

Abstract
Parkinson's disease (PD) is a progressive movement disorder resulting primarily from loss of nigrostriatal dopaminergic neurons. PD is characterized by the accumulation of protein aggregates, and evidence suggests that aberrant protein deposition in dopaminergic neurons could be related to the dysregulation of the lysosomal autophagy pathway. The therapeutic potential of autophagy modulators has been reported in experimental models of PD. Trehalose is a natural disaccharide that has been considered as a new candidate for the treatment of neurodegenerative diseases. It has a chaperone‐like activity, prevents protein misfolding or aggregation, and by promoting autophagy, contributes to the removal of accumulated proteins. In this review, we briefly summarize the role of aberrant autophagy in PD and the underlying mechanisms that lead to the development of this disease. We also discuss reports that used trehalose to counteract the neurotoxicity in PD, focusing particularly on the autophagy promoting, protein stabilization, and anti‐neuroinflammatory effects of trehalose.
Abbreviations
- ALP
autophagy–lysosome pathway
- AMPK
AMP‐dependent protein kinase
- Atg
autophagy‐related protein
- CMA
chaperone‐mediated autophagy
- HD
Huntington's disease
- LB
Lewy body
- LC3
microtubule‐associated protein 1 light chain 3
- mTOR
mechanistic target of rapamycin kinase
- PD
Parkinson's disease
- TFEB
transcription factor EB
- UPS
ubiquitin–proteasome system
- WT
wild‐type
1. INTRODUCTION
Parkinson's disease (PD) is the second most frequent, age‐related neurodegenerative disorder that is characterized by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (Mishra et al., 2015). Selective loss of nigral dopaminergic neurons leads to a dramatic decrease in dopamine levels in the striatum, which leads to the cardinal symptoms of PD like tremor, muscle rigidity, balance disturbances, and bradykinesia (Dawson & Dawson, 2003; Learmonth & Freitas, 2002).
Despite the great advances that have been made in the understanding of the molecular basis and neuronal pathways of PD over the last decades, the aetiology of PD remains unclear. About 90% of PD cases occur as a sporadic condition that is caused by an interplay between environmental and genetic risk factors (Ghavami et al., 2014). Although familial and sporadic forms of PD have different causes, they present similar phenotypic features suggesting that both forms may involve common pathophysiological mechanisms. It has been proposed that mitochondrial dysfunction, oxidative stress, and the mishandling of damaged proteins and mitochondria contribute to the progressive nature of PD (Kroemer, Mariño, & Levine, 2010).
An accumulation of intracytoplasmic protein inclusions known as Lewy bodies (LBs) and Lewy neurites are the histological hallmark of PD (Ebrahimi‐Fakhari, Saidi, & Wahlster, 2013; Singh, Patel, Dikshit, & Gupta, 2006). LBs are mainly composed of abnormal aggregates of α‐synuclein (Iwatsubo, 2003). Post‐mortem brains of patients with PD exhibit highly phosphorylated, ubiquitinated, and insoluble aggregates of α‐synuclein protein accumulated in LBs and Lewy neurites (Baba et al., 1998; Spillantini et al., 1998).
At present, there are no proven disease‐modifying or neuroprotective therapies for PD patients (AlDakheel, Kalia, & Lang, 2014), and treatment is initiated after functional impairment or social anxiety from the symptoms. Currently, the oral dopamine precursor levodopa is the most effective treatment for PD and provides good control of motor symptoms (Poskanzer, 1969). However, complications such as on–off phenomena and dyskinesia are the consequence of prolonged levodopa treatment that makes this drug less effective (Prashanth, Fox, & Meissner, 2011). To prevent the motor complications associated with levodopa, other medications such as dopamine agonists, amantadine, monoamine oxidase B (MAO‐B) inhibitors, anticholinergics, or β‐blockers are initiated first in patients with mild symptoms, patients with tremor as the only or most important symptom, and patients aged over 60 years (Connolly & Lang, 2014).
Deep brain stimulation has been developed for improving the motor complications in PD. Deep brain stimulation treats levodopa‐responsive symptoms, and the benefits of this therapy are durable for several years, but it is only indicated for patients having PD with disabling motor fluctuations and/or resistant tremor without significant cognitive or psychiatric disorders (Bronstein et al., 2011).
Recent clinical trials investigating the therapeutic effects of the dopamine agonist pramipexole, coenzyme Q10, creatine, pioglitazone, and adeno‐associated virus‐mediated gene therapy to increase the expression of neurturin did not provide evidence of their efficacy (NINDS Exploratory Trials in Parkinson Disease (NET‐PD) FS‐ZONE Investigators 2015; Bartus et al., 2013; Parkinson Study Group QE3 Investigators et al., 2014; Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET‐PD) Investigators et al., 2015; Schapira et al., 2013).
It has been demonstrated that toxic forms of α‐synuclein aggregates released into the extracellular space can be internalized and lead to the propagation of pathogenic α‐synuclein seeds (Hansen et al., 2011). To limit this propagation, both active and passive immunotherapies against α‐synuclein are currently being investigated in clinical trials (Oertel & Schulz, 2016). Clinical and experimental evidence indicate that a failure in the autophagy pathway caused by both genetic factors and environmental neurotoxins plays an important role in PD pathogenesis (Gan‐Or, Dion, & Rouleau, 2015; Pan, Kondo, Le, & Jankovic, 2008). Approaches such as the use of small molecules with the capacity to block or modulate α‐synuclein aggregation and compounds that up‐regulate cell‐intrinsic pathways such as the autophagy–lysosome pathway (ALP) to eliminate the α‐synuclein aggregation are under development. Rapamycin, also known as sirolimus, is used as an agent to induce macroautophagy, but its safety profile limits its potential as a therapeutic for PD (Kalia, Kalia, & Lang, 2015; Oertel & Schulz, 2016). The neuroprotective effects of agents such as trehalose, which enhances the clearance of abnormal protein aggregation, have been previously elucidated, and these agents should be considered as potential therapies for PD (Tanaka et al., 2004).
In this review, we summarize common mechanisms that may be involved in the therapeutic effects of trehalose and also provide the current state of the in vitro and in vivo evidence on the use of this agent for PD treatment.
2. AUTOPHAGY: INTRODUCTION AND PATHWAYS
There are two main protein degradation systems that regulate protein clearance including the ubiquitin–proteasome system (UPS) and the ALP (Pan et al., 2008). Under basal metabolic conditions, the UPS is the major route for degradation of short‐lived and misfolded proteins as well as the majority of cell constituents (Pan, Kondo, Le, & Jankovic, 2008). Although, the UPS acts as the first line of defence against misfolded proteins (Ebrahimi‐Fakhari et al., 2013; Lynch‐Day, Mao, Wang, Zhao, & Klionsky, 2012; Osellame & Duchen, 2014), large membrane proteins and oligomeric or aggregated proteins cannot penetrate the narrow pore of the proteasome barrel and are thus removed via ALP (Cuervo et al., 2005; Hideshima, Bradner, Chauhan, & Anderson, 2005; Levine & Klionsky, 2004). Autophagy is a major proteolytic system that culminates in the lysosomal degradation of cytoplasmic components, organelles, and proteins (Levine & Kroemer, 2008). Autophagy tightly modulates the normal balance between the formation and degradation of cellular proteins, and its specific roles in cell survival, differentiation, development, and homeostasis have been elucidated (Mizushima & Levine, 2010; Wu, Chen, et al., 2015). With respect to mechanisms by which a substrate enters the lysosome, ALP can be subdivided into macroautophagy, microautophagy, and chaperone‐mediated autophagy (CMA; Cuervo et al., 2005; Levine & Klionsky, 2004; Mizushima & Komatsu, 2011). Microautophagy contributes to the slow and continuous turnover of cytosolic proteins and occurs via invagination of lysosomal membrane and direct uptake of cytosolic materials by lysosomes, a process similar to pinocytosis (Pan, Kondo, Le, & Jankovic, 2008).
Macroautophagy, the most general form of autophagy, contributes to the degradation of large portions of the cytoplasm and even whole organelles (Xilouri, Brekk, & Stefanis, 2016). In this process, cytosolic constituents are sequestered within double‐membrane lipid structures known as autophagosomes, and subsequently, autophagosomes fuse with a lysosome to form autolysosomes, and their contents are then broken down into free amino acids by hydrolytic enzymes (Pan, Kondo, Le, & Jankovic, 2008; Xilouri et al., 2016). The Golgi complex, endosomes, endoplasmic reticulum, mitochondria, and the plasma membrane are known sources for the membrane to build the phagophore (Yang & Klionsky, 2010). Indeed, the formation of autophagosomes requires the function of autophagy‐related (Atg) proteins (Cao & Klionsky, 2007; Mizushima & Levine, 2010; Suzuki, Kubota, Sekito, & Ohsumi, 2007).
The Atg proteins are classified into six functional complexes among which the ULK1/2 (unc‐51 like autophagy activating kinase 1/2) complex is the most upstream of autophagosome formation. The ULK1/2 complex is the target of several cellular signalling pathways and contributes to autophagy coordination (Chen et al., 2017; Suzuki et al., 2007; Suzuki & Ohsumi, 2007). In mammalian cells, ULK1 activity is directly modulated by the AMP‐dependent protein kinase (AMPK) and the mechanistic target of rapamycin kinase (mTOR; Alers, Loffler, Wesselborg, & Stork, 2012; Kim, Kundu, Viollet, & Guan, 2011; Shang & Wang, 2011). mTOR is a serine/threonine protein kinase, which acts as a primary negative regulator of macroautophagy, and inactivation of mTOR results in the induction of macroautophagy (Pyo, Nah, & Jung, 2012). Mammalian cells have two distinct multiprotein mTOR complexes, named mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is highly sensitive to cellular nutrient content and rapamycin (Diaz‐Troya, Perez‐Perez, Florencio, & Crespo, 2008; Wullschleger, Loewith, & Hall, 2006) and regulates protein homeostasis and autophagy, whereas mTORC2 regulates cell survival and proliferation. mTORC1 can inhibit autophagy under nutrient‐rich conditions by phosphorylating the Atg13 subunit and disrupting the ULK1–ATG13 complex. In contrast, under glucose starvation, AMPK stimulates autophagy directly by phosphorylating ULK1 at the Ser317 and Ser777 residues (Kim et al., 2011). Also, the conjugation of microtubule‐associated protein 1 light chain 3 (LC3)‐I with phosphatidylethanolamine to form LC3‐II is a key step in the formation of autophagosomes such that the LC3‐positive puncta is an accepted marker for autophagosome formation (Fujita et al., 2008; Hanada et al., 2007; Klionsky et al., 2008). It has been well demonstrated that Atg5 plays a critical role in autophagosome formation by evoking the lipidation of LC3‐I to LC3‐II (Klionsky et al., 2008; Mizushima et al., 2001).
Proteins are the only cargo degraded via CMA and are selectively delivered into the lysosome (Wu, Chen, et al., 2015). In contrast to the other types of autophagy, the arrival of substrates to the lysosomal lumen does not require vesicle formation, or major changes in the lysosomal membrane, and proteins are directly transported across the lysosomal membrane (Arias & Cuervo, 2011; Xilouri et al., 2016). To be CMA substrates, proteins need to have a specific targeting motif (KFERQ‐like sequence) in their amino acid sequence, which is recognized by the cytosolic chaperone Hsp70 (Dice, 1990). The chaperone and substrate complex interacts with lysosome‐associated membrane protein type 2a (LAMP‐2A; Bandyopadhyay, Kaushik, Varticovski, & Cuervo, 2008), driving the transportation of proteins into the lysosomal lumen with the assistance of lysosomal hsc70 (Agarraberes, Terlecky, & Dice, 1997; Cuervo, Dice, & Knecht, 1997). It has been shown that the lysosomal mTORC2/PHLPP1/Akt axis participates in the regulation of CMA activity (Arias et al., 2015). This modulatory effect is mediated by changes in the kinetics of assembly and disassembly of the CMA translocation complex in the lysosomal membrane (Arias, 2015).
