

Abstract
Background and Purpose
A new class of dihydropyridine derivatives, which act as co‐inducers of heat shock protein but are devoid of calcium channel antagonist and vasodilator effects, has recently been developed with the purpose of selectively targeting neurodegeneration. Here, we evaluated the action of one of these novel compounds LA1011 on neurovascular coupling in the ischaemic rat cerebral cortex. As a reference, we applied nimodipine, a vasodilator dihydropyridine and well‐known calcium channel antagonist.
Experimental Approach
Rats were treated with LA1011 or nimodipine, either by chronic, systemic (LA1011), or acute, local administration (LA1011 and nimodipine). In the latter treatment group, global forebrain ischaemia was induced in half of the animals by bilateral common carotid artery occlusion under isoflurane anaesthesia. Functional hyperaemia in the somatosensory cortex was created by mechanical stimulation of the contralateral whisker pad under α‐chloralose anaesthesia. Spreading depolarization (SD) events were elicited subsequently by 1 M KCl. Local field potential and cerebral blood flow (CBF) in the parietal somatosensory cortex were monitored by electrophysiology and laser Doppler flowmetry.
Key Results
LA1011 did not alter CBF, but intensified SD, presumably indicating the co‐induction of heat shock proteins, and, perhaps an anti‐inflammatory effect. Nimodipine attenuated evoked potentials and SD. In addition to the elevation of baseline CBF, nimodipine augmented hyperaemia in response to both somatosensory stimulation and SD, particularly under ischaemia.
Conclusions and implications
In contrast to the CBF improvement achieved with nimodipine, LA1011 seems not to have discernible cerebrovascular effects but may up‐regulate the stress response.
Abbreviations
- 2VO
permanent, bilateral occlusion of the common carotid arteries (“two‐vessel occlusion”)
- CBF
cerebral blood flow
- DC
direct current
- EFP
evoked field potential
- eNOS
endothelial NOS
- Hsp
heat shock protein
- LDF
laser Doppler flowmetry
- LFP
local field potential
- MABP
mean arterial BP
- rSD
recurrent spreading depolarization
- SD
spreading depolarization
- SD1
the first spreading depolarization in a train of events
What is already known
LA1011, a dihydropyridine derivative and co‐inducer of heat shock protein, is neuroprotective in models of chronic neurodegeneration.
Nimodipine, a dihydropyridine derivative and L‐type voltage‐gated Ca2+ channel antagonist, decreases resting vascular tone.
What this study adds
LA1011 has no major effect on the cerebrovascular system but intensifies spreading depolarization, possibly by modulating potassium currents.
Nimodipine effectively improves neurovascular coupling, particularly under cerebral ischaemia.
What is the clinical significance
LA1011 may be considered to counteract neurodegeneration, with no concomitant impact on vascular tone.
1. INTRODUCTION
Treatment with the dihydropyridine compound nimodipine (isopropyl 2‐methoxyethyl 1,4‐dihydro‐2,6‐dimethyl‐4‐(3‐nitrophenyl)‐3,5‐pyridinedicarboxylate) has long been recommended in clinical practice to alleviate neurological symptoms of acute or chronic cerebrovascular diseases, such as subarachnoid haemorrhage or vascular dementia (Tomassoni, Lanari, Silvestrelli, Traini, & Amenta, 2008). Nimodipine has proven to be particularly relevant for the treatment of cerebrovascular disorders, because it achieves vasorelaxation by inhibiting calcium influx to vascular smooth muscle cells through L‐type voltage gated calcium channels, and, in addition, it also exerts neuroprotection by preventing neuronal calcium overload (Scriabine, Schuurman, & Traber, 1989). Although nimodipine was proposed to increase cerebral blood flow (CBF) by its selective affinity to cerebral arteries without altering systemic blood pressure (Freedman & Waters, 1987), the approved dose can still initiate undesired side effects (Kieninger et al., 2017) including hypotension in some patients (Diringer & Zazulia, 2017; Porchet, Chioléro, & de Tribolet, 1995).
Recently, a new class of dihydropyridine derivatives devoid of vasodilator and anti‐hypertensive action has been developed with the purpose of selectively targeting neurodegeneration (Fülöp et al., 2013; Kasza et al., 2016). One of these promising novel compounds is LA1011 (dimethyl 4‐(4‐trifluoromethylphenyl)‐2,6‐bis‐(2‐dimethylaminoethyl)‐1‐methyl‐1,4‐dihydropyridine‐3,5‐dicarboxylate dihydrochloride), which is known as a co‐inducer of heat shock protein (Hsp; especially Hsp27 and Hsp70) with no affinity for calcium channels (Kasza et al., 2016). Long‐term administration of LA1011 was shown to improve learning ability, counteract neuronal loss, increase dendritic spine density, and restrain tau pathology and amyloid plaque formation in a transgenic mouse model of Alzheimer's disease (Kasza et al., 2016), possibly by maintaining protein homeostasis (Penke et al., 2018). Taken that Hsp27 and Hsp70 activation has been suggested to be protective against ischaemic neurodegeneration (Kim, Han, Lee, & Yenari, 2018; van der Weerd et al., 2010; Yenari et al., 2005), it is of interest whether LA1011, a co‐inducer of Hsp, has the potential to preserve neuronal function under cerebral ischaemia. Hence, here, we set out to assess the impact of LA1011 treatment on neuronal activation in the acutely ischaemic rat cerebral cortex.
Cerebral ischaemia impairs neuronal function in part by compromising neurovascular coupling (Jackman & Iadecola, 2015). Neurovascular coupling is a vital feed‐forward control mechanism, which adjusts local CBF to the energy requirements of activated neurons (Attwell et al., 2010; Roy & Sherrington, 1890). Inefficient neurovascular coupling therefore deprives neurons of their essential nutrient supply and leads to failing neuronal function. Even though the cerebrovascular action of nimodipine has been studied in much detail, there is only indirect and ambiguous reference to its impact on neurovascular coupling (Suzuki, Ooi, & Seki, 2012). More importantly, it is still not known whether LA1011 may achieve neuroprotection by improving neurovascular coupling. The second goal of this study was, therefore, to assess the potentially beneficial action of LA1011 and nimodipine on the efficacy of neurovascular coupling during ischaemia and in the intact rat cerebral cortex.
As a final point, ischaemic brain injury gives rise to the spontaneous recurrence of spreading depolarization (SD), a wave of mass depolarization of neurons that propagates across the cerebral grey matter at a rate of 2–6 mm·min−1 (Hossmann, 1996; Leão, 1944; Nedergaard, 1996; Somjen, 2001). SD elicited experimentally in the intact cerebral cortex is known as an ischaemic preconditioning stimulus (Shen et al., 2016). Furthermore, SDs spontaneously recurring in the injured brain to exacerbate ischaemic brain injury and contribute to the expansion of the primary lesion by affecting the associated CBF response (Dreier, 2011; Hartings et al., 2017; Woitzik et al., 2013). Limiting SD occurrence in patients has thus emerged as a therapeutic approach to constrain ischaemia‐related, delayed neurological deficit (Sakowitz et al., 2009). Furthermore, nimodipine has been demonstrated to moderately impede SD (Richter, Ebersberger, & Schaible, 2002) and to convert the harmful vasoconstrictive CBF response to SD into hyperaemia in an experimental model of subarachnoid haemorrhage (Dreier et al., 2002). In anticipation of identifying favourable pharmacological effects, we investigated here the impact of LA1011 on SD occurrence and the development of the coupled CBF response, with nimodipine used as a reference compound.
2. METHODS
2.1. Surgical procedures
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. The experimental procedures were approved by the National Food Chain Safety and Animal Health Directorate of Csongrád County, Hungary. The procedures were performed according to the guidelines of the Scientific Committee of Animal Experimentation of the Hungarian Academy of Sciences (updated Law and Regulations on Animal Protection: 40/2013. [II. 14.] Gov. of Hungary), following the EU Directive 2010/63/EU on the protection of animals used for scientific purposes.
Young adult, male Sprague–Dawley rats (Charles River Laboratories, 323 ± 36 g, n = 62) were used in this study. Standard rodent chow and tap water were supplied ad libitum. The animals were housed under constant temperature, humidity, and lighting conditions (23°C, 12:12 hr light/dark cycle, lights on at 7 a.m.).
