

Abstract
A highly selective CuII‐catalyzed cross‐dehydrogenative ortho‐aminomethylation of phenols with aniline derivatives is described. The corresponding C(sp2)−C(sp3) coupling products were obtained in moderate to excellent yields under mild reaction conditions and with a broad substrate scope. A radical mechanism is proposed.
Keywords: aminomethylation, dehydrogenative coupling, Cu catalysis, Mannich reactions, radical cross-coupling
Carbon–carbon bond forming processes are at the heart of organic synthesis, since these typically allow the rapid construction of molecular complexity.1 In the past decade, direct transition‐metal‐catalyzed C−H bond functionalization has emerged as an important tool for the construction of various C−C bonds.2 The latter methods are increasingly popular due to atom‐ and step‐economy considerations, which often lends them an inherently sustainable character. Therein, the field of cross‐dehydrogenative couplings (CDCs) is particularly attractive because this concept avoids pre‐activation steps for both coupling partners.3 The development of useful intermolecular CDCs, however, is associated with considerable challenges, such as regioselectivity or undesired homocoupling processes. In order to intercept the oxidation of the most electron‐rich coupling partner into a true heterocoupling process, one can resort to substrate bias (i.e., sterics).4 Alternatively, one can utilize metal catalysis in order to control the various competing oxidation pathways, and thereby escape the narrow substrate specificity often imposed by metal‐free systems. Herein, we propose such a strategy through the CuII‐catalyzed cross‐dehydrogenative ortho‐aminomethylation of phenols5, 6 with aniline derivatives (Scheme 1 and Scheme 2).
Scheme 1.
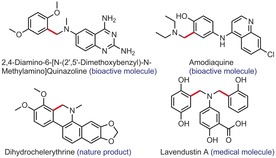
Aminomethylated phenols.
Scheme 2.
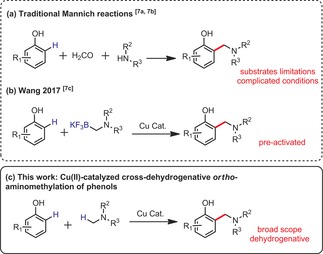
Ortho‐aminomethylation of phenols.
The ortho‐aminomethylation of phenols represents a useful retrosynthetic tool, because this particular motif is prevalent in natural products, medicines, and materials (Scheme 1). Thus, some synthetic methods7 have been reported to construct this strategic structural unit, such as the Mannich reaction (Scheme 2 a).7a,7b However, some drawbacks are usually associated with this synthetic approach. For instance, the substrate scope is usually limited to electron‐rich and/or fused polycyclic phenols on the one hand, and often to aliphatic amines on the other. Moreover, the reaction conditions must accommodate very reactive formaldehyde, or a derivative of it, which typically leads to relatively poor chemoselectivity. Indeed, Mannich reactions are often associated with further cyclization and/or over‐coupling events, which call for structural bias in the substrates (blocking/protecting undesired positions), as well as bias in the order, time, and speed of additions of the components, for example. In 2017, the Wang group reported a more concise way to achieve this process.7c However, this method requires pre‐functionalization of the amine coupling partner (Scheme 2 b). To the best of our knowledge, no CDC approach has ever been proposed (Scheme 2 c).
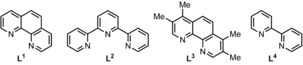
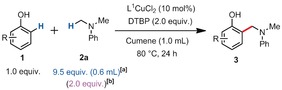
We started our reactivity investigations with dichloro(1,10‐phenanthroline)copper(II) (L1CuCl2) as a prospective catalyst, since this species has been recently found to be successful in some inspiring radical coupling reactions,8 notably by the Stahl group8a and independently by Liu group.8b We thus began our study by examining the reaction of 4‐t‐butylphenol (1 a) and N,N‐dimethylaniline (2 a) in the presence of a catalytic amount of L1CuCl2 (10 mol %) and di‐tert‐butyl peroxide (DTBP) as the oxidant to form coupling product 3 a (Table 1, Entry 1). Importantly, it was found that the N,N‐dimethylaniline loading considerably affects the yield. Eventually, a loading of 0.6 mL of 2 a (9.5 equiv) for 0.5 mmol of phenol 1 a led to an improved 55 % NMR yield (Table 1, entries 2, 3). When the amount of cumene solvent was reduced from 1.5 mL to 1.0 mL, the yield improved to 60 % NMR yield (62 % yield of isolated product, entries 4, 5). It should moreover be noted that no solvent performed better than cumene.4, 9 Conversely, benzene and tert‐butylbenzene are both tolerated as solvents, albeit with lower yields (Entries 6 and 8), thus indicating that the benzylic C−H position is not essential. The higher performance of the cumene solvent may suggest the ability of persistent cumyl radicals to act as radical reservoirs in the reaction. However, cumene and [D12]‐cumene afford the same initial reaction rate (KIE=k H/k D≈1, see the Supporting Information), such that this hypothesis cannot be confirmed at this stage.
