

Abstract
Background and Purpose
An alteration in the communication between the innate and adaptive immune cells is a hallmark of ulcerative colitis (UC). Semaphorin‐3E (SEMA3E), a secreted guidance protein, regulates various immune responses.
Experimental Approach
We investigated the expression of SEMA3E in colonic biopsies of active UC patients and its mechanisms in Sema3e −/− mice using an experimental model of UC.
Key Results
SEMA3E level was decreased in active UC patients and negatively correlated with pro‐inflammatory mediators. Colonic expression of SEMA3E was reduced in colitic Sema3e +/+ mice, and recombinant (rec‐) Plexin‐D1 treatment exacerbated disease severity. In vivo rec‐SEMA3E treatment restored SEMA3E level in colitic Sema3e +/+ mice. In Sema3e −/− mice, disease severity was increased, and rec‐SEMA3E ameliorated these effects. Lack of Sema3e increased the expression of CD11c and CD86 markers. Colitic Sema3e −/− splenocytes and splenic CD11c+ cells produced more IL‐12/23 and IFN‐γ compared to Sema3e +/+, and rec‐SEMA3E reduced their release as much as NF‐κB inhibitors, whereas an NF‐κB activator increased their production and attenuated the effect of rec‐SEMA3E. Colitic Sema3e −/− splenic CD11c+/CD4+CD25− T‐cell co‐cultures produced higher concentrations of IFN‐γ and IL‐17 when compared to colitic Sema3e +/+ splenic cell co‐cultures, and rec‐SEMA3E decreased these effects. In vitro, anti‐IL‐12p19 and ‐12p35 antibodies and rec‐IL‐12 and ‐23 treatment confirmed the crosstalk between CD11c+ and CD4+CD25− T‐cells.
Conclusion and Implications
SEMA3E is reduced in colitis and modulates colonic inflammation by regulating the interaction between CD11c+ and CD4+CD25− T‐cells via an NF‐κB‐dependent mechanism. Thus, SEMA3E could be a potential therapeutic target for UC patients.
Abbreviations
- APC
antigen‐presenting cells
- CD
cluster of differentiation
- DAI
disease activity index
- DSS
dextran sulfate sodium
- IBD
inflammatory bowel diseases
- rec
recombinant
- Sema3E
semaphorin‐3E
- UC
ulcerative colitis
1.
What is already known
Semaphorin‐3E (SEMA3E) is a secreted membrane‐bound protein, which regulates cell trafficking and immune cell‐to‐cell interactions.
IL‐12/23 genes are implicated in the pathogenesis of ulcerative colitis and may be a potential therapeutic target.
What this study adds
SEMA3E is expressed in the colonic mucosa and reduced in patients with active ulcerative colitis and in experimental‐induced colitis.
Pharmacological manipulations or deletion of Sema3e regulate experimental colitis by promoting pro‐inflammatory activity of CD11c+ cells via the NF‐κB‐dependent pathway.
What is the clinical significance
These findings may expedite the development of novel therapeutic strategies for UC patients.
Functional analysis of SEMA3E may lead to a better understanding of immune cell regulation mechanisms in human intestine.
2. INTRODUCTION
Inflammatory bowel diseases (IBD), including Crohn's disease and ulcerative colitis (UC), are idiopathic gastrointestinal diseases characterized by a chronic inflammation of the gastrointestinal tract. The incidence of IBD has become a growing problem with an increasing number of patients reported in Western and Asian countries (Kaplan, 2015).
Genome‐wide association studies have identified IL‐12 and IL‐23 genes as being involved in the pathogenesis of UC (Franke et al., 2010; Rioux et al., 2007). IL‐12 and IL‐23 are considered as early pro‐inflammatory signals in response to immune activation and are mainly produced by clusters of differentiation (CD)11c + cells, which are known to accumulate within the inflamed mucosa of patients with UC (Bates & Diehl, 2014; Chin & Parkos, 2006; Hart et al., 2005; Steinbach & Plevy, 2014; Woodruff, Masterson, Fillon, Robinson, & Furuta, 2011). IL‐12 and IL‐23 are composed of two subunits p40 and p35, and p40 and p19 respectively (Steinman, 2010). CD11c+‐IL‐12/23 production is a critical component of the innate and adaptive immune responses (Goodall et al., 2010); inappropriate CD11c+‐IL‐12/23 production favours pro‐inflammatory T‐cell responses with preferential priming and proliferation of effector T‐cells towards a Th1/Th17 profile (Kaiko, Horvat, Beagley, & Hansbro, 2008; Tas et al., 2005). Recently, the anti‐IL‐12p40 monoclonal antibody (Ustekinumab™) has demonstrated good clinical efficacy in a group of UC patients resistant to anti‐TNF therapy (Sandborn et al., 2012) demonstrating that blocking the communication between CD11c+ and T‐cells can result in a decrease in the activity of the IL‐12/23 pro‐inflammatory pathway (Fitzpatrick, 2012). Among various intracellular pathways that activate CD11c+ cell functions, NF‐κB pathway regulates IL‐12/23 production (Kaiko et al., 2008; Rescigno, Martino, Sutherland, Gold, & Ricciardi‐Castagnoli, 1998; Tas et al., 2005), and in active UC, activation of NF‐κB is increased in lamina propria mononuclear cells; therefore, inhibition of the NF‐κB pathway has been proposed as a therapeutic strategy (Eissa, Hussein, Kermarrec, Elgazzar, et al., 2017; Eissa & Ghia, 2015; Eissa, Hussein, Hendy, Bernstein, & Ghia, 2018).
Semaphorins (SEMA) are secreted and membrane‐bound proteins that regulate a wide range of biological functions, from tissue morphogenesis to immune response regulation (Kruger, Aurandt, & Guan, 2005). The semaphorin family is composed of eight classes including semaphorin‐3E (SEMA3E), which is involved in cell trafficking and immune cell‐to‐cell interactions (Choi et al., 2008; Takamatsu et al., 2010) and controls the functions of CD11c+ (Movassagh, Shan, Mohammed, et al., 2017). SEMA3E is also implicated in the pathogenesis of many chronic inflammatory diseases, including rheumatoid arthritis (Mangasser‐Stephan, Dooley, Welter, Mutschler, & Hanselmann, 1997), asthma (Movassagh, Shan, Mohammed, et al., 2017), atherosclerosis (Wanschel et al., 2013), adipose tissue inflammation, and insulin resistance (Shimizu et al., 2013). SEMA3E mediates anti‐proliferative and anti‐migratory functions by binding to the plexin‐D1 receptor (Gu et al., 2005), which is expressed by CD11c+ cells (Worzfeld & Offermanns, 2014).
Several animal models of IBD have been developed to characterize the molecular mechanisms of the pathophysiology of UC (Raper, 2000). Dextran sulfate sodium (DSS)‐induced colitis is widely used as an animal model of intestinal injury/repair (Eissa et al., 2016), as the DSS causes intestinal mucosal injury, exposing CD11c+ immune cells of the lamina propria and the spleen (Chassaing, Aitken, Malleshappa, & Vijay‐Kumar, 2014; Ji et al., 2014; Munyaka et al., 2014; Perše & Cerar, 2012) to various antigens to initiate the inflammatory process.
As yet, no studies have investigated the immune‐modulatory role of SEMA3E during the development of intestinal inflammation. We hypothesized that SEMA3E regulates intestinal inflammation through the modulation of the immune response. To verify our hypothesis, we investigated (a) clinically, SEMA3E levels and their correlation with various inflammatory mediators in colonic biopsies from UC patients and healthy individuals; (b) experimentally, using wild‐type (Sema3e +/+ ) or SEMA3E knockout mice (Sema3e −/−) and exogenous recombinant (rec‐) SEMA3E protein or rec‐PLXD1 soluble receptor treatment, the effect of SEMA3E during the development of DSS‐induced colitis; and (c) the effect of SEMA3E on the functions of splenic CD11c+ cell and CD4+CD25− T‐cells. We found that a decrease in SEMA3E level/activity in active UC and experimental UC colitis is associated with an alteration in the functions of splenic CD11c+ and CD4+CD25− T‐cell activity.
3. METHODS
3.1. UC patients and healthy individuals
This study was approved by the University of Manitoba Health Research Ethics Board (HS14878 [E]). Active UC patients (n = 7) and healthy individuals (n = 9) were recruited in the section of Gastroenterology at the University of Manitoba. The consent was obtained. The healthy individuals enrolled had not been diagnosed and/or treated for any inflammatory disease in the past. Colonic biopsies were taken within the active inflammatory areas of the distal colon in active UC patients and within non‐inflamed but comparable sites in healthy individuals. Status discrimination was established by colonoscopy and pathological assessment. Biopsies were used immediately for RNA extraction and further gene expression analyses. Inclusion criteria included a confirmed active UC endoscopic diagnosis and age ≥ 18 years. All concomitant IBD therapies, including 5‐amino salicylates, corticosteroids, and methotrexate, were permitted in active UC patients. Healthy control individuals had no history of abdominal afflictions especially IBD. The individuals in the control group were not on any regular medication at the time of the study. Mean age for control and active UC groups was 50.30 ± 12.86 and 39.90 ± 12.98 (P < 0.05) respectively. The biopsies were randomly collected and coded by Dr. Bernstein, and further mRNA analysis was performed blind using coded samples by Dr. Eissa.
