

Abstract
Fine-tuning of gene expression is crucial for protein expression and pathway construction, but it still faces formidable challenges due to the hierarchical gene regulation at multiple levels in a context-dependent manner. In this study, we defined the optimal targeting windows for CRISPRa and CRISPRi of the dCas9-α/ω system, and demonstrated that this system could act as a single master regulator to simultaneously activate and repress the expression of different genes by designing position-specific gRNAs. The application scope of dCas9-ω was further expanded by a newly developed CRISPR-assisted Oligonucleotide Annealing based Promoter Shuffling (OAPS) strategy, which could generate a high proportion of functional promoter mutants and facilitate the construction of effective promoter libraries in microorganisms with low transformation efficiency. Combing OAPS and dCas9-ω, the influences of promoter-based transcription, molecular chaperone-assisted protein folding and protease-mediated degradation on the expression of amylase BLA in Bacillus subtilis were systematically evaluated, and a 260-fold enhancement of BLA production was obtained. The success of the OAPS strategy and dCas9-ω for BLA production in this study thus demonstrated that it could serve as a powerful tool kit to regulate the expression of multiple genes multi-directionally and multi-dimensionally in bacteria.
INTRODUCTION
With the rapid technology development and advancement to dramatically reduce the cost of DNA reading and writing, synthetic biology has been widely applied to fabricate complex biological systems to deal with environmental pollution, energy dilemma and health issues (1,2). The construction of elaborated genetic circuits and achievement of balanced metabolic fluxes required the ability to implement precisely targeted changes in gene expression over a broad set of genes (3,4). Since cells have evolved robust regulatory networks, for the purpose of dealing with environmental changes and genetic disturbances, to control gene expression at distinct, yet interwoven, levels of regulation (5), it is challenging for strategies like promoter engineering (5) and RBS Calculator (6) to finely tune gene expression because they only execute one layer of expression control. Though increasing efforts have been implemented in complexity from regulating one layer of expression control to multi-component and multi-dimensional optimization (7,8), predictive rules about the effects of individual components such as molecular chaperones and proteases and their interactions on the expression of target genes have not yet been elucidated, due to each layer of expression control having more or less substrate specificity (9,10). Thus, it is still a trial and error process to iteratively optimize individual components to achieve a satisfactory expression level (11). In addition, multiple targets need to be coordinately regulated in constructing more sophisticated systems. Unfortunately, such regulation is often performed sequentially and with low throughput (12). These dilemmas highlight the requirement of developing molecular tool kits capable of modifying gene expression parallelly and multi-directionally.
The bacterial immune system-derived CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9) system has been demonstrated to be a robust, designable and multi-plexable tool for genome editing in diverse organisms (13,14). Interestingly, the catalytically inactive Cas9 mutant (dCas9) maintained its ability to bind target DNA with the guidance of gRNAs, which has been used in transcriptional engineering platforms for gene activation (CRISPRa), interference (CRISPRi) and modifications (15–19). In bacteria, dCas9 alone can efficiently interfere with RNA polymerase (RNAP) activity by forming a DNA bubble (20), which has been used to elucidate interconnections among core processes and to identify potential targets of uncharacterized antibiotics in Bacillus subtilis (21). Moreover, through fusing an RNAP recruiting domain to dCas9, sequence-specific RNA-guided transcription activation (CRISPRa) was achieved in Escherichia coli (20). The one-directional transcriptional regulation of dCas9 was also expanded to execute multi-directional transcriptional regulation. Deaner and Alper established a technique for fine-tuned, graded expression of pathway enzymes by modulating the targeting position of dCas9-VPR to the core promoter in Saccharomyces cerevisiae (19). Zalatan et al. extended the guide RNAs with modular RNA domains to recruit specific transcription effectors, thus achieving multi-directional transcriptional regulation in eukaryotes (22). In another study, Lian et al. developed a tri-functional CRISPR system for parallel gene regulation and deletion using orthogonal CRISPR systems in the S. cerevisiae (12). Furthermore, the truncated gRNA, which maintained the ability to guide Cas9 to target sequences but without introducing double-stranded breaks, was used to perform orthogonal gene knockout and transcriptional regulation in human cells (23,24). Recently, programmable control over multiple genes with simultaneous activation and repression in E. coli was accomplished by combination of sgRNA scaffold and bacterial transcriptional activators (25). However, the multi-directional regulation has not yet been reported in Gram-positive bacteria. And current multi-functional CRISPR systems have limitations of requiring purposely designed synthetic promoters for effectors (22), impaired binding affinity between Cas9 and the engineered gRNA (12), and a potential metabolic burden to the host cells caused by multiple CRISPR-associated proteins and transcriptional effectors.
In metabolic engineering practices, high-level expression of the rate-limiting enzymes is usually required to drive the metabolic fluxes for the biosynthesis pathways of target products. As the expression of rate-limiting enzymes is often tightly controlled at the transcriptional level (26), transcriptional activators like subunits of RNAP are unlikely to efficiently improve the expression of rate-limiting enzymes to maximize product yield. Synthetic promoters that can deregulate the cellular regulatory machinery may provide a valuable tool to enhance gene expression and to optimize metabolic flux. Thus, an alternative strategy to maximize the target biosynthetic flux is driving the expression of rate-limiting enzymes by strong synthetic promoters while coordinating the expression of other enzymes by a multi-directional transcription programing. Randomization of non-conserved sequences and error-prone PCR have been widely used to construct promoter libraries (27,28), where reporter genes, such as GFP, were commonly used for high-throughput screening of the enormous promoter mutants (28,29). Nevertheless, recent studies indicated that promoter strengths seem to be context-dependent with the variation of several orders of magnitude (30,31), highlighting a need of screening the promoter library using the target gene itself. Another constraint was that a massive library size was required due to the low proportion of functional mutants in the generated libraries, which poses great challenges on proteins lacking high-throughput screening methods and many industrially important strains with low transformation efficiency (32). Therefore, a novel promoter engineering strategy that can efficiently generate abundant sequence diversities with a high proportion of functional mutants is valuable to generate satisfactory expression level of target genes.
