ig)D (IA6‐2). NK cells were defined as CD4negative, CD19negative, CD8αhinegative or CD56positive. Plasma cells were defined as CD19lo, CD27hi or CD28hi, IgDnegative. CD11chi B cells were defined as CD19+CD11chi and CD95+ B cells were defined as CD19+ CD95+.
Gene expression analysis
Quantification of type I IFN genes was performed using microarray (Affymetrix HG‐U133 Plus 2.0; Thermo Fisher Scientific, Santa Clara, CA, USA). The type I IFN gene signature (IFNGS) was determined based on a set of 21 genes validated previously 33 The IFN gene score was calculated as follows: (1) calculate the mean signal across all healthy donors (HD) for the 21 probesets, (2) calculate the fold change between HD and SLE samples for each probeset = log2 (probeset for sample) – log2 (probeset HD mean) and (3) calculate median of fold change values for all probesets. A median of 2 (log2, which is fourfold of HD) is the cut‐off for positive versus negative score.
Serum cytokine assay
Serum IL‐15 was detected using human high sensitivity IL‐15 Magnetic Luminex Assay (R&D Systems, Minneapolis, MN, USA). The assay was performed as described by the manufacturer.
In‐vitro NK cell cultures
NK cells were isolated using the human NK cell enrichment kit (Stemcell Technologies, Vancouver, BC, Canada). NK cells were then cultured in 96‐well round bottomed plates for 14 days using 1 ng/ml recombinant human (rh) IL‐15 with or without 10 ng/ml IFN‐β or 10 ng/ml IFN‐γ (all PeproTech, Rocky Hill, NJ, USA). Medium plus cytokines were changed every 3 days.
Statistical analysis
Ki67hi samples were operationally defined as those samples that were above the first standard deviation from the mean (> 40% Ki67 positivity). All other samples were automatically placed into the Ki67mid/low NK group.
Results
Peripheral blood NK cells express markers of activation in patients with SLE
Peripheral blood samples collected from SLE patients and HD, matched for age and gender, were analyzed using multi‐color flow cytometry. Patient demographics and therapies are shown in Supporting information, Tables S1 and S2. NK cells were defined as CD56posCD4negCD8mid/neg, which were confirmed to contain few CD3‐positive cells in a separate staining panel (Supporting information, Figs. S1, S2a). Consistent with previous reports 18, 19, 22 both NK cell frequency as well as number per microliter of whole blood were decreased in our cohort of patients with SLE (mean = 104/μl) versus in HD (mean = 204/μl) (Supporting information, Fig. S3a). The distribution of CD56bright and CD56dim NK cell subsets was not significantly different between SLE and control samples (data not shown).
We next performed a more in‐depth analysis of the NK cell phenotype. Remarkably, Ki67, a nuclear protein expressed by proliferating cells, was significantly increased in NK cells from SLE patients (Fig. 1a,b). The small fraction of CD3pos cells in the NK cell gate was found to not express Ki67 (Supporting information, Fig. S2b). In contrast, the fraction of NK cells expressing the NK receptors (NKR) NKG2A, NKG2D, NKp30 or NKp46 was not statistically different between HD and SLE, with a small subset of SLE patients having enrichment of NKG2C+ NK cells (Fig. 1c). This may reflect HCMV infection in these patients, as NKG2C+ NK cells have been shown to be specifically expanded in HCMV+ donors 27, 28. HCMV status was not tested on these patients, as this pathogen is rarely a risk to patients with SLE.
Figure 1.
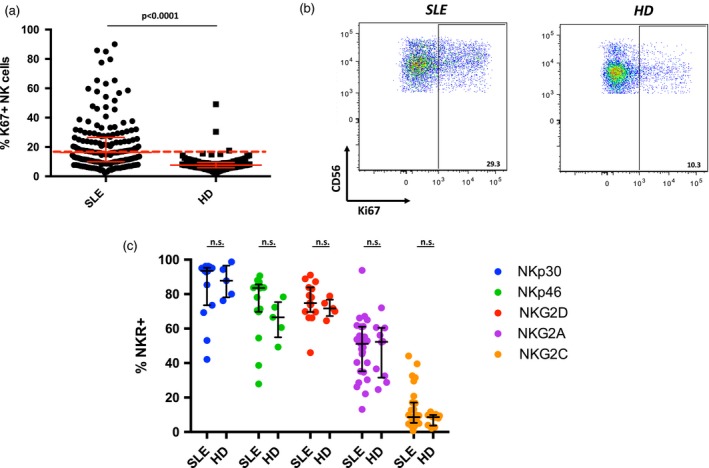
Natural killer (NK) cells expressing Ki67 are increased in systemic lupus erythematosus (SLE). (a) Scatter‐plot of the frequency of Ki67+ NK cells in the peripheral blood of SLE patients (n = 174, median = 16·3) and healthy donors (HD) (n = 99, median = 7·6). Dashed line indicates 2 standard deviations (s.d.) from HD mean (16·74%) Middle bar denotes median; surrounding bars denote first and second interquartile ranges. (b) Representative examples of Ki67 expression on NK cells from SLE patient and HD. (c) Scatter‐plot of the percentage of the indicated NK receptors (NKR) expressed on NK cells SLE (n = 15 or 27) and HD (n = 5 or 10). Middle bar denotes median; surrounding bars denote first and second interquartile range. Statistical analysis performed using the Mann–Whitney U‐test.