3.2. Animals and ethics statement
Mice heterozygous for SEMA3E expression (Sema3e +/−) on a mixed C57BL/6 background were used to generate Sema3e +/+ and Sema3e −/−, and Sema3e −/− was used to generate littermates. The immunophenotypic proprieties of Sema3e −/− have been described previously (Movassagh, Shan, Mohammed, et al., 2017). Male Sema3e −/− (20–25 g, 6–8 weeks old; IMSR Cat# KOMP:VG14098‐1‐Vlcg, RRID:IMSR_KOMP:VG14098‐1‐Vlcg) were obtained from the animal facility at the University of Manitoba and used according to the guidelines of the Canadian Council on Animal Care in science. Mice were housed in a cage (three mice per ventilated cage) at the specific pathogen‐free Basic Medical Science Building facility at the University of Manitoba. Mice were housed with free access to pellet food and water in plastic ventilated cages at 21 ± 2°C, 50–55% humidity and kept on a 12 h light/dark cycle under specific pathogen free conditions. All experiments were performed in strict accordance with the recommendations approved by the University of Manitoba Animal Ethics Committee (Protocol # 15‐010). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010), the editorial on reporting animal studies (McGrath & Lilley, 2015), and the recommendations made by the British Journal of Pharmacology.
3.3. Experimental models of colitis and treatment with recombinant mouse SEMA3E‐Fc chimera protein, CF (rec‐SEMA3E) or recombinant mouse PLXD1 soluble receptor (rec‐PLXD1)
Murine models of colitis are key tools used to investigate the pathophysiology of intestinal inflammation (Auletta et al., 2018; Rabbi et al., 2017). Dextran sodium sulfate (DSS)‐induced colitis is the most common animal model of IBD used with over 4,000 entries in PubMed (Eissa et al., 2016); therefore, DSS‐induced colitis was used in this study (PubMed, RRID:SCR_004846). Mice were randomly assigned to different experimental groups. Mice received daily i.p. injection of recombinant mouse SEMA3E‐Fc chimera protein (rec‐SEMA3E; Movassagh Shan, Duke‐Cohan, et al., 2017) or recombinant mouse PLXD1 soluble receptor (rec‐PLXD1; 10 μg·kg−1·day−1 in 1% sterile PBS [i.p.]; R&D Systems, Inc., MN, USA) for 6 days, while control groups received 1% PBS with added human IgG Fc protein (Vehicle; AG100, Millipore Sigma, USA; Baldo, 2015; Movassagh, Shan, Duke‐Cohan, et al., 2017). Twenty‐four hours after the first injection with rec‐SEMA3E or rec‐PLXD1 or vehicle, colitis was induced by adding 5% (wt:vol) of DSS (MW, 40 kDa; MP Biomedicals, Soho, OH) in the drinking water for 5 days (n = 6–8; Okayasu et al., 1990). Non‐colitic groups were time‐matched and received only regular drinking water (n = 6). Mean DSS consumption was noted per cage each day.
3.4. Assessment of DSS‐induced colitis severity
The severity of colitis was assessed by reporting weight loss (0: no loss; 1: 5–10%; 2: 10–15%; 3: 15–20%; and 4: ≥20% of weight loss), stool consistency (from 0: normal stool to 4: diarrhoea), and bleeding (from 0: no blood to 4: gross blood) evaluated with Hemoccult II test (Beckman Coulter, Oakville, Canada). These experimental inflammatory parameters were combined into a disease activity index (DAI), calculated every day starting from the day of induction up to the day the mice were killed (Cooper, Murthy, Shah, & Sedergran, 1993). After 6 days of treatment, mice were anaesthetized using isoflurane and then killed by cervical dislocation, the abdominal cavity was opened, and colonic distension, fluid content, hyperaemia, and erythema were recorded. The colon was removed, opened longitudinally to assess directly the macroscopic damage, and macroscopic scoring was evaluated as previously described (Cooper et al., 1993). To obtain histopathological scores, formalin‐fixed (Sigma, Mississauga, Canada) colon segments from the splenic flexure were paraffin‐embedded (Sigma, Mississauga, Canada), and 3‐mm sections were stained using haematoxylin–eosin (Sigma, Mississauga, Canada) as described previously(Eissa et al., 2018). The disease activity index (DAI) was not assessed blindly, while the histological score was carried out in a blinded manner by two investigators.
3.5. Protein assays and ELISA
The colonic samples were coded, and the levels of cytokines were quantified blindly. Briefly, the day the mice were killed, colonic samples were taken, coded, immediately frozen in liquid nitrogen, and then stored at −80°C for further experiments. The day of the quantification, 1 ml of cool lysis buffer was added per tube (stock solution containing 50 mM Tris PH 7.5, 200 mM NaCl, 1% Tween 20, 0.2% NP40, 1 mM NaF in ddH2O, supplemented freshly with 2 mM PMSF, 1 mM activated vanadate, 50 mM β‐glycerol PO4, and one tablet of Roche Complete Mini EDTA‐free protease inhibitors per 10 ml), and samples were homogenized in the buffer by sonication. After 30 min of digestion on ice (vortexing every 10 min), the tissue homogenates were centrifuged at maximum speed for 10 min at 4°C, and supernatants were collected for protein quantification by Bradford assay. Protein levels of colonic pro‐inflammatory cytokines (TNF‐α, IL‐6, IL‐1β and IL‐12p70, IL‐12p40, IL‐23, IL‐17, and IL‐4) were quantified using ELISA according to the manufacturer's instructions (R&D Systems). Briefly, ELISA plates were first coated with the primary antibody prepared in coating buffer (50 μl per well) overnight at 4°C. The day after, the blocking buffer was added to the plates (75 μl per well) for 2 hr at 37°C. After the plates had been washed, samples and standards prepared in dilution buffer were added to the wells overnight at 4°C. After a wash, the secondary antibody prepared in dilution buffer was added to the wells (50 μl per well). The plate was washed once before the addition of 50 μl of dilution buffer complemented with serum amyloid P component per well for 45 min at 37°C. After one wash, 50 μl of substrate buffer complemented with p‐nitrophenyl phosphate (one tablet for 5 ml) was added per well for 30 min up to 90 min before reading the absorbance with the Epoch Microplate Spectrophotometer (Biotek Instruments Inc., Winooski, VT, USA).
3.6. Splenocyte cultures and isolation of splenic CD11c+ cells
Spleens were collected from colitic or non‐colitic mice, immediately after they were killed, and cut into small pieces in 4 ml warmed RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 25 mg·ml−1 gentamicin, 2 mM l‐glutamin, and 10% deactivated FBS and with 2 mg·ml−1 collagenase D (Roche Diagnostics, Meylan, France) added for digestion 30 min at 37°C. For the last 5 min, 5 mM EDTA (Sigma) was added to disrupt the antigen‐presenting cells (APC)–T‐cell complexes (Kamath et al., 2000), and the cell lysate was filtered (100 μm) before digestion for 30 min at room temperature on a shaker with ammonium‐chloride‐potassium lysing buffer (150 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA; Life Technologies) to remove the red blood cells. After incubation or not with CD11c+ microbeads (Miltenyi Biotec, Auburn, CA) for 15 min at 48°C and one wash, cells were resuspended in a cell separation buffer (Dulbecco's PBS without Ca2+ and Mg2+ containing 2% FBS and 2‐mM EDTA; Life Technologies, California, US) and passed through two magnetic columns (Miltenyi Biotec, Bergisch Gladbach, Germany) consecutively for a positive selection. The yield and viability of CD11c+ were >95% as previously described (Merad, Sathe, Helft, Miller, & Mortha, 2013; Munyaka et al., 2014).
3.6.1. Release of cytokines in splenocytes and splenic CD11c+ cells co‐culture system
Splenocytes and splenic CD11c+ cells were isolated from the different groups and were cultured for 24 hr in complete RPMI in 12‐well plates at 1 × 106 cells·well−1 in a humidified 5% CO2 incubator at 37°C. BAY 11–7082 (NF‐κB inhibitor) or betulinic acid (NF‐κB activator; R&D Systems) was added, or not, to splenic CD11c+ cells culture for 1.5 and 4 hr, respectively, at 10 μM. Measurement of IL‐12p70, 40, IL‐23, and IFN‐γ in the supernatants of splenocytes and splenic CD11c+ cells culture was performed by ELISA (R&D Systems).
3.6.2. Co‐culture of splenic CD11c+ cells with CD4+CD25− T‐cells and protein measurement
Naïve splenic CD4 + T‐cells were isolated from non‐colitic mice. The yield of CD4+CD25− cells was >93%. CD11c+ cells isolated from colitic mice receiving the different treatments were cultured for 24 hr before being washed with medium and co‐cultured with CD4+CD25− T‐cells isolated from naïve non‐colitic mice at a ratio of 1∶10 (CD11c+: T‐cells) in plates coated with 10 μg·ml−1 of anti‐CD3 (R&D Systems Cat# MAB4841, RRID:AB_358426) and 2 μg·ml−1 of anti‐CD28 (R&D Systems Cat# MAB4831, RRID:AB_2073857; Goudin et al., 2016; Wong, Chen, Gau, Yen, & Suen, 2016). In neutralization experiments using monoclonal antibodies (mAb), cells were treated with 10 μg·ml−1 of mouse anti‐IL‐12p35 (AF1619, R&D Systems) or anti‐IL‐23p19 (MAB6688, R&D Systems), neutralizing mAb to block the endogenous sources of these cytokines. In the stimulation experiments, cells were treated with 25 ng·ml−1 recombinant mouse IL‐23 protein (1887‐ML‐010, R&D Systems) or recombinant mouse IL‐12p70 (419‐ML‐010, R&D Systems). Supernatants were collected after 24 hr, and IFN‐γ, IL‐4, and IL‐17 levels were quantified using ELISA (R&D Systems).
3.7. Gene expression analysis
RNA was extracted from tissues using TRIzol® Plus RNA Purification Kit (Ambion, NY, USA) and reverse transcribed into cDNA using SuperScript VILO cDNA Synthesis Master Mix (Invitrogen, Grand Island, NY, USA). Quantitative real‐time PCR in the Roche Light Cycler 96 Real‐Time System (Roche Diagnostics, Laval, Québec, Canada) was used to measure gene expression, using SYBR green master mix (Life Technologies, Burlington, ON). Relative gene expression was calculated using the differences in the threshold cycle (ΔCt) number between the reference gene and the optimal target gene (Eissa et al., 2016; Eissa, Kermarrec, et al., 2017). Differences in the threshold cycle (ΔCt) number between the target genes and mouse eukaryotic elongation factor 2 and human TATA‐box binding protein (optimal reference genes) were used to normalize expression (Eissa et al., 2018). Human and mouse primer sequences for the target gene markers are provided in Tables 1 and 2.