3. AUTOPHAGY IN PARKINSON'S DISEASE
Autophagy plays a key role in the basal maintenance of protein homeostasis in the nervous system. Because neurons are postmitotic cells, redistribution of damaged constituents to the daughter cells does not occur (Pan & Yue, 2014), and autophagic dysfunction can trigger a cascade of events that lead to neuronal degeneration (Kochergin & Zakharova, 2016). Furthermore, ageing is an important element associated with dysfunctions in the protein control machinery. The activities of the UPS and autophagy are decreased during ageing (Cuervo et al., 2005; Keller et al., 2004; Pan, Kondo, Le, & Jankovic, 2008), resulting in the accumulation and aggregation of proteins and possible neurodegeneration as seen in PD (Cuervo et al., 2005; Wong & Cuervo, 2010). Deficiences in the ALP are implicated in the progression of several human neurodegenerative disorders such as Alzheimer's disease, PD, and Huntington's disease (HD; Crews et al., 2010; Wu, Chen, et al., 2015). In neurodegenerative diseases, defects in the autophagic machinery can occur at different stages including autophagosome formation, cargo recognition, autophagosome fusion with the lysosome, autophagosome clearance, and cargo degradation (Ghavami et al., 2014).
In the field of PD, several defective genes encoding α‐synuclein, LRRK2, Parkin, PINK1, ATP13A2, Rab7L, and VPS35 are associated with autophagy pathways (Gusdon, Zhu, Van Houten, & Chu, 2012; Hyttinen, Niittykoski, Salminen, & Kaarniranta, 2013; Lynch‐Day et al., 2012; Zavodszky et al., 2014). α‐Synuclein, the main component of LBs, is an aggregation‐prone protein. Two main mutations in this protein (A53T and A30P) have been linked to the early onset of PD (Recchia et al., 2004). Studies have shown that wild‐type (WT) α‐synuclein is degraded by the CMA pathway due to the presence of a CMA‐targeting motif in its sequences (Cuervo, Stefanis, Fredenburg, Lansbury, & Sulzer, 2004). However, the mutant or overexpressed form of α‐synuclein undergoes a posttranslational modification and binds abnormally to lysosome‐associated membrane protein type 2a (LAMP‐2A), leading to blockage of their translocation into the lysosomal lumen (Lee & Lee, 2002; Xilouri, Vogiatzi, Vekrellis, Park, & Stefanis, 2009). Moreover, oxidation of dopamine to aminochrome induces the formation of α‐synuclein protofibril and α‐ and β‐tubulin adducts, which could inactivate the CMA pathway. Also, adducts could prevent the formation of microtubules essential for the fusion of autophagosome with the lysosome (Munoz, Huenchuguala, Paris, & Segura‐Aguilar, 2012). A reduction of CMA activity increases the accumulation of more aggregated proteins and augments degeneration of dopaminergic neurons (Alvarez‐Erviti et al., 2010; Cuervo et al., 2004). In this regard, an impairment of the CMA pathway was found to stimulate macroautophagy as a compensatory response to maintain cellular homeostasis. Previous studies demonstrated that macroautophagy plays a key role in the elimination of aggregated proteins, including mutant huntingtin, α‐synuclein, and ataxin (Weissmann et al., 2001; Williams et al., 2006). An accumulation of autophagosomes in post‐mortem PD brains has been largely linked to macroautophagy activation (Vila, Bove, Dehay, Rodriguez‐Muela, & Boya, 2011). Moreover, an overexpression of mutant α‐synuclein in mice brain increased autophagy markers, including LC3‐II and Beclin‐1 (Bossy, Perkins, & Bossy‐Wetzel, 2008). Induction of autophagy in cellular and mouse models of PD via rapamycin treatment enhances autophagic degradation of misfolded proteins (Pan, Kondo, Zhu, et al., 2008). However, it should be noted that macroautophagy is also disrupted during disease progression (Mishra et al., 2015). It was shown that α‐synuclein oligomers, upon direct interaction with the lipid membrane and vesicles of an organelle, damage their regular functions. This leads to disruption of the lysosomal integrity and abnormal permeabilization of lysosomal membranes, defective clearance, and subsequent accumulation of autophagosome (Sulzer, 2010). In vivo evidence indicates an accumulation of autophagosomes and depletion of lysosomes in PD post‐mortem brains. Moreover, LBs in PD brains were strongly positive for autophagosome markers, suggesting that these inclusions might have derived from nondegraded autophagosomes (Vila et al., 2011). In support of these findings, it was also shown that genetic or pharmacological activation of transcription factor EB (TFEB) induces lysosomal biogenesis, prevents the accumulation of autophagosomes, and protects dopaminergic neurons (Dehay et al., 2010). Also, it has been revealed that 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) exposure in dopaminergic BE‐M17 neuroblastoma cells induces abnormal permeabilization of lysosomal membranes, whereas an overexpression of TFEB in these cells induces lysosomal biogenesis and reduces cell death (Vila et al., 2011). Collectively, increased autophagy in samples from PD patients suggests the neurons' effort to withstand cellular stress (Macchi et al., 2015).
4. TREHALOSE: WHAT IT IS AND THERAPEUTIC EFFECTS
Trehalose is a nonreducing and naturally occurring disaccharide that is widely distributed in many organisms including bacteria, yeast, fungi, insects, invertebrates, and plants. Trehalose acts as a bioprotectant and protects cells against a wide variety of environmental conditions including heat, cold, desiccation, dehydration, and oxidation (Chen & Haddad, 2004). Trehalose contains no reducing end hydroxyl group, which makes it a stable molecule unable to take part in glycation reactions and prevents it from being metabolized (Chen & Haddad, 2004; Ohtake & Wang, 2011). Therefore, it acts in the form of a disaccharide in peripheral tissues and cells. In healthy humans, trehalose ingestion des not induce rapid changes in blood glucose (Yoshizane et al., 2017). As such, after oral administration, trehalose was detected in the peripheral circulation in mice (DeBosch et al., 2016; Mayer et al., 2016; Yoshizane et al., 2017). The non‐toxic nature of trehalose makes it a favourable pharmacological agent, and no adverse effects were determined for doses up to 10% in safety studies in humans (Richards et al., 2002). Currently, trehalose is regarded as a safe food ingredient for humans by the European regulation system following approval by the U.S. Food and Drug Administration in 2000 (Richards et al., 2002).
Having many unique physical and chemical properties, trehalose has been the focus of a wide range of studies (Ohtake & Wang, 2011). Several lines of experimental evidence confirm the inhibitory effect of trehalose on amyloid formation. For instance, it was shown that trehalose diminishes amyloid‐β aggregation, the neuropathology underlying Alzheimer's disease (Du, Liang, Xu, Sun, & Wang, 2013; Liu, Barkhordarian, Emadi, Park, & Sierks, 2005), and prevents insulin‐induced amyloid formation in vitro (Arora, Ha, & Park, 2004). In addition, it attenuates brain pathology in the R6/2 mouse model of HD (Tanaka et al., 2004) by directly binding to the expanded polyQ region, which results in apparent stabilization of partially unfolded mutant proteins. In spite of many supporting data, the exact mechanisms that induce this protective effect of trehalose are not clear. There is evidence that trehalose stabilizes protein folding as a chemical chaperon via direct interaction with protein (Welch & Brown, 1996). Three theories have been proposed to explain the mechanism by which trehalose stabilizes the protein structure. The preferential exclusion theory implies that an increased stability of protein in the presence of trehalose is associated with the removal of a solvation layer around the protein, which limits its motility and increases its compactness. The water replacement theory states that trehalose replaces water and forms hydrogen bonds that provide the major stabilization for protein. According to the vitrification theory, trehalose forms a glassy matrix and immobilizes proteins inside the matrix, thus making them more stable to deal with stress conditions (Jain & Roy, 2009; Liu, Ji, Dong, & Sun, 2009). Experimental evidence supports a preferential hydration hypothesis, which postulates that protein may be stabilized by preferential exclusion of cosolvent molecules from the close proximity of the protein (Arakawa & Timasheff, 1982; Timasheff, 2002). A molecular dynamics simulation study of trehalose's interaction with a protein demonstrated that trehalose covers the protein (Lins, Pereira, & Hünenberger, 2004) and competes with it in forming hydrogen bonds with the overlaying thin layer of water around the protein molecule. Therefore, a reduction in the number of protein and water hydrogen bonds decreases conformational fluctuations in the protein and contributes towards greater stability (Ganea & Harding, 2005). Furthermore, an investigation into the antiamyloidogenic effect of trehalose on hen egg white lysozyme through docking studies revealed that trehalose interacts with amyloidogenic regions in hen egg white lysozyme mainly via hydrogen and hydrophobic interactions and blocking aggregation (Chatterjee, Kolli, & Sarkar, 2017).
Although there are several studies investigating the activation of autophagy by trehalose, the underlying mechanism by which it induces autophagy remains unclear. It has been proposed that trehalose can act as an autophagy inducer through an mTOR‐independent pathway and enhances the clearance of aggregate‐prone proteins like α‐synuclein, mutant huntingtin, and prion protein (PrPSc; Aguib et al., 2009; Beranger, Crozet, Goldsborough, & Lehmann, 2008; Sarkar, Davies, Huang, Tunnacliffe, & Rubinsztein, 2007). Moreover, in the cellular model of PD and HD, trehalose has been shown to protect cells against secondary pro‐apoptotic insults in an autophagy‐dependent manner (Hosseinpour‐Moghaddam, Caraglia, & Sahebkar, 2018; Sarkar et al., 2007). In a recent report, trehalose induced autophagy in hepatocytes through an AMPK‐dependent pathway, downstream of GLUT8 (Mayer et al., 2016). However, as the GLUT8 transporter is tissue‐specific and neurons do not contain SLC2A8 (GLUT8) in their plasma membrane, this pathway may not occur in the brain tissue (Yoon et al., 2017). Also, trehalose can activate TFEB to enhance ALP by decreasing Akt activity (Palmieri et al., 2017).
Trehalose might induce protective actions through its ability to protect cells from inflammation; trehalose has been shown to have an anti‐inflammatory effect against endotoxic shock both in vivo and in vitro (Echigo et al., 2012; Minutoli et al., 2007; Minutoli et al., 2008). Almost all of the evidence suggests that trehalose suppresses inflammatory responses by inhibiting the NF‐κB pathway (Echigo et al., 2012; Minutoli et al., 2007; Minutoli et al., 2008).