On the day of the experiments, animals were anaesthetized with 1.5–2% isoflurane in N2O:O2 (3:2) and were allowed to breathe spontaneously through a head cone during surgical interventions. Two series of experiments were designed on the basis of pharmacological treatment strategy (Figure 1; Table 1). In Series 1, isoflurane anaesthesia was maintained throughout the experimental protocol. In Series 2, isoflurane was substituted with α‐chloralose (dissolved in saline at a concentration of 7 mg·ml−1 and administered either as a bolus injection of 40 mg·kg−1 per 15 min for initiation and 40 mg·kg−1·hr−1 for maintenance, i.v., or as a bolus injection of 50 mg·kg−1 for initiation and 30 mg·kg−1·hr−1 for maintenance, i.p.) for the period of actual data acquisition. Body temperature was kept at 37.2°C by a feedback controlled homeothermic blanket (Harvard Apparatus, Holliston, MA, USA). In order to avoid the production of airway mucus, atropine was administered (0.1%, 0.05 ml; i.m.) shortly before surgical procedures. The mean arterial blood pressure (MABP) was continuously monitored via a left femoral artery catheter. The adjacent femoral vein was also cannulated for the administration of the anaesthetic agent, whenever relevant.
Figure 1.
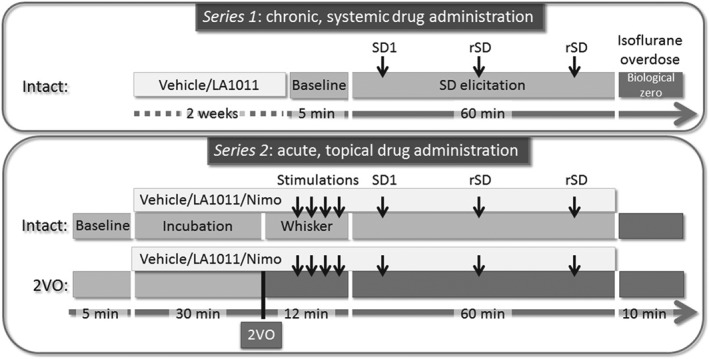
Time lines of the protocols implemented. 2VO: permanent, bilateral occlusion of the common carotid arteries (“two‐vessel occlusion”); Nimo: nimodipine; rSD: recurrent spreading depolarization; SD: spreading depolarization; SD1: first spreading depolarization in a train of events
Table 1.
Physiological variables and number of animals included in each experimental group
| Series | Pharmacological treatment | Ischaemia induction | n | Body weight (g) | Arterial blood pH | Arterial pO2 (mmHg) | Arterial pCO2 (mmHg) | MABP prior to drug/vehicle administration (mmHg) | MABP after drug/vehicle administration (mmHg) |
|---|---|---|---|---|---|---|---|---|---|
| Series 1 | Vehicle | Intact | 6 | 342 ± 14 | 7.43 ± 0.04 | 123.7 ± 6.4 | 37.4 ± 12.0 | N/A | N/A |
| LA1011 | 6 | 336 ± 23 | 7.40 ± 0.04 | 100.6 ± 18.3 | 36.5 ± 7.8 | N/A | N/A | ||
| Series 2 | Vehicle | Intact | 9 | 335 ± 32 | 7.33 ± 0.07 | 140.5 ± 29.6 | 47.4 ± 11.4 | 92 ± 14 | 100 ± 22 |
| 2VO | 8 | 326 ± 48 | 7.33 ± 0.03 | 127.0 ± 32.8 | 39.9 ± 6.3 | 100 ± 1 | 102 ± 8 | ||
| LA1011 | Intact | 6 | 310 ± 40 | 7.28 ± 0.02 | 127.8 ± 30.9 | 47.8 ± 5.7 | 102 ± 10 | 102 ± 12 | |
| 2VO | 9 | 305 ± 45 | 7.35 ± 0.05 | 117.8 ± 34.8 | 38.9 ± 8.6 | 93 ± 11 | 100 ± 22 | ||
| Nimodipine | Intact | 6 | 331 ± 31 | 7.39 ± 0.06 | 103.4 ± 16.1 | 44.8 ± 8.1 | 103 ± 18 | 106 ± 15 | |
| 2VO | 6 | 313 ± 25 | 7.30 ± 0.05 | 99.8 ± 4.4 | 56.2 ± 11.2 | 89 ± 12 | 92 ± 15 |
Note: Data are given as mean ± stdev. 2VO, permanent, bilateral occlusion of the common carotid arteries (“two‐vessel occlusion”); MABP, mean arterial blood pressure.
For the later initiation of incomplete global forebrain ischaemia, a midline incision was made in the neck, and both common carotid arteries were carefully separated from the vagal nerves. Lidocaine (1%) was administered topically before opening the skin and each tissue layer underneath. A surgical thread used as occluder was looped around each artery for later occlusion of the arteries. Control rats (i.e., those with intact cerebral perfusion) were either sham operated or naïve. All rats were placed prone in a stereotaxic apparatus (Stoelting Co., Wood Dale, IL, USA). The skin above the skull and temporal muscle were retracted from the underlying right parietal and temporal bones. Two cranial windows (~3 × 3 mm) 1 mm apart were prepared over the right parietal cortex (3 mm caudal from bregma and 5 mm lateral from the sagittal suture). A high‐precision electrical drill (Technobox, Bien Air 810; Switzerland) was used to thin the bone under saline cooling, and the remaining soft bone layer was gently peeled away to reveal the dura surface. The dura was then carefully opened in each craniotomy. The rostral window was later used for data acquisition (i.e., electrophysiology and CBF measurement) and topical drug administration, while the caudal window served SD elicitation. The cranial windows were constantly kept moist by artificial cerebrospinal fluid (aCSF; mM concentrations: 126.6 NaCl, 3 KCl, 1.5 CaCl2, 1.2 MgCl2, 24.5 NaHCO3, 6.7 urea, 3.7 glucose bubbled with 95% O2 and 5% CO2 to achieve a constant pH of 7.4). Experiments were terminated at the end of the experimental protocol by an overdose of the anaesthetic agent.
2.2. Recording of electrophysiological variables
A saline‐filled glass capillary electrode (20 μm outer tip diameter) was inserted 700 μm deep into the cerebral cortex at the rostral window for the synchronous recording of local field potential (LFP) and slow cortical or direct current (DC) potential. An Ag/AgCl reference electrode was implanted under the skin of the animal's neck. The electrophysiological signals were recorded via a high input impedance pre‐amplifier (NL102GH, NeuroLog System, Digitimer Ltd., UK), connected to a differential amplifier (NL106, NeuroLog System, Digitimer Ltd.) with associated filter and conditioner systems (NL125, NL530, NeuroLog System, Digitimer Ltd.). Potential line frequency noise (50 Hz) was removed by a high‐quality noise eliminator (HumBug, Quest Scientific Instruments Inc., Canada) without any signal attenuation. Analogue to digital conversion was performed by a dedicated A/D converter card (NI USB‐6008/6009, National Instruments, Austin, TX, USA) controlled through a custom‐made software, written in LabView (National Instruments; RRID:SCR_014325).
2.3. Monitoring of local CBF
Laser Doppler flowmetry (LDF) was used to record changes in local CBF in response to somatosensory (whisker) stimulation and SD. A laser Doppler needle probe (Probe 403 connected to PeriFlux 5000; Perimed AB, Sweden) was positioned above the barrel cortex, which is supplied by arterial blood arriving via penetrating arterioles branching off the pial arterioles carrying blood from the middle cerebral artery (Wu et al., 2016). The ideal position of the LDF probe was identified prior to the actual experimental protocol: The probe was positioned over the region where the amplitude of functional hyperaemia in response to whisker stimulation proved to be the greatest. The LDF signal was digitalized and acquired, together with the DC potential and LFP, essentially as described above.
2.4. Pharmacological treatment
Animals were assigned to pharmacological treatments randomly. In Series 1, LA1011 (property of Gedeon Richter Plc., kindly provided at the time of experimentation by LipidArt Ltd., Szeged, Hungary; 1 mg·kg−1 body weight in 0.4 ml saline) or its vehicle (i.e., saline) was administered systemically (i.p.) in equal volume, twice a day for 2 weeks before surgical intervention. In Series 2, the rostral cranial window was incubated with either LA1011 (100 μM, in aCSF) or nimodipine (Sigma‐Aldrich, 100 μM in 0.1% DMSO in aCSF) following a 5 min baseline recording. Drug solutions were refreshed every 10 min to maintain efficacy until the termination of each experiment. In other rats, rinsing the cranial window with vehicle served as a control for pharmacological treatments.
2.5. Experimental protocol
In Series 1, three SD events were triggered in each animal (n = 12) by placing a 1 M KCl‐soaked cotton ball in the caudal craniotomy. The cotton ball was removed, and the craniotomy was rinsed with aCSF after each successful SD elicitation, to allow the development of a single SD in response to each trigger. SDs were provoked at an inter‐SD interval of at least 15 min (Figure 1).