Bacillus subtilis has long been used as a model organism for molecular research as well as an industrial workhorse for the production of valuable enzymes (33). However, its application in the fields of synthetic biology and metabolic engineering has not been widely explored due to the complicated cellular regulatory networks (26). In this study, new effective transcriptional regulation strategies were developed in B. subtilis, which could promote the basic research and biotechnological applications in B. subtilis. First, the dCas9-ω/α system was established to be used as a single master regulator in B. subtilis to simultaneously activate and repress the expression of different target genes through designing locus-specific sgRNAs. Through this strategy, the influence of transcription, protein folding and protease degradation on target gene expression was easily evaluated, which sequentially guided the rational refactor of the cellular protein quality control system to multi-dimensionally tune the expression of target genes. In addition, a new promoter engineering strategy named Oligonucleotide Annealing based Promoter Shuffling (OAPS) was developed to expand the application scope of dCas9-ω/α, which utilized the naturally occurring genetic diversity of promoters as the driving force for promoter evolution and could generate a high proportion of functional promoter mutants. Finally, through the combination of OAPS and dCas9-ω, the production of amylase BLA was improved by 260-fold compared to the commonly used strong promoter P43 in B. subtilis (Scheme 1).
Scheme 1.

Schematic diagram of multi-dimensional regulation for fine-tuning gene expression. (I) Promoter with desired strength was generated by promoter shuffling to control target gene expression at the transcriptional level. (II) The effects of molecular chaperones on target protein folding were interrogated by dCas9-ω based CRISPR-activation system, and the expression of functional molecular chaperones were specifically enhanced by dCas9-ω to improve target protein folding. (III) Extracellular proteases were individually inhibited by dCas9-ω mediated CRISPR-interference system, and proteases exhibited degradative activities on target protein were simultaneously repressed.
MATERIALS AND METHODS
Strains, media and chemicals
Escherichia coli DH5α (Invitrogen, USA) was used for routine cloning. Bacillus subtilis SCK6 (BGSCID: 1A976) (34) was used as host for promoter engineering. Bacillus subtilis and Escherichia coli were routinely grown in LB medium supplemented with the corresponding antibiotics to final concentrations of 100 μg/ml ampicillin for E. coli, or 100 μg/ml spectinomycin, 15 μg/ml tetracycline and 10 μg/ml chloromycetin for B. subtilis. Phusion® polymerase, restriction endonucleases and T4 DNA Ligase (high concentration) were purchased from New England Biolabs (NEB, USA), DNA polymerase was obtained from Yeasen Biotech Co., Ltd (Shanghai, China). The synthesis of oligonucleotides and DNA sequencing were performed by Sangon Biotech (Shanghai) Co., Ltd (Wuhan, China). All sequences and primers used in this study were listed in Supplementary Table S1.
Construct design for multi-directional transcriptional regulation in B. subtilis
To construct CRISPRa system in B. subtilis, the α and ω subunits of B. subtilis RNA polymerase was cloned into pHT01 to produce pHT01-ω and pHT01-α, respectively. Then, the catalytically dead Cas9 (dCas9) was amplified from plasmid pET-dCas9-VP64-6 × His and ligated into pHT01-ω and pHT01-α to generate fusion protein dCas9-ω and dCas9-α, respectively. To evaluate their transcriptional activation efficiency on different target genes, two expression cassettes of P43-GFP and P43-BLA were used as reporters. Five sgRNAs that target to different regions upstream of P43 promoter were designed for constitutive expression. After 12 h induction of dCas9-α or dCas9-ω with 0.1 mM isopropyl-β-d-thiogalactoside (IPTG), the relative GFP fluorescence or relative amylase activities of strains containing each sgRNAs were determined and normalized to strain expressing a non-targeting sgRNA.
To assess the ability of dCas9-ω to activate endogenous gene expression, the sgRNA guiding dCas9-ω to 224-bp upstream of amyE start codon was constructed. To analyze the position effects of dCas9-α and dCas9-ω on transcriptional regulation, different sgRNAs tiling P43-GFP expression cassette were designed and the relative fluorescence was assayed as mentioned above. To define the rules of dCas9-ω mediated transcriptional regulation, the strongest promoter P566-TA isolated from BLA specific library was used as the target. Similarly, sgRNAs guiding dCas9-ω to different positions of P566-TA-BLA were designed, and the relative amylase activities were assayed as mentioned above.
Construct designs for multivalent recruitment and multiplexed targeting
To analyze the influence of RNAP subunits valency on transcriptional activation, we fused the α and ω subunits to the C-terminal of dCas9 simultaneously with different range, generating fusion protein dCas9-α-ω and dCas9-ω-α, respectively. The fluorescence of SCK6/amyE::P43-GFP expressing sgRNAs (G9∼G13) and fusion proteins (dCas9-α-ω and dCas9-ω-α) was determined and normalized to strain expressing a non-targeting sgRNA. To analyze the activation efficiency of multiple sgRNAs that targeted to the same gene, G9 and G13 were assembled by BioBrick to produce G9+G13. The relative GFP fluorescence intensity of strains expressing G9+G13, dCas9-α or dCas9-ω was measured.
Construction of promoter libraries via OAPS
The integrative plasmid pDG1730 (BGSCID: ECE115, kindly as a gift from Prof. Sun Ming from Huazhong Agriculture University) was selected as backbone for constructing the promoter probe vector, which could circumvent the influence of plasmid copy number variation on library characterization. The fragment T1T2-BamHI-HindIII-T0 (Supplementary Table S2), which contains two strong terminators to avoid the influence of neighboring genetic contexts on promoter strengths, was synthesized and cloned into pDG1730, yielding pDGT. Then, GFP and BLA were cloned into pDGT to produce the probe vector pDGT-GFP and pDGT-BLA, respectively. For OAPS strategy, the characteristic consensus sequences (-35 and -10 motifs) of prokaryotic promoters were used as overlapping homologous sequences to divide each parent promoter into three modules. Each promoter module was individually synthesized by annealing two complementary single-stranded 5′ phosphorylated oligonucleotides in TE buffer according to the protocol described elsewhere (https://tools.thermofisher.com/content/sfs/brochures/TR0045-Anneal-oligos.pdf). As modules from different promoters have compatible overhangs, they could be randomly assembled to produce a library of combinatorial promoters.