Trehalose also has antioxidant activity and can scavenge free radicals (Oku et al., 2005). It is worth noting that oxidative damage is one of the potential drivers of cell death in a variety of human diseases like disorders associated with ageing and neurodegenerative diseases (Gandhi & Abramov, 2012). Trehalose exerts a stabilizing effect on membrane structure and function (Elbein, Pan, Pastuszak, & Carroll, 2003). As reported previously, it prevents lipid peroxidation induced by ROS in yeast (Herdeiro, Pereira, Panek, & Eleutherio, 2006). In another in vivo study on rat femoral artery model, trehalose suppressed the production of lipid peroxides in cerebral vasospasm induced by blood through direct interaction with the membrane (Echigo et al., 2012). Taken together, trehalose is able to exert a protective action against environmental stress through a variety of different mechanisms. Hence, due to its ability to suppress aggregation and enhance autophagy, and its anti‐inflammatory properties it could be an excellent candidate to treat amyloid‐related disorders and neurodegenerative diseases (Casarejos et al., 2011). Trehalose has been shown to have a neuroprotective effect in both in vivo and in vitro PD models. In the following section, we will present experimental evidence on the use of trehalose as a relevant therapeutic agent in PD (Tables 1 and 2).
Table 1.
Neuroprotective effects of trehalose in in vitro PD models
| Reference | Cell line | Model | Trehalose doses | Main finding |
|---|---|---|---|---|
| Lan et al. (2012) | PC12 cell line | Overexpressing WT or A53T mutant α‐synuclein | 10, 50, and 100 mM |
PC12 cells overexpressing A53T α‐synuclein: enhanced macroautophagy and reduced the level of A53T α‐synuclein via up‐regulation of macroautophagy PI3K pathway. PC12 cells overexpressing WT α‐synuclein: macroautophagy activation with no significant effects on WT α‐synuclein degradation. |
| Casarejos et al. (2011) | NB69 human neuroblastoma cells | Epoxomicin‐induced accumulation of proteins | 50 and 100 mM | Increase in the number of autophagosomes and markers of autophagy and prevents the necrosis of cells, reduction in the accumulation of polyubiquitinated proteins, total and phosphorylated τ, p‐GSK‐3, and α‐synuclein, as well as the α‐synuclein intracellular aggregates in a dose‐ and time‐dependent manner. Reduction in the p‐ERK and chaperone HSP‐70 activation. |
| He et al. (2014) | PC12 cell line | The conditioned medium of BV‐2 cells (LPS induced) was applied to PC12 neurons | 10 and 50 mM | Protect PC12 neurons by suppressing NF‐κB and AP‐1 and production of inflammatory mediators. |
| Redmann, Wani, Volpicelli‐Daley, Darley‐Usmar, and Zhang (2017) | Primary cortical mouse neurons | Treated with α‐synuclein preformed fibrils | 1, 10, and 25 mM | Trehalose alone increased cell viability (10 and 25 mM) and LC3‐II levels and remained elevated with PFF exposure. |
| Zhao, Zhi, Pan, and Zhou (2017) | PC12 cell line | Transduced cells expressed A53T α‐synuclein | 0.2, 0.5, 1.5, 5, and 10 mM | At concentrations lower than 1 mM, trehalose decreased the A53T α‐synuclein expression. |
| Yu et al. (2012) | — | A53T α‐synuclein | 10 and 100 mM |
At low concentration: Trehalose disaggregates preformed A53T α‐synuclein protofibrils and fibrils into small aggregates or even random coil structure. At high concentration: Trehalose slows down the structural transition into β‐sheet structure and completely prevents the formation of mature A53T α‐synuclein fibrils. |
| Naik, Kardani, and Roy (2016) | — | Recombinant human α‐synuclein | 0.5 M | Drive α‐synuclein towards aggregation by re‐arranging the pathway intermediates into a non‐native conformation. |
| Sarkar et al. (2007) | COS‐7, SKN–SH, HeLa, stable HeLa cells expressing EGFP‐LC3, and WT Atg5 (Atg5+/+) and Atg5‐deficient (Atg5−/−) mouse, MEFs, inducible PC12 stable cell lines expressing EGFP‐HDQ74 or EGFP‐HDQ23, and HA‐tagged A30P or A53T α‐synuclein, and T‐REx 293 | 100 mM | Induction of autophagy and increase the clearance mutant huntingtin and the A30P and A53T mutants of α‐synuclein. | |
| Bussi et al. (2017) | BV2 microglial cell line and N2A neuronal cell line | Treatment with LPS and α‐synuclein fibres and monomers | 30 mM | Induction of autophagy by reducing the release of pro‐inflammatory molecules in response to LPS and α‐synuclein. |
| Jiang et al. (2013) | PC12 cell line | Overexpression of WT α‐synuclein | 0.2, 0.5, 1.0, 5.0, and 10.0 mM | WT α‐synuclein assembled into the large amorphous protein aggregates and after long‐term incubation could be disassembled into the small amorphous particles or random coil structure. |
| Recombinant WT α‐synuclein coincubated with trehalose | 100 mM | Trehalose at low concentration inhibited the overexpression of WT α‐synuclein and at high concentration enhanced the overexpression and decreased cell viability. | ||
| Yoon et al. (2017) | SH–SY5Y cell line and rat cortical neurons | Transduction with recombinant adenoviral vectors (adeno/α‐synuclein or adeno/tfLC3) | 0, 4, 20, and 100 mM | Accumulation of lipidated LC3 (LC3‐II), p62, and autophagosomes, decrease in the autolysosomes, and increase in α‐synuclein aggregation and secretion. In contrast, cell viability was not affected upon treatment with trehalose. |
| Wu, Xu, et al. (2015) | PC12 cell line | Treatment with rotenone (500 nM) | 100 mM | Decrease in LC3‐II and α‐synuclein accumulation, induction of nuclear translocation of TFEB, restoration of lysosomal levels, decrease cell death, recover LAMP2 levels, and increase clearance of autophagosomes. |
LC3: microtubule‐associated protein 1 light chain 3; MEFs: mouse embryonic fibroblasts; PD: Parkinson's disease; PFF: preformed fibril; WT: wild‐type.
Table 2.
Neuroprotective effects of trehalose in in vivo PD models
| Reference | Animal model | Route of administration | Doses | Treatment duration | Main finding |
|---|---|---|---|---|---|
| Ferguson, Law, and Sarkar (2015) | Adult male C57Bl/6 mice (MPTP‐induced PD) | In drinking water | 1% (1.90–2.34 g·kg−1) | 38 days |
No improvement in the MPTPp‐induced behavioural alterations. Attenuation in the striatal reduction of DA, DOPAC, HVA, and 5‐HIAA levels. |
| Sarkar et al. (2014) | Adult male C57Bl/6 mice (MPTP‐induced PD) | In drinking water | 2% | Twice a week for 5 weeks and started at 3 days prior to the start of MPTP + probenecid administration | Decrease in the loss of TH and DA transporter in the ventral midbrain SNpc and CPu and dopamine levels in CPu. Reduction in microglial activation and astrocytic hypertrophy, amelioration of motor deficits, protecting ZO‐1 and occluding (two tight junctional proteins), and glucose transporter‐1 in brain endothelial cells from MPTP‐induced down‐regulation. |
| He et al. (2016) | Female SD rats (injected with AAV1/2 expressing human A53T α‐synuclein) | In drinking water | 5, 2, and 0.5% | 6 weeks |
0.5% trehalose: fail to improve the behavioural and neurochemical deficits. 5% and 2% trehalose: reduction in α‐synuclein‐mediated deficits in motor asymmetry and DA neurodegeneration and α‐synuclein aggregation in the nigrostriatal, enhancement of autophagy in the striatum by increasing formation of LC3‐II. |
| Rodríguez‐Navarro et al. (2010) | PK−/−/TauVLW transgenic mice | In drinking water | 1% |
Twice a week for 2.5 months (in 3‐month‐old mice) 4 months (in 3‐month‐old mice) 3‐week treatment (in 14‐month‐old mice) |
2.5 months: improvement in the motor and cognitive behaviours, increase in the number of dopamine neurons in the ventral midbrain and the dopamine‐related protein levels in the midbrain and striatum, and decrease in the number of phosphorylated tau‐positive neuritic plaques, the levels of phosphorylated tau protein, and the astrogliosis. 4 months: maintained the amelioration of the tau pathology and astrogliosis and failed to revert DA‐related pathology in the striatum. 3 weeks (at the limit of their life expectancy): improving in the motor behaviour and anxiety and reducing in the levels of phosphorylated tau and the number of murine β‐amyloid plaques. |
| Tanji et al. (2015) | LBD model mice (overexpressed A53T α‐synuclein) | Oral intake and intraperitoneal injection | 2% (wt/vol) |
Short period (1‐week intake) Long period (3‐ or 12‐week intake) |
I.p. injection: no effect on autophagy in the brain. Oral intake: (a) in short period: induction of autophagy and increase in the levels of a heat shock protein, HSP90, and an ER stress chaperone, SigmaR1. (b) In long period: The effect of trehalose was obscured. |
| Kaur and Nazir (2015) | Transgenic Caenorhabditis elegans model (expressing human α‐synuclein) | Seeded onto the nematode growth medium plates | 5 and 10 mM | 48 hr |
5 mM concentration: no reduction in α‐synuclein levels. 10 mM concentration: reduction in ROS and α‐synuclein levels, increase in motility and dopamine levels, and up‐regulation of autophagic and chaperonic genes bec‐1, lgg‐1, epg‐8, hsp‐60, and hsp‐4. |
| Wu, Xu, et al. (2015) | Male Lewis rats (rotenone‐induced PD) | In drinking water | 2% (wt/vol) | Daily, for 56 days | Reduction in the loss of SNpc DA neurons. |
5‐HIAA: 5‐hydroxyindoleacetic acid; CPu: caudate putamen; DA: dopamine; DOPAC: 3,4‐dihydroxyphenylacetic acid; HVA: homovanillic acid; LBD: Lewy body disease; MPTP: 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine; PD: Parkinson's disease; SD: Sprague–Dawley; SNpc: substantia nigra pars compacta; WT: wild‐type.
4.1. Modulation of autophagy
A progressive accumulation of specific protein aggregates is the common neurodegenerative pathology that impairs neuronal function. Effective therapeutic approaches to amyloid‐related disorders are achieved when protein self‐assembly into amyloid fibrils is controlled. PD is a proteopathic disorder in which failure of proteolytic systems can initiate the formation of LB‐like aggregates (Xilouri, Brekk, & Stefanis, 2013; Figure 1). Existing literature supports the likelihood that the therapeutic effect of trehalose against neurodegeneration is mainly through the induction of the autophagy pathway (Liu et al., 2005; Sarkar et al., 2007; Tien, Karaca, Tamboli, & Walter, 2016). Several studies in animal and in vitro models of PD have reported the ability of trehalose to reduce the toxicity of aggregated proteins; these are summarized below.
Figure 1.