In Series 2, after drug incubation for 30 min, both common carotid arteries were permanently occluded (two‐vessel occlusion [2VO]) by pulling on the occluder lines until resistance was felt and then securing the occluders in place (n = 23). Successful 2VO was confirmed by a sharp drop in the LDF signal. Rats with no occlusion served as control for 2VO (i.e., intact, n = 21). Fifteen minutes after 2VO onset, whisker stimulation involving the entire left whisker pad was performed mechanically. Stimulation frequency was set at 2–3 Hz, each stimulation lasting for 25 s. The stimulation was repeated 4 times with 2 min intermissions. Subsequently, exactly as in Series 1, three SD events were triggered by placing a 1 M KCl‐soaked cotton ball in the caudal craniotomy. Considering the combination of 2VO and pharmacological manipulations, eight experimental groups were established (Table 1).
The ischaemia model has been widely used (Farkas, Luiten, & Bari, 2007); rodents are the typical model organisms to study neurovascular coupling or SD (Hartings et al., 2017; Iadecola, 2017). The experimenter was not blinded to the procedure, because she had to create ischaemia by a surgical intervention (thus, it was clear which animal was allocated to the ischaemic group).
2.6. Data analysis
All variables (i.e., DC potential, LDF signal, and MABP) were simultaneously acquired, displayed live, and stored using a personal computer equipped with a custom‐made software in LabView (National Instruments). Data analysis was assisted by the inbuilt instructions of the software AcqKnowledge 4.2 for MP 150 (Biopac Systems, Inc., Goleta, USA). Raw LDF recordings were downsampled to 1 Hz and then expressed relative to baseline by using the average CBF value of the first 5 min (100%) and the biological zero obtained after terminating each experiment (0%) as reference points. Evoked field potentials (EFPs) during whisker stimulation were analysed in the original LFP recordings acquired at 2000 Hz, while raw DC recordings were also downsampled to 1 Hz.
Blinding data analysis was intended by assigning codes to files and recordings, which do not reveal the experimental condition (i.e., date of the experiment). All recordings were first screened for events suitable for comprehensive analysis. For EFPs, it was required that the segment under analysis (10 s) was completely free of harmonic network oscillations (~50 Hz, produced by the feedback controlled on–off switch of the heating blanket) or any accidental noise produced by manipulations by the experimenter. For SD analysis, it was essential that an SD elicited propagated to the recording window. The evaluation of CBF responses was achieved when hyperaemia evolved in response to stimulation (functional hyperaemia in response to whisker stimulation remained undetectable on several occasions, and SDs in one animal gave rise to inverse neurovascular coupling) and if the CBF responses to subsequent events were clearly separated.
In the case of whisker stimulation, the peak amplitude of evoked potentials was assessed to reveal drug effect, if any. The efficacy of hyperaemia was characterized by measuring the maximum amplitude of the CBF response (mean of 15 s at the plateau of the hyperaemia).
For SD events, peak amplitude, duration at half amplitude, and the rate of repolarization were measured on the DC potential trace. The hyperaemic element of the CBF response was first sorted on the basis of the presence of late hyperaemia. Type 1 was noted whenever peak hyperaemia was obvious, but late hyperaemia remained undetectable. The CBF response was labelled Type 2 when both peak and late hyperaemia could be identified confidently. The SD‐associated hyperaemia was then characterized by the peak amplitude, and the duration of hyperaemia (peak and late elements together), taken from deflection from and return to baseline. Data analysis was shared among four independent investigators.
Unequal group sizes were achieved because some of the completed experiments did not qualify for comprehensive data analysis due to high arterial blood pCO2 and low arterial blood pH values (n = 4), unsuccessful SD elicitation (n = 1), or an overdose of anaesthesia during the experimental protocol (n = 1). However, pilot experiments, which met all the criteria of a successful experiment (e.g., anaesthesia protocol yielding physiological arterial blood gas values, uninterrupted monitoring of mean arterial pressure, appropriate concentration of drug solutions, successful somatosensory stimulation, and SD elicitation; n = 6), were incorporated in the final groups to utilize all relevant data produced.
Data are given as mean ± stdev. The results were statistically analysed with the software SPSS (IBM SPSS Statistics for Windows, Version 22.0, IBM Corp., USA; RRID:SCR_002865). The distribution of the data was tested with the Shapiro–Wilk normality test. Outliers were filtered with Grubbs test. Homogeneity of the variances was checked by Levene's test. Two‐ or three‐way ANOVA models were used, dependent on the type of data obtained. Levels of significance were defined as *P < 0.05. Tukey's HSD or Games–Howell post hoc test was used for group comparisons, whenever applicable. Non‐parametric data were statistically evaluated with a Pearson chi‐square test for association. All relevant statistical methods are given in each figure legend. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology.
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Kelly et al., 2017; Alexander, Striessnig et al., 2017).
3. RESULTS
3.1. Physiological variables and baseline variations of CBF
Physiological variables (MABP, arterial blood pH, and partial arterial pressure of O2 and CO2) are presented in Table 1. Statistical analysis did not reveal any significant ischaemia‐ or treatment‐related difference across experimental groups.
Typical, original recordings of the DC potential and CBF variations over the full experimental protocol of Series 2 are shown in Figure 2.
Figure 2.
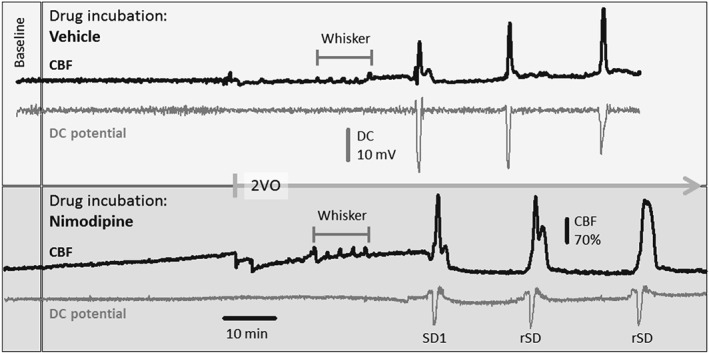
Representative direct current (DC) potential and cerebral blood flow (CBF) traces of the full experimental protocol including common carotid artery occlusion (2VO) in Series 2 under vehicle (upper box) or nimodipine (lower box) treatment. The traces were downsampled to 1‐Hz frequency (original sampling frequency: 500 or 2000 Hz), and the CBF traces were smoothed by median filtering for 6 data points. Note that nimodipine gradually elevated baseline perfusion, increased the amplitude of functional hyperaemia to whisker stimulation, and enlarged hyperaemia in response to the first (SD1) and recurrent spreading depolarizations (rSD). Also, nimodipine reduced SD amplitude. Note also the strikingly different size of the CBF response to whisker stimulation and SD within the same preparation
Variation of baseline CBF was assessed at selected time points of the experimental protocol, in order to evaluate the impact of pharmacological treatments (Figure 3). Ischaemia induction in Series 2 obviously caused a marked reduction of CBF (53 ± 23%), which stabilized in the vehicle‐treated group at 74 ± 11% prior to first SD (SD1) and at 67 ± 15% prior to recurrent SDs (rSDs; Figure 3b,c). The occurrence of SD produced long‐lasting oligaemia, the final element of the CBF response to SD, which was apparent between SD events in the intact groups, as well (Figure 3c). Neither chronic nor acute treatment with LA1011 altered baseline CBF at any time point or condition considered. In contrast, nimodipine treatment increased baseline CBF significantly by the time of ischaemia induction with respect to the pretreatment CBF level (Figure 3a). Further, nimodipine counteracted the reduction in CBF evoked by 2VO (Figure 3b) and prevented the development of post‐SD oligaemia (Figure 3c).
Figure 3.
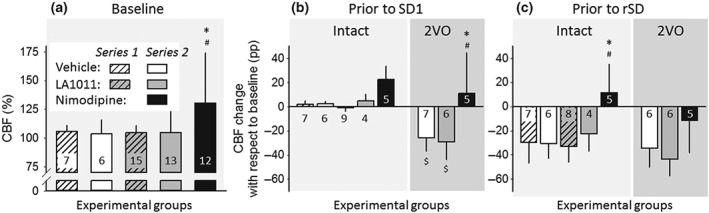
Variation of baseline cerebral blood flow (CBF; i.e., inbetween stimulations) with respect to pharmacological treatments. (a) CBF prior to the first experimental intervention (i.e., production of spreading depolarization [SD] in Series 1, or induction of ischaemia in Series 2). In Series 2, the intact group and the group of animals later undergoing bilateral common carotid artery occlusion (2VO) are merged, because they received identical handling until this point of the experimental protocol. (b) CBF before the production of the first SD (SD1). Data are expressed as change with respect to the corresponding baseline. (c) CBF before the production of recurrent SD events (rSD). Data are given as mean ± stdev; sample size (i.e., number of animals) is indicated in each bar. Statistical analysis relied on a two‐way ANOVA paradigm (factors: acute/chronic and treatment), Panel (a), or a three‐way ANOVA (acute/chronic, ischaemia, and treatment), Panels (b) and (c). The level of significance was defined as *P < 0.05. Tukey's HSD post hoc test was applied for group comparisons (*P < 0.05 vs. vehicle; # P < 0.05 vs. LA1011; $ P < 0.05 vs. respective intact)
3.2. Somatonsensory stimulation
For the estimation of drug effect on physiological neurovascular coupling, somatosensory EFPs and the associated CBF response provoked by whisker stimulation were evaluated in the somatosensory barrel cortex in Series 2 (Figure 4). The amplitude of EFPs was considerably attenuated during ischaemia (in the presence of 2VO) with respect to the intact condition (Figure 4b). The application of LA1011 exerted no effect on EFPs; however, nimodipine dramatically decreased the amplitude of evoked potentials in the intact cortex (Figure 4a,b). The relative amplitude of the hyperaemic response was notably smaller during ischaemia (2VO) than in the intact brain (Figure 4c,d). Treatment with LA1011 did not improve functional hyperaemia significantly, but nimodipine restored the relative amplitude of the CBF response to the intact level (Figure 4d). In addition to the elevation of baseline CBF achieved by nimodipine, the further improvement in the CBF response to whisker stimulation in the ischaemic cortex was very apparent (Figure 4d).