Table 1.
Optimization of reaction conditions.
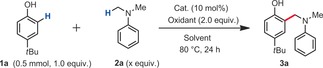
| Entry | x | Catalyst | Oxidant | Solvent | Yield [%][a] |
|---|---|---|---|---|---|
| 1 | 1.6 | L1CuCl2 | DTBP | Cumene 1.5 mL | 20 |
| 2 | 4.8 | L1CuCl2 | DTBP | Cumene 1.5 mL | 38 |
| 3 | 9.5 | L1CuCl2 | DTBP | Cumene 1.5 mL | 55 |
| 4 | 9.5 | L1CuCl2 | DTBP | Cumene 1.0 mL | 60(62) |
| 5 | 9.5 | L1CuCl2 | DTBP | Cumene 0.5 mL | 55 |
| 6 | 9.5 | L1CuCl2 | DTBP | Benzene 1.0 mL | 34 |
| 7 | 9.5 | L1CuCl2 | DTBP | Toluene 1.0 mL | 58 |
| 8 | 9.5 | L1CuCl2 | DTBP | t Bu‐benzene 1.0 mL | 29 |
| 9 | 9.5 | L1CuCl2 | TBHP | Cumene 1.0 mL | trace |
| 10 | 9.5 | L1CuCl2 | TBPB | Cumene 1.0 mL | trace |
| 11 | 9.5 | Cu(TC) [c] | DTBP | Cumene 1.0 mL | 34 |
| 12[b] | 9.5 | CuF2+L1 | DTBP | Cumene 1.0 mL | 42 |
| 13[b] | 9.5 | CuCl2+L2 | DTBP | Cumene 1.0 mL | 12 |
| 14[b] | 9.5 | CuCl2+L3 | DTBP | Cumene 1.0 mL | 29 |
| 15[b] | 9.5 | CuCl2+L4 | DTBP | Cumene 1.0 mL | 12 |
| |||||
[a] The yield was determined by 1H NMR analysis of the crude reaction mixture using 1,3,5‐trimethoxybenzene as an internal standard. [b] The amount of ligand was 15 mol %. [c] thiophene‐2‐carboxylate.
It should be noted that in some cases, homocoupling byproducts derived from 2 a as well as unidentified byproducts were detected. Importantly, no phenol homocoupling byproducts (so‐called binols) could be detected under those reaction conditions by MS analysis of the crude mixture. Moreover, unreacted phenol is usually detected at the end of the reaction, thus indicating that in spite of our best efforts at maximizing the yield of this reaction (Table 1, entry 4), full conversion is typically not reached. The reason for this became clearer when studying the regioselectivity of this reaction. Indeed, when testing unfunctionalized phenol substrate 1 b under the optimized conditions, the ortho‐selective product 3 b was obtained in 47 % yield of isolated product, as well as a small amount of double‐functionalized byproduct 3 b′ (Scheme 3).
Scheme 3.
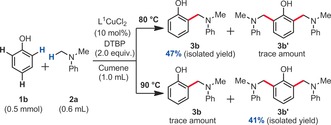
Selectivity study.
To our delight, no para‐functionalized product was detected. Interestingly, when the reaction temperature was raised to 90 °C, 3 b′ actually became the major product with a 41 % yield of isolated product and still no para‐functionalization detected (Scheme 3). This competing double‐ortho‐functionalization process explains why the temperature has to be kept at a moderate 80 °C, and consequently why the reaction cannot be pushed to full conversion. The reaction scope was then investigated (Table 2 and Table 3, see the Supporting Information for a detailed description).