As Ki67 is a marker of proliferation, we measured whether induction of Ki67 correlates with an increase in NK cell numbers. However, the percentage of Ki67+ NK cells was inversely related to the absolute count of NK cells (Supporting information, Fig. S3b). Notably, neither the increase in Ki67 expression nor the decrease in NK cell counts were correlated with overall leukopenia (Supporting information, Fig. S3c).
As shown in Fig. 1b, the increase in Ki67 expression in SLE patient NK cells was characterized by high expression on a distinct subset of NK cells rather than increased expression on the whole population. The CD56bright subset appears to account at least partially for this observation, as these NK cells have a significant increase in Ki67 expression over CD56dim NK cells (Fig. 2a). NK cells from SLE donors expressing NKG2A, ‐2C, ‐2D or NKp30 or NKp46 did not preferentially express Ki67 (Fig. 2b). The small number of CD3+ T cells present in the gated NK cell population did not express Ki67 (Supporting information, Fig. S2b).
Figure 2.
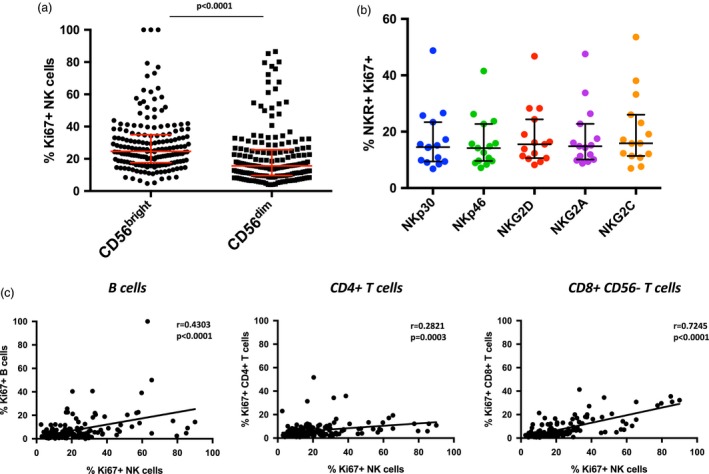
The distribution of Ki67+ cells in systemic lupus erythematosus (SLE). (a) Scatter‐plot of the frequency of Ki67+ CD56bright (n = 174, median = 24·7) and CD56dim (n = 174, median = 15·7). Middle bar denotes median; surrounding bars denote first and second interquartile ranges. (b) Scatter‐plot of the percentage of the indicated natural killer receptor (NKR) expressing Ki67 in SLE (n = 15) and healthy donors (HD) (n = 5). Middle bar denotes median; surrounding bars denote first and second interquartile range. Statistical tests for (a) and (b) performed using the Mann–Whitney U‐test. (c) Linear regression analyses of Ki67+ NK cells with the indicated lymphocyte populations. Statistical analyses performed using Pearson’s correlation. CD8+CD56+ cells could not be analyzed because of overlap with NK cells.
Correlation of Ki67 expression on other lymphocyte subsets revealed that B cells, CD4+ T cells and CD8+CD56– T cells can also express Ki67, but on average these subsets expressed lower frequencies of Ki67 compared to NK cells (Fig. 2c). There was a significant correlation of Ki67+ NK cells with Ki67+ B cells, CD4 T cells and CD56–CD8+ T cells, with the highest correlation found between NK cells and CD8+ T cells.
Ki67+ NK cells are associated with increased autoantibody production, low complement levels and nephritis in SLE patients
To investigate the clinical significance of Ki67 expression on NK cells in SLE, we correlated the percentage of Ki67+ NK cells with the SLEDAI‐2 K score measured on the day of sample collection. We observed a significant positive correlation between increased NK cell expression of Ki67+ and the SLEDAI score (Fig. 3a). Additional analysis binning patients based on SLEDAI score revealed significant increases in the percentage of Ki67+ NK cells in SLE patients over healthy donors in all categories of SLEDAI, even those with no discernable disease activity (SLEDAI of 0). Moreover, increasing percentages of Ki67+ NK cells were found in those with increasing disease activity (Fig. 3b). Although relatively few patients had a SLEDAI score of 10 or more, NK cells in this group had the highest frequency of Ki67+ expression.
Figure 3.
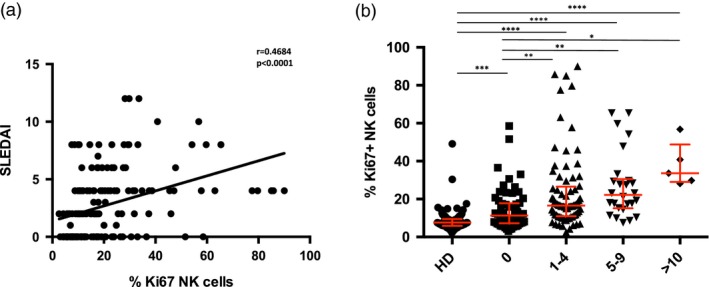
High frequencies of Ki67+ natural killer (NK) cells are associated with increased disease severity. (a) Linear regression analysis of the frequency of Ki67+ NK cells and Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) (n = 174). (b) Scatter‐plot of binned healthy donor (HD) and SLE patients according to disease severity comparing the frequency of Ki67+ NK cells. Middle bar denotes median; surrounding bars denote first and second interquartile ranges. Statistical analysis performed using Spearman’s ranked correlation and Kruskal–Wallis test. ***P < 0·0001.