Figure 4.
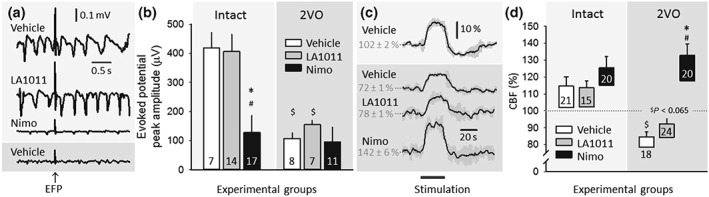
The impact of ischaemia or pharmacological treatments on evoked potentials and the coupled cerebral blood flow (CBF) response during whisker stimulation in Series 2. (a) Representative local field potential (LFP) traces display somatosensory evoked field potentials (EFPs) during whisker stimulation. Traces in front of light grey background were taken from intact animals, while the trace in front of dark grey background represents an animal with bilateral common carotid artery occlusion (2VO). (b) The peak amplitude of evoked potentials. (c) Traces (each is the mean of four stimulations in an animal representative of each condition) show the CBF response to whisker stimulation. (d) The amplitude of the CBF response to whisker stimulation. The base of each bar in the chart is set to the CBF level directly preceding whisker stimulation. In (b) and (d), data are given as mean ± stdev; sample size (i.e., the number of events analysed) is indicated in each bar. Statistical analysis relied on a two‐way ANOVA paradigm (factors: ischaemia and treatment). The level of significance was defined as *P < 0.05. A Games–Howell post hoc test was applied for group comparisons (*P < 0.05 vs. vehicle; # P < 0.05 vs. LA1011; $ P < 0.05 vs. respective intact)
3.3. Spreading depolarization
Experimentally‐evoked SDs were exploited for the investigation of drug effect on non‐physiological neural depolarization and the associated CBF response (Figures 5 and 6). The SD1 was taken apart from rSD events for analysis, because of known dissimilarities between SD1 and rSDs (Farkas, Pratt, Sengpiel, & Obrenovitch, 2008). Eventually, detailed data analysis was completed on rSDs. As expected, the amplitude of SD events was not altered (Series 2), while their duration was extended (Series 2) during ischaemia with respect to the intact condition (Figure 5a–c). Chronic or acute application of LA1011 consistently intensified SD amplitude, irrespective of ischaemia (Series 2; Figure 5b). In contrast, acutely applied nimodipine significantly decreased SD amplitude (Series 2). In addition, nimodipine shortened SD duration in the ischaemic cortex to its intact level (Series 2; Figure 5c).
Figure 5.
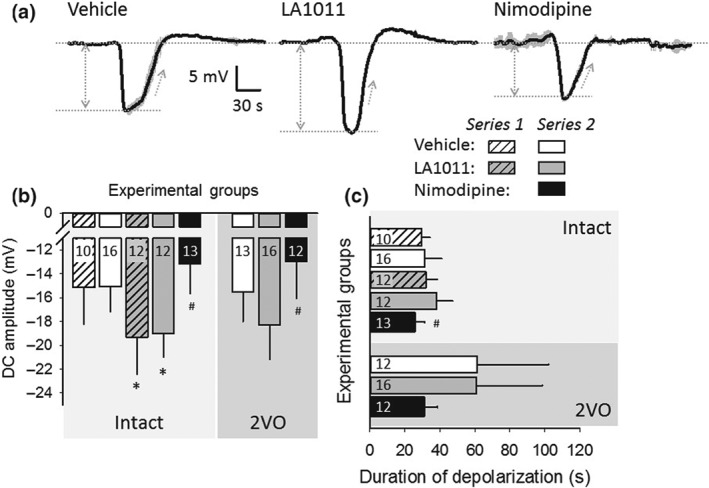
The impact of ischaemia or pharmacological treatments on recurrent spreading depolarization (rSD). (a) Traces (each is the mean of rSDs in an animal representative of each condition) demonstrate the negative DC potential shift indicative of rSDs, triggered after the bilateral occlusion of the common carotid arteries (2VO). (b) Amplitude of the negative DC potential shift with rSDs. (c) Duration of the negative DC potential shift with rSDs. (d) Rate of repolarization of the negative DC potential shift with rSDs. In (b) to (d), data are given as mean ± stdev; sample size (i.e., the number of events analysed) is indicated in each bar. Statistical analysis relied on a three‐way ANOVA paradigm (factors: acute/chronic, ischaemia, and treatment). The level of significance was defined as *P < 0.05. Tukey's HSD (b) or a Games–Howell (c) post hoc test was applied for group comparisons (*P < 0.05 vs. vehicle; # P < 0.05 vs. LA1011)
Figure 6.
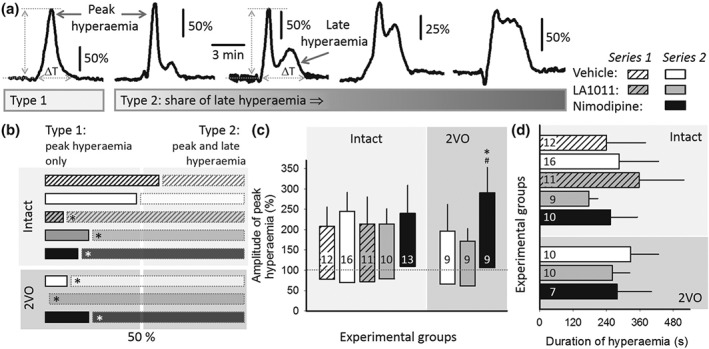
The impact of ischaemia or pharmacological treatments on the cerebral blood flow (CBF) response to recurrent spreading depolarization (rSD) events. (a) Representative traces demonstrate the spectrum of the kinetics of the CBF response considering the weight of late hyperaemia in the signature. The CBF response was classified as Type 1 whenever peak hyperaemia was obvious, with late hyperaemia being undetectable. The CBF response was labelled Type 2, when both peak and late hyperaemia could be identified. (b) Occurrence of Type 1 and Type 2 CBF responses is depicted with respect to all of the CBF responses analysed being taken as 100% in each experimental group. Horizontal bars with black outline (left) depict the share of Type 1, while bars with grey outline (right) represent the ratio of Type 2 CBF responses. Note that the two types were represented equally (i.e., near 50% indicated by a vertical white band) in the vehicle‐treated, intact groups. In the animals undergoing bilateral common carotid artery occlusion (2VO) and receiving LA1011 treatment, only Type 2 CBF responses were observed. Pearson chi‐square test for association indicated a significant effect (value: 16.996, P < 0.017). Calculating column proportion by z test with Bonferroni correction identified a significant shift in Type 1 and Type 2 ratios for groups labelled (*). (c) Amplitude of peak hyperaemia. The base of each bar in the chart is set to the CBF level preceding rSD events. (d) Duration of hyperaemia, that is, ΔT in Panel (a). In (b) to (d), data are given as mean ± stdev; sample size (i.e., the number of events analysed) is indicated in each bar. Statistical analysis relied on a three‐way ANOVA paradigm (factors: acute/chronic, ischaemia, and treatment). The level of significance was defined as *P < 0.05. Tukey's HSD test was applied for group comparisons (*P < 0.05 vs. vehicle; # P < 0.05 vs. LA1011)
The CBF response to SD consists of four elements, starting with a transient hypoperfusion, followed by a peak and then a late hyperaemia, and concluded by a long‐lasting oligaemia (Ayata & Lauritzen, 2015). The share of the individual elements in the CBF response to SD is variable across animal species and anaesthesia protocols and appears to change markedly according to the actual metabolic status of the tissue (Ayata & Lauritzen, 2015).