Table 1.
Human primers sequences
| Gene name | Forward | Reverse |
|---|---|---|
| IL6 | CCTGAACCTTCCAAAGATGGC | TTCACCAGGCAAGTCTCCTCA |
| IL‐1β | TTCGACACATGGGATAACGAGG | TTTTTGCTGTGAGTCCCGGAG |
| TNF‐α | GAGGCCAAGCCCTGGTATG | CGGGCCGATTGATCTCAGC |
| SEMA3E | GTTTGCTGGACTCTACAGTGAC | CTTTCAACAGACGCTCATCGT |
| CD86 | CAGACCACATTCCTTGGATCA | CCGCTTCTTCTTCTTCCATTTC |
| TBP | CCCGAAACGCCGAATATAATCC | AATCAGTGCCGTGGTTCGTG |
| CD11C | ACTCAGATCGGCTCCTACTT | TCGGGTCTGCTCGTAGTAAT |
| IL12A | ATTCCAGAGAGACCTCTTTCATAAC | CCACCTGGTACATCTTCAAGTC |
| IL23A | CAGGTCACTATTCAATGGGATGC | GCAGTTCTTAATTGCTGCTTGG |
Table 2.
Mouse primers sequences
| Gene | Forward | Reverse |
|---|---|---|
| Sema3e | TCAGTGACGGCTACAGAGAGA | CACACTCATTTGCGTCTTTTCC |
| CD86 | TTACGGAAGCACCCACGATG | ACTACCAGCTCACTCAGGCT |
| CD11c | CCAAGACATCGTGTTCCTGATT | ACAGCTTTAACAAAGTCCAGCA |
| Eef2 | TGTCAGTCATCGCCCATGTG | CATCCTTGCGAGTGTCAGTGA |
3.8. Statistical analysis
Data are expressed as mean ± SEM. Data distribution was verified, and data with a non‐parametric distribution were log‐transformed prior to analyses. Student's unpaired t test, one‐way and two‐way ANOVA followed by multiple comparisons post hoc test were applied (F value < 0.05) to compare between the experimental groups. A post hoc test was run only if F achieved P < 0.05 and there was no significant variance inhomogeneity (Curtis et al., 2015; Curtis et al., 2018). Correlation analysis was performed using Spearman's correlation test. The significance level was adjusted at 0.05, and values for P < 0.05 were considered statistically significant. GraphPad Prism (version 7.0a MAC; GraphPad Software, Inc., La Jolla, CA) was used for statistical analysis and creating figures (GraphPad Prism, RRID:SCR_002798). www.dssresearch.com was used to calculate the appropriate number of animals (8) with 85% of statistical power. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2015; Curtis et al., 2018).
3.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017; Alexander, Kelly et al., 2017).
4. RESULTS
4.1. SEMA3E is reduced in active UC patients and negatively correlated with pro‐inflammatory markers
To investigate whether SEMA3E level is altered in patients with active UC, we quantified its mRNA levels in colonic biopsies. We found that the mRNA expression levels of SEMA3E were significantly reduced by 14.6‐fold in the patients with active UC compared to the healthy individuals (Figure 1a). Furthermore, SEMA3E was negatively correlated with pro‐inflammatory mediators IL6 (r = −0.59, P < 0.05), TNF‐α (r = −0.51, P < 0.05), and IL1β (r = −0.45, P < 0.05; Figure 1b). Moreover, SEMA3E was negatively correlated with the APC markers CD86 (r = −0.57, P < 0.05) and IL23 (r = −0.51, P < 0.05). A trend was also detected for the cytokine, IL12 (r = −0.45, P > 0.05), but no changes were visible for CD11c (r = −0.3607, P > 0.05; Figure 1c).
Figure 1.
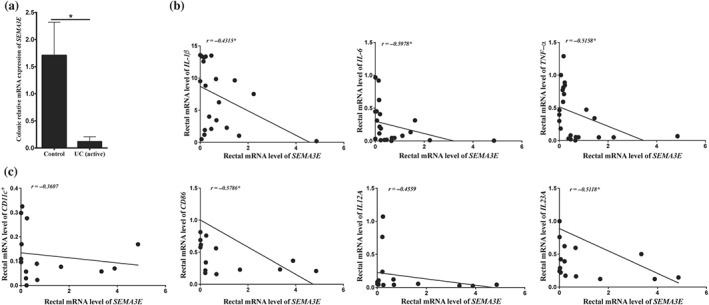
SEMA3E is reduced in patients with active ulcerative colitis (UC) patients and negatively correlated with immune markers. (a) mRNA levels of SEMA3E in active UC patients (n = 7) and healthy individuals (n = 9). Each value represents mean ± SEM. (b) Correlation between SEMA3E and pro‐inflammatory cytokines (IL1β, IL6, and TNF‐α). (c) Correlation between SEMA3E, CD11c + and CD86, or IL12A and IL23A. mRNA levels were quantified using RT‐qPCR. Student's t test and Spearman's correlation were applied (*P < 0.05)
4.2. Sema3e is reduced during the progression of colitis and associated with the expression of CD11c+ maturation markers
In order to confirm our observation in humans, an active UC experimental colonic inflammation was induced in wild‐type (Sema3e +/+) and Sema3e‐knockout (Sema3e −/−) mice by administration of DSS in the drinking water. Consistently with UC patients (Figure 1a), DSS‐induced colitis in Sema3e +/+ mice resulted in a reduction in Sema3e relative mRNA level by 3.5‐fold change when compared to non‐colitic Sema3e +/+ mice (Figure 2a). As expected, the relative Sema3e mRNA level was equal to zero in either non‐colitic or colitic Sema3e −/− mice. Furthermore, the lack of SEMA3E (Sema3e −/−) in colitic mice increased the colonic mRNA expression of CD86 and CD11c by approximately fivefold (Figure 2b,c).
Figure 2.
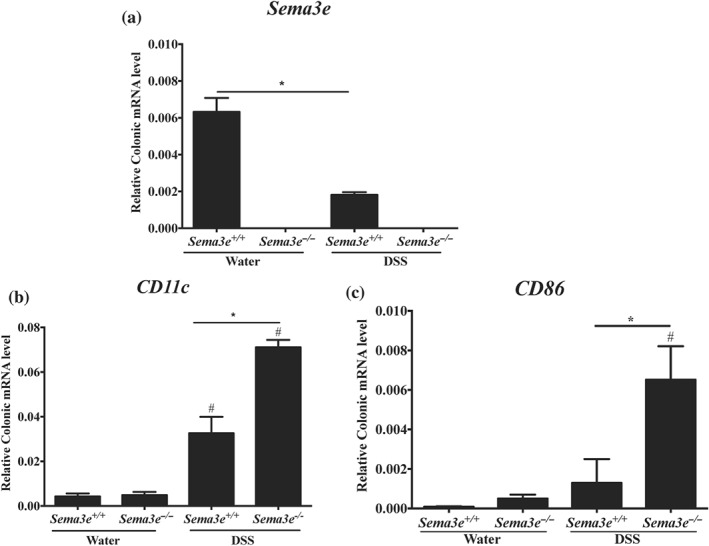
Sema3e is reduced during the progression of acute colitis and is associated with the expression of immune markers. Colon samples were obtained from Sema3e +/+ and Sema3e −/− C57Bl/6 mice. Colitic mice were treated with 5% dextran sulfate sodium (DSS) in the drinking water for 5 days; non‐colitic mice received only drinking water and served as control groups. Colonic mRNAs levels of (a) SEMA3E, (b) CD11c, and (c) CD86. mRNA levels were quantified using RT‐qPCR. Each value represents mean ± SEM from n = 6 per group (*P < 0.05). # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied
4.2.1. Reduced availability of SEMA3E in the gut increases the severity of experimental colitis
To investigate further the potential effect of the depletion of SEMA3E during the progression of colonic inflammation, we decreased the availability of SEMA3E through the administration of rec‐PLXD1 in a model of DSS‐induced colitis Sema3e +/+ mice. We found that administration of rec‐PLXD1 significantly aggravated the onset and severity of DSS‐induced colitis and was associated with an increase in DAI (Figure 3a), macroscopic score (Figure 3b), pro‐inflammatory mediators (IL‐6, IL‐1β, and TNF‐α; Figure 3c–e), and histological scores (Figure 4a–b) when compared with non‐colitic groups. No significant changes were observed between the non‐colitic mice groups.
Figure 3.
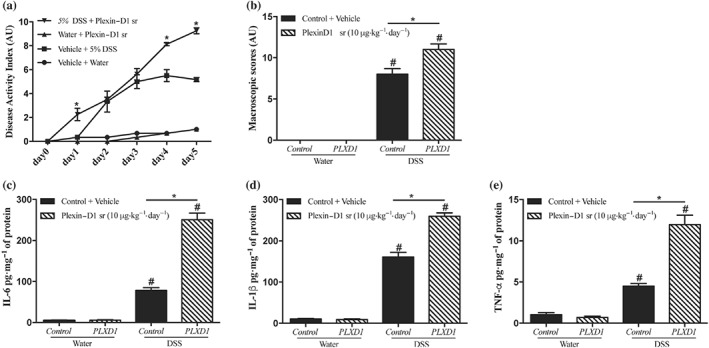
Reduced availability of SEMA3E in the gut increases the severity of experimental colitis. Colon were obtained from C57Bl/6 colitic mice treated with dextran sulfate sodium (DSS) 5% in the drinking water for 5 days or from non‐colitic mice (water). Sema3e +/+ mice received daily i.p. injection of plexin‐D1 soluble receptor (PLXD1; 10 μg·kg−1·day−1, R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) starting 1 day before DSS administration and lasted for 6 days. (a) Disease activity index and (b) macroscopic score. Colonic protein levels of pro‐inflammatory cytokines (c) IL‐6, (d) IL‐1β, and (e) TNF‐α. Data represent mean ± SEM (n = 6). *P < 0.05 and # refers to a significance when compared with control groups. One‐way ANOVA followed by multiple comparison test was applied
Figure 4.