Autoantibody production and B cell activation are key features of SLE and certain autoantibody specificities, such as anti‐dsDNA, can be predictive of disease flares. To determine the relationship between NK cell activation and these features of disease in SLE, we measured the frequency of Ki67+ NK cells in patients with and without anti‐dsDNA and anti‐extractable nuclear antigen (ENA) antibodies. As expected, most SLE patients had measurable quantities of these autoantibodies (Fig. 4a). However, we found that patients with dsDNA or ENA autoantibodies had significantly higher Ki67+ NK cells compared to those without these serum autoantibodies (Fig. 4a).
Figure 4.
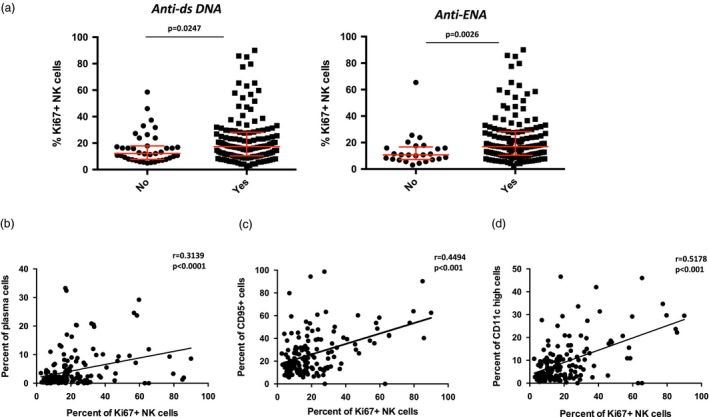
High frequencies of Ki67+ natural killer (NK) cells are associated with autoantibody production and B cell expansions. (a) Frequency of Ki67+ NK cells in systemic lupus erythematosus (SLE) patients grouped based on the presence (yes) or absence (no) of the indicated autoantibody [anti‐extractable nuclear antigen (ENA)]. Middle bar denotes median; surrounding bars denote first and second interquartile ranges. (b,d) Linear regression analysis of the frequency of plasma cells (b), CD95+ B cells (c), CD11chi B cells versus the frequency of Ki67+ NK cells in SLE patients. Statistical analyses performed using Pearson’s correlation.
Antibody‐producing plasma cells are expanded in SLE patients and are thought to participate in the production of autoantibodies, as treatments which achieve a certain degree of plasma cell depletion result in a significant decrease in anti‐dsDNA titers 29, 30. Correlation analysis of circulating plasma cell frequencies with Ki67+ NK cells showed a positive association of Ki67+ NK cells with plasma cell expansion (Fig. 4b). Similarly, we found that Ki67+ NK cells significantly correlated with B cells expressing CD95, a marker of germinal center B cells (Fig. 4c). Recently, we have described that CD11chi B cells are significantly increased in the peripheral blood of SLE patients and are able to differentiate into autoreactive plasma cells 31. We examined this population and found that the frequency of CD11chi B cells significantly correlated with Ki67+ NK cells (Fig. 4d). Taken together, these data link NK cell Ki67 expression to features of B cell activation in lupus.
Complement levels are among the best biomarkers for disease activity in SLE, and high percentages of Ki67+ NK cells strongly correlated with reduced C3 and C4 levels in serum analyzed concurrently (Fig. 5a). As the distribution of Ki67 expression on NK cells from SLE patients is non‐Gaussian, we divided patients into Ki67hi and Ki67mid/low NK cell groups using 1 standard deviation (s.d.) (40%) above the mean percentage of Ki67 + NK cells in SLE donors as a cut‐off. Patients with high frequencies of Ki67+ NK cells were significantly more likely to have low serum complement levels with a relative risk of 4.19 [95% confidence interval (CI) = 2·16–6·72; Fig. 5b). Patients with > 40% frequency of Ki67+ NK cells were also much more likely to have active nephritis, defined as active renal sediment, worsening renal function or new‐onset proteinuria, with a relative risk of 4.3 (95% CI = 2·48–7·48; Fig. 5c). Active renal disease was the only clinical component that correlated with elevated Ki67+ NK cells, although it should be noted that the frequency of organ manifestations of SLE, such as neurological, skin or vascular complications, were relatively uncommon in the cohort (Supporting information, Table S1).
Figure 5.