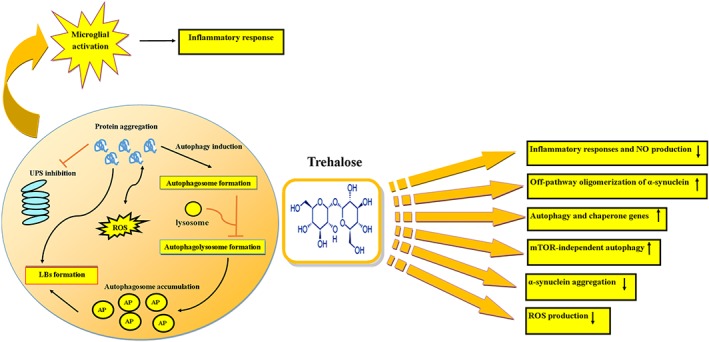
Protective effects of trehalose in Parkinson's disease (PD). The ubiquitin–proteasome system (UPS) and autophagy pathways are blocked (red arrow) in the pathological state of PD resulting in protein accumulation and Lewy body (LB) formation. Trehalose protects through different mechanisms. AP: autophagosome
Previously, an in vitro study of neuronal cell lines showed that trehalose accelerated the clearance of both A53T and A30P α‐synuclein mutants through activation of autophagy in a mTOR‐independent manner (Sarkar et al., 2007; Table 1). Trehalose also increased the removal of α‐synuclein in an AVV1/2 A53T α‐synuclein rat model of PD. Indeed, it activated autophagy in the striatum and significantly reduced the amount of α‐synuclein aggregates, enhanced the survival of dopaminergic neurons, and improved the motor deficits (He et al., 2016; Table 2). A study on PC12 cells overexpressing WT or A53T α‐synuclein demonstrated that trehalose enhanced the clearance of A53T α‐synuclein, and that this effect was mediated by an up‐regulation of the macroautophagy PI3K pathway. However, trehalose had no significant effect on WT α‐synuclein clearance (Lan et al., 2012; Table 1). This is consistent with the observation that macroautophagy is the predominant pathway for clearance of A53T α‐synuclein due to its propensity to aggregate and WT α‐synuclein can be efficiently cleared by the UPS pathway (Ravikumar, Duden, & Rubinsztein, 2002; Webb, Ravikumar, Atkins, Skepper, & Rubinsztein, 2003). Inhibition of UPS function by epoxomicin results in the accumulation of polyubiquitinated proteins such as total and phosphorylated tau, p‐GSK‐3, and α‐synuclein, as well as the α‐synuclein intracellular aggregates in NB69 human neuroblastoma cells. After treatment with trehalose, autophagosomes and autophagy markers increased in these cells in a dose‐ and time‐dependent manner (Casarejos et al., 2011; Table 1). In transgenic Caenorhabditis elegans model expressing human α‐synuclein, treatment with trehalose reduced ROS and α‐synuclein levels, whereas motility and dopamine levels were significantly increased. Also, trehalose induced autophagy, and mRNA expression of genes related to autophagy and heat‐shock system was increased in the transgenic worms, indicating that trehalose has a synergistic interaction with the cellular machinery to tackle the proteotoxic load of aggregated proteins. Other osmolytes including sorbitol and xylitol exert many opposite effects in the C. elegans model of Parkinsonism (Kaur & Nazir, 2015; Table 2).
A recent study has demonstrated that trehalose decreased the A53T α‐synuclein expression level in transduced PC12 cells at low concentration (lower than 1 mM). In contrast, at a high concentration (higher than 1 mM), trehalose induced the expression of A53T α‐synuclein and attenuated cell viability through stabilization of A53T α‐synuclein oligomers (Zhao et al., 2017; Table 1). Likewise, a high concentration of trehalose induced cytotoxic effects, an effect also observed in previous studies (Lan et al., 2012; Table 1). Along these same lines, trehalose did not efficiently induce autophagy in the brain of mice after long‐term treatment (3 or 12 weeks; Tanji et al., 2015; Table 2). Short‐term treatment with trehalose ameliorates dopamine‐related pathology in the mouse tauopathy model by induction of mitophagy and improvement in the redox status. However, long‐term treatment leads to energy deficits due to the uncontrolled activation of the autophagic pathway. The detrimental long‐term effect of trehalose is more noticeable in cerebral regions associated with higher metabolic activity and oxidative stress like the striatum, which has a high content of dopamine (Table 2; Rodríguez‐Navarro et al., 2010). As shown by Zhao et al. (2017), a high trehalose concentration in PC12 cells affected mitochondrial morphology and reduced the mitochondrial membrane potential (Table 1). Therefore, it appears that the lowest effective dose and duration of maximum neuroprotective effect need to be defined to avoid the adverse effects of a high dosage.
Despite the general consensus regarding the autophagy‐inducing activity of trehalose, an in vitro study on PD cell culture models showed that it blocked autophagy, thus resulting in the accumulation of lipidated LC3 (LC3‐II), p62, and autophagosomes, whereas a decreased number of autolysosomes were observed. According to the results of this study, trehalose disturbed lysosomal membrane integrity and inhibited autophagosome–lysosome fusion. Nevertheless, although α‐synuclein aggregation was significantly increased, cell viability did not change, suggesting the neuroprotective function of trehalose was mediated via an autophagy‐independent mechanism (Table 1; Yoon et al., 2017). Similarly, neuropathological analysis of brains from a mouse model of LB disease by Tanji et al. revealed that trehalose increased the accumulation of the autophagic marker LC3‐II in the brain with no apparent effect on the abnormal aggregation of α‐synuclein, but it decreased the level of insoluble α‐synuclein in mice. Moreover, they demonstrated that trehalose can act through modulation of different chaperone molecules such as HSP90 and SigmaR1 (Table 2; Tanji et al., 2015). Various studies have shown that exogenous application of α‐synuclein preformed fibrils (PFFs) promotes the formation of protein aggregation in cells (Luk et al., 2012; Volpicelli‐Daley et al., 2011). Interestingly, it has been shown that when primary cortical neurons are treated with trehalose alone, the LC3‐II levels increase and remained elevated on treatment with both trehalose and α‐synuclein PFFs, whereas no significant changes were observed in the levels of aggregated α‐synuclein, indicating that PFFs were resistant to degradation even with augmentation of macroautophagy (Table 1; Redmann et al., 2017).
In a rotenone‐induced PD model, rotenone‐treated PC12 cells exhibited a significant increase in the levels of aggregated α‐synuclein, LC3‐II, Beclin‐1, and autophagic substrate p62, and trehalose treatment attenuated this accumulation of LC3‐II and α‐synuclein. Trehalose also increased the nuclear translocation of TFEB, an enhancer of lysosomal biogenesis, and elevated the clearance of autophagosomes (Wu, Xu, et al., 2015). Decressac and Bjorklund in an in vivo study showed that excess cellular concentration of α‐synuclein in nigral dopaminergic neurons caused cytoplasmic sequestration of TFEB and effectively blocked its nuclear translocation. Genetic or pharmacological activation of TFEB improved the clearance of toxic α‐synuclein aggregates. Similar alterations also occur in the human PD brain, and TFEB colocalizes with α‐synuclein in Lewy bodies (Decressac & Bjorklund, 2013).
4.2. Stabilization of protein
The ability of trehalose to fold denatured proteins into their native structures has been defined in previous studies (Melo et al., 2003). Although the precise mechanism is not clear, it seems plausible that trehalose acts as a chaperone and enhances protein folding via direct interaction with the protein (Jain & Roy, 2009; Welch & Brown, 1996). Conformational transition of α‐synuclein using synchrotron radiation circular dichroism spectroscopy showed that trehalose can interact with α‐synuclein and block in vitro polymerization in a dose‐dependent manner (Ruzza et al., 2015). An assessment of the inhibitory effect of trehalose on α‐synuclein aggregation revealed that a low trehalose concentration disaggregates preformed mutant α‐synuclein (A53T) protofibrils and fibrils into small aggregates or dissolves them into disordered structures. However, at a higher concentration, the structural transition of A53T α‐synuclein into a β‐sheet was blunted, and the formation of mature fibrils was completely prevented (Table 1; Yu et al., 2012). In another study, it was revealed that early coincubation of WT α‐synuclein with trehalose leads to the formation of amorphous aggregates, and after a longer incubation large amorphous aggregates are remoulded into small amorphous particles or into the random coil conformer (Jiang et al., 2013). In contrast, results from another in vitro study indicated that fibrillization of α‐synuclein is clearly accelerated in the presence of trehalose (Naik et al., 2016). In this latter study, it was proposed that the inverse effect is due to the intrinsically disordered structure of α‐synuclein, which in the presence of trehalose re‐arranged into a partially folded conformation and the interaction of these structures initiated aggregation (Table 1; Naik et al., 2016). In a more recent study, Katyal, Agarwal, Sen, Kumar, and Deep (2018) found that trehalose reduces potentially toxic fibril load by driving the aggregation route into the formation of off‐pathway amorphous aggregates, which are unable to nucleate the formation of amyloid fibrils. Protofibrils and oligomer species, as nucleation‐prone species, could contribute to neurotoxicity in the dopaminergic neurons (Goldberg & Lansbury, 2000; Volles et al., 2001). Therefore, agents that could block protein oligomerization might represent a potential therapy for PD.
4.3. Anti‐inflammatory effect of trehalose
Neuroinflammation is proposed to be central to the progressive nature of several neurodegenerative disorders and occurs in virtually all lysosomal storage diseases with neurological involvement (Farfel‐Becker et al., 2011). Nowadays, microglia are a critical target in many PD therapeutic trials aiming at the attenuation of inflammation and suppression of the self‐perpetuating ROS production cycle (Glass, Saijo, Winner, Marchetto, & Gage, 2010). There is overwhelming support for the ability of trehalose to inhibit inflammatory response in PD models. For example, treatment of a chronic MPTP‐intoxicated mouse model of PD with trehalose resulted in the inhibition of both microglial and astroglial activation, which in turn s at present attenuates the release of pro‐inflammatory and neurotoxic molecules. Interestingly, the disruption of the integrity of the blood–brain barrier and also endothelial cell “cluster” formation, which occur in MPTP‐intoxicated mice, were diminished by trehalose through its ability to restore the levels of two tight junctional proteins (ZO‐1 and occludin). The subsequent impact of trehalose on the MPTP mouse model was devoid of motor deficits (Table 2; Sarkar et al., 2014). The link between microglial activation and neural damage was confirmed in an in vitro study. NF‐κB pathway activation increased the release of cytokines including IL‐1β, IL‐6, TNF‐α, and NO in BV‐2 microglial cells that were challenged with LPS. Trehalose suppressed apoptosis in PC12 cells and protected these cells against neurotoxicity evoked by microglial activation (Table 1; He et al., 2014). Moreover, trehalose efficiently attenuated NF‐κB activation by down‐regulation of toll‐like receptor 4 (TLR4) and protected the PD animal model from LPS‐mediated neuroinflammation (Minutoli et al., 2008). In a recent study, Bussi et al. (2017) showed that exposure to both rapamycin and trehalose efficiently promoted autophagy and decreased the release of pro‐inflammatory mediators, including NO, in response to LPS and α‐synuclein in BV2 microglial cells (Table 1). Autophagy may play a critical role in the control of inflammatory responses through inhibition of spurious inflammasome activation and down‐regulation of immune response, induced by the activated inflammasome (Lupfer et al., 2013; Nakahira et al., 2011; Saitoh et al., 2008), and inhibition of type I interferon responses either directly or indirectly (Konno, Konno, & Barber, 2013; Liang et al., 2014; Saitoh et al., 2009). Therefore, therapeutic strategies that prevent neuroinflammation by targeting autophagy might be helpful in finding effective treatments for neurodegenerative diseases (Ghavami et al., 2014).