For promoter library using GFP as reporter, four parent promoters Pveg, Pveg2, Pn26 and PlepA were selected for shuffling, namely a total of 12 pairs of 5′ phosphorylated oligonucleotides were synthesized (Supplementary Table S1). To construct amylase BLA specific promoter library, two additional promoters PtrnQ and PserA were added, and the -10 motif was designed as TAwwAT (w = a or t). To construct the CRISPR/Cas9 system, the Streptococcus pyogenes cas9 was cloned into pHT01 to generate an IPTG-inducible Cas9 expression cassette. The DNA fragments of P43/P242-BsmBI/BsmBI-sgRNA were synthesized and ligated into pHY300PLK to produce pHY-P43/P242-sgRNA. To assemble the sgRNA plasmids, 20-bp complementary primers were annealed and the double-stranded fragments were cloned into BsmBI digested pHYT-P43/p242-sgRNA. Because promoter P43 was a widely used strong promoter in B. subtilis, it has also been widely used as a reference promoter to evaluate the strengths of newly isolated or engineered promoters. Thus, P43 was used as a reference promoter in this study, allowing the evaluation of the relative activities of promoter mutants isolated in our study.
Characterization of promoter library
For promoter library using GFP as reporter, transformants were inoculated into 200 μl of LB media containing spectinomycin (100 μg/ml) in black-wall, clear-bottom 96-well plates (Corning) and cultivated at 37°C, 850 rpm. The relative fluorescence of the ratio of fluorescence intensity at 488/511 to optical density (OD600) was determined at 2-h intervals. Colonies exhibited higher relative fluorescence than reference strain containing P43-GFP were inoculated into shake flasks. The promoter strengths of isolated colonies were determined by a previously published dynamic model (P = fSS [μ (1 + μ/m) + D (2 + μ/m)]) (28,35).
To characterize the α-amylase BLA specific promoter library, transformants produce larger halos on the substrate LB agar plate were inoculated in 96 deep-well plates containing 100 μl of LB medium supplemented with spectinomycin (100 μg/ml). After 4-h cultivation, cell supernatants were harvested by centrifugation, 10 μl of supernatants were transferred to another 96 deep-well plates containing 80 μl of 1% soluble starch in 20 mM phosphate buffer (pH 6.0). After incubation at 70°C for 20 min, the reaction was stopped at 100°C for 5 min, then 150 μl of DNS was added for continuing 5-min incubation at 100°C. The relative amylase activity of each colony was calculated by the ratio of amylase activity (OD540) to its corresponding cell density (OD600). Colonies have higher relative activity than P43 were selected and sequenced. The intracellular GFP concentration of isolated colonies was determined by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE).
Construct designs to regulate protein quality control system
The strain producing the highest amylase activity from the BLA specific promoter library was named 6-102, which contains a genome-integrated P556-TA-BLA expression cassette. To analyze the effects of protein folding and protease degradation on BLA production in 6-102, two chaperone genes groES (Swiss-Prot: P28599) and prsA (Swiss-Prot: P24327) and six well-known extracellular protease genes (bpr, Swiss-Prot: P16397; vpr, Swiss-Prot: P29141; nprB, Swiss-Prot: P39899; epr, Swiss-Prot: P16396; mpr, Swiss-Prot: P39790; wprA, Swiss-Prot: P54423) were selected as targets. Two sgRNAs that targeted to different sites of each target were designed (Supplementary Table S1). The relative amylase activities of strains expressing these sgRNAs were determined and normalized to strain expressing a non-targeting sgRNA after 14-h induction of dCas9-ω. To assess the synergistic effect of multi-dimensional regulation, the effective sgRNAs were assembled into one plasmid by BioBrick method. The specific activity of α-amylase (OD540/OD600) was determined as mentioned above.
Quantitative real-time PCR (qRT-PCR)
Bacillus subtilis cells were harvested at the exponential growth phase for total RNA extraction using E.Z.N.A.® Bacterial RNA Kit (Omega Biotek, Norcross, GA, USA). DNA contamination was eliminated by DNase I treatment, and RNA qualities were checked by formaldehyde denatured agarose gel. After RNA concentration was quantified by absorbance at 260 nm, qRT-PCR was conducted to determine the relative mRNA expression of target genes to internal reference gene rpsj (ribosomal protein S10 coding gene). The value of the quantification cycle (Cq), which is defined as the cycle when the reporter fluorescence is distinguishable from the background in the extension phase of the PCR reaction, was averaged with triplicates.
RESULTS
dCas9-ω/α can activate the expression of heterologous and endogenous genes in B. subtilis
Since the CRISPR activation system in Gram-positive bacteria has not yet been reported, we first constructed the CRISPR activation system in B. subtilis by fusing the ω subunit of RNAP to the C-terminal of dCas9 (Figure 1A). Then B. subtilis strain with a genome-integrated GFP reporter gene driven by the widely used strong promoter P43 was constructed to evaluate the activation efficiency of dCas9-ω. Five sgRNAs were designed to guide the dCas9-ω to sites located at 267, 289, 331, 351 and 415 bp upstream of the transcriptional start site (TSS) of P43-GFP expression cassette, which resulted in the increase of GFP fluorescence ranging 2∼3 folds dependent on the targeting positions (Figure 1B). The activation efficacy on different targets was tested by guiding dCas9-ω to another expression cassette P43-BLA using three sgRNAs mentioned above. However, the relative amylase activity was only improved by 1.5-fold. Because similar transcriptional activation of GFP (2.4- to 3.8- fold, Figure 1B) and BLA (2.2- to 5-fold, Figure 1C) by dCas9-ω was observed, suggesting the varied correlations between the relative fluorescence/activity and transcripts level were mainly caused at post-transcriptional levels, including translation efficiency, protein folding and protease-mediated degradation. Namely, the increased transcripts of GFP and BLA were not equally converted to proteins.
Figure 1.

dCas9-α/ω mediated activation of reporter and endogenous genes in Bacillus subtilis. (A) Schematic description of dCas9-α and dCas9-ω mediated gene expression activation. (B) The activation efficiency of dCas9-ω on P43-GFP expression cassette. The relative fluorescence of GFP was measured after guiding the dCas9-ω to different upstream regions of promoter P43. (C) The activation efficiency of dCas9-ω on P43-BLA expression cassette. (D) The activation effect of dCas9-ω on endogenous gene by guiding dCas9-ω to site 224-bp upstream of amyE. (E) The activation efficiency of dCas9-α on P43-GFP expression cassette. The relative transcription level represents the value of 2−ΔΔCq that was measured by qRT-PCR. The x-axis indicates the distances of target sites to transcription start site. - represents upstream region. C represents control where sgRNA without target sequence was used. White and black bars represent the relative fluorescence intensity and relative transcription respectively.