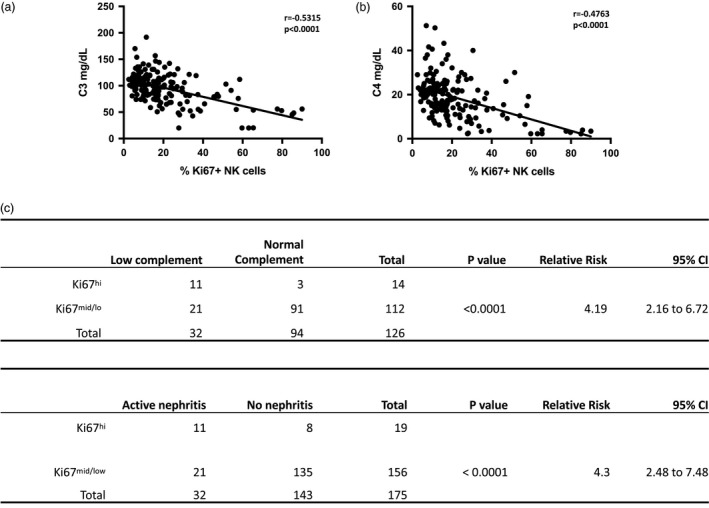
High frequencies of Ki67+ natural killer (NK) cells are present in patients with decreased levels of complement as well as active nephritis. (a) Linear regression analysis of serum C3 and C4 proteins in systemic lupus erythematosus (SLE) patients. Statistical analyses performed using Pearson’s correlation. (b) Top: contingency table of patients ranked based on Ki67hi NK cells (> 40% Ki67+ NK cells) or Ki67mid/lo (< 40% Ki67+ NK cells) comparing serum complement as either low (< 90 mg/dL C3 and < 10 mg/dL C4) or normal at the time of visit and blood draw. Sensitivity and specificity of test = 34·3 and 96·8%. Positive predictive value = 78·5%. Bottom: contingency table of patients ranked based on Ki67hi NK cells (> 40% Ki67+) or Ki67mid/lo (< 40% Ki67+ NK cells) comparing active or no nephritis at the time of visit and blood draw. Sensitivity and specificity of test = 34·3 and 94·4%. Positive predictive value = 57·9%. Statistical analysis was performed using Fisher’s exact test.
Role of IFNs and IL‐15 in NK cell up‐regulation of Ki67
PBMCs from patients with SLE exhibit increased transcription of genes induced by type I IFNs 32, 33. To determine whether up‐regulation of Ki67 on NK cells may be influenced by type I IFN, we measured IFN‐induced genes in the peripheral blood of SLE patients and correlated this with the frequency of Ki67 on NK cells. We found a strong correlation between the IFNGS score and the frequency of Ki67+ NK cells, where all patients with Ki67hi NK cells had an IFN signature score greater than 3 (Fig. 6a). These results are consistent with the known association of type I IFN‐induced gene expression to increased clinical activity in SLE, and suggest that IFNs may have a role in the induction of Ki67 on NK cells 34.
Figure 6.
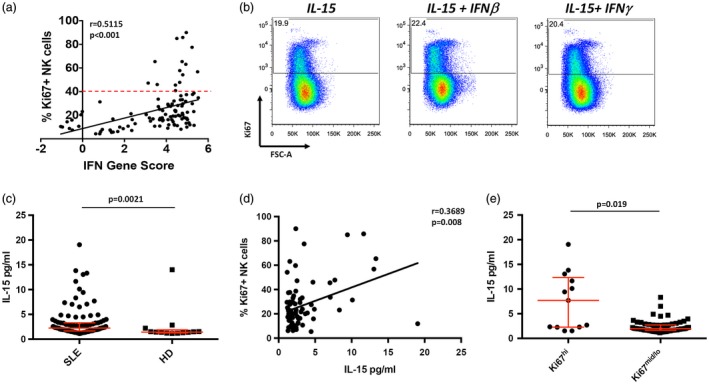
High frequencies of Ki67+ natural killer (NK) cells correlate with a type I interferon (IFN) gene signature and serum interleukin (IL)‐15 levels. (a) Linear regression analysis of IFN gene score with percentage of Ki67 + NK cells in systemic lupus erythematosus (SLE) patients. Statistical analysis was performed using Spearman’s ranked correlation. Dashed line indicates 1 standard deviation (s.d.) from mean (40%). (b) Flow cytometry dot plots of peripheral blood NK cells cultured with the indicated stimuli. Data are representative of four individual healthy donors (HD). Cells shown are NK cells within the viable lymphocyte gate. (c) Scatter‐plot analysis of serum IL‐15 levels. Serum concentrations of IL‐15 in SLE (n = 105, median = 2·24) and HD (n = 13, median = 1·43). Middle bar denotes mean; surrounding bars denote first and second interquartile ranges. Statistical analyses performed using the Mann–Whitney U‐test. (d) Linear regression analysis of serum IL‐15 levels with percentage of Ki67+ NK cells in SLE patients. Statistical analysis was performed using Pearson’s correlation. (e) Serum IL‐15 levels in SLE patients in Ki67hi and Ki67mid/lo NK cell groups.
To test whether IFNs can directly induce the expression of Ki67 on NK cells, we purified NK cells from HD and cultured them in vitro for 14 days with IFN‐β or IFN‐γ, in addition to low‐dose IL‐15, to maintain cell viability 35. Strikingly, IL‐15 alone markedly induced Ki67 on NK cells, while the addition of either IFN‐β or IFN‐γ did not increase Ki67 expression further (Figs. 1a and 6b). These data suggest that IFNs do not directly induce Ki67 expression on NK cells in SLE but, rather, indicate a role for IL‐15.