Table 2.
Phenol scope.
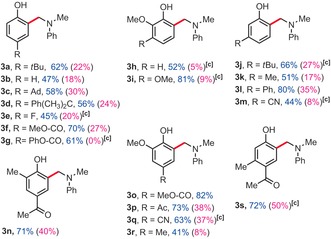
|
[a] Yields of isolated product. [b] 1H NMR yields, 1,3,5‐trimethoxybenzene as an internal standard, 80 °C, 48 h. [c] 90 °C, 24 h.
Table 3.
Methylamine scope.
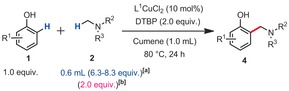
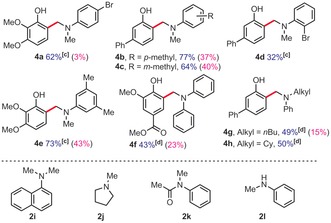
|
[a] Yields of isolated product. [b] 1H NMR yields, 1,3,5‐trimethoxybenzene as an internal standard, 80 °C, 48 h. [c] 48 h. [d] 32 h.
In order to characterize the largest drawback of this method, which is arguably the excess of amine coupling partner, we also re‐performed some of the most representative examples with only two equivalents of amine (yields in parentheses, Table 2 & Table 2). Clearly, this has a severe impact on the yields. Indeed, under those conditions, the highest yield does not exceed 43 %, for product 4 e. However, it was also found that a significant portion of the excess of amine coupling partner could be recovered. Out of five tested examples, 64 %, 66 %, 70 %, and 72 % of the initial amount of coupling partner 2 a could be recovered from entries 3 e, 3 f, 3 g, and 3 o, respectively. Moreover, 67 % of the initial coupling partner 2 c could be recovered from entry 4 c.
A series of mechanistic experiments were then performed. First, the addition of TEMPO completely suppresses product formation (Scheme 4 a), thus suggesting a pronounced radical character to the reaction. Moreover, a normal KIE (k H/k D) of 1.7 was observed between phenol 1 b and labelled phenol [D6]‐1 b in two parallel experiments (t=2 h, Scheme 4 b). Importantly, GCMS analysis shows complete preservation of the deuterium labels in the starting material (t=2 h). Because the KIE lies significantly beneath 2, the phenolic C(sp2)−H bond cleavage may not be rate limiting.10 In contrast, a rare inverse secondary KIE of circa 0.9 was observed upon comparing the initial rates of 2 a and [D6]‐2 a (Scheme 4 c). This modest secondary KIE may suggest a C(sp2)‐to‐C(sp3) rate‐determining step, which may thus correspond to the intermolecular C−C bond formation step. Alternatively, it might also accommodate a sterically encumbered rate‐determining Cu−N bond formation, prior to C−C bond formation, which would be in good agreement with the required excess of aniline.11 Finally, we also tested phenol 1 t, for which both ortho positions are blocked with methoxy groups (Scheme 4 d), and which was chosen for its structural and electronic resemblance with successful phenol 1 i (Table 2). Under standard conditions, the expected aminomethylation product could not be detected, thus confirming the exclusive ortho‐selectivity of the reaction.
Scheme 4.
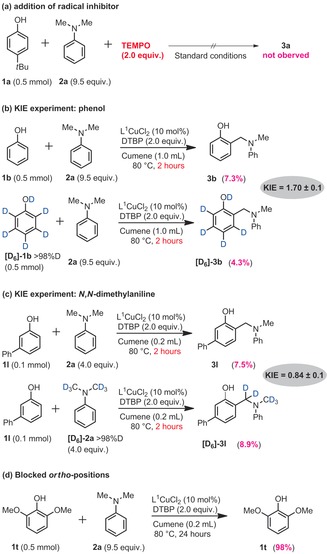
Mechanistic experiments, 1H NMR yields, 1,3,5‐trimethoxybenzene as an internal standard.