Furthermore, to determine whether endogenous genes could be regulated by dCas9-ω, a single sgRNA was designed to guide the dCas9-ω to the site 224-bp upstream of the start codon of endogenous amylase gene amyE, a carbohydrate hydrolytic enzyme that was regulated by the cellular regulatory systems. This dCas9-ω mediated regulation resulted in a 3.2- and 4-fold increase in amylase activity and amyE transcripts, respectively (Figure 1D). These results combined together demonstrated that dCas9-ω was capable of activating endogenous gene transcription in the Gram-positive bacterium B. subtilis.
To further investigate whether other subunits of RNAP could also function as transcriptional activator, the α subunit was fused to dCas9, followed by characterization using P43-GFP expression cassette as the reporter system. A similar position-dependent activation pattern was observed between the α and ω subunits (Figure 1E), in which the maximum activation efficiency of 3.3-fold increase in GFP expression was obtained when the dCas9-α was guided to the site 289 bp upstream of TSS. And it turned weaker along with the increase of distance.
dCas9-ω/α can mediate multi-directional transcriptional regulation
The ability to simultaneously activate and repress multiple genes promises to control a full spectrum of expression profiles, which is highly appreciated in biotechnological applications where fine-tuning gene expression is required. Previous studies demonstrated that dCas9 was able to directly silence proximal regulatory elements through steric hindrance (20,36). To investigate whether dCas9-ω/α still maintain the silencing ability, 13 sgRNAs with different targeting sites of P43-GFP cassette were designed to examine the targeting effects on transcriptional regulation (Figure 2). Transcriptional activation was obtained when the dCas9-α/ω was guided to a window of DNA from 250 to 415 bp upstream of TSS. Interestingly, GFP expression was nearly abolished when dCas9-ω bound to the TSS or the 5′ end of template strand, suggesting that the fusion proteins could block transcriptional initiation and elongation efficiently (Figure 2). When the dCas9-α/ω was guided to sites G5-G8 (38–208 bp upstream of TSS), GFP expression was only slightly influenced. It might be speculated that the steric hindrance caused by dCas9 was offset by the activation effects of α/ω subunits. These results suggested that the transcriptional regulation mediated by dCas9-α/ω is highly target-position-dependent, thus they can act as a single master regulator to execute multi-directional transcriptional regulation by designing specific sgRNAs.
Figure 2.

dCas9-α/ω mediated multi-directional transcriptional regulation. The position effects of dCas9-ω (A) and dCas9-α (B) on transcription regulation of P43-GFP. The relative transcription level represents the value of 2−ΔΔCq that was measured by qRT-PCR. TSS means transcription start site of P43-GFP expression cassette. The distances of target sites to the TSS of P43-GFP expression cassette: G1, 88; G2, 78; G3, 42; G4, 24; G5, -28; G6, -77; G7, - 108; G8, -208; G9, -267; G10, -289; G11, -331; G12, -351; G13, -415.
Multivalent recruitment and multiplexed targeting did not further improve the activation efficiency
To investigate the synergistic effect of the recruitment of multiple activators in dCas9-ω/α mediated transcriptional regulation system, we fused the α and ω subunits to dCas9 simultaneously in varied arrays, producing fusion proteins of dCas9-α-ω and dCas9-ω-α. Different from observations that the recruitment of multiple activators was able to efficiently further activate gene transcription in eukaryotic cells (37,38), decreased activation efficiencies were observed for both dCas9-α-ω and dCas9-ω-α, their maximal activation on P43-GFP was lower than 1.8-fold (Figure 3A), compared to 3.3-fold of dCas9-α and 2.7-fold of dCas9-ω, respectively (Figure 2A).
Figure 3.

The effects of multiplexed targeting and multivalent recruitment on transcription activation. (A) The activation efficiency of simultaneous fusion of RNAP α and ω subunits to dCas9. (B) The activation efficiency of guiding dCas9-α/ω to two sites G9 and G13. The error bars indicate the standard deviations of biological triplicates.
In addition, we evaluated the effect of multiplexed targeting on activation efficiency by simultaneously targeting the dCas9-ω to two sites upstream of TSS. Contrast to the add-on effects multiplexed targeting in eukaryotic cells (15), lowered transcription activation was observed (Figure 3B), which might attribute to the different action mechanisms of activators in bacteria and eukaryotic cells, i.e., direct attachment of activators to RNA polymerase in bacteria versus indirect chromatin modification of activation domains in eukaryotic cells (25).
OAPS strategy can generate a high proportion of functional promoter mutants efficiently
The rate-limiting enzymes in metabolic pathways are often transcriptionally controlled by host regulatory networks. Synthetic promoters generated by promoter engineering could deregulate the cellular regulatory machinery and promise to fully sample the host’s transcriptional capacity, which would expand the application scope of dCas9-α/ω mediated transcriptional program in metabolic engineering. Since two characteristic consensus motifs of prokaryotic promoters are necessary to bind RNA polymerase, the existing promoter engineering strategies were normally designed to introduce variations in non-conserved proximal regions in a completely random manner (27,28), which inevitably produce a small fraction of functional mutants. We hypothesized that a high proportion of functional mutants can be generated by utilizing the naturally occurring genetic diversity of native promoters as the driving force for promoter evolution. Thus, we developed a new strategy named OAPS to shuffle the non-conserved promoter regions with the consensus -35 and -10 motifs as overlapping homologous sequence (Figure 4A).
Figure 4.

Construction of promoter libraries via OAPS strategy. (A) Scheme of oligonucleotide-annealing based promoter shuffling (OAPS). Each native promoter was divided into three modules with the well conserved -35 and -10 motifs as joints. The modules were synthesized by oligonucleotide annealing and randomly assembled to produce combinatorial promoters. (B) Promoters stronger than reference promoter P43 in library using GFP as reporter. Four parent promoters Pveg, Pveg2, Pn26 and PlepA were used for shuffling. Bars represent the relative fluorescence intensity. Line represents the promoter strength (PS), which was calculated by a dynamic model (P = fSS [μ (1 + μ/m) + D (2 + μ/m)]). (C) Promoters stronger than P43 for amylase BLA expression in B. subtilis. The bars represent the relative amylase activity of each promoter, line indicates the relative mRNA concentration as determined by qPCR. (D) SDS-PAGE analysis of BLA expression level of isolated promoters. The error bars indicate the standard deviations of biological triplicates.