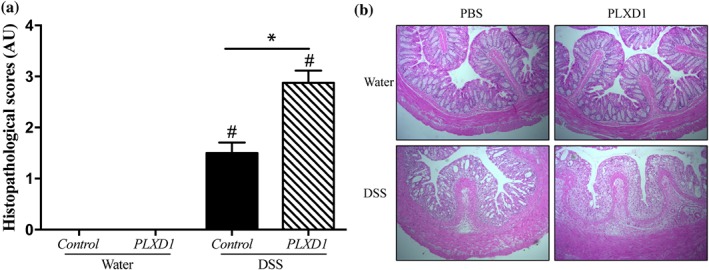
Reduced availability of SEMA3E increases the colonic histological damage. Colon were obtained from C57Bl/6 colitic mice treated with dextran sulfate sodium (DSS) 5% in the drinking water for 5 days or from non‐colitic mice (water). Sema3e +/+ mice received daily i.p. injection of plexin‐D1 soluble receptor (PLXD1; 10 μg·kg−1·day−1, R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) starting 1 day before DSS administration and lasted for 6 days. (a) Histological score; (b) haematoxylin–eosin staining of colonic tissues at 100× magnification power. Data represent mean ± SEM (n = 6). *P < 0.05 and # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied
4.3. Lack of SEMA3E exacerbates disease severity and the release of colonic pro‐inflammatory mediators in experimental colitis
Then, we investigated the functional consequences of the reduced availability of SEMA3E during DSS‐induced colitis using Sema3e −/− mice compared to Sema3e +/+ mice (Figure 5a). Consistent with the colonic mRNA level (Figure 2a), we observed that experimental colonic inflammation in Sema3e +/+ mice resulted in a reduction in colonic Sema3e protein levels (Figure 5a). Then, deletion of Sema3e increased the onset and severity of DSS colitis as indicated by an increase in the DAI, macroscopic, and histological scores in Sema3e −/− mice when compared with Sema3e +/+ mice (Figures 5b,c and 6a,b). Furthermore, pro‐inflammatory cytokines (IL‐6, IL‐1β, and TNF‐α [Figure 5d–f] and IL‐12p40, IL‐12p70, IL‐23, INF‐γ, and IL‐17 [Figure 7a–e]) were significantly increased in colitic Sema3e −/− mice when compared with Sema3e +/+ mice. No changes were reported in IL‐4 levels (Figure 7f). The colonic pro‐inflammatory cytokines, IL‐12p40, IL‐12p70, IL‐23, INF‐γ, and IL‐17 levels, did not show any difference in non‐colitic groups that received drinking water (Figures 5d–f and 7a–e).
Figure 5.
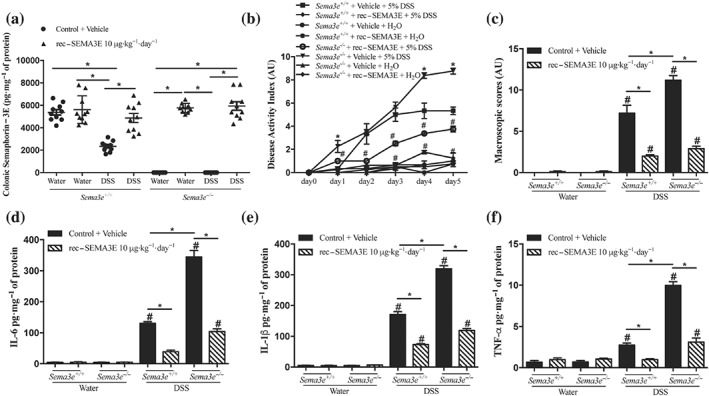
The lack of SEMA3E aggravates acute dextran sulfate sodium (DSS)‐induced colitis, and treatment with SEMA3E improves disease severity. Sema3e +/+ and Sema3e −/− mice received 5% DSS in the drinking water to induce colitis, or drinking water, and were killed on day 5 post‐DSS. Mice received daily i.p. injection of SEMA3E recombinant protein (rec‐SEMA3E; 10 μg·kg−1·day−1; R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) for 6 days, starting 1 day before colitis induction. (a) Colonic protein levels of SEMA3E, (b) disease activity index, and (c) macroscopic score. Colonic expression of pro‐inflammatory cytokines (d) IL‐6, (e) IL‐1β, and (f) TNF‐α. Data represent mean ± SEM (n = 6, *P < 0.05). # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied
Figure 6.
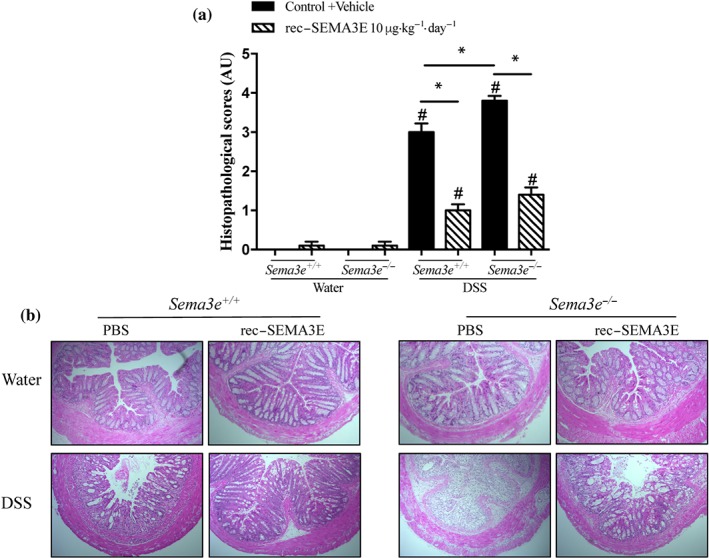
The lack of SEMA3E aggravates acute dextran sulfate sodium (DSS)‐induced colitis and treatment with SEMA3E improves disease severity. Sema3e +/+ and Sema3e −/− mice received 5% DSS in the drinking water to induce colitis, or drinking water, and were killed on day 5 post‐DSS. Mice received daily i.p. injection of SEMA3E recombinant protein (rec‐SEMA3E; 10 μg·kg−1·day−1; R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) for 6 days, starting 1 day before colitis induction. (a) histological scores (Arbitrary Unit, AU) and (b) Haematoxylin–eosin staining of colonic tissues at 100× magnification power. Data represent mean ± SEM (n = 6, *P < 0.05). # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied
Figure 7.
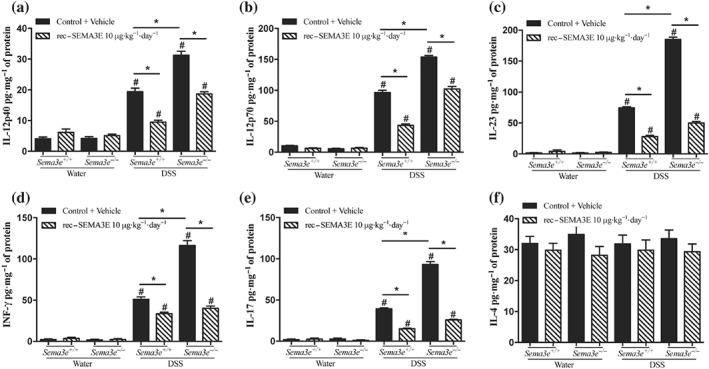
In vivo SEMA3E treatment regulates colonic expression of cytokines in acute dextran sulfate sodium (DSS)‐induced colitis. Sema3e +/+ and Sema3e −/− mice received 5% DSS in the drinking water to induce colitis, or water control, and were killed on day 5 post‐DSS. Mice received daily i.p. injection of recombinant (rec‐) SEMA3E protein (10 μg·kg−1·day−1; R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) for 6 days, starting 1 day before colitis induction. Colonic protein levels of (a) IL‐12p40, (b) IL‐12p70, (c) IL‐23, (d) INF‐γ, (e) IL‐17, and (f) IL‐4. Colonic cytokines were quantified using ELISA. Data represent mean ± SEM (n = 6, *P < 0.05). # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied at significance level of 0.05
4.4. Administration of rec‐SEMA3E ameliorates intestinal inflammation and decreases the release of colonic pro‐inflammatory mediators in experimental colitis
To verify that the exacerbation of the inflammatory cascades in colitic Sema3e −/− mice was a result of the absence of Sema3e, Sema3e −/− mice were administered rec‐SEMA3E to restore Sema3e level. rec‐SEMA3E treatment significantly increased the colonic levels of SEMA3E in colitic Sema3e −/− and Sema3e +/+ mice (Figure 5a), and this treatment reduced the progression and the severity of DSS‐induced colitis in Sema3e −/− and Sema3e +/+ mice (Figures 5b,c and 6a,b). Rec‐SEMA3E treatment significantly decreased colonic pro‐inflammatory cytokines (IL‐6, IL‐1β, and TNF‐α [Figure 5d–f] and IL‐12p40, IL‐12p70, IL‐23, INF‐γ, and IL‐17 [Figure 7a,e]) in colitic Sema3e −/− and Sema3e +/+ mice. No changes were reported in IL‐4 levels (Figure 7f). In non‐colitic mice groups treated with rec‐SEMA3E, no significant changes were reported in terms of disease severity (data not shown) and levels of pro‐inflammatory mediators (Figures 5c–f, 6a,b, and 7a–e).