A proposed mechanism12 is shown in Scheme 5. First, the low‐valence copper species I would donate an electron to DTBP to generate copper species II and the tert‐butoxy radical. The tert‐butoxy radical would then abstract a hydrogen atom from either phenol 1, to form phenoxy radical intermediate III, and/or methylamine derivative 2 to generate the aminomethyl radical intermediate IV, a well‐documented process.13 The latter process is moreover expected to be a relatively facile and non‐rate‐limiting step considering the relatively low oxidative potentials of dimethylanilines.13 Copper phenolate intermediate V could otherwise form by proton exchange from phenol and the tert‐butanolate‐copper species II. The intermediacy of phenoxy radicals III is moreover realistic in consideration of the relatively low and similar bond dissociation energies (BDEs) of phenols (88 kcal mol−1 for phenol),14 in comparison to that of dimethylaniline (92 kcal mol−1).15 The C−C bond forming and possibly rate‐determining step would then occur within the coordination sphere of the copper catalyst to yield coupling product 3 and the lower‐valence copper species I. The strong ortho‐selective character of the herein presented reaction is in good agreement with a copper‐centered cyclic organometallic transition state. The intermediacy of iminium ions12i cannot be excluded, but seems less likely because of the absence of para‐functionalization (Scheme 4 d).
Scheme 5.
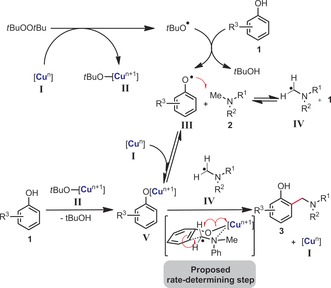
Mechanism proposal.
The utility of this cross‐dehydrogenative ortho‐aminomethylation reaction was then examined in the derivatization of a precious steroid natural product, Estrone 1 u smoothly affording coupling product 3 u (Scheme 6 a). In addition, the reaction was found to be easily scalable (Scheme 6 b). Indeed, 1.74 g of coupling product 3 l could be obtained in a single batch (60 % yield). Moreover, with a published method,16 a cyclization reaction was achieved from the aminomethylation product 3 l to a 7‐membered ring product 5 in excellent yield, thus increasing the scope of that method.
Scheme 6.
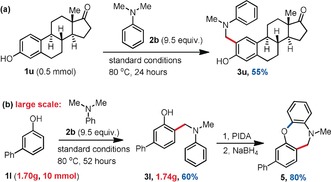
Synthetic applications of the CDC reaction, isolated in yields.
In summary, we have developed a CuII‐catalyzed ortho‐selective aminomethylatio of phenols by direct intermolecular CDC reaction. Moreover, a relatively broad variety of functional groups were tolerated. This method represents a rare case of C(sp2)−C(sp3) CDC with phenols.6 This unusual dehydrogenative process is anticipated to lead to the development of other general classes of C−C bond forming CDC reactions. Further mechanistic investigations may be necessary in order to rationally achieve those objectives.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The research of FWP is supported by the DFG‐funded transregional collaborative research center SFB/TRR 88 “Cooperative effects in homo and heterometallic complexes” (http://3MET.de), DFG funded project PA 2395/2‐1, COST Action CA15106 (CHAOS), and since March 2017: by ERC project 716136: “2O2ACTIVATION.”