As a proof-of-concept study, four parent promoters of Pveg, Pveg2, Pn26 and PlepA with typical σA recognized consensus motifs and different strengths were shuffled, which produce a theoretically library size of 64 (4 × 4 × 4) synthetic promoter variants. Approximately 300 transformants were obtained to achieve over 95% probability of mutant coverage for library characterization. The strength of synthetic promoter variants spanning two orders of magnitude was observed (Supplementary Figure S1), among which 11 different promoters stronger than the reference promoter P43 were isolated, with P223 being the strongest promoter to exhibit a 16-fold enhanced fluorescence intensity comparing to that of P43 (Figure 4A). Our results thus suggested that OAPS can generate a high proportion of functional mutants with two orders of magnitude of transcriptional capabilities.
CRISPR-assisted transformation enabled to construct promoter library for arbitrary genes via OAPS
The remaining challenge of constructing promoter library with the target gene is the existence of endogenous isoenzymes in the host, which would interfere library characterization. To solve this problem, we constructed the CRISPR/Cas9 system in B. subtilis to inactivate the endogenous isoenzyme, and found that the double-strand breaks (DSBs) introduced by CRISPR/Cas9 could significantly improve homologous recombination efficiency. Through CRISPR-assisted transformation and OAPS strategy, a promoter library for amylase BLA was directly constructed in B. subtilis. Specifically, the endogenous isoenzyme AmyE was taken as the integration locus of library fragments and DSB was introduced at amyE by CRISPR/Cas9 during transformation.
To further increase sequence diversity of this library, we added two additional parent promoters of PtrnQ and PserA and designed the -10 motif as TAwwAT (w = a or t) (Supplementary Table S3). With a theoretical library size of 864 (6 × 6 × 6 × 4), over 2400 transformants were obtained to achieve 95% probability of mutant coverage. A total of 34 different promoters that are apparently stronger than the reference promoter P43 for BLA expression were obtained (Figure 4C), representing 8.8 % (34/864) of total transformants. Among these isolated promoters, the relative activity of the strongest promoter P556-TA was over 1000-fold higher than that of the weakest promoter P111-TA, and 100-fold of that of promoter P43. Real-time PCR and SDS-PAGE analysis demonstrated that the BLA expression levels were correlated well with its production (Figure 4C and D). In addition, comparative analysis of the isolated strong promoter sets for GFP and BLA suggested that there are obvious context effects on promoter activities. For example, strong promoters like P222 and P313 for BLA expression are absent from the set of GFP, while P223 and P333 are the opposite.
Rules for optimal CRISPRa and CRISPRi sgRNA design of dCas9-ω
To better understand the functions of dCas9-ω mediated multi-directional regulator, the strongest promoter P556-TA obtained from the BLA specific promoter library was used, and 13 sgRNAs tiling the promoter region of P556-TA-BLA cassette were designed. Similar to the transcriptional regulation pattern obtained on P43-GFP, dCas9-ω was able to activate and repress the expression of P566-AT-BLA in a position-dependent manner (Figure 5). When dCas9-ω bound to the transcription initiation site (B3) or 5′ end of coding region (B2), ∼90% of amylase activity loss was observed. More importantly, the BLA expression could be slightly enhanced by 1.2- to 1.5-fold when dCas9-ω was guided to a window of DNA from 250 to 350 bp upstream of TSS, a close activation window of 250 to 415 bp upstream of TSS was observed for P43-GFP. The highly similar distance windows of dCas9-ω mediated transcription activation and repression for both P43-GFP and P566-AT-BLA suggested that they could serve as a guidance for optimal CRISPRa and CRISPRi sgRNAs design.
Figure 5.
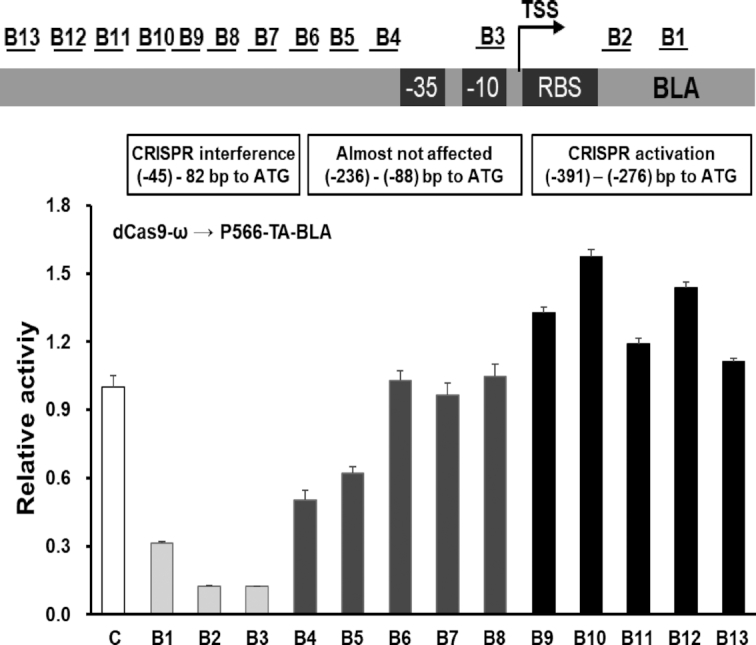
The position effects of dCas9-ω on transcription regulation of P556-TA-BLA. C represents control where sgRNA without target sequence was used. ATG means the translation initiation site of BLA. The distance of target site to TSS of P556-TA-BLA: B1, 102; B2, 32; B3, 9; B4, -68; B5, -90; B6, -132; B7, -152; B8, -216; B9, -256; B10, -283; B11, -303; B12, -348; B13, -371. - represents upstream region. The error bars indicate the standard deviations of biological triplicate.
Redirection of the intricate protein quality control system to fine-tune gene expression
Protein quality control system plays a vital role in maintaining the cellular protein homeostasis (39), which regulates gene expression at multi-dimensional levels, including transcription, translation and protein degradation. As dCas9-ω has proved its efficacy to execute different modes of transcriptional regulation, we hypothesized that dCas9-ω can be utilized to redirect the protein quality control system for fine-tuning gene expression. Using B. subtilis strain 6-102 containing P566-AT-BLA cassette as a case study, we first interrogated the effects of molecular chaperone-assisted protein folding and protease-caused degradation on BLA production by individually activating the expression of molecular chaperones and repressing extracellular proteases. We found that protein folding has an obvious influence on BLA production in strain 6-102, guiding dCas9-ω to site 254-bp upstream of the initial codon of extracellular chaperon PrsA resulted in a 1.7-fold increase in BLA expression (Figure 6A). However, improving the intracellular chaperon did not affect BLA production, which was consistent with the secretory expression pattern of BLA in this study (Figure 6A).