4.5. In vivo SEMA3E treatment regulates splenocytes’ cytokine production and splenic CD11c+ cells through the NF‐κB pathway
Splenic CD11c+ antigen presenting cells (APC) play a critical role in T‐cell activation during the development of intestinal inflammation (Ji et al., 2014; Munyaka et al., 2014). To gain mechanistic insight into how in vivo rec‐SEMA3E treatment regulates the development of colitis, we next investigated the immunomodulatory role of SEMA3E on the functions of splenocytes and splenic CD11c+ cells. Because, under water, the inflammatory markers in the colonic samples and splenocytes of Sema3e +/+ mice were equivalent to those in Sema3e −/− mice, we then focused on the colitic mice. Splenocytes isolated from colitic Sema3e −/− mice demonstrated a significant increase in the release of IFN‐γ and IL‐12p40 when compared with splenocytes isolated from colitic Sema3e +/+ mice (Figure 8a,b), and in vivo rec‐SEMA3E treatment decreased the release of IFN‐γ and IL‐12p40 in colitic Sema3e −/− and Sema3e +/+ mice (Figure 8a,b).
Figure 8.
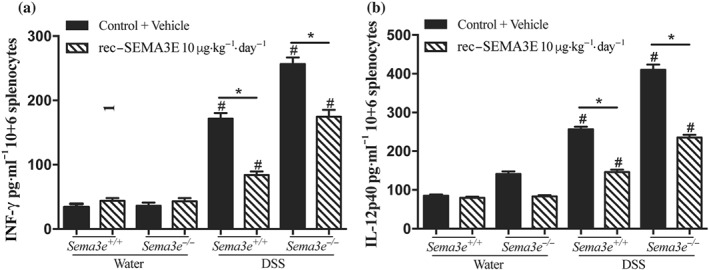
In vivo SEMA3E treatment regulates splenocytes cytokines production in acute dextran sulfate sodium (DSS)‐induced colitis. Sema3e +/+ and Sema3e −/− mice received 5% DSS in the drinking water to induce colitis, or water vehicle, and were killed on day 5 post‐DSS. Mice received daily i.p. injections of recombinant (rec‐) SEMA3E protein (10 μg·kg−1·day−1; R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) for 6 days, starting 1 day before colitis induction. Supernatants were harvested, and cytokine levels were quantified using ELISA. (a) IFN‐γ; (b) IL‐12p40. Data represent mean ± SEM (n = 6, *P < 0.05). # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied at significance level 0.05
Splenic CD11c+ cells isolated from colitic Sema3e −/− mice released more IL‐12p70, IL‐12p40, and IL‐23 when compared to splenic CD11c+ cells isolated from colitic Sema3e +/+ mice (Figure 9a–c), and in vivo rec‐SEMA3E treatment decreased the release of IL‐12p70, IL‐12p40, and IL‐23. BAY 11‐7082‐treated CD11c+ cells isolated from colitic Sema3e −/− and Sema3 +/+ mice produced less IL‐12p70, IL‐12p40, and IL‐23 when compared with controls (Figure 9a–c). Conversely, betulinic acid increased the production of these cytokines in Sema3e +/+ mice, but no further effect was detected in Sema3e −/− mice in relation to IL‐12p40 and IL‐23 levels (Figure 9a,b). In colitic Sema3e −/− and Sema3e +/+ mice, the beneficial effect of the in vivo rec‐SEMA3E treatment was completely abolished in the presence of betulinic acid, with a level of cytokine comparable to betulinic acid alone (Figure 9). Treatments did not show any effect in non‐colitic mice (data not shown), and the proliferation of splenic CD11c+ cells was not affected (Figure 9d).
Figure 9.
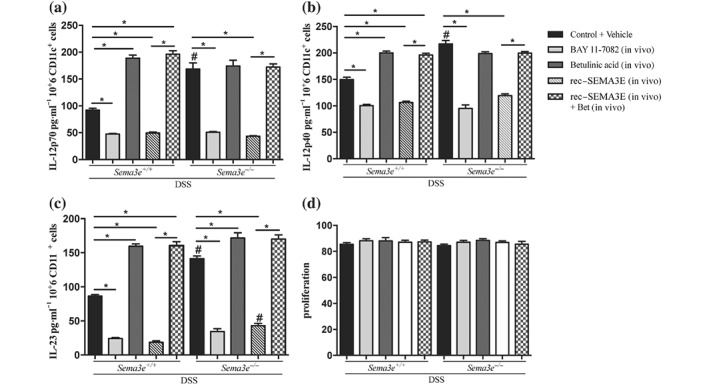
In vivo SEMA3E treatment regulates the production of cytokines by splenic CD11c+ cells through the NF‐κB‐dependent pathway in acute dextran sulfate sodium (DSS)‐induced colitis. Sema3e +/+ and Sema3e −/− mice received 5% DSS solution in the drinking water to induce colitis and were killed on day 5 post‐DSS. Mice received daily i.p. injection of recombinant SEMA3E protein (rec‐SEMA3E; 10 μg·kg−1·day−1; R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) for 6 days, starting 1 day before colitis induction. Splenic CD11c+ cells were isolated and treated or not with NF‐κB activator (betulinic acid 10 μM; R&D Systems) for 4 hr or NF‐κB inhibitor (BAY‐11‐7082, 10 μM) for 1.5 hr. Supernatants were harvested, and cytokine levels were quantified using ELISA. (a) IL‐12p70, (b) IL‐12p40, (c) IL‐23, and (d) CD11c+ cell proliferation. Data represent mean ± SEM (n = 6, *P < 0.05). # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied at significance level of 0.05
4.6. SEMA3E regulates the interactions between splenic CD11c+ and CD4+CD25− T‐cells
As shown in Figure 7d,e, the lack of Sema3e in mice resulted in an increase in the colonic expression of IFN‐γ and IL‐17 and as in vivo rec‐SEMA3E treatment regulates IL‐12p70 and IL‐23 splenic CD11c+ cells cytokines production (Figure 7a–c), we next investigated the ability of CD11c+ cells isolated from Sema3e −/− mice to interact with T‐cells interaction by co‐culturing splenic CD11c+ with CD4+CD25− T‐cells.
We found that CD11c+ cells isolated from colitic Sema3e −/− mice significantly increased the level of IFN‐γ and IL‐17 in the medium when co‐cultured with CD4+CD25− T‐cells when compared with CD11c+ cells isolated from colitic Sema3e +/+ mice (Figure 10a,b). CD11c+ cells isolated from rec‐SEMA3E‐treated Sema3e +/+ and Sema3e −/− colitic mice demonstrated a significant decrease in IFN‐γ and IL‐17 in the co‐culture medium (Figure 10a,b). To investigate whether the lack of SEMA3E or the in vivo treatment with rec‐SEMA3E influenced T‐cells interaction through the release of IL‐12 or IL‐23, CD11c+/CD4+CD25− T‐cells were treated with a mAb directed against IL‐12p35 or IL‐23p19, neutralizing IL‐12 and IL‐23 respectively. In CD11c+/CD4+CD25− T‐cells co‐cultures with CD11c+ cells isolated from colitic Sema3e +/+ and Sema3e −/− mice, the presence of anti‐IL‐12p35 and anti‐IL‐23p19 resulted in a significant decrease in the levels of IFN‐γ and IL‐17 respectively (Figure 10a,b). Anti‐IL‐12p35 and anti‐IL‐23p19 treatments did not have an additional effect on CD11c+ cells isolated from colitic rec‐SEMA3E‐treated Sema3e +/+ and Sema3e −/− mice (Figure 10a,b). Conversely, the addition of rec‐IL‐12p70 or rec‐IL‐23 proteins restored the level of IFN‐γ and IL‐17 in CD4+CD25− T‐cells co‐cultured with splenic CD11c+ cells isolated from rec‐SEMA3E‐treated colitic mice (Figure 10c,d) respectively. No significant effect on T‐cell proliferation was observed within a 24‐hr period of time; mean proliferation was 86 ± 2% between groups.
Figure 10.
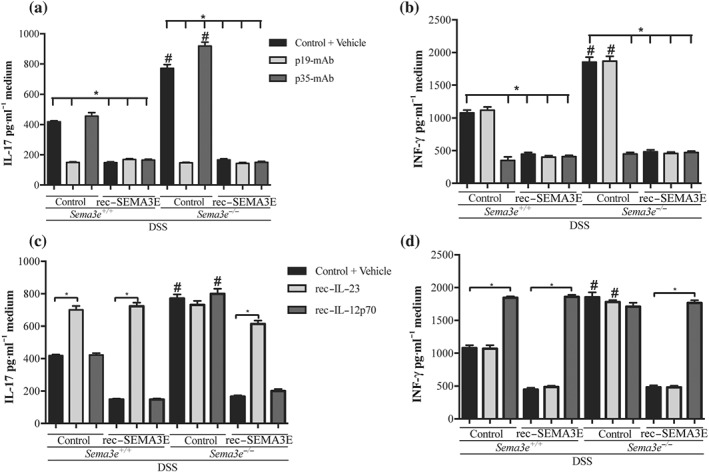
In vivo SEMA3E treatment regulates in vitro interactions between splenic CD11c+ isolated from colitic mice and naïve CD4+CD25− T‐cells. Sema3e +/+ and Sema3e −/− mice received 5% dextran sulfate sodium (DSS) in the drinking water to induce colitis and were killed on day 5 post‐DSS. Mice received daily i.p. injection of recombinant (rec‐) SEMA3E protein (10 μg·kg−1·day−1; R&D Systems) or 1% PBS with added human IgG Fc protein (Vehicle, Control) for 6 days, starting 1 day before colitis induction. Splenic CD11c+ were isolated from the different groups and co‐cultured with naive CD4+CD25− T‐cells in the presence or absence of p19 or p35 monoclonal antibody (mAb) (10 μg·ml−1) for 6 hr or with rec‐IL‐23 or rec‐IL‐12p70 (25 mg·ml−1). (a, c) IFN‐γ and (b, d) IL‐17 levels in the medium were assessed. Data represent mean ± SEM (n = 6, *P < 0.05). # refers to a significance when compared with control. One‐way ANOVA followed by multiple comparison test was applied at significance level of 0.05
5. DISCUSSION
UC is characterized by excessive immune responses associated with hyperactivation of APC and T‐cells resulting in a production of pro‐inflammatory cytokines in the inflamed colonic mucosa. Over the past few years, several studies have highlighted the immune‐regulatory properties of the SEMA family, which are newly emerging inflammatory regulators (Neurath, 2014); however, to our knowledge, this is the first study that has investigated their role in the context of colonic inflammation. Herein, using colonic samples of patients with active UC and an acute model of DSS‐experimental colitis, we have examined the role SEMA3E may play during colonic inflammation. For the first time, we provide evidence of the existence of an immune‐regulatory role of SEMA3E during the progression of colitis.