C. Yu, F. W. Patureau, Angew. Chem. Int. Ed. 2018, 57, 11807.
References
- 1. Corey E. J., Cheng X., The Logic of Chemical Synthesis, Wiley, New York, 1989, pp. 1–91. [Google Scholar]
- 2.Selected recent reviews:
- 2a. Leitch J. A., Frost C. G., Chem. Soc. Rev. 2017, 46, 7145; [DOI] [PubMed] [Google Scholar]
- 2b. Xue X., Ji P., Zhou B., Cheng J., Chem. Rev. 2017, 117, 8622; [DOI] [PubMed] [Google Scholar]
- 2c. Shang R., Ilies L., Nakamura E., Chem. Rev. 2017, 117, 9086; [DOI] [PubMed] [Google Scholar]
- 2d. Ma W., Gandeepan P., Li J., Ackermann L., Org. Chem. Front. 2017, 4, 1435; [Google Scholar]
- 2e. Gensch T., Hopkinson M. N., Glorius F., Wencel-Delord J., Chem. Soc. Rev. 2016, 45, 2900; [DOI] [PubMed] [Google Scholar]
- 2f. Li S., Qin L., Dong L., Org. Biomol. Chem. 2016, 14, 4554; [DOI] [PubMed] [Google Scholar]
- 2g. Sandtorv A. H., Adv. Synth. Catal. 2015, 357, 2403; [Google Scholar]
- 2h. Topczewski J., Sanford M. S., Chem. Sci. 2015, 6, 70; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2i. Thirunavukkarasu V. S., Kozhushkov S. I., Ackermann L., Chem. Commun. 2014, 50, 29; [DOI] [PubMed] [Google Scholar]
- 2j. Wencel-Delord J., Glorius F., Nat. Chem. 2013, 5, 369; [DOI] [PubMed] [Google Scholar]
- 2k. Neufeldt S. R., Sanford M. S., Acc. Chem. Res. 2012, 45, 936; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2l. Arockiam P. B., Bruneau C., Dixneuf P. H., Chem. Rev. 2012, 112, 5879; [DOI] [PubMed] [Google Scholar]
- 2m. Song G., Wang F., Li X., Chem. Soc. Rev. 2012, 41, 3651; [DOI] [PubMed] [Google Scholar]
- 2n. Colby D. A., Tsai A. S., Bergman R. G., Ellman J. A., Acc. Chem. Res. 2012, 45, 814; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2o. Engle K. M., Mei T.-S., Wasa M., Yu J.-Q., Acc. Chem. Res. 2012, 45, 788; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2p. McMurray L., Hara F. O., Gaunt M. J., Chem. Soc. Rev. 2011, 40, 1885; [DOI] [PubMed] [Google Scholar]
- 2q. Xu L.-M., Li B.-J., Yang Z., Shi Z.-J., Chem. Soc. Rev. 2010, 39, 712. [DOI] [PubMed] [Google Scholar]
- 3.Selected recent reviews on CDC reactions:
- 3a. Guo S.-R., Kumar P. S., Yang M., Adv. Synth. Catal. 2017, 359, 2; [Google Scholar]
- 3b. Varun B. V., Dhineshkumar J., Bettadapur K. R., Siddaraju Y., Alagiri K., Prabhu K. R., Tetrahedron Lett. 2017, 58, 803; [Google Scholar]
- 3c. Henry M., Mostafa M. A. B., Sutherland A., Synthesis 2017, 49, 4586; [Google Scholar]
- 3d. Lakshman M. K., Vuram P. K., Chem. Sci. 2017, 8, 5845; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Lv L., Li Z., Top. Curr. Chem. 2016, 374, 38; [DOI] [PubMed] [Google Scholar]
- 3f. Chen T., Zhang J.-S., Han L.-B., Dalton Trans. 2016, 45, 1843; [DOI] [PubMed] [Google Scholar]
- 3g. Batra A., Singh P., Singh K. N., Eur. J. Org. Chem. 2016, 4927; [Google Scholar]
- 3h. Miao J., Ge H., Eur. J. Org. Chem. 2015, 7859; [Google Scholar]
- 3i. Krylov L. B., Vil’ V. A., Terent'ev A. O., Beilstein J. Org. Chem. 2015, 11, 92; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3j. Girard S. A., Knauber T., Li C.-J., Angew. Chem. Int. Ed. 2014, 53, 74; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 76; [Google Scholar]
- 3k. Wu Y., Wang J., Mao F., Kwong F. K., Chem. Asian J. 2014, 9, 26; [DOI] [PubMed] [Google Scholar]
- 3l. Zhang C., Tang C., Jiao N., Chem. Soc. Rev. 2012, 41, 3464; [DOI] [PubMed] [Google Scholar]