5. CONCLUSIONS
Extensive data derived from animal models indicate that trehalose, via its effect on autophagy, has therapeutic potential in various proteopathic disorders. There are four main mechanisms for autophagy regulation in eukaryote cells: PI3K/Akt/mTOR, AMPK/ULK1/mTOR, Bcl‐2/Beclin‐1, and TFEB pathways (Wang et al., 2017). At present most of the evidence suggests that trehalose can activate autophagy via each of these pathways, depending on the type of disease and cell. Nevertheless, there is controversy surrounding the effect of trehalose on autophagy activation in PD treatment. Although in many studies an mTOR‐independent pathway is proposed as the main mechanism of action, recent studies have revealed that trehalose drives ALP through activation of TFEB function (Evans, Jeong, Zhang, Sergin, & Razani, 2018; Figure 2). Lysosomal depletion and autophagosome accumulation in PD neurons corroborate the notion that TFEB may be a promising target for the development of novel therapies for PD. Also, on the basis of the experimental evidence, trehalose could affect protein aggregation by stabilizing proteins. In PD, trehalose drives the aggregation of α‐synuclein into amorphous aggregates, which are non‐toxic. However, dose and time‐dependency are major factors affecting the efficacy of trehalose in PD treatment as no effect was observed with high‐dose and long‐term use of this disaccharide. Therefore, determination of the optimal dose and treatment duration with trehalose may be an important direction for future studies. Above all, it should also be noted that in animal models of PD treatment with trehalose has been demonstrated to be effective in improving the manifestation of symptoms associated with parkinsonism (He et al., 2016; Kaur & Nazir, 2015; Rodríguez‐Navarro et al., 2010; Sarkar et al., 2014). Even with short‐term treatment, the number of Aβ‐positive plaques and motor deficits were reduced in old PK−/−/TauVLW mice, which were at the limit of their life expectancy (Rodríguez‐Navarro et al., 2010). Despite the beneficial effects of trehalose in animal models, no clinical trials designed to assess the efficacy of this disaccharide in human PD subjects have been done. Such trials should take into consideration the limited intestinal absorption of trehalose in humans that justifies the need for alternative routes of administration or the use of tailored formulations allowing systemic absorption of this disaccharide. In this context, intravenous trehalose administration has been suggested to tackle the problem of intestinal degradation of trehalose by trehalase. There is currently evidence from phase I and II trials supporting the efficacy and safety of long‐term intravenous trehalose administration in patients with oculopharyngeal muscular dystrophy and spinocerebellar ataxia type 3 (Argov, Vornovitsky, Blumen, & Caraco, 2015). Given these convincing data, proof‐of‐concept clinical trials are recommended to be initiated in patients with neurodegenerative diseases including PD to assess if intravenous trehalose administration can improve the symptoms of the disease.
Figure 2.
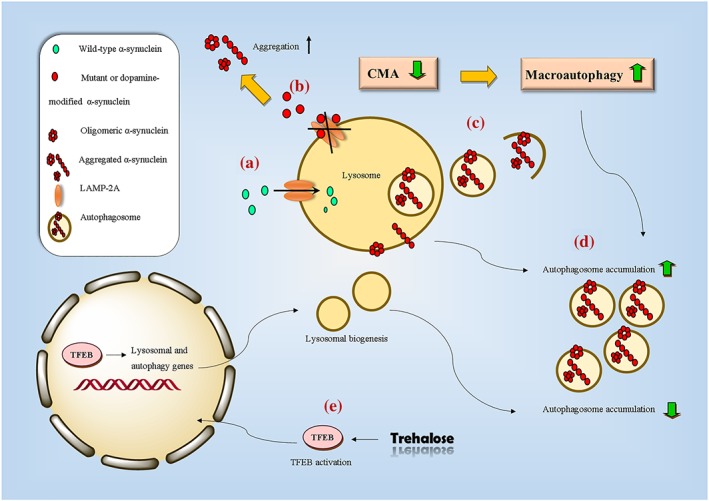
A schematic presentation of the autophagy–lysosome pathway in Parkinson's disease. (a) Under physiological conditions, chaperone‐mediated autophagy (CMA) is the main route for α‐synuclein degradation. Wild‐type α‐synuclein is internalized to the lysosomal lumen through binding to lysosome‐associated membrane protein type 2a (LAMP‐2A) receptor. (b) In the pathological condition, mutant or dopamine‐modified α‐synuclein strongly binds to the LAMP‐2A receptor and impairs the CMA pathway. CMA inactivation leads to aberrant accumulation of toxic α‐synuclein. (c) In response to blockage of CMA, clearance of aggregated α‐synuclein occurs by a compensatory mechanism that is up‐regulation of macroautophagy. (d) Up‐regulation of macroautophagy might increase autophagosome accumulation. Moreover, the interaction of oligomeric α‐synuclein with organelle lipid bilayers could disrupt and permeabilize the lysosomal membrane, causing the leakage of lysosomal hydrolases to the cytosol and accumulation of autophagic vacuoles. (e) Pharmacological activation of the transcription factor EB (TFEB) promotes lysosomal biogenesis and alleviates autophagosome accumulation
5.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017; Alexander, Kelly, Marrion, Peters, Faccenda, Harding, Pawson, Sharman, Southan, Buneman et al., 2017a; Alexander, Kelly, Marrion, Peters, Faccenda, Harding, Pawson, Sharman, Southan, Davies et al., 2017b).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Khalifeh M, Barreto GE, Sahebkar A. Trehalose as a promising therapeutic candidate for the treatment of Parkinson's disease. Br J Pharmacol. 2019;176:1173–1189. 10.1111/bph.14623
REFERENCES
- Agarraberes, F. A. , Terlecky, S. R. , & Dice, J. F. (1997). An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. The Journal of Cell Biology, 137, 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguib, Y. , Heiseke, A. , Gilch, S. , Riemer, C. , Baier, M. , Schatzl, H. M. , & Ertmer, A. (2009). Autophagy induction by trehalose counteracts cellular prion infection. Autophagy, 5, 361–369. 10.4161/auto.5.3.7662 [DOI] [PubMed] [Google Scholar]
- AlDakheel, A. , Kalia, L. V. , & Lang, A. E. (2014). Pathogenesis‐targeted, disease‐modifying therapies in Parkinson disease. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics, 11, 6–23. 10.1007/s13311-013-0218-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alers, S. , Loffler, A. S. , Wesselborg, S. , & Stork, B. (2012). Role of AMPK‐mTOR‐Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Molecular and Cellular Biology, 32, 2–11. 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(S1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Overview. British Journal of Pharmacology, 174(S1), S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174(S1), S360–S446. 10.1111/bph.13883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Erviti, L. , Rodriguez‐Oroz, M. C. , Cooper, J. M. , Caballero, C. , Ferrer, I. , Obeso, J. A. , & Schapira, A. H. (2010). Chaperone‐mediated autophagy markers in Parkinson disease brains. Archives of Neurology, 67, 1464–1472. 10.1001/archneurol.2010.198 [DOI] [PubMed] [Google Scholar]
- Arakawa, T. , & Timasheff, S. N. (1982). Stabilization of protein structure by sugars. Biochemistry, 21, 6536–6544. 10.1021/bi00268a033 [DOI] [PubMed] [Google Scholar]
- Argov, Z. , Vornovitsky, H. , Blumen, S. , & Caraco, Y. (2015). First human use of high dose IV trehalose: Safety, tolerability and pharmacokinetic results from the oculopharyngeal muscular dystrophy (OPMD) therapy trial (P7.068). Neurology, 84, P7.068. [Google Scholar]
- Arias, E. (2015). Lysosomal mTORC2/PHLPP1/Akt axis: A new point of control of chaperone‐mediated autophagy. Oncotarget, 6, 35147–35148. 10.18632/oncotarget.5903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias, E. , & Cuervo, A. M. (2011). Chaperone‐mediated autophagy in protein quality control. Current Opinion in Cell Biology, 23, 184–189. 10.1016/j.ceb.2010.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias, E. , Koga, H. , Diaz, A. , Mocholi, E. , Patel, B. , & Cuervo, A. M. (2015). Lysosomal mTORC2/PHLPP1/Akt regulate chaperone‐mediated autophagy. Molecular Cell, 59, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora, A. , Ha, C. , & Park, C. B. (2004). Inhibition of insulin amyloid formation by small stress molecules. FEBS Letters, 564, 121–125. 10.1016/S0014-5793(04)00326-6 [DOI] [PubMed] [Google Scholar]
- Baba, M. , Nakajo, S. , Tu, P. H. , Tomita, T. , Nakaya, K. , Lee, V. M. , … Iwatsubo, T. (1998). Aggregation of alpha‐synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. The American Journal of Pathology, 152, 879–884. [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay, U. , Kaushik, S. , Varticovski, L. , & Cuervo, A. M. (2008). The chaperone‐mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Molecular and Cellular Biology, 28, 5747–5763. 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus, R. T. , Baumann, T. L. , Siffert, J. , Herzog, C. D. , Alterman, R. , Boulis, N. , … Olanow, C. W. (2013). Safety/feasibility of targeting the substantia nigra with AAV2‐neurturin in Parkinson patients. Neurology, 80, 1698–1701. 10.1212/WNL.0b013e3182904faa [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beranger, F. , Crozet, C. , Goldsborough, A. , & Lehmann, S. (2008). Trehalose impairs aggregation of PrPSc molecules and protects prion‐infected cells against oxidative damage. Biochemical and Biophysical Research Communications, 374, 44–48. 10.1016/j.bbrc.2008.06.094 [DOI] [PubMed] [Google Scholar]
- Bossy, B. , Perkins, G. , & Bossy‐Wetzel, E. (2008). Clearing the brain's cobwebs: The role of autophagy in neuroprotection. Current Neuropharmacology, 6, 97–101. 10.2174/157015908784533897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein, J. M. , Tagliati, M. , Alterman, R. L. , Lozano, A. M. , Volkmann, J. , Stefani, A. , … DeLong, M. R. (2011). Deep brain stimulation for Parkinson disease: An expert consensus and review of key issues. Archives of Neurology, 68, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi, C. , Ramos, J. M. P. , Arroyo, D. S. , Gaviglio, E. A. , Gallea, J. I. , Wang, J. M. , … Iribarren, P. (2017). Autophagy down regulates pro‐inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha‐synuclein induced neuronal cell death. Scientific Reports, 7, 43153 10.1038/srep43153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, Y. , & Klionsky, D. J. (2007). Physiological functions of Atg6/Beclin 1: A unique autophagy‐related protein. Cell Research, 17, 839–849. 10.1038/cr.2007.78 [DOI] [PubMed] [Google Scholar]
- Casarejos, M. J. , Solano, R. M. , Gómez, A. , Perucho, J. , De Yébenes, J. G. , & Mena, M. A. (2011). The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochemistry International, 58, 512–520. [DOI] [PubMed] [Google Scholar]
- Chatterjee, R. , Kolli, V. , & Sarkar, N. (2017). Trehalose and magnesium chloride exert a common anti‐amyloidogenic effect towards hen egg white lysozyme. Protein Journal, 36, 138–146. 10.1007/s10930-017-9705-2 [DOI] [PubMed] [Google Scholar]
- Chen, L. , Su, Z. Z. , Huang, L. , Xia, F. N. , Qi, H. , Xie, L. J. , … Chen, Q. F. (2017). The AMP‐activated protein kinase KIN10 is involved in the regulation of autophagy in Arabidopsis . Frontiers in Plant Science, 8, 1201 10.3389/fpls.2017.01201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , & Haddad, G. G. (2004). Role of trehalose phosphate synthase and trehalose during hypoxia: From flies to mammals. The Journal of Experimental Biology, 207, 3125–3129. 10.1242/jeb.01133 [DOI] [PubMed] [Google Scholar]