It is important to note that patients with high Ki67+ NK cells and active renal disease were taking cyclophosphamide. However, our data suggest that cyclophosphamide is not a driver of Ki67 expression, as >50% of patients with Ki67hi NK cells were not receiving this immunosuppressive therapy, and the percentage of Ki67+ NK cells was not linked to leukopenia, which is found at the nadir of the hematological response to cyclophosphamide (Supporting information, Fig. S4a and data not shown). Additionally, analysis of the percentage of Ki67+ NK cells in patients based taking steroid treatment revealed that those not taking steroids lacked high frequencies of Ki67+ NK cells. There was also a trend towards higher levels of Ki67 expression in patients taking steroid therapy (Supporting information, Fig. S4b).
To investigate the possibility that IL‐15 may promote NK cell expression of Ki67 in vivo, we correlated serum IL‐15 levels with the frequency of Ki67+ NK cells from SLE patients measured from peripheral blood drawn on the same day. In concordance with previous reports, serum IL‐15 was elevated in patients with SLE (Fig. 6c) 25, 26, 36. IL‐2, another serum cytokine which is a potent activator of NK cell proliferation and function, was not detectable in the same sera (data not shown). Expression of Ki67 on NK cells was positively correlated with serum IL‐15 (Fig. 6d). Although IL‐15 was not detectable in all samples, the individuals with the highest levels of IL‐15 were those with high frequencies of Ki67+ NK cells (Fig. 6e). Taken together, these results suggest that IL‐15 may play a role in up‐regulating NK cell Ki67 expression in SLE.
Discussion
Herein, we describe that phenotypical alterations in NK cells are associated with SLE disease activity, data which provide insight into this autoimmune pathology. We found that in the majority of SLE patients, NK cell Ki67 expression was increased above what was observed in HD. Individuals with high frequencies of Ki67+ NK cells correlated not only with increased overall disease activity in SLE, but specifically with low serum complement and active LN. The frequencies of Ki67+ NK cells were inversely correlated with NK cell numbers but its expression is probably not driven solely by homeostatic proliferation of NK cells. Rather, lower NK cell numbers could result from migration of cells to affected tissues such as the kidney in LN. Alternatively, the inflammatory environment in SLE may promote NK cells to undergo activation‐induced cell death and shorten their life‐span. The generation of Ki67+ NK cells may be a result of circulating IL‐15, given that IL‐15 can induce the expression of Ki67 on NK cells in vitro, and Ki67 expression on NK cells is positively associated with serum IL‐15 levels.
NK cell expression of Ki67 is strikingly associated with SLE disease activity, and in particular with LN. Analysis of patients with high (> 40%) levels of NK cells expressing Ki67 revealed that that the majority (57.8%) had active LN at the time of the blood collection, which is in stark contrast to those patients with lower levels of Ki67, of whom only a small (12.3%) portion presented with concurrent LN. Available biopsy data indicated that Ki67+ NK cells were not associated with a particular LN disease stage, with the majority of the patients in this cohort presenting with diffuse proliferative glomerulonephritis, which is the most common form of LN. This suggests that our sample cohort was representative of the general population of SLE patients who present with LN. These results extend previous observations of reduced numbers of circulating NK cells in patients with lupus‐related renal disease 18.
While the etiology of SLE is unclear, B cells are a critical component of disease pathogenesis 37. In turn, high titers of dsDNA autoantibodies have been shown to correlate with renal involvement 38, 39. Here we show that activated NK cells correlate significantly with the presence of CD11chi B cells, plasma cells and serum anti‐dsDNA autoantibodies. Further work will be required to determine if expansion of Ki67+ NK cells contribute to the pathogenesis of these features of lupus or are a part of counter‐regulatory mechanisms.
These results provide novel insights into the cytokines which promote expression of Ki67 on NK cells in SLE. The frequency of Ki67+ NK cells correlated with up‐regulation of type I interferon genes in PBMC. However, IFN‐β or IFN‐γ alone could not directly induce the expression of Ki67 on NK cells from HD in vitro. Rather, IL‐15, a cytokine which can support the proliferation of NK cells in vitro, is positively associated with Ki67 on NK cells in SLE PBMC. IL‐15 plays an essential role in NK cell development and survival, as targeted mutations in the genes encoding IL‐15 or its receptor IL‐15Rα result in NK cell deficiency 40, 41, 42. IL‐15 is also required for memory CD8 T cell homeostatic proliferation 43. Interestingly, our data suggest that CD8+ T cells and NK cells, but not B cells or CD4+ T cells in SLE, are induced to express Ki67 by the same factor, which may indeed be IL‐15. IL‐15 infusions in patients with cancer can induce both NK cells as well as CD8 T cells to express Ki67 15. Moreover, numerous publications have shown that IL‐15 is a strong inducer of NK cell proliferation 14, 44, 45. However, a subset of SLE PBMC shows substantial frequencies of Ki67+ NK cells in the absence of high IL‐15 levels, which suggest that cytokines in addition to IL‐15 may play a role in Ki67 induction. Candidate factors include IL‐18 and IL‐12, which can promote NK cell proliferation and have been shown to be increased in SLE 46.