Figure 6.

Improve BLA expression in B. subtilis 6-102 by simultaneously activating molecular chaperone expression and repressing proteases expression by dCas9-ω. (A) Effects of activation of individual molecular chaperone and repression of individual protease on BLA production. GroES (intracellular molecular chaperone, Swiss-Prot: P28599), PrsA (extracellular molecular chaperone, Swiss-Prot: P24327) and six well-known extracellular protease genes (Bpr, bacillopeptidase F, Swiss-Prot: P16397; Vpr, extracellular serine protease, Swiss-Prot: P29141; NprB, extracellular neutral protease B, Swiss-Prot: P39899; Epr, extracellular serine protease, Swiss-Prot: P16396; WprA, cell wall-associated protein precursor, Swiss-Prot: P54423) were selected. (B) The relative expression of prsA, bpr, vpr and nprB measured by qPCR after expressing the dCas9-ω and sgRNAs. The strain 6-102/pHT01-dCas9-ω expressing sgRNA without target sequence was served as the control (C). (C) The synergistic effects of effective molecular chaperone and proteases on BLA production. The error bars indicate the standard deviations of biological triplicates.
As two extracellular proteases NprE and AprE have already been inactivated in SCK6, the remaining six extracellular proteases NprB (neutral protease B), Epr (serine protease), Mpr (metalloprotease), Bpr (bacillopeptidase F), WprA (cell wall-associated protein precursor) and Vpr (serine protease) were transcriptionally repressed by guiding dCas9-ω to the RBS and 5′ coding region, respectively. The results showed that the inhibition of protease-encoding genes of nprB (with sgRNA targeting to 20-bp downstream of initial codon), bpr (with sgRNA targeting to 210-bp downstream of initial codon) and vpr (with sgRNA targeting to 10-bp upstream of initial codon) improved the BLA yield by 110%, 45% and 60%, respectively (Figure 6A), indicating that they were the primary attributors for BLA degradation in the medium. In contrast, proteases Epr, Mpr and WprA had no or marginal proteolytical activities on BLA (Figure 6A). qRT-PCR showed that the concentration of prsA transcripts was increased by 1.26-fold while proteases vpr, bpr and nprB transcripts were significantly repressed with a 95%, 78% and 98% reduction respectively, after the dCas9-ω targeting (Figure 6B).
To test the efficacy of dCas9-ω to modulate the protein quality control system multi-dimensionally to further improve BLA production, the effective components including chaperone PrsA and proteases of NprB, Bpr and Vpr were simultaneously targeted by dCas9-ω in various combinations. The results showed that simultaneous repression of Bpr and Vpr could significantly improve BLA production, indicating a synergistic effect between Bpr and Vpr on BLA production. Upon which, simultaneously improving the expression of molecular chaperone PrsA produced a 2.6-fold increase in BLA production compared to the strain 6-102 (Figure 6C). Thus, through combinational OAPS and dCas9-ω strategies, the expression of amylase BLA were improved by 260-fold compared to the commonly used strong promoter P43 in strain SCK6.
DISCUSSION
The ability to fine-tune gene expression exhibits inspiring potentials in many biotechnological applications such as genetic circuit design, flux balance and expression system optimization. Here, we demonstrated that dCas9-ω/α could execute different modes of transcriptional regulations in a target-position-dependent manner in B. subtilis, which enabled the semi-quantitative characterization of the effect of each component on target gene expression in a high-throughput manner. For example, previous studies showed that protein folding and protease degradation could significantly affect BLA production in B. subtilis (40,41). In this study, we identified that extracellular chaperone PrsA and proteases NprB, Vpr and Bpr are effective components on amylase BLA production in B. subtilis 6-102. Moreover, their synergistic effects on BLA production were evaluated by guiding the dCas9-ω to these effective components with various combinations. We observed that they did not always produce add-on effects, indicating that interactions between these components would exist.
As dCas9-ω/α can act as a single master regulator to modify target gene expression in different directions, simultaneous activation and repression of multiple genes expression can be precisely and temporally controlled by controlling the timing of dCas9-ω expression. Thus, dCas9-ω/α mediated multi-directional transcriptional program has several advantages over other transcriptional programs. First, similar to dCas9, dCas9-ω/α remained the ability to silence proximal regulatory elements by steric hindrance in bacteria. It can act as a single master regulator for both transcription activation and repression, which brings minimal metabolic burdens on host cell growth compared to programs using multiple CRISPR-associated proteins and effectors. Second, because the regulation mode of dCas9-ω/α functions in a target-position-dependent manner, simultaneous activation and repression of multi-locus can be achieved by designing sgRNAs that targeted to certain positions, thus circumventing the complicated sgRNA engineering and the decreased regulation efficiency due to the lower binding affinity between Cas9 and engineered sgRNAs (12). Finally, the multi-directional regulation can be precisely controlled spatiotemporally by controlling the timing of dCas9-ω/α expression, without requiring purposely designed promoters to express different effectors. Theoretically, the dCas9-ω based transcriptional programming could play roles in any situations requiring simultaneous activation and repression of multiple genes, like deciphering cell fate decision (42), probing cellular responses to environmental stress (43) and rewiring the complex cellular metabolism for biotechnological applications (19,44,45).
In contrast to the observations that the recruitment of multiple activators could greatly enhance activation efficiency in eukaryotic cells (15), the multivalent recruitment of α and ω subunits to dCas9 results in the decreased activation efficiency. There are two possible explanations for this contradiction: one is interactions between ω and α may impaired their ability to recruit other subunits of RNAP, and the other is the physical connection of ω and α subunits by a linker may destroyed the proper assembly of functional RNA polymerase holoenzyme, suggesting the requirement of orthogonal transcriptional activators for greater activation efficiency. Actually, effective transcriptional activators stronger than ω subunit of RNAP have been identified in E. coli (25), while the versatilities of these activators on different contexts and the effectiveness of their homologs in B. subtilis still need to be evaluated.