Our finding demonstrate that SEMA3E is down‐regulated in active UC patients and in DSS‐experimental colitis and this correlated negatively with inflammation and CD11c+ markers. Intestinal inflammation is characterized by structural and functional abnormalities of the enteric neurons and enteric glial cells (Lakhan & Kirchgessner, 2010; Lomax, Fernandez, & Sharkey, 2005; Zhou et al., 2013). In the brain, SEMA3E is mainly produced by specific neurons (Gu & Giraudo, 2013; Schmidt & Moore, 2013; Zhou, Gunput, & Pasterkamp, 2008), therefore, it is possible that the destruction of enteric neurons during the colonic inflammation (Lakhan & Kirchgessner, 2010; Lomax et al., 2005; Zhou et al., 2013) can result in the reduction of colonic levels of SEMA3E. Furthermore, reduced levels of SEMA3E are associated with various inflammatory conditions (Movassagh, Saati, et al., 2017; Movassagh, Shan, Mohammed, et al., 2017), and genetic ablation of Sema3e in a mouse model of allergic asthma results in an aggravated inflammation associated with a massive recruitment of immune cells into the inflamed tissue (Movassagh, Shan, Mohammed, et al., 2017). Furthermore, we reported that colonic Sema3E level is not higher in colitic Sema3e +/+ mice compared to Sema3e −/− mice in the same condition; this could be explained by the possibility that the recombinant protein reached a maximum level in the colon and passed into the blood stream. In the present study, we demonstrated that both pharmacological blockades using a rec‐PLXD1 soluble receptor and genetic deletion of Sema3e using knockout mice exacerbated the severity of colitis and enhanced the release of colonic pro‐inflammatory mediators. Interestingly, rec‐SEMA3E treatment ameliorated the development of colitis and decreased the level of colonic pro‐inflammatory cytokines not only in colitic Sema3e −/− but also in colitic Sema3e +/+ mice. Our findings are consistent with a recent study that showed an improved allergic airway inflammation in mice treated with exogenous rec‐SEMA3E (Movassagh, Saati, et al., 2017; Movassagh, Shan, Duke‐Cohan, et al., 2017; Movassagh, Shan, Mohammed, et al., 2017). Therefore, the present study highlights the immune‐modulatory role of SEMA3E during the pathogenesis of colitis.
IL12/IL23 cytokines, including the IL‐12p40 subunit, are mainly produced by CD11c+ cells (Ahern et al., 2010; Bates & Diehl, 2014; Hart et al., 2005; Maloy & Kullberg, 2008; Steinbach & Plevy, 2014) and are the main cytokine implicated in T‐cell interactions (Gutcher & Becher, 2007). We demonstrated that deletion of Sema3e increased colonic expression of IL‐12p40, IL‐12p70, and IL‐23, suggesting a role for SEMA3E as an immune‐regulatory protein. This set of data corroborated with those obtained in our study in patients with active UC, where we demonstrated that SEMA3E was negatively correlated with cells expressing CD11C and maturation markers IL12/IL23p19. These findings are in agreement with previous data showing that a deficiency in SEMA3E receptors (plexin‐D1 and plexin‐B2) is associated with a negative regulation of IL‐12/IL‐23p40 in CD11C‐dendritic cells (DC; Holl et al., 2012). Moreover, Movassagh, Shan, Mohammed, et al. (2017) depicted that the deletion of Sema3e can enhance the recruitment of pulmonary DC and can result in a worsening of the allergic airway inflammation, associated with an increase in T‐cell activation. These findings, along with the observation of an increase of CD11c, CD86 markers in the absence of SEMA3E, suggest that Sema3E has an important role in regulating potential DC activation and the associated T‐cell response during colonic inflammation. A previous study already highlighted a role for SEMA3E in the regulation of a particular T‐cell population in IBD (Vadasz et al., 2015), but nothing is known about SEMA3E in this context. However, this proves the importance of the SEMA family in T‐cell response, and further exploratory works will be undertaken to confirm our hypothesis with SEMA3E.
During the development of the inflammatory cascade, IL‐12/23 production by CD11c+ cells is dependent on the activation of NF‐κB, which is an important intracellular inflammatory pathway (Rescigno et al., 1998; Tas et al., 2005). This is demonstrated in our experiments by the high production of IL‐12/23 by the CD11c+ cells isolated from both control and Sema3e‐deficient colitic mice and stimulated with the NF‐κB activator, betulinic acid. In the context of colitis, its blockade results in amelioration of the severity (Eissa, Hussein, Kermarrec, Elgazzar, et al., 2017), confirming previous published data where the SEMA family has been implicated in the regulation of NF‐κB activation (Gu & Giraudo, 2013). In agreement with these concepts, we found that CD11c+ cells isolated from colitic Sema3e‐deficient mice produced more IL‐12p40, IL‐12p70, and IL‐23 and that rec‐SEMA3E suppressed this cytokine release through an NF‐κB‐dependent mechanism. Interestingly, the production of IL‐23 by the cells of Sema3e‐deficient mice is maximal, whereas the production of IL‐12 can still be boosted, as demonstrated by an increase in its production by betulinic acid alone. It is possible that we reached the maximum capacity of the cells when we stimulated them directly with betulinic acid. Taken together, these data suggest that SEMA3E regulates colonic inflammation through an NF‐κB‐dependent pathway in CD11c+ cells.
In a translational work, it has been demonstrated that UC is characterized by hyperactivation of colonic and splenic CD11c+ cells (Hart et al., 2005) that proliferate, differentiate, and activate T‐cells subsets (Th1, Th2, and Th17) through the release of a wide range of effector cytokines (IL12, IL23, and IL‐4) (Glimcher & Murphy, 2000; Mosmann & Sad, 1996; Neurath, Finotto, & Glimcher, 2002). Here, we found that when CD11c+ cells isolated from colitic Sema3e‐deficient mice were co‐cultured with CD4+CD25− T‐cells, a significant increased level of IFN‐γ and IL‐17 was detected in the medium. Moreover, CD11c+ cells isolated from colitic Sema3e‐deficient mice and treated in vivo with rec‐SEMA3E released less IFN‐γ and IL‐17, confirming the specificity of SEMA3E. Our in vitro findings also demonstrated that anti‐IL‐12p35 and anti‐IL‐23p19 decreased the levels of IFN‐γ and IL‐17 but did not prevent the benefits of the rec‐SEMA3E treatment. Moreover, naïve Sema3e +/+ and Sema3e −/− mice demonstrated low expression of inflammatory markers produced by the colonic mucosa or splenocytes, with no significant difference between Sema3e +/+ and Sema3e −/− mice, focusing, therefore, our main interest to inflammatory conditions. These data are in agreement with those from a previous study demonstrating that blockade of IL‐12/IL‐23 can reduce the intestinal inflammation by suppressing Th1 and Th17 activities (Feng et al., 2011). However, at this stage, we did not demonstrate a direct effect on T‐cells or other regulatory T‐cell activation, regulation that remains to be elucidated. Our findings suggest that SEMA3E can be a potential target towards a novel therapeutic strategy to alter intestinal inflammation, by regulating the interactions of CD11c+ and CD4+CD25− T‐cells. However, further studies are required to investigate the functional consequences of Sema3E on the functions of other immune cells such as macrophages, which play critical roles during colitis (Eissa et al., 2017; Eissa, Hussein, Mesgna, et al., 2018), epithelial cells, and angiogenesis, all implicated in the pathogenesis of colitis and also the exact source of Sema3E. Moreover, we acknowledge that CD11C isolated from the laminar propria need to be studied, and new studies should define if DC expressing CD11C markers are the main target of SEMA3E.
In conclusion, we demonstrated that Sema3E plays an essential immune‐regulatory role in intestinal inflammation. We found that SEMA3E is reduced in patients with active UC and DSS‐induced colitis and is associated with an increased production of pro‐inflammatory mediators. The absence of Sema3E led to higher colonic inflammation in colitic mice, which was associated with exacerbated levels of CD11c+‐ associated cytokines through a NF‐κB‐dependent mechanism. Sema3e deficiency resulted in uncontrollable activation of CD11c+/CD4+CD25− T‐cells resulting in enhanced Th1/Th17 profiles. This previously unknown role for Sema3E in colonic inflammation may help to broaden research done by other groups on the overall effect of SEMA3E on immune regulation in the context of IBD, leading to the development of future novel therapeutic strategies in IBD and other inflammatory disorders.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
C.N.B., J.E.G., L.K., and N.E. conceived and designed the experiments. H.W., N.E., L.K., K.P., A.D. performed the experiments. C.N.B., J.‐E.G., L.K., and N.E. analysed the data. A.S.G. and J.‐E.G. contributed reagents/materials/analysis tools. C.N.B., J.‐E.G., L.K., and N.E. wrote the paper.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
This study was supported by grants from Crohn's and Colitis Canada (CCC) and the Canadian Institutes of Health Research (CIHR) to J.‐E.G.; Children's Hospital Research Institute of Manitoba, Research Manitoba, University of Manitoba, Health Science Foundation – Mindel and Tom Olenick Research Excellence Award in Immunology, and Canadian Institutes of Health Research (CIHR) (Grant 395678) to N.E.; MITACS to L.K.; and N.E.; C.N.B. is supported in part by the Bingham Chair in Gastroenterology.