NK cells can be directly activated by IFNs, which induce downstream effector functions such as cytotoxicity, but as we and others have shown, this cytokine is not sufficient to promote NK cell proliferation 14. Interestingly, exposure to type I IFN causes dendritic cells to up‐regulate IL‐15 and IL‐15Rα, a mechanism known for its ability to prime NK cells for expansion and effector functions 14, 45, 47, 48. This suggests that the IFNs up‐regulated in SLE could indirectly activate NK cells via IL‐15. Our data showing IFNGS and serum IL‐15 correlate with Ki67 expression on NK cells in SLE, as well as the ability of IL‐15 to up‐regulate Ki67 on peripheral blood NK cells, and support a role for the IFN/IL‐15 axis in the activation of NK cells in SLE.
Our results suggest that more detailed investigations of NK cells in SLE is warranted. Particularly, studies to determine how the IFN/IL‐15 axis controls NK cell function will be of interest. Under some conditions, IL‐15 can stimulate NK cell cytotoxicity and IFN‐γ production 48. As such, IL‐15 could enhance NK cell function as effector cells in autoimmunity. Mechanistically, these effector functions could be pathogenic through including antibody‐dependent cellular cytotoxicity (ADCC) of opsonized target cells, but could also play a regulatory role through killing of CD4+ T cells which, interestingly, has been shown to be negatively regulated by type I IFNs 6, 9. Understanding whether Ki67 expression drives more pathogenic or protective functions of NK cells in SLE will shed light on disease pathogenesis and possibly identify new avenues of therapy through harnessing the regulatory power of NK cells.
Disclosures
R. E., M. A. S. and S. W. were employees at Medimmune and own AstraZeneca stock. R. E., S. W. and M. A. S. are currently employees at and shareholders of Viela Bio. J. W., K. A. C., M. P., N. W., K. Z., J. R., B. W., G. B., B. R, B, N, R, G., G. C., R. K. S. R. are full‐time employees and shareholders of MedImmune/AstraZeneca. R. M. S. has been an employee of Novartis since June 2018.
Supporting information
Fig. S1. Flow cytometry of peripheral blood lymphocytes with the indicated markers. NK cell gating strategy for flow cytometry. NK cells were selected using the indicated Boolean gating strategy.
Fig. S2. Example flow cytometric plots of peripheral blood lymphocytes with the indicated markers. (a) Cells were gated as in Fig. S1. CD3 expression in CD56posCD4negCD8αhighnegative population. (b) Cells were gated on as in Fig. S1. Expression of Ki67 on CD56+ (left) and CD3+ (right) in the CD56posCD4negCD8αhineg population. Data is representative of 2 healthy and 2 SLE donors.
Fig. S3. NK cells are decreased in SLE and inversely correlate with frequencies of Ki67 + NK cells. (a) Left, scatter plot of the frequency of NK cells in the peripheral blood of SLE patients (n = 174, median = 8.8) and HD (n = 99, median = 13.3). Right, absolute numbers of NK cells in the peripheral blood of SLE patients (n = 142, median = 87.9) and HD (n = 85, median = 185.9). (b) Linear regression analysis of NK cell count and percent Ki67 + NK cells in SLE patients. (c) Scatter plot of the frequency of white blood cells in the peripheral blood of SLE patients (n = 112, median = 4890) and HD (n = 33, median = 5140). Middle bar in scatter plots denotes median; surrounding bars denote 1st and 2nd interquartile ranges Statistical analyses performed using Mann‐Whitney U test and Pearson’s correlation. NK cells were selected as on Fig. S1a.
Fig. S4. NK cell expression of Ki67 in relation to immunosuppressive therapy administration. (a) Percentage of Ki67hi patients receiving cyclophosphamide (Cy Yes) or not (Cy No) at the time of blood draw. (b) Frequency of Ki67 expression on NK cells from patients grouped based on high dose steroids (High; >15mg; n = 32, median = 23.9), Low dose steroids (Low; <10mg; n = 118, median = 16.05) and No (no steroids; n = 25, median = 11.3). Middle bar denotes median; surrounding bars denote 1st and 2nd interquartile range. Statistical analysis performed using Mann‐Whitney U test. ***, P < 0·0001
Table S1. Patient demography*. *Patient and Healthy donor characteristics at time of blood draw
Table S2. Therapies*. * Therapies from SLE patients, taking at the time of blood draw
Acknowledgement
This study was supported by Medimmune and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), National Institutes of Health.
This article has been contributed to by US Government employees and their work is in the public domain in the USA.