- Connolly, B. S. , & Lang, A. E. (2014). Pharmacological treatment of Parkinson disease: A review. Jama, 311, 1670–1683. [DOI] [PubMed] [Google Scholar]
- Crews, L. , Spencer, B. , Desplats, P. , Patrick, C. , Paulino, A. , Rockenstein, E. , … Masliah, E. (2010). Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of α‐synucleinopathy. PLoS ONE, 5, e9313 10.1371/journal.pone.0009313 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cuervo, A. M. , Bergamini, E. , Brunk, U. T. , Droge, W. , Ffrench, M. , & Terman, A. (2005). Autophagy and aging: The importance of maintaining “clean” cells. Autophagy, 1, 131–140. 10.4161/auto.1.3.2017 [DOI] [PubMed] [Google Scholar]
- Cuervo, A. M. , Dice, J. F. , & Knecht, E. (1997). A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. The Journal of Biological Chemistry, 272, 5606–5615. 10.1074/jbc.272.9.5606 [DOI] [PubMed] [Google Scholar]
- Cuervo, A. M. , Stefanis, L. , Fredenburg, R. , Lansbury, P. T. , & Sulzer, D. (2004). Impaired degradation of mutant α‐synuclein by chaperone‐mediated autophagy. Science (New York, NY), 305, 1292–1295. [DOI] [PubMed] [Google Scholar]
- Dawson, T. M. , & Dawson, V. L. (2003). Molecular pathways of neurodegeneration in Parkinson's disease. Science (New York, NY), 302, 819–822. [DOI] [PubMed] [Google Scholar]
- DeBosch, B. J. , Heitmeier, M. R. , Mayer, A. L. , Higgins, C. B. , Crowley, J. R. , Kraft, T. E. , et al. (2016). Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Science Signaling, 9, ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac, M. , & Bjorklund, A. (2013). TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy, 9, 1244–1246. 10.4161/auto.25044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay, B. , Bove, J. , Rodriguez‐Muela, N. , Perier, C. , Recasens, A. , Boya, P. , & Vila, M. (2010). Pathogenic lysosomal depletion in Parkinson's disease. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 30, 12535–12544. 10.1523/JNEUROSCI.1920-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Troya, S. , Perez‐Perez, M. E. , Florencio, F. J. , & Crespo, J. L. (2008). The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy, 4, 851–865. 10.4161/auto.6555 [DOI] [PubMed] [Google Scholar]
- Dice, J. F. (1990). Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends in Biochemical Sciences, 15, 305–309. 10.1016/0968-0004(90)90019-8 [DOI] [PubMed] [Google Scholar]
- Du, J. , Liang, Y. , Xu, F. , Sun, B. , & Wang, Z. (2013). Trehalose rescues Alzheimer's disease phenotypes in APP/PS1 transgenic mice. The Journal of Pharmacy and Pharmacology, 65, 1753–1756. [DOI] [PubMed] [Google Scholar]
- Ebrahimi‐Fakhari, D. , Saidi, L. J. , & Wahlster, L. (2013). Molecular chaperones and protein folding as therapeutic targets in Parkinson's disease and other synucleinopathies. Acta Neuropathologica Communications, 1, 79 10.1186/2051-5960-1-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echigo, R. , Shimohata, N. , Karatsu, K. , Yano, F. , Kayasuga‐Kariya, Y. , Fujisawa, A. , … Sasaki, N. (2012). Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. Journal of Translational Medicine, 10, 80 10.1186/1479-5876-10-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein, A. D. , Pan, Y. T. , Pastuszak, I. , & Carroll, D. (2003). New insights on trehalose: A multifunctional molecule. Glycobiology, 13, 17r–27r. [DOI] [PubMed] [Google Scholar]
- Evans, T. D. , Jeong, S. J. , Zhang, X. , Sergin, I. , & Razani, B. (2018). TFEB and trehalose drive the macrophage autophagy–lysosome system to protect against atherosclerosis. Autophagy, 14, 724–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel‐Becker, T. , Vitner, E. B. , Pressey, S. N. , Eilam, R. , Cooper, J. D. , & Futerman, A. H. (2011). Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Human Molecular Genetics, 20, 1375–1386. [DOI] [PubMed] [Google Scholar]
- Ferguson, S. A. , Law, C. D. , & Sarkar, S. (2015). Chronic MPTP treatment produces hyperactivity in male mice which is not alleviated by concurrent trehalose treatment. Behavioural Brain Research, 292, 68–78. [DOI] [PubMed] [Google Scholar]
- Fujita, N. , Itoh, T. , Omori, H. , Fukuda, M. , Noda, T. , & Yoshimori, T. (2008). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Molecular Biology of the Cell, 19, 2092–2100. 10.1091/mbc.e07-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, S. , & Abramov, A. Y. (2012). Mechanism of oxidative stress in neurodegeneration. Oxidative Medicine and Cellular Longevity, 2012, 1–11. 10.1155/2012/428010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganea, E. , & Harding, J. J. (2005). Trehalose and 6‐aminohexanoic acid stabilize and renature glucose‐6‐phosphate dehydrogenase inactivated by glycation and by guanidinium hydrochloride. Biological Chemistry, 386, 269–278. [DOI] [PubMed] [Google Scholar]
- Gan‐Or, Z. , Dion, P. A. , & Rouleau, G. A. (2015). Genetic perspective on the role of the autophagy–lysosome pathway in Parkinson disease. Autophagy, 11, 1443–1457. 10.1080/15548627.2015.1067364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami, S. , Shojaei, S. , Yeganeh, B. , Ande, S. R. , Jangamreddy, J. R. , Mehrpour, M. , … Łos, M. J. (2014). Autophagy and apoptosis dysfunction in neurodegenerative disorders. Progress in Neurobiology, 112, 24–49. 10.1016/j.pneurobio.2013.10.004 [DOI] [PubMed] [Google Scholar]
- Glass, C. K. , Saijo, K. , Winner, B. , Marchetto, M. C. , & Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell, 140, 918–934. 10.1016/j.cell.2010.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, M. S. , & Lansbury, P. T. Jr. (2000). Is there a cause‐and‐effect relationship between α‐synuclein fibrillization and Parkinson's disease? Nature Cell Biology, 2, E115–E119. 10.1038/35017124 [DOI] [PubMed] [Google Scholar]
- Gusdon, A. M. , Zhu, J. , Van Houten, B. , & Chu, C. T. (2012). ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiology of Disease, 45, 962–972. 10.1016/j.nbd.2011.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada, T. , Noda, N. N. , Satomi, Y. , Ichimura, Y. , Fujioka, Y. , Takao, T. , … Ohsumi, Y. (2007). The Atg12–Atg5 conjugate has a novel E3‐like activity for protein lipidation in autophagy. The Journal of Biological Chemistry, 282, 37298–37302. 10.1074/jbc.C700195200 [DOI] [PubMed] [Google Scholar]
- Hansen, C. , Angot, E. , Bergström, A.‐L. , Steiner, J. A. , Pieri, L. , Paul, G. , … Brundin, P. (2011). α‐Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. The Journal of Clinical Investigation, 121, 715–725. 10.1172/JCI43366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Q. , Koprich, J. B. , Wang, Y. , Yu, W. B. , Xiao, B. G. , Brotchie, J. M. , & Wang, J. (2016). Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV α‐synuclein rat model of Parkinson's disease. Molecular Neurobiology, 53, 2258–2268. 10.1007/s12035-015-9173-7 [DOI] [PubMed] [Google Scholar]
- He, Q. , Wang, Y. , Lin, W. , Zhang, Q. , Zhao, J. , Liu, F. T. , … Wang, J. (2014). Trehalose alleviates PC12 neuronal death mediated by lipopolysaccharide‐stimulated BV‐2 cells via inhibiting nuclear transcription factor NF‐κB and AP‐1 activation. Neurotoxicity Research, 26, 430–439. 10.1007/s12640-014-9487-7 [DOI] [PubMed] [Google Scholar]
- Herdeiro, R. S. , Pereira, M. D. , Panek, A. D. , & Eleutherio, E. C. (2006). Trehalose protects Saccharomyces cerevisiae from lipid peroxidation during oxidative stress. Biochimica et Biophysica Acta, 1760, 340–346. 10.1016/j.bbagen.2006.01.010 [DOI] [PubMed] [Google Scholar]
- Hideshima, T. , Bradner, J. E. , Chauhan, D. , & Anderson, K. C. (2005). Intracellular protein degradation and its therapeutic implications. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 11, 8530–8533. [DOI] [PubMed] [Google Scholar]
- Hosseinpour‐Moghaddam, K. , Caraglia, M. , & Sahebkar, A. (2018). Autophagy induction by trehalose: Molecular mechanisms and therapeutic impacts. Journal of Cellular Physiology, 233, 6524–6543. 10.1002/jcp.26583 [DOI] [PubMed] [Google Scholar]
- Hyttinen, J. M. , Niittykoski, M. , Salminen, A. , & Kaarniranta, K. (2013). Maturation of autophagosomes and endosomes: A key role for Rab7. Biochimica et Biophysica Acta, 1833, 503–510. [DOI] [PubMed] [Google Scholar]
- Iwatsubo, T. (2003). Aggregation of α‐synuclein in the pathogenesis of Parkinson's disease. Journal of Neurology, 250(Suppl 3), Iii11–Iii14. [DOI] [PubMed] [Google Scholar]
- Jain, N. K. , & Roy, I. (2009). Effect of trehalose on protein structure. Protein Science, 18, 24–36. 10.1002/pro.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, T. , Yu, W. B. , Yao, T. , Zhi, X. L. , Pan, L. F. , Wang, J. , & Zhou, P. (2013). Trehalose inhibits wild‐type α‐synuclein fibrillation and overexpression and protects against the protein neurotoxicity in transduced PC12 cells. RSC Advances, 3, 9500–9508. 10.1039/c3ra40600h [DOI] [Google Scholar]
- Kalia, L. V. , Kalia, S. K. , & Lang, A. E. (2015). Disease‐modifying strategies for Parkinson's disease. Movement Disorders: Official Journal of the Movement Disorder Society, 30, 1442–1450. 10.1002/mds.26354 [DOI] [PubMed] [Google Scholar]
- Katyal, N. , Agarwal, M. , Sen, R. , Kumar, V. , & Deep, S. (2018). Paradoxical effect of trehalose on the aggregation of α‐synuclein: Expedites onset of aggregation yet reduces fibril load. ACS Chemical Neuroscience, 9(6), 1477–1491. 10.1021/acschemneuro.8b00056 [DOI] [PubMed] [Google Scholar]
- Kaur, S. , & Nazir, A. (2015). Potential role of protein stabilizers in amelioration of Parkinson's disease and associated effects in transgenic Caenorhabditis elegans model expressing alpha‐synuclein. RSC Advances, 5, 77706–77715. 10.1039/C5RA13546J [DOI] [Google Scholar]
- Keller, J. N. , Dimayuga, E. , Chen, Q. , Thorpe, J. , Gee, J. , & Ding, Q. (2004). Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. The International Journal of Biochemistry & Cell Biology, 36, 2376–2391. 10.1016/j.biocel.2004.05.003 [DOI] [PubMed] [Google Scholar]
- Kim, J. , Kundu, M. , Viollet, B. , & Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology, 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D. J. , Abeliovich, H. , Agostinis, P. , Agrawal, D. K. , Aliev, G. , Askew, D. S. , … Deter, R. L. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy, 4, 151–175. 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochergin, I. , & Zakharova, M. (2016). The role of autophagy in neurodegenerative diseases. Neurochemical Journal, 10, 7–18. 10.1134/S1819712416010098 [DOI] [Google Scholar]