Kermarrec L, Eissa N, Wang H, et al. Semaphorin‐3E attenuates intestinal inflammation through the regulation of the communication between splenic CD11C+ and CD4+CD25− T‐cells. Br J Pharmacol. 2019;176:1235–1250. 10.1111/bph.14614
REFERENCES
- Chin, A. C. , & Parkos, C. A. (2006). Neutrophil transepithelial migration and epithelial barrier function in IBD. Annals of the New York Academy of Sciences, 1072, 276–287. [DOI] [PubMed] [Google Scholar]
- Ahern, P. P. , Schiering, C. , Buonocore, S. , McGeachy, M. J. , Cua, D. J. , Maloy, K. J. , & Powrie, F. (2010). Interleukin‐23 drives intestinal inflammation through direct activity on T cells. Immunity, 33, 279–288. 10.1016/j.immuni.2010.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174(S1), S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Overview. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auletta, S. , Bonfiglio, R. , Wunder, A. , Varani, M. , Galli, F. , Borri, F. , … Bonanno, E. (2018). Animal models for the study of inflammatory bowel diseases: A meta‐analysis on modalities for imaging inflammatory lesions. The Quarterly Journal of Nuclear Medicine and Molecular Imaging: Official Publication of the Italian Association of Nuclear Medicine (AIMN)[and] the International Association of Radiopharmacology (IAR), [and] Section of the Society of Radiopharmaceutical Chemistry and Biology, 62, 78–100. [DOI] [PubMed] [Google Scholar]
- Baldo, B. A. (2015). Chimeric fusion proteins used for therapy: Indications, mechanisms, and safety. Drug Safety, 38, 455–479. 10.1007/s40264-015-0285-9 [DOI] [PubMed] [Google Scholar]
- Bates, J. , & Diehl, L. (2014). Dendritic cells in IBD pathogenesis: An area of therapeutic opportunity? The Journal of Pathology, 232, 112–120. 10.1002/path.4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing, B. , Aitken, J. D. , Malleshappa, M. , & Vijay‐Kumar, M. (2014). Dextran sulfate sodium (DSS)‐induced colitis in mice. Current Protocols in Immunology, 15(25), 11–15.25.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y. I. , Duke‐Cohan, J. S. , Ahmed, W. B. , Handley, M. A. , Mann, F. , Epstein, J. A. , … Reinherz, E. L. (2008). PlexinD1 glycoprotein controls migration of positively selected thymocytes into the medulla. Immunity, 29, 888–898. 10.1016/j.immuni.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, H. S. , Murthy, S. , Shah, R. , & Sedergran, D. (1993). Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Laboratory Investigation; a Journal of Technical Methods and Pathology, 69, 238–249. [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , … McGrath, J. C. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP. British Journal of Pharmacology, 172, 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa, N. , & Ghia, J. (2015). Immunomodulatory effect of ghrelin in the intestinal mucosa. Neurogastroenterology & Motility, 27, 1519–1527. 10.1111/nmo.12703 [DOI] [PubMed] [Google Scholar]
- Eissa, N. , Hussein, H. , Hendy, G. N. , Bernstein, C. N. , & Ghia, J.‐E. (2018). Chromogranin‐A and its derived peptides and their pharmacological effects during intestinal inflammation. Biochemical Pharmacology, 152, 315–326. 10.1016/j.bcp.2018.04.009 [DOI] [PubMed] [Google Scholar]
- Eissa, N. , Hussein, H. , Kermarrec, L. , Ali, A. Y. , Marshall, A. , Metz‐Boutigue, M.‐H. , … Ghia, J. E. (2018). Chromogranin‐A regulates macrophage function and the apoptotic pathway in murine DSS colitis. Journal of Molecular Medicine, 96, 183–198. 10.1007/s00109-017-1613-6 [DOI] [PubMed] [Google Scholar]
- Eissa, N. , Hussein, H. , Kermarrec, L. , Elgazzar, O. , Metz‐Boutigue, M.‐H. , Bernstein, C. N. , & Ghia, J. E. (2017). Chromofungin (CHR: CHGA47‐66) is downregulated in persons with active ulcerative colitis and suppresses pro‐inflammatory macrophage function through the inhibition of NF‐κB signaling In Biochemical pharmacology (Vol. 145) (pp. 102–113). 10.1016/j.bcp.2017.08.013 [DOI] [PubMed] [Google Scholar]
- Eissa, N. , Hussein, H. , Kermarrec, L. , Grover, J. , Metz‐Boutigue, M.‐H. E. , Bernstein, C. N. , & Ghia, J. E. (2017). Chromofungin ameliorates the progression of colitis by regulating alternatively activated macrophages. Frontiers in Immunology, 8, 1131 10.3389/fimmu.2017.01131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa, N. , Hussein, H. , Mesgna, R. , Bonin, S. , Hendy, G. , Metz‐Boutigue, M.‐H. , … Ghia, J. E. (2018). Catestatin regulates epithelial cell dynamics to improve intestinal inflammation. Vaccine, 6, 67 10.3390/vaccines6040067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa, N. , Hussein, H. , Wang, H. , Rabbi, M. F. , Bernstein, C. N. , & Ghia, J.‐E. (2016). Stability of reference genes for messenger RNA quantification by real‐time PCR in mouse dextran sodium sulfate experimental colitis. PLoS ONE, 11, e0156289 10.1371/journal.pone.0156289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa, N. , Kermarrec, L. , Hussein, H. , Bernstein, C. N. , & Ghia, J.‐E. (2017). Appropriateness of reference genes for normalizing messenger RNA in mouse 2,4‐dinitrobenzene sulfonic acid (DNBS)‐induced colitis using quantitative real time PCR. Scientific Reports, 7 10.1038/srep42427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, T. , Qin, H. , Wang, L. , Benveniste, E. N. , Elson, C. O. , & Cong, Y. (2011). Th17 cells induce colitis and promote Th1 cell responses through IL‐17 induction of innate IL‐12 and IL‐23 production. The Journal of Immunology, 186, 6313–6318. 10.4049/jimmunol.1001454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick, L. R. (2012). Novel pharmacological approaches for inflammatory bowel disease: Targeting key intracellular pathways and the IL‐23/IL‐17 axis. International Journal of Inflammation, 2012, 1–8. 10.1155/2012/389404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke, A. , McGovern, D. P. , Barrett, J. C. , Wang, K. , Radford‐Smith, G. L. , Ahmad, T. , … Parkes, M. (2010). Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nature Genetics, 42, 1118–1125. 10.1038/ng.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher, L. H. , & Murphy, K. M. (2000). Lineage commitment in the immune system: The T helper lymphocyte grows up. Genes & Development, 14, 1693–1711. [PubMed] [Google Scholar]
- Goodall, J. C. , Wu, C. , Zhang, Y. , McNeill, L. , Ellis, L. , Saudek, V. , & Gaston, J. S. H. (2010). Endoplasmic reticulum stress‐induced transcription factor, CHOP, is crucial for dendritic cell IL‐23 expression. Proceedings of the National Academy of Sciences, 107, 17698–17703. 10.1073/pnas.1011736107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudin, N. , Chappert, P. , Mégret, J. , Gross, D.‐A. , Rocha, B. , & Azogui, O. (2016). Depletion of regulatory T Cells induces high numbers of dendritic cells and unmasks a subset of anti‐tumour CD8+ CD11c+ PD‐1lo effector T cells. PLoS ONE, 11, e0157822 10.1371/journal.pone.0157822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, C. , & Giraudo, E. (2013). The role of semaphorins and their receptors in vascular development and cancer. Experimental Cell Research, 319, 1306–1316. 10.1016/j.yexcr.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, C. , Yoshida, Y. , Livet, J. , Reimert, D. V. , Mann, F. , Merte, J. , … Ginty, D. D. (2005). Semaphorin 3E and plexin‐D1 control vascular pattern independently of neuropilins. Science, 307, 265–268. 10.1126/science.1105416 [DOI] [PubMed] [Google Scholar]
- Gutcher, I. , & Becher, B. (2007). APC‐derived cytokines and T cell polarization in autoimmune inflammation. Journal of Clinical Investigation, 117, 1119–1127. 10.1172/JCI31720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–d1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart, A. L. , Al‐Hassi, H. O. , Rigby, R. J. , Bell, S. J. , Emmanuel, A. V. , Knight, S. C. , … Stagg, A. J. (2005). Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology, 129, 50–65. 10.1053/j.gastro.2005.05.013 [DOI] [PubMed] [Google Scholar]
- Holl, E. K. , Roney, K. E. , Allen, I. C. , Steinbach, E. , Arthur, J. C. , Buntzman, A. , … Ting, J. P. Y. (2012). Plexin‐B2 and Plexin‐D1 in dendritic cells: Expression and IL‐12/IL‐23p40 production. PLoS ONE, 7, e43333 10.1371/journal.pone.0043333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, H. , Rabbi, M. F. , Labis, B. , Pavlov, V. A. , Tracey, K. J. , & Ghia, J.‐E. (2014). Central cholinergic activation of a vagus nerve‐to‐spleen circuit alleviates experimental colitis. Mucosal Immunology, 7, 335–347. 10.1038/mi.2013.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko, G. E. , Horvat, J. C. , Beagley, K. W. , & Hansbro, P. M. (2008). Immunological decision‐making: How does the immune system decide to mount a helper T‐cell response? Immunology, 123, 326–338. 10.1111/j.1365-2567.2007.02719.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath, A. T. , Pooley, J. , O'Keeffe, M. A. , Vremec, D. , Zhan, Y. , Lew, A. M. , … Shortman, K. (2000). The development, maturation, and turnover rate of mouse spleen dendritic cell populations. The Journal of Immunology, 165, 6762–6770. 10.4049/jimmunol.165.12.6762 [DOI] [PubMed] [Google Scholar]