The overall metabolic fluxes of biosynthesis pathway were typically limited by one or two steps, and enzymes catalyzing these steps were often transcriptionally regulated by host feedback control system in B. subtilis (26). Thus, it required not only the coordinate activation of enzymes in the biosynthetic pathway while repression of enzymes in the competing pathways, but also the deregulation of the feedback inhibition on rate-limiting enzymes to maximize product productivities while maintaining host fitness. Synthetic promoters deregulate the control of host transcriptional regulatory networks, and can provide a wide range of transcriptional activities, thus representing a good complement of dCas9-ω/α to drive the expression of rate-limiting enzymes and to relieve the feedback inhibition at transcriptional levels.
In order to develop strong synthetic promoters in B. subtilis for biotechnological applications, we initially attempted to construct a promoter library via the strategy of randomization of non-conserved promoter sequences (46). Despite we improved the transformation efficiency of B. subtilis competent cells by two orders of magnitude (47), unfortunately, we still failed to construct the promoter library with enough transformants to cover the theoretical library size. Actually, there has no report as we know of constructing a promoter library directly in B. subtilis until now. To our limited knowledge, there are only two studies about promoter engineering in B. subtilis (8,48). In both studies, the promoter libraries were first transformed into E. coli, and mixed plasmids were extracted and re-transformed into B. subtilis, which would inevitably introduce sequence bias introduced by host restriction-modification systems (49). In addition, it was disappointing to find that most isolated promoter mutants have low transcriptional activities. For example, Liu et al. isolated 214 functional promoter variants from the library generated by randomizing the non-conserved promoter sequences, but all these isolated promoters are weaker than the reference promoter P43 (48). The other study (8) constructed several promoter libraries by saturated mutagenesis at several nucleotides in the promoters, while most promoter variants have significantly decreased transcriptional activities.
Inspired by DNA shuffling for protein evolution, the OAPS strategy was developed in this work to exploit the naturally occurring genetic diversity of native promoters as the driving force for promoter engineering. One of the most important features of OAPS is the ability to generate a high proportion of functional mutants, meaning that a wide range of promoter activities could be obtained within a small library size. This makes OAPS suitable to construct promoter libraries with proteins lacking high-throughput screening methods and in industrially important strains with low transformation efficiency. One possible limitation of OAPS is the requirement of consensus motifs for promoter shuffling, while the characteristic -35 and -10 motifs are not intact even in promoters recognized by same σ factor (Supplementary Table S3). To analyze whether more potent promoter variants can be created by introducing variations in the consensus motifs of native promoters, we mutated the relatively conserved sites in -10 motif of six native promoters. As shown in Supplementary Figure S3, stronger mutants were observed for promoters PserA, Pveg2 and PlepA. Therefore, promoters with different motifs can be shuffled by introducing degenerate bases at the relative conserved site(s).
In summary, dCas9-ω was proved to act as a single regulator to simultaneously activate and repress multiple genes expression by simply designing location-specific sgRNAs, which provided powerful tools to regulate gene expression at multi-dimensional levels. In addition, a novel promoter engineering strategy OAPS with the ability of generating a high proportion of functional mutants was developed, which provided a good complement of dCas9-ω to improve the expression of rate-limiting enzymes and deregulate the feedback inhibition in pathway optimization. Combining dCas9-ω mediated multi-directional transcriptional program and OAPS, a 260-fold increase in the production of amylase BLA in B. subtilis was achieved. Theoretically, the strategies developed in this study can be applicable in other bacteria.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31670069]; Technical Innovation Special Fund of Hubei Province [2017ACA171]; 2016 Wuhan Yellow Crane Talents (Science) Program; Distinguished Young Scholars of Hubei Province [2018CFA042]; State Key Laboratory of Biocatalysis and Enzyme Engineering. Funding for open access charge: National Natural Science Foundation of China [31670069].
Conflict of interest statement. None declared.
REFERENCES
- 1. Khalil A.S., Collins J.J.. Synthetic biology: applications come of age. Nat. Rev. Genet. 2010; 11:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keasling J.D. Synthetic biology and the development of tools for metabolic engineering. Metab. Eng. 2012; 14:189–195. [DOI] [PubMed] [Google Scholar]
- 3. Brophy J.A.N., Voigt C.A.. Principles of genetic circuit design. Nat. Methods. 2014; 11:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jarboe L.R., Zhang X., Wang X., Moore J.C., Shanmugam K.T., Ingram L.O.. Metabolic engineering for production of biorenewable fuels and chemicals: contributions of synthetic biology. J. Biomed. Biotechnol. 2010; 2010:761042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blazeck J., Alper H.S.. Promoter engineering: recent advances in controlling transcription at the most fundamental level. Biotechnol. J. 2013; 8:46–58. [DOI] [PubMed] [Google Scholar]
- 6. Salis H.M., Mirsky E.A., Voigt C.A.. Automated design of synthetic ribosome binding sites to precisely control protein expression. Nat. Biotechnol. 2009; 27:946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kosuri S., Goodman D.B., Cambray G., Mutalik V.K., Gao Y., Arkin A.P., Endy D., Church G.M.. Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:14024–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guiziou S., Sauveplane V., Chang H.-J., Clerté C., Declerck N., Jules M., Bonnet J.. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 2016; 44:7495–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mayer M.P., Bukau B.. Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005; 62:670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hendrick J.P., Hartl F.-U.. Molecular chaperone functions of heat-shock proteins. Ann. Rev. Biochem. 1993; 62:349–384. [DOI] [PubMed] [Google Scholar]
- 11. Wu S.-C., Yeung J.C., Duan Y., Ye R., Szarka S.J., Habibi H.R., Wong S.-L.. Functional production and characterization of a fibrin-specific single-chain antibody fragment from Bacillus subtilis: Effects of molecular chaperones and a wall-bound protease on antibody fragment production. Appl. Environ. Microbiol. 2002; 68:3261–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lian J., HamediRad M., Hu S., Zhao H.. Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat. Commun. 2017; 8:1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsu Patrick D., Lander Eric S., Zhang F.. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014; 157:1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shao Y., Lu N., Wu Z., Cai C., Wang S., Zhang L.-L., Zhou F., Xiao S., Liu L., Zeng X. et al.. Creating a functional single-chromosome yeast. Nature. 2018; 560:331–335. [DOI] [PubMed] [Google Scholar]