- Konno, H. , Konno, K. , & Barber, G. N. (2013). Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell, 155, 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer, G. , Mariño, G. , & Levine, B. (2010). Autophagy and the integrated stress response. Molecular Cell, 40, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan, D. M. , Liu, F. T. , Zhao, J. , Chen, Y. , Wu, J. J. , Ding, Z. T. , … Wang, J. (2012). Effect of trehalose on PC12 cells overexpressing wild‐type or A53T mutant α‐synuclein. Neurochemical Research, 37, 2025–2032. 10.1007/s11064-012-0823-0 [DOI] [PubMed] [Google Scholar]
- Learmonth, D. A. , & Freitas, A. P. (2002). Chemical synthesis and characterization of conjugates of a novel catechol‐O‐methyltransferase inhibitor. Bioconjugate Chemistry, 13, 1112–1118. 10.1021/bc0200327 [DOI] [PubMed] [Google Scholar]
- Lee, H. J. , & Lee, S. J. (2002). Characterization of cytoplasmic α‐synuclein aggregates. Fibril formation is tightly linked to the inclusion‐forming process in cells. The Journal of Biological Chemistry, 277, 48976–48983. 10.1074/jbc.M208192200 [DOI] [PubMed] [Google Scholar]
- Levine, B. , & Klionsky, D. J. (2004). Development by self‐digestion: Molecular mechanisms and biological functions of autophagy. Developmental Cell, 6, 463–477. [DOI] [PubMed] [Google Scholar]
- Levine, B. , & Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell, 132, 27–42. 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Q. , Seo, G. J. , Choi, Y. J. , Kwak, M. J. , Ge, J. , Rodgers, M. A. , … Jung, J. U. (2014). Crosstalk between the cGAS DNA sensor and Beclin‐1 autophagy protein shapes innate antimicrobial immune responses. Cell Host & Microbe, 15, 228–238. 10.1016/j.chom.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lins, R. D. , Pereira, C. S. , & Hünenberger, P. H. (2004). Trehalose–protein interaction in aqueous solution. Proteins: Structure, Function, and Bioinformatics, 55, 177–186. 10.1002/prot.10632 [DOI] [PubMed] [Google Scholar]
- Liu, F. F. , Ji, L. , Dong, X. Y. , & Sun, Y. (2009). Molecular insight into the inhibition effect of trehalose on the nucleation and elongation of amyloid β‐peptide oligomers. The Journal of Physical Chemistry B, 113, 11320–11329. 10.1021/jp905580j [DOI] [PubMed] [Google Scholar]
- Liu, R. , Barkhordarian, H. , Emadi, S. , Park, C. B. , & Sierks, M. R. (2005). Trehalose differentially inhibits aggregation and neurotoxicity of beta‐amyloid 40 and 42. Neurobiology of Disease, 20, 74–81. 10.1016/j.nbd.2005.02.003 [DOI] [PubMed] [Google Scholar]
- Luk, K. C. , Kehm, V. , Carroll, J. , Zhang, B. , O'Brien, P. , Trojanowski, J. Q. , & Lee, V. M. Y. (2012). Pathological α‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science (New York, NY), 338, 949–953. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupfer, C. , Thomas, P. G. , Anand, P. K. , Vogel, P. , Milasta, S. , Martinez, J. , … Kanneganti, T. D. (2013). Receptor interacting protein kinase 2‐mediated mitophagy regulates inflammasome activation during virus infection. Nature Immunology, 14, 480–488. 10.1038/ni.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch‐Day, M. A. , Mao, K. , Wang, K. , Zhao, M. , & Klionsky, D. J. (2012). The role of autophagy in Parkinson's disease. Cold Spring Harbor Perspectives in Medicine, 2, a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchi, B. , Di Paola, R. , Marino‐Merlo, F. , Felice, M. R. , Cuzzocrea, S. , & Mastino, A. (2015). Inflammatory and cell death pathways in brain and peripheral blood in Parkinson's disease. CNS & Neurological Disorders Drug Targets, 14, 313–324. 10.2174/1871527314666150225124928 [DOI] [PubMed] [Google Scholar]
- Mayer, A. L. , Higgins, C. B. , Heitmeier, M. R. , Kraft, T. E. , Qian, X. , Crowley, J. R. , … DeBosch, B. J. (2016). SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose‐induced autophagy. Scientific Reports, 6, 38586 10.1038/srep38586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo, E. P. , Chen, L. , Cabral, J. M. , Fojan, P. , Petersen, S. B. , & Otzen, D. E. (2003). Trehalose favors a cutinase compact intermediate off‐folding pathway. Biochemistry, 42, 7611–7617. 10.1021/bi034267x [DOI] [PubMed] [Google Scholar]
- Minutoli, L. , Altavilla, D. , Bitto, A. , Polito, F. , Bellocco, E. , Lagana, G. , … Squadrito, F. (2008). Trehalose: A biophysics approach to modulate the inflammatory response during endotoxic shock. European Journal of Pharmacology, 589, 272–280. 10.1016/j.ejphar.2008.04.005 [DOI] [PubMed] [Google Scholar]
- Minutoli, L. , Altavilla, D. , Bitto, A. , Polito, F. , Bellocco, E. , Lagana, G. , … Squadrito, F. (2007). The disaccharide trehalose inhibits proinflammatory phenotype activation in macrophages and prevents mortality in experimental septic shock. Shock (Augusta, Ga), 27, 91–96. 10.1097/01.shk.0000235092.76292.bc [DOI] [PubMed] [Google Scholar]
- Mishra, A. K. , ur Rasheed, M. S. , Shukla, S. , Tripathi, M. K. , Dixit, A. , & Singh, M. P. (2015). Aberrant autophagy and parkinsonism: Does correction rescue from disease progression? Molecular Neurobiology, 51, 893–908. 10.1007/s12035-014-8744-3 [DOI] [PubMed] [Google Scholar]
- Mizushima, N. , & Komatsu, M. (2011). Autophagy: Renovation of cells and tissues. Cell, 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Mizushima, N. , & Levine, B. (2010). Autophagy in mammalian development and differentiation. Nature Cell Biology, 12, 823–830. 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N. , Yamamoto, A. , Hatano, M. , Kobayashi, Y. , Kabeya, Y. , Suzuki, K. , … Yoshimori, T. (2001). Dissection of autophagosome formation using Apg5‐deficient mouse embryonic stem cells. The Journal of Cell Biology, 152, 657–668. 10.1083/jcb.152.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz, P. , Huenchuguala, S. , Paris, I. , & Segura‐Aguilar, J. (2012). Dopamine oxidation and autophagy. Parkinson's Disease, 2012, 920953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik, V. , Kardani, J. , & Roy, I. (2016). Trehalose‐induced structural transition accelerates aggregation of α‐synuclein. Molecular Biotechnology, 58, 251–255. 10.1007/s12033-016-9923-4 [DOI] [PubMed] [Google Scholar]
- Nakahira, K. , Haspel, J. A. , Rathinam, V. A. , Lee, S. J. , Dolinay, T. , Lam, H. C. , … Choi, A. M. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology, 12, 222–230. 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (NINDS Exploratory Trials in Parkinson Disease (NET‐PD) FS‐ZONE Investigators 2015). Pioglitazone in early Parkinson's disease: A phase 2, multicentre, double‐blind, randomised trial. The Lancet Neurology, 14, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel, W. , & Schulz, J. B. (2016). Current and experimental treatments of Parkinson disease: A guide for neuroscientists. Journal of Neurochemistry, 139(Suppl 1), 325–337. 10.1111/jnc.13750 [DOI] [PubMed] [Google Scholar]
- Ohtake, S. , & Wang, Y. J. (2011). Trehalose: Current use and future applications. Journal of Pharmaceutical Sciences, 100, 2020–2053. 10.1002/jps.22458 [DOI] [PubMed] [Google Scholar]
- Oku, K. , Kurose, M. , Kubota, M. , Fukuda, S. , Kurimoto, M. , Tujisaka, Y. , … Sakurai, M. (2005). Combined NMR and quantum chemical studies on the interaction between trehalose and dienes relevant to the antioxidant function of trehalose. The Journal of Physical Chemistry B, 109, 3032–3040. 10.1021/jp045906w [DOI] [PubMed] [Google Scholar]
- Osellame, L. D. , & Duchen, M. R. (2014). Quality control gone wrong: Mitochondria, lysosomal storage disorders and neurodegeneration. British Journal of Pharmacology, 171, 1958–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri, M. , Pal, R. , Nelvagal, H. R. , Lotfi, P. , Stinnett, G. R. , Seymour, M. L. , … Sardiello, M. (2017). mTORC1‐independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nature Communications, 8, 14338 10.1038/ncomms14338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, P. Y. , & Yue, Z. (2014). Genetic causes of Parkinson's disease and their links to autophagy regulation. Parkinsonism & Related Disorders, 20(Suppl 1), S154–S157. [DOI] [PubMed] [Google Scholar]
- Pan, T. , Kondo, S. , Le, W. , & Jankovic, J. (2008). The role of autophagy–lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain: A Journal of Neurology, 131, 1969–1978. 10.1093/brain/awm318 [DOI] [PubMed] [Google Scholar]
- Pan, T. , Kondo, S. , Zhu, W. , Xie, W. , Jankovic, J. , & Le, W. (2008). Neuroprotection of rapamycin in lactacystin‐induced neurodegeneration via autophagy enhancement. Neurobiology of Disease, 32, 16–25. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group QE3 Investigators , Beal, M. F. , Oakes, D. , Shoulson, I. , Henchcliffe, C. , Galpern, W. R. , … Boyar, K. (2014). A randomized clinical trial of high‐dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurology, 71, 543–552. 10.1001/jamaneurol.2014.131 [DOI] [PubMed] [Google Scholar]
- Poskanzer DC (1969). l‐Dopa in Parkinson's syndrome. New England Journal of Medicine, 280, 382–383. [DOI] [PubMed] [Google Scholar]
- Prashanth, L. K. , Fox, S. , & Meissner, W. G. (2011). l‐Dopa‐induced dyskinesia—Clinical presentation, genetics, and treatment. International Review of Neurobiology, 98, 31–54. [DOI] [PubMed] [Google Scholar]
- Pyo, J.‐O. , Nah, J. , & Jung, Y.‐K. (2012). Molecules and their functions in autophagy. Experimental & Molecular Medicine, 44, 73 10.3858/emm.2012.44.2.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar, B. , Duden, R. , & Rubinsztein, D. C. (2002). Aggregate‐prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Human Molecular Genetics, 11, 1107–1117. 10.1093/hmg/11.9.1107 [DOI] [PubMed] [Google Scholar]
- Recchia, A. , Debetto, P. , Negro, A. , Guidolin, D. , Skaper, S. D. , & Giusti, P. (2004). α‐Synuclein and Parkinson's disease. The FASEB Journal, 18, 617–626. 10.1096/fj.03-0338rev [DOI] [PubMed] [Google Scholar]
- Redmann, M. , Wani, W. Y. , Volpicelli‐Daley, L. , Darley‐Usmar, V. , & Zhang, J. (2017). Trehalose does not improve neuronal survival on exposure to alpha‐synuclein pre‐formed fibrils. Redox Biology, 11, 429–437. 10.1016/j.redox.2016.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, A. B. , Krakowka, S. , Dexter, L. B. , Schmid, H. , Wolterbeek, A. P. , Waalkens‐Berendsen, D. H. , … Kurimoto, M. (2002). Trehalose: A review of properties, history of use and human tolerance, and results of multiple safety studies. Food and Chemical Toxicology: An International Journal Published for the British Industrial Biological Research Association, 40, 871–898. 10.1016/S0278-6915(02)00011-X [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Navarro, J. A. , Rodríguez, L. , Casarejos, M. J. , Solano, R. M. , Gómez, A. , Perucho, J. , … Mena, M. A. (2010). Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiology of Disease, 39, 423–438. 10.1016/j.nbd.2010.05.014 [DOI] [PubMed] [Google Scholar]