The analysis of the CBF response here predominantly focused on the hyperaemia phase. The kinetics of the observed CBF responses exhibited a spectrum considering the weight of late hyperaemia in the signature (Figure 6a). Further, the presence of the late hyperaemic element served as the basis for CBF response classification to distinguish CBF response Type 1 characterized by peak hyperaemia only, from CBF response Type 2 that included late hyperaemia in addition to the peak hyperaemia (Figure 6a). A semi‐quantitative approach of ours indicated that the likelihood for Type 1 and Type 2 CBF responses to evolve was near equal in the vehicle‐treated, intact condition. Conversely, ischaemia, or treatment with LA1011 or nimodipine, allowed late hyperaemia to emerge at a clearly higher incidence (Figure 6b). The amplitude of peak hyperaemia was conserved over experimental groups, except for nimodipine treatment in the ischaemic condition, which augmented peak hyperaemia amplitude ( Figure 6c). The duration of hyperaemia (i.e., peak and late hyperaemia together) was not significantly altered by ischaemia or the treatments (Series 2; Figure 6d).
4. DISCUSSION
The presented work investigated the potential neuroprotective effect of LA1011, an Hsp27 and Hsp70 co‐inducer dihydropyridine derivative with no affinity for L‐type voltage‐gated calcium channels (Kasza et al., 2016), with reference to nimodipine, a well‐known dihydropyridine compound that blocks L‐type voltage‐gated calcium channels (Scriabine et al., 1989). In the incomplete global forebrain ischaemia model used, we first assessed drug effect on physiological neuronal activation (i.e., achieved by somatosensory stimulation) and the coupled functional hyperaemic response. Next, we focused on the impact of the pharmacological treatments on SD and the associated CBF response. SD is an ischaemic preconditioning stimulus when triggered in intact tissue (Shen et al., 2016) and represents a pathophysiological process as it occurs due to extracellular potassium and glutamate accumulation in ischaemic brain injury (Dreier, 2011; Pietrobon & Moskowitz, 2014).
The cerebral ischaemia model has been selected to test the neuroprotective efficacy of LA1011, because the drug has been shown to co‐induce Hsp27 and Hsp70 (Kasza et al., 2016), both recognized to mediate anti‐inflammatory and anti‐apoptotic mechanisms in ischaemic brain injury (Franklin, Krueger‐Naug, Clarke, Arrigo, & Currie, 2005; Nowak & Jacewicz, 1994; van der Weerd et al., 2010; Yenari et al., 2005). The dose of the drugs applied in the present study was justified by previous experimental results and our pilot experiments (Kasza et al., 2016; Richter et al., 2002).
Here, we show in the anaesthetized rat that baseline CBF remained unchanged after LA1011 administration but was increased by nimodipine, as expected. Cerebrovascular smooth muscle cells are enriched by L‐type voltage‐gated calcium channels. Nimodipine inhibits calcium influx through these channels, leading to the hyperpolarization and relaxation of smooth muscle cells, and the reduction of vascular tone (Scriabine & van den Kerckhoff, 1988).
The current data demonstrate that LA1011 does not alter neuronal excitability in the intact cortex, which is consistent with the view that LA1011 increases Hsp expression only under stress conditions (Kasza et al., 2016). Further, LA1011 treatment did not restore EFP amplitude compromised by ischaemia and did not improve the hyperaemic response impaired by ischaemia, either. These results indicate that acutely administered LA1011 does not protect neuronal excitability or neurovascular coupling during the early phase of cerebral ischaemia. On the other hand, it is a novel finding of the presented work, that, in addition to baseline CBF elevation, nimodipine profoundly augmented functional hyperaemia in response to somatosensory stimulation (Figure 3c,d), without enhancing EFP amplitude under ischaemia. Still, nimodipine remarkably decreased EFP amplitude in the intact cortex, while the relative magnitude of the flow response was maintained. Both observations imply that the enhancement of functional hyperaemia by nimodipine is disproportionate with respect to EFP amplitude. This suggests that nimodipine augmented the amplitude of the CBF response (irrespective of the intensity of neuronal activation initiated by somatosensory stimulation), possibly by potentiating the release of vasodilator substances or the efficacy of vasodilator signalling cascades. Since vasodilator PGs and epoxyeicosatrienoic acids are produced by astrocytes during neurovascular coupling (Attwell et al., 2010), and L‐type voltage‐gated calcium channels are present in the astrocyte plasma membrane (Cheli et al., 2016), astrocytes may be involved in the nimodipine‐related enlargement of functional hyperaemia, without neuronal contribution being proportionally increased in the first place. Further details concerning this suggestion are given as Supporting Information to the paper.
Even though LA1011 treatment did not achieve a detectable change in neuronal activity with somatosensory stimulation in our experiments, it greatly enhanced the amplitude of SD, consistent in all experimental conditions created. The amplitude of SD appears to be fairly conserved, since neither ischaemia nor age can alter the SD‐related DC potential amplitude (Menyhárt et al., 2015; Menyhárt et al., 2017). However, several pharmacological manipulations have proven to reduce the amplitude of SD. As such, topical application of the pro‐inflammatory cytokine TNF‐α, the inhibition of the P2X7/pannexin 1 pore by the drug A438079, the blockade of large‐conductance Ca2+‐activated potassium channels by paxilline, or the uncoupling of postsynaptic density protein 95 from NMDA receptors by UCCB01‐144 (Tat‐N‐dimer) were all found to reduce the amplitude of SD; TNF‐α, and A438079 even in a dose‐dependent manner (Chen et al., 2017; Kucharz, Søndergaard Rasmussen, Bach, Strømgaard, & Lauritzen, 2017; Menyhárt et al., 2018; Richter et al., 2014). Two of these studies have pointed out that the amplitude of SD stands in a strong, positive correlation with the extracellular concentration of potassium (Chen et al., 2017; Menyhárt et al., 2018), which would infer that treatment with LA1011, an Hsp co‐inducer, may support cellular (possibly neuronal) potassium efflux with SD. Indeed, heat shock preconditioning preserved the diminishing potassium peak of recurrent depolarizations triggered by intermittent anoxia, and Hsp70 expression modulated potassium homeostasis throughout oxygen withdrawal in the drosophila brain (Armstrong et al., 2011). Although it remains to be determined which potassium channels may be tuned by Hsp70 in the mammalian nervous system, the chaperon has been shown to activate potassium channels linked to the rise of intracellular calcium near the plasma membrane in cultured human promonocytes (Negulyaev, Vedernikova, Kinev, & Voronin, 1996) and to be involved in the assembly of specific voltage‐gated potassium channels located in cardiac muscle (Ficker, Dennis, Wang, & Brown, 2003).
As listed above, TNF‐α is also known to reduce the amplitude of SD in a dose‐dependent manner (Richter et al., 2014). Interestingly, the overexpression of Hsp70—a target of LA1011—was shown to suppress pro‐inflammatory cytokine mRNA levels, including TNF‐α in cultured, oxygen–glucose‐deprived astrocytes (Kim, Yenari, & Lee, 2015). Furthermore, up‐regulation of Hsp70 decreased TNF‐α expression and improved neurological outcome in a rodent model of intracerebral haemorrhage (Manaenko et al., 2010). On the basis of these observations, it would be attractive to postulate that LA1011 increased SD amplitude possibly by the co‐induction of Hsp70 to cause the down‐regulation of TNF‐α, which would normally limit the magnitude of SD. The mechanistic link between LA1011 treatment and SD remains, however, speculative, and the temporal profile of Hsp induction in the model used is also obscure. Therefore, further investigations are needed to explore the LA1011‐related signalling that causes increasing SD amplitude.
In contrast to LA1011, nimodipine application reduced SD size, in agreement with previous reports applying nimodipine at the concentration used here (Menyhárt et al., 2018; Richter et al., 2002). Neurons express L‐type voltage‐gated calcium channels, which have been implicated in the modification of neuronal excitability, and are a well‐known target of nimodipine (Scriabine et al., 1989). Taken together, the opposite actions of LA1011 and nimodipine on SD amplitude correspond reliably to the different cellular targets of the two molecules.
Although pharmacologically decreased SD amplitude is often interpreted as a sign of protection (Richter et al., 2014), the lack of a clear‐cut association between SD amplitude and histological or neurological damage imposed creates persistent controversies. On the other hand, the number of rSDs, the cumulative duration of SDs, and the inability of the tissue to recover from SD (i.e., long SD duration) have been shown to correlate with or be a sign of injury progression (Dijkhuizen et al., 1999; Dreier et al., 2006; Nedergaard, 1996). In this context, LA1011 proved to be ineffective, since SD duration was not altered under LA1011 treatment (Figure 4c). Nimodipine, however, showed a tendency to shorten SD duration (Figure 4c), which may be accepted as a sign of its protective potential. SD is associated with neuronal calcium loading, in part via ionotropic glutamate receptors, such as the NMDA receptor (Pietrobon & Moskowitz, 2014). The reduction of the SD‐associated calcium influx has thus emerged as a promising target to achieve SD inhibition. For example, low‐dose ketamine (an NMDA receptor blocker) applied to brain slices has recently been shown to reduce calcium load and to facilitate the recovery from SD (Reinhart & Shuttleworth, 2018). In our study, nimodipine is thought to have inhibited calcium influx to neurons via L‐type voltage‐gated calcium channels, which also caused the more rapid recovery from SD. Together, these results suggest that the attenuation of neuronal calcium load (either via NMDA receptor blockade or via L‐type calcium channel inhibition) shortens SD duration.