Contributor Information
R. Ettinger, Email: ettingerr@vielabio.com
R. M. Siegel, Email: siegelr@nih.gov.
Autoimmunity Molecular Team:
M. Parker, N. White, K. Zerrouki, J. Riggs, B. Ward, G. Bhat, B. Rajan, B. Naiman, R. Grady, and C. Groves
References
- 1. Liu Z, Davidson A. Taming lupus – a new understanding of pathogenesis is leading to clinical advances. Nat Med 2012; 18:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schwartz N, Goilav B, Putterman C. The pathogenesis, diagnosis and treatment of lupus nephritis. Curr Opin Rheumatol 2014; 26:502–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hahn BH, McMahon MA, Wilkinson A et al American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken) 2012; 64:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reyes‐Thomas J, Blanco I, Putterman C. Urinary biomarkers in lupus nephritis. Clin Rev Allergy Immunol 2011; 40:138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vivier E, Tomasello E, Baratin M et al Functions of natural killer cells. Nat Immunol 2008; 9:503–10. [DOI] [PubMed] [Google Scholar]
- 6. Crouse J, Bedenikovic G, Wiesel M et al Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR6. Immunity 2014; 40:961–73. [DOI] [PubMed] [Google Scholar]
- 7. Lu L, Ikizawa K, Hu D et al Regulation of activated CD4+ T cells by NK cells via the Qa‐1‐NKG2A inhibitory pathway. Immunity 2007; 26:593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Waggoner SN, Cornberg M, Selin LK et al Natural killer cells act as rheostats modulating antiviral T cells. Nature 2011; 481:394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu HC, Grusdat M, Pandyra A et al Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 2014; 40:949–60. [DOI] [PubMed] [Google Scholar]
- 10. French AR, Yokoyama WM. Natural killer cells and autoimmunity. Arthritis Res Ther 2004; 6:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robbins SH, Tessmer MS, Mikayama T et al Expansion and contraction of the NK cell compartment in response to murine cytomegalovirus infection. J Immunol 2004; 173:259–66. [DOI] [PubMed] [Google Scholar]
- 12. Lugli E, Goldman CK, Smedley J et al Transient and persistent effects of IL‐15 on lymphocyte homeostasis in nonhuman primates. Blood 2010; 116:3238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morishima C, Paschal DM, Wang CC et al Decreased NK cell frequency in chronic hepatitis C does not affect ex vivo cytolytic killing. Hepatology 2006; 43:573–80. [DOI] [PubMed] [Google Scholar]
- 14. Nguyen KB, Salazar‐Mather TP, Dalod MY et al Coordinated and distinct roles for IFN‐alpha beta, IL‐12, and IL‐15 regulation of NK cell responses to viral infection. J Immunol 2002; 169:4279–87. [DOI] [PubMed] [Google Scholar]
- 15. Conlon KC, Lugli E, Welles HC et al Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first‐in‐human clinical trial of recombinant human interleukin‐15 in patients with cancer. J Clin Oncol 2015; 33:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Green MRJ, Kennell ASM, Larche MJ et al Natural killer cell activity in families of patients with systemic lupus erythematosus: demonstration of a killing defect in patients. Clin Exp Immunol 2005; 141:165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park YW, Kee S‐J, Cho Y‐N et al Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum 2009; 60:1753–63. [DOI] [PubMed] [Google Scholar]
- 18. Erkeller‐Yuksel FM, Lydyard PM, Isenberg DA. Lack of NK cells in lupus patients with renal involvement. Lupus 1997; 6:708–12. [DOI] [PubMed] [Google Scholar]
- 19. Riccieri V, Spadaro A, Parisi G et al Down‐regulation of natural killer cells and of gamma/delta T cells in systemic lupus erythematosus. Does it correlate to autoimmunity and to laboratory indices of disease activity? Lupus 2000; 9:333–7. [DOI] [PubMed] [Google Scholar]
- 20. Li W‐X, Pan H‐F, Hu J‐L et al Assay of T‐ and NK‐cell subsets and the expression of NKG2A and NKG2D in patients with new‐onset systemic lupus erythematosus. Clin Rheumatol 2010; 29:315–23. [DOI] [PubMed] [Google Scholar]
- 21. Huang Z, Fu B, Zheng SG et al Involvement of CD226+ NK cells in immunopathogenesis of systemic lupus erythematosus. J Immunol 2011; 186:3421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hervier B, Beziat V, Haroche J et al Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon‐gamma production in patients with active disease. Arthritis Rheum 2011; 63:1698–706. [DOI] [PubMed] [Google Scholar]
- 23. Puxeddu I, Bongiorni F, Chimenti S et al Cell surface expression of activating receptors and co‐receptors on peripheral blood NK cells in systemic autoimmune diseases. Scand J Rheumatol 2012; 41:298–304. [DOI] [PubMed] [Google Scholar]
- 24. Huntington ND. The unconventional expression of IL‐15 and its role in NK cell homeostasis. Immunol Cell Biol 2014; 92:210–3. [DOI] [PubMed] [Google Scholar]
- 25. Baranda L, de la Fuente H, Layseca‐Espinoza E et al IL‐15 and IL‐15R in leucocytes from patients with systemic lupus erythematosus. Rheumatology (Oxf) 2005; 44:1507–13. [DOI] [PubMed] [Google Scholar]
- 26. Lin SJ, Kuo ML, Hsaio HS, Huang JL. Activating and inhibitory receptors on natural killer cells in patients with systemic lupus erythematosis‐regulation with interleukin‐15. PLOS ONE 2017; 12:e0186223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guma M, Angelo A, Vilches C et al Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004; 104:3664–71. [DOI] [PubMed] [Google Scholar]