- Ruzza, P. , Hussain, R. , Biondi, B. , Calderan, A. , Tessari, I. , Bubacco, L. , & Siligardi, G. (2015). Effects of trehalose on thermodynamic properties of alpha‐synuclein revealed through synchrotron radiation circular dichroism. Biomolecules, 5, 724–734. 10.3390/biom5020724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh, T. , Fujita, N. , Hayashi, T. , Takahara, K. , Satoh, T. , Lee, H. , … Akira, S. (2009). Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proceedings of the National Academy of Sciences of the United States of America, 106, 20842–20846. 10.1073/pnas.0911267106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh, T. , Fujita, N. , Jang, M. H. , Uematsu, S. , Yang, B. G. , Satoh, T. , … Akira, S. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1β production. Nature, 456, 264–268. 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- Sarkar, S. , Chigurupati, S. , Raymick, J. , Mann, D. , Bowyer, J. F. , Schmitt, T. , … Paule, M. G. (2014). Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP‐induced Parkinson's disease mouse model. Neurotoxicology, 44, 250–262. 10.1016/j.neuro.2014.07.006 [DOI] [PubMed] [Google Scholar]
- Sarkar, S. , Davies, J. E. , Huang, Z. , Tunnacliffe, A. , & Rubinsztein, D. C. (2007). Trehalose, a novel mTOR‐independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α‐synuclein. Journal of Biological Chemistry, 282, 5641–5652. 10.1074/jbc.M609532200 [DOI] [PubMed] [Google Scholar]
- Schapira, A. H. , McDermott, M. P. , Barone, P. , Comella, C. L. , Albrecht, S. , Hsu, H. H. , … Marek, K. (2013). Pramipexole in patients with early Parkinson's disease (PROUD): A randomised delayed‐start trial. The Lancet Neurology, 12, 747–755. 10.1016/S1474-4422(13)70117-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, L. , & Wang, X. (2011). AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy, 7, 924–926. 10.4161/auto.7.8.15860 [DOI] [PubMed] [Google Scholar]
- Singh, M. P. , Patel, S. , Dikshit, M. , & Gupta, Y. K. (2006). Contribution of genomics and proteomics in understanding the role of modifying factors in Parkinson's disease. Indian Journal of Biochemistry & Biophysics, 43, 69–81. [PubMed] [Google Scholar]
- Spillantini, M. G. , Crowther, R. A. , Jakes, R. , Cairns, N. J. , Lantos, P. L. , & Goedert, M. (1998). Filamentous α‐synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neuroscience Letters, 251, 205–208. 10.1016/S0304-3940(98)00504-7 [DOI] [PubMed] [Google Scholar]
- Sulzer, D. (2010). Clues to how alpha‐synuclein damages neurons in Parkinson's disease. Movement Disorders: Official Journal of the Movement Disorder Society, 25(Suppl 1), S27–S31. 10.1002/mds.22639 [DOI] [PubMed] [Google Scholar]
- Suzuki, K. , Kubota, Y. , Sekito, T. , & Ohsumi, Y. (2007). Hierarchy of Atg proteins in pre‐autophagosomal structure organization. Genes to Cells: Devoted to Molecular & Cellular Mechanisms, 12, 209–218. 10.1111/j.1365-2443.2007.01050.x [DOI] [PubMed] [Google Scholar]
- Suzuki, K. , & Ohsumi, Y. (2007). Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae . FEBS Letters, 581, 2156–2161. [DOI] [PubMed] [Google Scholar]
- Tanaka, M. , Machida, Y. , Niu, S. , Ikeda, T. , Jana, N. R. , Doi, H. , … Nukina, N. (2004). Trehalose alleviates polyglutamine‐mediated pathology in a mouse model of Huntington disease. Nature Medicine, 10, 148–154. 10.1038/nm985 [DOI] [PubMed] [Google Scholar]
- Tanji, K. , Miki, Y. , Maruyama, A. , Mimura, J. , Matsumiya, T. , Mori, F. , … Wakabayashi, K. (2015). Trehalose intake induces chaperone molecules along with autophagy in a mouse model of Lewy body disease. Biochemical and Biophysical Research Communications, 465, 746–752. 10.1016/j.bbrc.2015.08.076 [DOI] [PubMed] [Google Scholar]
- Tien, N. T. , Karaca, I. , Tamboli, I. Y. , & Walter, J. (2016). Trehalose alters subcellular trafficking and the metabolism of the Alzheimer‐associated amyloid precursor protein. Journal of Biological Chemistry, 291, 10528–10540. 10.1074/jbc.M116.719286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timasheff, S. N. (2002). Protein‐solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proceedings of the National Academy of Sciences of the United States of America, 99, 9721–9726. 10.1073/pnas.122225399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila, M. , Bove, J. , Dehay, B. , Rodriguez‐Muela, N. , & Boya, P. (2011). Lysosomal membrane permeabilization in Parkinson disease. Autophagy, 7, 98–100. 10.4161/auto.7.1.13933 [DOI] [PubMed] [Google Scholar]
- Volles, M. J. , Lee, S. J. , Rochet, J. C. , Shtilerman, M. D. , Ding, T. T. , Kessler, J. C. , & Lansbury, P. T. (2001). Vesicle permeabilization by protofibrillar α‐synuclein: Implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry, 40, 7812–7819. 10.1021/bi0102398 [DOI] [PubMed] [Google Scholar]
- Volpicelli‐Daley, L. A. , Luk, K. C. , Patel, T. P. , Tanik, S. A. , Riddle, D. M. , Stieber, A. , … Lee, V. M. Y. (2011). Exogenous α‐synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron, 72, 57–71. 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. Y. , Liu, J. Y. , Yang, C. B. , Malampati, S. , Huang, Y. Y. , Li, M. X. , … Song, J. X. (2017). Neuroprotective natural products for the treatment of Parkinson's disease by targeting the autophagy–lysosome pathway: A systematic review. Phytotherapy Research: PTR, 31, 1119–1127. 10.1002/ptr.5834 [DOI] [PubMed] [Google Scholar]
- Webb, J. L. , Ravikumar, B. , Atkins, J. , Skepper, J. N. , & Rubinsztein, D. C. (2003). α‐Synuclein is degraded by both autophagy and the proteasome. Journal of Biological Chemistry, 278, 25009–25013. 10.1074/jbc.M300227200 [DOI] [PubMed] [Google Scholar]
- Weissmann, C. , Raeber, A. J. , Montrasio, F. , Hegyi, I. , Frigg, R. , Klein, M. A. , & Aguzzi, A. (2001). Prions and the lymphoreticular system. Philosophical Transactions of the Royal Society B: Biological Sciences, 356, 177–184. 10.1098/rstb.2000.0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, W. J. , & Brown, C. R. (1996). Influence of molecular and chemical chaperones on protein folding. Cell Stress & Chaperones, 1, 109–115. 10.1379/1466-1268(1996)001<0109:IOMACC>2.3.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, A. , Jahreiss, L. , Sarkar, S. , Saiki, S. , Menzies, F. M. , Ravikumar, B. , & Rubinsztein, D. C. (2006). Aggregate‐prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Current Topics in Developmental Biology, 76, 89–101. 10.1016/S0070-2153(06)76003-3 [DOI] [PubMed] [Google Scholar]
- Wong, E. , & Cuervo, A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nature Neuroscience, 13, 805–811. 10.1038/nn.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET‐PD) Investigators , Kieburtz, K. , Tilley, B. C. , Elm, J. J. , Babcock, D. , Hauser, R. , … Wills, A. M. (2015). Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: A randomized clinical trial. Jama, 313, 584–593. 10.1001/jama.2015.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Xu, H. D. , Guan, J. J. , Hou, Y. S. , Gu, J. H. , Zhen, X. C. , & Qin, Z. H. (2015). Rotenone impairs autophagic flux and lysosomal functions in Parkinson's disease. Neuroscience, 284, 900–911. 10.1016/j.neuroscience.2014.11.004 [DOI] [PubMed] [Google Scholar]
- Wu, H. , Chen, S. , Ammar, A. B. , Xu, J. , Wu, Q. , Pan, K. , … Hong, Y. (2015). Crosstalk between macroautophagy and chaperone‐mediated autophagy: Implications for the treatment of neurological diseases. Molecular Neurobiology, 52, 1284–1296. 10.1007/s12035-014-8933-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger, S. , Loewith, R. , & Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell, 124, 471–484. [DOI] [PubMed] [Google Scholar]
- Xilouri, M. , Brekk, O. R. , & Stefanis, L. (2013). α‐Synuclein and protein degradation systems: A reciprocal relationship. Molecular Neurobiology, 47, 537–551. 10.1007/s12035-012-8341-2 [DOI] [PubMed] [Google Scholar]
- Xilouri, M. , Brekk, O. R. , & Stefanis, L. (2016). Autophagy and alpha‐synuclein: Relevance to Parkinson's disease and related synucleopathies. Movement Disorders: Official Journal of the Movement Disorder Society, 31, 178–192. 10.1002/mds.26477 [DOI] [PubMed] [Google Scholar]
- Xilouri, M. , Vogiatzi, T. , Vekrellis, K. , Park, D. , & Stefanis, L. (2009). Abberant α‐synuclein confers toxicity to neurons in part through inhibition of chaperone‐mediated autophagy. PLoS ONE, 4, e5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , & Klionsky, D. J. (2010). Eaten alive: A history of macroautophagy. Nature Cell Biology, 12, 814–822. 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, Y. S. , Cho, E. D. , Jung Ahn, W. , Won Lee, K. , Lee, S. J. , & Lee, H. J. (2017). Is trehalose an autophagic inducer? Unraveling the roles of non‐reducing disaccharides on autophagic flux and alpha‐synuclein aggregation. Cell Death & Disease, 8, e3091 10.1038/cddis.2017.501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizane, C. , Mizote, A. , Yamada, M. , Arai, N. , Arai, S. , Maruta, K. , … Fukuda, S. (2017). Glycemic, insulinemic and incretin responses after oral trehalose ingestion in healthy subjects. Nutrition Journal, 16, 9 10.1186/s12937-017-0233-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, W. B. , Jiang, T. , Lan, D. M. , Lu, J. H. , Yue, Z. Y. , Wang, J. , & Zhou, P. (2012). Trehalose inhibits fibrillation of A53T mutant alpha‐synuclein and disaggregates existing fibrils. Archives of Biochemistry and Biophysics, 523, 144–150. 10.1016/j.abb.2012.04.021 [DOI] [PubMed] [Google Scholar]
- Zavodszky, E. , Seaman, M. N. , Moreau, K. , Jimenez‐Sanchez, M. , Breusegem, S. Y. , Harbour, M. E. , & Rubinsztein, D. C. (2014). Mutation in VPS35 associated with Parkinson's disease impairs WASH complex association and inhibits autophagy. Nature Communications, 5, 3828 10.1038/ncomms4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Zhi, X. , Pan, L. , & Zhou, P. (2017). Trehalose inhibits A53T mutant α‐synuclein overexpression and neurotoxicity in transduced PC12 cells. Molecules, 22, 1293 10.3390/molecules22081293 [DOI] [PMC free article] [PubMed] [Google Scholar]