Before the detailed discussion of drug effects, it must be appreciated that hyperaemia in response to SD was markedly accentuated under α‐chloralose anaesthesia used here (relative amplitude: 191 ± 61 and 151 ± 96%, intact and under ischaemia, respectively), with respect to that seen under isoflurane anaesthesia in our previous work (e.g., 51 ± 38 and 21 ± 11%, intact and under ischaemia, respectively, Varga et al., 2016; 76 ± 12 and 21 ± 9%, intact and under ischaemia, respectively, Menyhárt et al., 2017). Moreover, late hyperaemia in the CBF response to SD was revealed very often under α‐chloralose anaesthesia as seen here (Figures 2 and 6), while it was seldom encountered in many of our previous studies using isoflurane (Menyhárt et al., 2015; Menyhárt et al., 2017; Varga et al., 2016). Finally, the global ischaemia model we routinely use produces a considerable drop of CBF following 2VO under isoflurane anaesthesia (e.g., to 27 ± 13%, Varga et al., 2016, to 41 ± 9%; Menyhárt et al., 2015)—this drop proved to be considerably more moderate under α‐chloralose anaesthesia here (Figure 3b). Therefore, we propose that the anaesthetic procedure used in this study allowed the frequent evolution of large hyperaemia in response to SD, also when SD was elicited after the occlusion of the common carotid arteries.
The experiments presented here show that the kinetics of SD‐related hyperaemia was transformed discernibly by ischaemia and LA1011 or nimodipine treatment. In other words, the experimental manipulations accentuated the element of late hyperaemia in the SD‐associated CBF response (Figure 5a,b). On the basis of an original finding that L‐arginine administration supported late hyperaemia following SD (Fabricius, Akgoren, & Lauritzen, 1995), late hyperaemia was proposed to be NO dependent (Ayata & Lauritzen, 2015). Since baseline CBF elevation by L‐arginine can be linked to increased endothelial NOS (eNOS) activity (Yamada et al., 2000), and ischaemia apparently potentiates eNOS expression (Bolaños & Almeida, 1999), it is conceivable that the increased share of late hyperaemia in the CBF response to SD was supported by increased eNOS activity in our ischaemic condition. Interestingly, Hsps, particularly Hsp90, have been implicated in the adjustment of vascular tone via modulating the production of the vasodilator NO (García‐Cardeña et al., 1998), and a recent report has shown LA1011 to bind Hsp90 and activate the ATPase activity of Hsp90 (Roe et al., 2018). A potential interaction between LA1011 and NO release to unravel late hyperaemia in the SD‐related CBF response may thus be mediated via Hsp90; however, the signalling cascade remains hypothetical due to the paucity of explicit experimental data.
In conclusion, the current study appears to confirm the Hsp co‐inducer and possibly anti‐inflammatory potential of LA1011 during stress to the nervous tissue. Furthermore, the work demonstrates that L‐type voltage‐gated calcium channel inhibition augments functional hyperaemia in response to somatosensory stimulation especially under ischaemia, in addition to achieving a general, constitutive vasodilator effect. A more detailed understanding of the molecular pathways involved either in LA1011 or in nimodipine action assessed here requires further investigation. Overall, previous data (Kasza et al., 2016; Penke et al., 2018; Roe et al., 2018) and those of ours together suggest that LA1011 may be considered to counteract neurodegeneration, with no major, concomitant impact on vascular tone or neurovascular coupling.
CONFLICT OF INTEREST
A.H and Z.T. are employees of LipidArt Ltd. The authors Z.T., I.H., and L.V. are founding members of LipidArt Ltd.
AUTHOR CONTRIBUTIONS
Í.S. performed the acquisition of data, analysed and interpreted the data, drafted the article, and approved the final version to be submitted. O.M.T. performed the acquisition of data, analysed and interpreted the data, revised the article critically for important intellectual content, and approved the final version to be submitted. Z.T. designed the study, revised the article critically for important intellectual content, and approved the final version to be submitted. D.P.V. analysed and interpreted the data, and approved the final version to be submitted. Á.M. analysed and interpreted the data, revised the article critically for important intellectual content, and approved the final version to be submitted. R.F. performed the acquisition of data, analysed and interpreted the data, and approved the final version to be submitted. D.H. performed the acquisition of data and approved the final version to be submitted. I.H. and F.B. revised the article critically for important intellectual content and approved the final version to be submitted. L.V. designed the study, revised the article critically for important intellectual content, and approved the final version to be submitted. E.F. designed the study, analysed and interpreted the data, drafted the article, approved the final version to be submitted.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Figure S1.
Time line of the protocols implemented. Abbreviations: 2VO, permanent, bilateral occlusion of the common carotid arteries (“two‐vessel occlusion”); Fluoro: fluorocitrate; Nimo, nimodipine; rSD, recurrent spreading depolarization; SD, spreading depolarization; SD1, first spreading depolarization in a train.
Figure S2. The impact of pharmacological treatments on evoked potentials and the coupled cerebral blood flow (CBF) response during whisker stimulation after bilateral common carotid artery occlusion (2VO) in Series 2. A, Representative local field potential (LFP) traces display somatosensory evoked field potentials (EFPs) during whisker stimulation. Traces in front of light gray background were taken from animals control to fluorocitrate treatment, while the trace in front of dark gray background represents EFPs under fluorocitrate treatment. B, The peak amplitude of evoked potentials. C, Traces (each is the mean of 4 stimulations in an animal representative of each condition) show the CBF response to whisker stimulation. D, The amplitude of the CBF response to whisker stimulation. The base of each bar in the chart is set to the CBF level directly preceding whisker stimulation. In B and D, data are given as mean ± stdev; sample size (i.e. the number of events analyzed) is indicated in each bar. Statistical analysis relied on a two‐way analysis of variance (ANOVA) paradigm (factors: 2VO and treatment). The level of significance was defined as p < 0.05*. A Games‐Howell post hoc test was applied for group comparisons (p < 0.05* vs. vehicle; p < 0.05# vs. control). Significance symbols under the bars represent differences of baseline, while symbols over the bars stand for the difference between functional hyperemia amplitude.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Research, Development and Innovation Office of Hungary (Grants K111923 and K120358); the Szeged Scientists Academy Program of the Foundation for the Future of Biomedical Sciences in Szeged, implemented with the support of the Ministry of Human Capacities of Hungary (34232‐3/2016/INTFIN, to D.H.); the Economic Development and Innovation Operational Programme in Hungary (Ministry of National Economy of Hungary) co‐financed by the European Union and the European Regional Development Fund (GINOP‐2.3.2‐15‐2016‐00060, GINOP‐2.3.2‐15‐2016‐00040, and GINOP‐2.1.7‐15‐2016‐02085); and the EU‐funded Hungarian (Grant EFOP‐3.6.1‐16‐2016‐00008).