- 15. Dominguez A.A., Lim W.A., Qi L.S.. Beyond editing: repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2015; 17:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. et al.. CRISPR-mediated modular rna-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H., La Russa M., Qi L.S.. CRISPR/Cas9 in genome editing and beyond. Ann. Rev. Biochem. 2016; 85:227–264. [DOI] [PubMed] [Google Scholar]
- 18. Shimatani Z., Kashojiya S., Takayama M., Terada R., Arazoe T., Ishii H., Teramura H., Yamamoto T., Komatsu H., Miura K. et al.. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat. Biotechnol. 2017; 35:441–451. [DOI] [PubMed] [Google Scholar]
- 19. Deaner M., Alper H.S.. Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae. Metab. Eng. 2017; 40:14–22. [DOI] [PubMed] [Google Scholar]
- 20. Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L.A.. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013; 41:7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peters J.M., Colavin A., Shi H., Czarny T.L., Larson M.H., Wong S., Hawkins J.S., Lu C.H.S., Koo B.-M., Marta E. et al.. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell. 2016; 165:1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zalatan J.G., Lee M.E., Almeida R., Gilbert L.A., Whitehead E.H., La Russa M., Tsai J.C., Weissman J.S., Dueber J.E., Qi L.S.. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015; 160:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dahlman J.E., Abudayyeh O.O., Joung J., Gootenberg J.S., Zhang F., Konermann S.. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015; 33:1159–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kiani S., Chavez A., Tuttle M., Hall R.N., Chari R., Ter-Ovanesyan D., Qian J., Pruitt B.W., Beal J., Vora S. et al.. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods. 2015; 12:1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong C., Fontana J., Patel A., Carothers J.M., Zalatan J.G.. Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat. Commun. 2018; 9:2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sierro N., Makita Y., de Hoon M., Nakai K.. DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res. 2008; 36:93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jensen P.R., Hammer K.. The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl. Environ. Microbiol. 1998; 64:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alper H., Fischer C., Nevoigt E., Stephanopoulos G.. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:12678–12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong L., Engel J., Jin E., Holdridge B., Xu P.. YaliBricks, a versatile genetic toolkit for streamlined and rapid pathway engineering in Yarrowia lipolytica. Metab. Eng. Commun. 2017; 5:68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nie L., Wu G., Zhang W.. Correlation of mRNA expression and protein abundance affected by multiple sequence features related to translational efficiency in Desulfovibrio vulgaris: a quantitative analysis. Genetics. 2006; 174:2229–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu C., Jeffries T.. Shuffling of promoters for multiple genes to optimize xylose fermentation in an engineered Saccharomyces cerevisiae strain. Appl. Environ. Microbiol. 2007; 73:6072–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rud I., Jensen P.R., Naterstad K., Axelsson L.. A synthetic promoter library for constitutive gene expression in Lactobacillus plantarum. Microbiology. 2006; 152:1011–1019. [DOI] [PubMed] [Google Scholar]
- 33. Liu Y., Li J., Du G., Chen J., Liu L.. Metabolic engineering of Bacillus subtilis fueled by systems biology: recent advances and future directions. Biotechnol. Adv. 2017; 35:20–30. [DOI] [PubMed] [Google Scholar]
- 34. Zhang X.Z., Zhang Y.. Simple, fast and high-efficiency transformation system for directed evolution of cellulase in Bacillus subtilis. Microb. Biotechnol. 2011; 4:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leveau J.H.J., Lindow S.E.. Predictive and interpretive simulation of green fluorescent protein expression in reporter bacteria. J. Bacteriol. 2001; 183:6752–6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. La Russa M.F., Qi L.S.. The new state of the art: CRISPR for gene activation and repression. Mol. Cell. Biol. 2015; 35:3800–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanenbaum M.E., Gilbert L.A., Qi L.S., Weissman J.S., Vale R.D.. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014; 159:635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W., Rangarajan S., Shivalila C.S., Dadon D.B., Jaenisch R.. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013; 23:1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen B., Retzlaff M., Roos T., Frydman J.. Cellular strategies of protein quality control. Cold Spring Harbor Perspect. Biol. 2011; 3:a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu Y.-H., Lu F.-P., Li Y., Yin X.-B., Wang Y., Gao C.. Characterisation of mutagenised acid-resistant alpha-amylase expressed in Bacillus subtilis WB600. Appl. Microbiol. Biotechnol. 2008; 78:85–94. [DOI] [PubMed] [Google Scholar]
- 41. Bolhuis A., Tjalsma H., Smith H.E., de Jong A., Meima R., Venema G., Bron S., van Dijl J.M.. Evaluation of bottlenecks in the late stages of protein secretion in Bacillus subtilis. Appl. Environ. Microbiol. 1999; 65:2934–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Norman T.M., Lord N.D., Paulsson J., Losick R.. Memory and modularity in cell-fate decision making. Nature. 2013; 503:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamaguchi-Shinozaki K., Shinozaki K.. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Ann. Rev. Plant Biol. 2006; 57:781–803. [DOI] [PubMed] [Google Scholar]
- 44. Liu Y., Zhu Y., Li J., Shin H.D., Chen R.R., Du G., Liu L., Chen J.. Modular pathway engineering of Bacillus subtilis for improved N-acetylglucosamine production. Metab. Eng. 2014; 23:42–52. [DOI] [PubMed] [Google Scholar]
- 45. Maeder M.L., Linder S.J., Cascio V.M., Fu Y., Ho Q.H., Joung J.K.. CRISPR RNA–guided activation of endogenous human genes. Nat. Methods. 2013; 10:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hammer K., Mijakovic I., Jensen P.R.. Synthetic promoter libraries-tuning of gene expression. Trends Biotechnol. 2006; 24:53–55. [DOI] [PubMed] [Google Scholar]
- 47. Li X., Lu Z., Zhou Y., Li S., Zhang G.. Preparation and transformation optimization for supercompetent B. subtilis SCK6 cells. Chinese J. Biotechnol. 2017; 33:692–698. [DOI] [PubMed] [Google Scholar]
- 48. Liu D., Mao Z., Guo J., Wei L., Ma H., Tang Y., Chen T., Wang Z., Zhao X.. Construction, model-based analysis, and characterization of a promoter library for fine-tuned gene expression in Bacillus subtilis. ACS Synth. Biol. 2018; 7:1785–1797. [DOI] [PubMed] [Google Scholar]
- 49. Kusano K., Naito T., Handa N., Kobayashi I.. Restriction-modification systems as genomic parasites in competition for specific sequences. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:11095–11099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.