- Kaplan, G. G. (2015). The global burden of IBD: from 2015 to 2025. Nature Reviews Gastroenterology & Hepatology, 12, 720–727. 10.1038/nrgastro.2015.150 [DOI] [PubMed] [Google Scholar]
- Kruger, R. P. , Aurandt, J. , & Guan, K.‐L. (2005). Semaphorins command cells to move. Nature Reviews Molecular Cell Biology, 6, 789–800. 10.1038/nrm1740 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhan, S. E. , & Kirchgessner, A. (2010). Neuroinflammation in inflammatory bowel disease. Journal of Neuroinflammation, 7, 37 10.1186/1742-2094-7-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax, A. , Fernandez, E. , & Sharkey, K. (2005). Plasticity of the enteric nervous system during intestinal inflammation. Neurogastroenterology & Motility, 17, 4–15. 10.1111/j.1365-2982.2004.00607.x [DOI] [PubMed] [Google Scholar]
- Maloy, K. , & Kullberg, M. (2008). IL‐23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunology, 1, 339–349. 10.1038/mi.2008.28 [DOI] [PubMed] [Google Scholar]
- Mangasser‐Stephan, K. , Dooley, S. , Welter, C. , Mutschler, W. , & Hanselmann, R. G. (1997). Identification of human semaphorin E gene expression in rheumatoid synovial cells by mRNA differential display. Biochemical and Biophysical Research Communications, 234, 153–156. 10.1006/bbrc.1997.6607 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad, M. , Sathe, P. , Helft, J. , Miller, J. , & Mortha, A. (2013). The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annual Review of Immunology, 31, 563–604. 10.1146/annurev-immunol-020711-074950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann, T. R. , & Sad, S. (1996). The expanding universe of T‐cell subsets: Th1, Th2 and more. Immunology Today, 17, 138–146. 10.1016/0167-5699(96)80606-2 [DOI] [PubMed] [Google Scholar]
- Movassagh, H. , Saati, A. , Nandagopal, S. , Mohammed, A. , Tatari, N. , Shan, L. , … Gounni, A. S. (2017). Chemorepellent semaphorin 3E negatively regulates neutrophil migration in vitro and in vivo. The Journal of Immunology, 198, 1023–1033. 10.4049/jimmunol.1601093 [DOI] [PubMed] [Google Scholar]
- Movassagh, H. , Shan, L. , Duke‐Cohan, J. S. , Halayko, A. J. , Uzonna, J. E. , & Gounni, A. S. (2017). Semaphorin 3E alleviates hallmarks of house dust mite‐induced allergic airway disease. The American Journal of Pathology, 187, 1566–1576. 10.1016/j.ajpath.2017.03.008 [DOI] [PubMed] [Google Scholar]
- Movassagh, H. , Shan, L. , Mohammed, A. , Halayko, A. J. , & Gounni, A. S. (2017). Semaphorin 3E deficiency exacerbates airway inflammation, hyperresponsiveness, and remodeling in a mouse model of allergic asthma. The Journal of Immunology, 198, 1805–1814. 10.4049/jimmunol.1601514 [DOI] [PubMed] [Google Scholar]
- Munyaka, P. , Rabbi, M. F. , Pavlov, V. A. , Tracey, K. J. , Khafipour, E. , & Ghia, J.‐E. (2014). Central muscarinic cholinergic activation alters interaction between splenic dendritic cell and CD4+ CD25‐T cells in experimental colitis. PLoS ONE, 9, e109272 10.1371/journal.pone.0109272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath, M. F. (2014). Cytokines in inflammatory bowel disease. Nature Reviews Immunology, 14, 329–342. 10.1038/nri3661 [DOI] [PubMed] [Google Scholar]
- Neurath, M. F. , Finotto, S. , & Glimcher, L. H. (2002). The role of Th1/Th2 polarization in mucosal immunity. Nature Medicine, 8, 567–573. 10.1038/nm0602-567 [DOI] [PubMed] [Google Scholar]
- Okayasu, I. , Hatakeyama, S. , Yamada, M. , Ohkusa, T. , Inagaki, Y. , & Nakaya, R. (1990). A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology, 98, 694–702. 10.1016/0016-5085(90)90290-H [DOI] [PubMed] [Google Scholar]
- Perše, M. , & Cerar, A. (2012). Dextran sodium sulphate colitis mouse model: Traps and tricks. BioMed Research International, 2012, 13, 718617 10.1155/2012/718617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbi, M. F. , Eissa, N. , Munyaka, P. M. , Kermarrec, L. , Elgazzar, O. , Khafipour, E. , … Ghia, J. E. (2017). Reactivation of intestinal inflammation is suppressed by catestatin in a murine model of colitis via M1 macrophages and not the gut microbiota. Frontiers in Immunology, 8, 985 10.3389/fimmu.2017.00985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper, J. A. (2000). Semaphorins and their receptors in vertebrates and invertebrates. Current Opinion in Neurobiology, 10, 88–94. 10.1016/S0959-4388(99)00057-4 [DOI] [PubMed] [Google Scholar]
- Rescigno, M. , Martino, M. , Sutherland, C. L. , Gold, M. R. , & Ricciardi‐Castagnoli, P. (1998). Dendritic cell survival and maturation are regulated by different signaling pathways. Journal of Experimental Medicine, 188, 2175–2180. 10.1084/jem.188.11.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux, J. D. , Xavier, R. J. , Taylor, K. D. , Silverberg, M. S. , Goyette, P. , Huett, A. , … Brant, S. R. (2007). Genome‐wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature Genetics, 39, 596–604. 10.1038/ng2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandborn, W. J. , Gasink, C. , Gao, L.‐L. , Blank, M. A. , Johanns, J. , Guzzo, C. , … CERTIFI Study Group (2012). Ustekinumab induction and maintenance therapy in refractory Crohn's disease. New England Journal of Medicine, 367, 1519–1528. 10.1056/NEJMoa1203572 [DOI] [PubMed] [Google Scholar]
- Schmidt, A. M. , & Moore, K. J. (2013). The semaphorin 3E/PlexinD1 axis regulates macrophage inflammation in obesity. Cell Metabolism, 18, 461–462. 10.1016/j.cmet.2013.09.011 [DOI] [PubMed] [Google Scholar]
- Shimizu, I. , Yoshida, Y. , Moriya, J. , Nojima, A. , Uemura, A. , Kobayashi, Y. , & Minamino, T. (2013). Semaphorin3E‐induced inflammation contributes to insulin resistance in dietary obesity. Cell Metabolism, 18, 491–504. 10.1016/j.cmet.2013.09.001 [DOI] [PubMed] [Google Scholar]
- Steinbach, E. C. , & Plevy, S. E. (2014). The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflammatory Bowel Diseases, 20, 166–175. 10.1097/MIB.0b013e3182a69dca [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman, L. (2010). Mixed results with modulation of TH‐17 cells in human autoimmune diseases. Nature Immunology, 11, 41–44. 10.1038/ni.1803 [DOI] [PubMed] [Google Scholar]
- Takamatsu, H. , Takegahara, N. , Nakagawa, Y. , Tomura, M. , Taniguchi, M. , Friedel, R. H. , … Kumanogoh, A. (2010). Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nature Immunology, 11, 594–600. 10.1038/ni.1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas, S. W. , de Jong, E. C. , Hajji, N. , May, M. J. , Ghosh, S. , Vervoordeldonk, M. J. , & Tak, P. P. (2005). Selective inhibition of NF‐κB in dendritic cells by the NEMO‐binding domain peptide blocks maturation and prevents T cell proliferation and polarization. European Journal of Immunology, 35, 1164–1174. 10.1002/eji.200425956 [DOI] [PubMed] [Google Scholar]
- Vadasz, Z. , Rainis, T. , Nakhleh, A. , Haj, T. , Bejar, J. , Halasz, K. , & Toubi, E. (2015). The involvement of immune semaphorins in the pathogenesis of inflammatory bowel diseases (IBDs). PLoS ONE, 10, e0125860 10.1371/journal.pone.0125860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanschel, A. , Seibert, T. , Hewing, B. , Ramkhelawon, B. , Ray, T. D. , van Gils, J. M. , … Moore, K. J. (2013). Neuroimmune guidance cue semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention significance. Arteriosclerosis, Thrombosis, and Vascular Biology, 33, 886–893. 10.1161/ATVBAHA.112.300941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, T.‐H. , Chen, H.‐A. , Gau, R.‐J. , Yen, J.‐H. , & Suen, J.‐L. (2016). Heme oxygenase‐1‐expressing dendritic cells promote Foxp3+ regulatory t cell differentiation and induce less severe airway inflammation in murine models. PLoS ONE, 11, e0168919 10.1371/journal.pone.0168919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff, S. A. , Masterson, J. C. , Fillon, S. , Robinson, Z. D. , & Furuta, G. T. (2011). Role of eosinophils in inflammatory bowel and gastrointestinal diseases. Journal of Pediatric Gastroenterology and Nutrition, 52, 650–661. 10.1097/MPG.0b013e3182128512 [DOI] [PubMed] [Google Scholar]
- Worzfeld, T. , & Offermanns, S. (2014). Semaphorins and plexins as therapeutic targets. Nature Reviews Drug Discovery, 13, 603–621. 10.1038/nrd4337 [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Gunput, R.‐A. F. , & Pasterkamp, R. J. (2008). Semaphorin signaling: Progress made and promises ahead. Trends in Biochemical Sciences, 33, 161–170. 10.1016/j.tibs.2008.01.006 [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Yang, J. , Watkins, D. J. , Boomer, L. A. , Matthews, M. A. , Su, Y. , & Besner, G. E. (2013). Enteric nervous system abnormalities are present in human necrotizing enterocolitis: Potential neurotransplantation therapy. Stem Cell Research & Therapy, 4, 157 10.1186/scrt387 [DOI] [PMC free article] [PubMed] [Google Scholar]