Szabó Í, M. Tóth O, Török Z, et al. The impact of dihydropyridine derivatives on the cerebral blood flow response to somatosensory stimulation and spreading depolarization. Br J Pharmacol. 2019;176:1222–1234. 10.1111/bph.14611
REFERENCES
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Overview. British Journal of Pharmacology, 174(S1), S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017. Dec). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174(Suppl 1), S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, G. A. , Xiao, C. , Krill, J. L. , Seroude, L. , Dawson‐Scully, K. , & Robertson, R. M. (2011). Glial Hsp70 protects K+ homeostasis in the Drosophila brain during repetitive anoxic depolarization. PLoS ONE, 6(12), e28994 10.1371/journal.pone.0028994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell, D. , Buchan, A. M. , Charpak, S. , Lauritzen, M. , Macvicar, B. A. , & Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature, 468, 232–243. 10.1038/nature09613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata, C. , & Lauritzen, M. (2015. Jul). Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiological Reviews, 95(3), 953–993. 10.1152/physrev.00027.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolaños, J. P. , & Almeida, A. (1999. May 5). Roles of nitric oxide in brain hypoxia‐ischemia. Biochimica et Biophysica Acta, 1411(2–3), 415–436. 10.1016/S0005-2728(99)00030-4 [DOI] [PubMed] [Google Scholar]
- Cheli, V. T. , Santiago González, D. A. , Smith, J. , Spreuer, V. , Murphy, G. G. , & Paez, P. M. (2016. Aug). L‐type voltage‐operated calcium channels contribute to astrocyte activation in vitro. Glia, 64(8), 1396–1415. 10.1002/glia.23013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. P. , Qin, T. , Seidel, J. L. , Zheng, Y. , Eikermann, M. , Ferrari, M. D. , … Eikermann‐Haerter, K. (2017. Jun 1). Inhibition of the P2X7‐PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain, 140(6), 1643–1656. 10.1093/brain/awx085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weerd, L. , Tariq Akbar, M. , Aron Badin, R. , Valentim, L. M. , Thomas, D. L. , Wells, D. J. , … de Belleroche, J. S. (2010). Overexpression of heat shock protein 27 reduces cortical damage after cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism, 30(4), 849–856. 10.1038/jcbfm.2009.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen, R. M. , Beekwilder, J. P. , van der Worp, H. B. , Berkelbach van der Sprenkel, J. W. , Tulleken, K. A. , & Nicolay, K. (1999. Sep 4). Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Research, 840(1–2), 194–205. 10.1016/S0006-8993(99)01769-2 [DOI] [PubMed] [Google Scholar]
- Diringer, M. N. , & Zazulia, A. R. (2017. Dec). Aneurysmal subarachnoid hemorrhage: Strategies for preventing vasospasm in the intensive care unit. Seminars in Respiratory and Critical Care Medicine, 38(6), 760–767. [DOI] [PubMed] [Google Scholar]
- Dreier, J. P. (2011. Apr). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nature Medicine, 17(4), 439–447. 10.1038/nm.2333 [DOI] [PubMed] [Google Scholar]
- Dreier, J. P. , Windmüller, O. , Petzold, G. , Lindauer, U. , Einhäupl, K. M. , & Dirnagl, U. (2002. Dec). Ischemia triggered by red blood cell products in the subarachnoid space is inhibited by nimodipine administration or moderate volume expansion/hemodilution in rats. Neurosurgery, 51(6), 1457–1465. discussion 1465‐7. 10.1097/00006123-200212000-00017 [DOI] [PubMed] [Google Scholar]
- Dreier, J. P. , Woitzik, J. , Fabricius, M. , Bhatia, R. , Major, S. , Drenckhahn, C. , … Strong, A. J. (2006. Dec). Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain, 129(Pt 12), 3224–3237. 10.1093/brain/awl297 [DOI] [PubMed] [Google Scholar]
- Fabricius, M. , Akgoren, N. , & Lauritzen, M. (1995. Jul). Arginine‐nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. The American Journal of Physiology, 269(1 Pt 2), H23–H29. [DOI] [PubMed] [Google Scholar]
- Farkas, E. , Luiten, P. G. , & Bari, F. (2007. Apr). Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion‐related neurodegenerative diseases. Brain Research Reviews, 54(1), 162–180. 10.1016/j.brainresrev.2007.01.003 [DOI] [PubMed] [Google Scholar]
- Farkas, E. , Pratt, R. , Sengpiel, F. , & Obrenovitch, T. P. (2008. Feb). Direct, live imaging of cortical spreading depression and anoxic depolarisation using a fluorescent, voltage‐sensitive dye. Journal of Cerebral Blood Flow and Metabolism, 28(2), 251–262. 10.1038/sj.jcbfm.9600569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker, E. , Dennis, A. T. , Wang, L. , & Brown, A. M. (2003. Jun 27). Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circulation Research, 92(12), e87–e100. [DOI] [PubMed] [Google Scholar]
- Franklin, T. B. , Krueger‐Naug, A. M. , Clarke, D. B. , Arrigo, A. P. , & Currie, R. W. (2005). The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. International Journal of Hyperthermia, 21(5), 379–392. 10.1080/02656730500069955 [DOI] [PubMed] [Google Scholar]
- Freedman, D. D. , & Waters, D. D. (1987. Nov). ‘Second generation’ dihydropyridine calcium antagonists. Greater vascular selectivity and some unique applications. Drugs, 34(5), 578–598. 10.2165/00003495-198734050-00005 [DOI] [PubMed] [Google Scholar]
- Fülöp F, Vigh, L. , Torok Z., Penke, B. , Horvath, I. , Balogh, G. , … Hunya Á (2013). 1,4 dihydropyridine derivatives with HSP modulating activity. PCT. Int. Appl., WO2013076516 A1.
- García‐Cardeña, G. , Fan, R. , Shah, V. , Sorrentino, R. , Cirino, G. , Papapetropoulos, A. , & Sessa, W. C. (1998. Apr 23). Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature, 392(6678), 821–824. 10.1038/33934 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018. Jan 4). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings, J. A. , Shuttleworth, C. W. , Kirov, S. A. , Ayata, C. , Hinzman, J. M. , Foreman, B. , … Dreier, J. P. (2017. May). The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão's legacy. Journal of Cerebral Blood Flow and Metabolism, 37(5), 1571–1594. 10.1177/0271678X16654495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann, K. A. (1996). Periinfarct depolarizations. Cerebrovascular and Brain Metabolism Reviews, 8(3), 195–208. [PubMed] [Google Scholar]
- Iadecola, C. (2017. Sep 27). The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron, 96(1), 17–42. 10.1016/j.neuron.2017.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman, K. , & Iadecola, C. (2015. Jan 10). Neurovascular regulation in the ischemic brain. Antioxidants & Redox Signaling, 22(2), 149–160. 10.1089/ars.2013.5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasza, Á. , Hunya, Á. , Frank, Z. , Fülöp, F. , Török, Z. , Balogh, G. , … Penke, B. (2016. May 7). Dihydropyridine derivatives modulate heat shock responses and have a neuroprotective effect in a transgenic mouse model of Alzheimer's disease. Journal of Alzheimer's Disease, 53(2), 557–571. 10.3233/JAD-150860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieninger, M. , Flessa, J. , Lindenberg, N. , Bele, S. , Redel, A. , Schneiker, A. , … Silbereisen, V. (2017. Side effects of long‐term continuous intra‐arterial nimodipine infusion in patients with severe refractory cerebral vasospasm after subarachnoid hemorrhage. Neurocritical Care, 28, 65–76. 10.1007/s12028-017-0428-1. [Epub ahead of print]). [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. Y. , Han, Y. , Lee, J. E. , & Yenari, M. A. (2018. Feb 15). The 70‐kDa heat shock protein (Hsp70) as a therapeutic target for stroke. Expert Opinion on Therapeutic Targets, 22(3), 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. Y. , Yenari, M. A. , & Lee, J. E. (2015. Feb 12). Regulation of inflammatory transcription factors by heat shock protein 70 in primary cultured astrocytes exposed to oxygen‐glucose deprivation. Neuroscience, 286, 272–280. 10.1016/j.neuroscience.2014.11.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharz, K. , Søndergaard Rasmussen, I. , Bach, A. , Strømgaard, K. , & Lauritzen, M. (2017). PSD‐95 uncoupling from NMDA receptors by Tat‐ N‐dimer ameliorates neuronal depolarization in cortical spreading depression. Journal of Cerebral Blood Flow and Metabolism, 37(5), 1820–1828. 10.1177/0271678X16645595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão, A. A. P. (1944). Spreading depression of activity in the cerebral cortex. Journal of Neurophysiology, 7, 359–390. 10.1152/jn.1944.7.6.359 [DOI] [PubMed] [Google Scholar]
- Manaenko, A. , Fathali, N. , Chen, H. , Suzuki, H. , Williams, S. , Zhang, J. H. , & Tang, J. (2010. Dec). Heat shock protein 70 upregulation by geldanamycin reduces brain injury in a mouse model of intracerebral hemorrhage. Neurochemistry International, 57(7), 844–850. 10.1016/j.neuint.2010.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menyhárt, Á. , Farkas, A. E. , Varga, D. P. , Frank, R. , Tóth, R. , Bálint, A. R. , … Farkas, E. (2018). Large‐conductance Ca2+‐activated potassium channels are potently involved in the inverse neurovascular response to spreading depolarization. Neurobiology of Disease, 119, 41–52. 10.1016/j.nbd.2018.07.026 [DOI] [PubMed] [Google Scholar]