- 28. Lopez‐Verges S, Milush JM, Schwartz BS et al Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci USA 2011; 108:14725–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Illei GG, Shirota Y, Yarboro CH et al Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open‐label phase I dosage‐escalation study. Arthritis Rheum 2010; 62:542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexander T, Safert R, Klotsche J et al The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis 2015; 74:1474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang S, Wang J, Kumar V et al Role and regulation of CD11chi T‐bet+ B cells in SLE. Nat Commun 2018; 9:1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Furie R, Khamashta M, Merrill JT et al Anifrolumab, an anti‐interferon‐alpha receptor monoclonal antibody, moderate‐to‐severe systemic lupus erythematosus. arthritis. Rheumatol 2017; 69:376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yao Y, Higgs BW, Morehouse C et al Development of potential pharmacodynamic and diagnostic markers for anti‐IFN‐alpha monoclonal antibody trials in systemic lupus erythematosus. Hum Genom Proteom 2009; 2009:374312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol 2014; 192:5459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romee R, Schneider SE, Leong JW et al Cytokine activation induces human memory‐like NK cells. Blood 2012; 120:4751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aringer M, Stummvoll GH, Steiner G et al Serum interleukin‐15 is elevated in systemic lupus erythematosus. Rheumatology (Oxf) 2001; 40:876–81. [DOI] [PubMed] [Google Scholar]
- 37. Dorner T, Giesecke C, Lipsky PE. Mechanisms of B cell autoimmunity in SLE. Arthritis Res Ther 2011; 13:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Compagno M, Rekvig OP, Bengtsson AA et al Clinical phenotype associations with various types of anti‐dsDNA antibodies in patients with recent onset of rheumatic symptoms. Results from a multicentre observational study. Lupus Sci Med 2014; 1:e000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haddon DJ, Diep KD, Price JV et al Autoantigen microarrays reveal autoantibodies associated with proliferative nephritis and active disease in pediatric systemic lupus erythematosus. Arthritis Res Ther 2015; 17:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kennedy MK, Glaccum M, Brown SN et al Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15‐deficient mice. J Exp Med 2000; 191:771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carson WE, Fehniger TA, Haldar S et al A potential role for interleukin‐15 in the regulation of human natural killer cell survival. J Clin Invest 1997; 99:937–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huntington ND, Legrand N, Alves NL et al IL‐15 trans‐presentation promotes human NK cell development and differentiation in vivo . J Exp Med 2009; 206:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Becker TC, Wherry EJ, Boone D et al Interleukin 15 is required for proliferative renewal of virus‐specific memory CD8 T cells. J Exp Med 2002; 195:1541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dunne J, Lynch S, O’Farrelly C et al Selective expansion and partial activation of human NK cells and NK receptor‐positive T cells by IL‐2 and IL‐15. J Immunol 2001; 167:3129–38. [DOI] [PubMed] [Google Scholar]
- 45. Lucas M, Scachterle W, Oberle K et al Dendritic cells prime natural killer cells by trans‐presenting interleukin 15. Immunity 2007; 26:503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lauwerys BR, Renauld JC, Houssiau FA. Synergistic proliferation and activation of natural killer cells by interleukin 12 and interleukin 18. Cytokine 1999; 11:822–30. [DOI] [PubMed] [Google Scholar]
- 47. Ferlazzo G, Pack M, Thomas D et al Distinct roles of IL‐12 and IL‐15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci USA 2004; 101:16606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zanoni I, Spreafico R, Bodio C et al IL‐15 cis presentation is required for optimal NK cell activation in lipopolysaccharide‐mediated inflammatory conditions. Cell Rep 2013; 4:1235–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Flow cytometry of peripheral blood lymphocytes with the indicated markers. NK cell gating strategy for flow cytometry. NK cells were selected using the indicated Boolean gating strategy.
Fig. S2. Example flow cytometric plots of peripheral blood lymphocytes with the indicated markers. (a) Cells were gated as in Fig. S1. CD3 expression in CD56posCD4negCD8αhighnegative population. (b) Cells were gated on as in Fig. S1. Expression of Ki67 on CD56+ (left) and CD3+ (right) in the CD56posCD4negCD8αhineg population. Data is representative of 2 healthy and 2 SLE donors.
Fig. S3. NK cells are decreased in SLE and inversely correlate with frequencies of Ki67 + NK cells. (a) Left, scatter plot of the frequency of NK cells in the peripheral blood of SLE patients (n = 174, median = 8.8) and HD (n = 99, median = 13.3). Right, absolute numbers of NK cells in the peripheral blood of SLE patients (n = 142, median = 87.9) and HD (n = 85, median = 185.9). (b) Linear regression analysis of NK cell count and percent Ki67 + NK cells in SLE patients. (c) Scatter plot of the frequency of white blood cells in the peripheral blood of SLE patients (n = 112, median = 4890) and HD (n = 33, median = 5140). Middle bar in scatter plots denotes median; surrounding bars denote 1st and 2nd interquartile ranges Statistical analyses performed using Mann‐Whitney U test and Pearson’s correlation. NK cells were selected as on Fig. S1a.
Fig. S4. NK cell expression of Ki67 in relation to immunosuppressive therapy administration. (a) Percentage of Ki67hi patients receiving cyclophosphamide (Cy Yes) or not (Cy No) at the time of blood draw. (b) Frequency of Ki67 expression on NK cells from patients grouped based on high dose steroids (High; >15mg; n = 32, median = 23.9), Low dose steroids (Low; <10mg; n = 118, median = 16.05) and No (no steroids; n = 25, median = 11.3). Middle bar denotes median; surrounding bars denote 1st and 2nd interquartile range. Statistical analysis performed using Mann‐Whitney U test. ***, P < 0·0001
Table S1. Patient demography*. *Patient and Healthy donor characteristics at time of blood draw
Table S2. Therapies*. * Therapies from SLE patients, taking at the time of blood draw