- Menyhárt, Á. , Makra, P. , Szepes, B. É. , Tóth, O. M. , Hertelendy, P. , Bari, F. , & Farkas, E. (2015. Dec). High incidence of adverse cerebral blood flow responses to spreading depolarization in the aged ischemic rat brain. Neurobiology of Aging, 36(12), 3269–3277. 10.1016/j.neurobiolaging.2015.08.014 [DOI] [PubMed] [Google Scholar]
- Menyhárt, Á. , Zölei‐Szénási, D. , Puskás, T. , Makra, P. , Bari, F. , & Farkas, E. (2017. Aug 1). Age or ischemia uncouples the blood flow response, tissue acidosis, and direct current potential signature of spreading depolarization in the rat brain. American Journal of Physiology. Heart and Circulatory Physiology, 313(2), H328–H337. 10.1152/ajpheart.00222.2017 [DOI] [PubMed] [Google Scholar]
- Nedergaard, M. (1996). Spreading depression as a contributor to ischemic brain damage. Advances in Neurology, 71, 75–83. discussion 83‐4 [PubMed] [Google Scholar]
- Negulyaev, Y. A. , Vedernikova, E. A. , Kinev, A. V. , & Voronin, A. P. (1996. Jun 13). Exogenous heat shock protein hsp70 activates potassium channels in U937 cells. Biochimica et Biophysica Acta, 1282(1), 156–162. 10.1016/0005-2736(96)00055-7 [DOI] [PubMed] [Google Scholar]
- Nowak, T. S. Jr. , & Jacewicz, M. (1994. Jan). The heat shock/stress response in focal cerebral ischemia. Brain Pathology, 4(1), 67–76. [DOI] [PubMed] [Google Scholar]
- Penke, B. , Bogár, F. , Crul, T. , Sántha, M. , Tóth, M. E. , & Vígh, L. (2018. Jan 22). Heat shock proteins and autophagy pathways in neuroprotection: From molecular bases to pharmacological interventions. International Journal of Molecular Sciences, 19(1). 10.3390/ijms19010325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon, D. , & Moskowitz, M. A. (2014. Jun). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nature Reviews. Neuroscience, 15(6), 379–393. 10.1038/nrn3770 [DOI] [PubMed] [Google Scholar]
- Porchet, F. , Chioléro, R. , & de Tribolet, N. (1995). Hypotensive effect of nimodipine during treatment for aneurysmal subarachnoid haemorrhage. Acta Neurochirurgica, 137(1–2), 62–69. 10.1007/BF02188783 [DOI] [PubMed] [Google Scholar]
- Reinhart, K. M. , & Shuttleworth, C. W. (2018. Jul). Ketamine reduces deleterious consequences of spreading depolarizations. Experimental Neurology, 305, 121–128. 10.1016/j.expneurol.2018.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter, F. , Ebersberger, A. , & Schaible, H. G. (2002. Dec 13). Blockade of voltage‐gated calcium channels in rat inhibits repetitive cortical spreading depression. Neuroscience Letters, 334(2), 123–126. 10.1016/S0304-3940(02)01120-5 [DOI] [PubMed] [Google Scholar]
- Richter, F. , Lütz, W. , Eitner, A. , Leuchtweis, J. , Lehmenkühler, A. , & Schaible, H. G. (2014. Jul). Tumor necrosis factor reduces the amplitude of rat cortical spreading depression in vivo. Annals of Neurology, 76(1), 43–53. 10.1002/ana.24176 [DOI] [PubMed] [Google Scholar]
- Roe, M. S. , Wahab, B. , Török, Z. , Horváth, I. , Vigh, L. , & Prodromou, C. (2018. Jun 7). Dihydropyridines allosterically modulate Hsp90 providing a novel mechanism for heat shock protein co‐induction and neuroprotection. Frontiers in Molecular Biosciences, 5, 51 10.3389/fmolb.2018.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, C. S. , & Sherrington, C. S. (1890). On the regulation of the blood‐supply of the brain. The Journal of Physiology, 85‐158, 11. 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowitz, O. W. , Kiening, K. L. , Krajewski, K. L. , Sarrafzadeh, A. S. , Fabricius, M. , Strong, A. J. , … Dreier, J. P. (2009. Aug). Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke, 40(8), e519–e522. 10.1161/STROKEAHA.109.549303 [DOI] [PubMed] [Google Scholar]
- Scriabine, A. , Schuurman, T. , & Traber, J. (1989. May). Pharmacological basis for the use of nimodipine in central nervous system disorders. The FASEB Journal, 3(7), 1799–1806. 10.1096/fasebj.3.7.2565839 [DOI] [PubMed] [Google Scholar]
- Scriabine, A. , & van den Kerckhoff, W. (1988). Pharmacology of nimodipine. A review. Annals of the New York Academy of Sciences, 522, 698–706. 10.1111/j.1749-6632.1988.tb33415.x [DOI] [PubMed] [Google Scholar]
- Shen, P. P. , Hou, S. , Ma, D. , Zhao, M. M. , Zhu, M. Q. , Zhang, J. D. , … Feng, J.‐c. (2016. Nov). Cortical spreading depression‐induced preconditioning in the brain. Neural Regeneration Research, 11(11), 1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen, G. G. (2001. Jul). Mechanisms of spreading depression and hypoxic spreading depression‐like depolarization. Physiological Reviews, 81(3), 1065–1096. 10.1152/physrev.2001.81.3.1065 [DOI] [PubMed] [Google Scholar]
- Suzuki, T. , Ooi, Y. , & Seki, J. (2012. Apr). Infrared thermal imaging of rat somatosensory cortex with whisker stimulation. Journal of Applied Physiology (Bethesda, MD: 1985), 112(7), 1215–1222. 10.1152/japplphysiol.00867.2011 [DOI] [PubMed] [Google Scholar]
- Tomassoni, D. , Lanari, A. , Silvestrelli, G. , Traini, E. , & Amenta, F. (2008. Nov). Nimodipine and its use in cerebrovascular disease: Evidence from recent preclinical and controlled clinical studies. Clinical and Experimental Hypertension, 30(8), 744–766. 10.1080/10641960802580232 [DOI] [PubMed] [Google Scholar]
- Varga, D. P. , Puskás, T. , Menyhárt, Á. , Hertelendy, P. , Zölei‐Szénási, D. , Tóth, R. , … Farkas, E. (2016). Contribution of prostanoid signaling to the evolution of spreading depolarization and the associated cerebral blood flow response. Scientific Reports, 6, 31402 10.1038/srep31402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woitzik, J. , Hecht, N. , Pinczolits, A. , Sandow, N. , Major, S. , Winkler, M. K. , … COSBID study group (2013). Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology, 80(12), 1095–1102. 10.1212/WNL.0b013e3182886932 [DOI] [PubMed] [Google Scholar]
- Wu, J. , Guo, C. , Chen, S. , Jiang, T. , He, Y. , Ding, W. , … Gong, H. (2016). Direct 3D analyses reveal barrel‐specific vascular distribution and cross‐barrel branching in the mouse barrel cortex. Cerebral Cortex, 26(1), 23–31. 10.1093/cercor/bhu166 [DOI] [PubMed] [Google Scholar]
- Yamada, M. , Huang, Z. , Dalkara, T. , Endres, M. , Laufs, U. , Waeber, C. , … Moskowitz, M. A. (2000. Apr). Endothelial nitric oxide synthase‐dependent cerebral blood flow augmentation by L‐arginine after chronic statin treatment. Journal of Cerebral Blood Flow and Metabolism, 20(4), 709–717. 10.1097/00004647-200004000-00008 [DOI] [PubMed] [Google Scholar]
- Yenari, M. A. , Liu, J. , Zheng, Z. , Vexler, Z. S. , Lee, J. E. , & Giffard, R. G. (2005. Aug). Antiapoptotic and anti‐inflammatory mechanisms of heat‐shock protein protection. Annals of the New York Academy of Sciences, 1053, 74–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Time line of the protocols implemented. Abbreviations: 2VO, permanent, bilateral occlusion of the common carotid arteries (“two‐vessel occlusion”); Fluoro: fluorocitrate; Nimo, nimodipine; rSD, recurrent spreading depolarization; SD, spreading depolarization; SD1, first spreading depolarization in a train.
Figure S2. The impact of pharmacological treatments on evoked potentials and the coupled cerebral blood flow (CBF) response during whisker stimulation after bilateral common carotid artery occlusion (2VO) in Series 2. A, Representative local field potential (LFP) traces display somatosensory evoked field potentials (EFPs) during whisker stimulation. Traces in front of light gray background were taken from animals control to fluorocitrate treatment, while the trace in front of dark gray background represents EFPs under fluorocitrate treatment. B, The peak amplitude of evoked potentials. C, Traces (each is the mean of 4 stimulations in an animal representative of each condition) show the CBF response to whisker stimulation. D, The amplitude of the CBF response to whisker stimulation. The base of each bar in the chart is set to the CBF level directly preceding whisker stimulation. In B and D, data are given as mean ± stdev; sample size (i.e. the number of events analyzed) is indicated in each bar. Statistical analysis relied on a two‐way analysis of variance (ANOVA) paradigm (factors: 2VO and treatment). The level of significance was defined as p < 0.05*. A Games‐Howell post hoc test was applied for group comparisons (p < 0.05* vs. vehicle; p < 0.05# vs. control). Significance symbols under the bars represent differences of baseline, while symbols over the bars stand for the difference between functional hyperemia amplitude.