- 3m. Liu C., Zhang H., Shi W., Lei A., Chem. Rev. 2011, 111, 1780; [DOI] [PubMed] [Google Scholar]
- 3n. Yeung C. S., Dong V. M., Chem. Rev. 2011, 111, 1215; [DOI] [PubMed] [Google Scholar]
- 3o. Scheuermann C. J., Chem. Asian J. 2010, 5, 436; [DOI] [PubMed] [Google Scholar]
- 3p. Li C.-J., Acc. Chem. Res. 2009, 42, 335. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Louillat-Habermeyer M.-L., Jin R., Patureau F. W., Angew. Chem. Int. Ed. 2015, 54, 4102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4175; [Google Scholar]
- 4b. Goswami M., Konkel A., Rahimi M., Louillat-Habermeyer M.-L., Kelm H., Jin R., de Bruin B., Patureau F. W., Chem. Eur. J. 2018, 10.1002/chem.201800730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Yamaguchi M. in The Chemistry of Phenols (Ed.: Z. Rappoport), Wiley, Chichester: 2003, pp. 661–712; [Google Scholar]
- 5b. González C., Castedo L. in The Chemistry of Phenols (Ed.: Z. Rappoport), Wiley, Chichester: 2003, pp. 395–489. [Google Scholar]
- 6.Some CDCs with phenols (most of them represent C(sp2)−C(sp2) bond formation):
- 6a. Shalit H., Libman A., Pappo D., J. Am. Chem. Soc. 2017, 139, 13404; [DOI] [PubMed] [Google Scholar]
- 6b. Eisenhofer A., Hioe J., Gschwind R. M., König B., Eur. J. Org. Chem. 2017, 2194; [Google Scholar]
- 6c. Morimoto K., Sakamoto K., Ohnishi Y., Dohi T., Kita Y., Angew. Chem. Int. Ed. 2016, 55, 3652; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3716; [Google Scholar]
- 6d. Libman A., Shalit H., Vainer Y., Narute S., Kozuch S., Pappo D., J. Am. Chem. Soc. 2015, 137, 11453; [DOI] [PubMed] [Google Scholar]
- 6e. Gaster E., Vainer Y., Regev A., Narute S., Sudheendran K., Werbeloff A., Shalit H., Pappo D., Angew. Chem. Int. Ed. 2015, 54, 4198; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4272; [Google Scholar]
- 6f. Elsler B., Eiebe A., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Chem. Eur. J. 2015, 21, 12321; [DOI] [PubMed] [Google Scholar]
- 6g. Pandita R. P., Leea Y. R., Adv. Synth. Catal. 2014, 356, 3171; [Google Scholar]
- 6h. Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2014, 53, 5210; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5311; [Google Scholar]
- 6i. Morimoto K., Sakamoto K., Ohnishi Y., Miyamoto T., Ito M., Dohi T., Kita Y., Chem. Eur. J. 2013, 19, 8726; [DOI] [PubMed] [Google Scholar]
- 6j. Dohi T., Kamitanaka T., Watanabe S., Hu Y., Washimi N., Kita Y., Chem. Eur. J. 2012, 18, 13614; [DOI] [PubMed] [Google Scholar]
- 6k. Kirste A., Elsler B., Schnakenburg G., Waldvogel S. R., J. Am. Chem. Soc. 2012, 134, 3571; [DOI] [PubMed] [Google Scholar]
- 6l. Kirste A., Schnakenburg G., Stecker F., Fischer A., Waldvogel S. R., Angew. Chem. Int. Ed. 2010, 49, 971; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 983; [Google Scholar]
- 6m. Li X., Hewgley J. B., Mulrooney C. A., Yang J., Kozlowski M. C., J. Org. Chem. 2003, 68, 5500; [DOI] [PubMed] [Google Scholar]
- 6n. Yamamoto K., Yumioka H., Okamoto Y., Chikamatsu H., J. Chem. Soc. Chem. Commun. 1987, 168. [Google Scholar]
- 7.
- 7a. Tramontini M., Synthesis 1973, 703; [Google Scholar]
- 7b. Kim S., Hong S. H., Adv. Synth. Catal. 2017, 359, 798; [Google Scholar]
- 7c. Dai J.-L., Shao N.-Q., Zhang J., Jia R.-P., Wang D.-H., J. Am. Chem. Soc. 2017, 139, 12390; [DOI] [PubMed] [Google Scholar]
- 7d. Hwang D.-R., Uang B.-J., Org. Lett. 2002, 4, 463; [DOI] [PubMed] [Google Scholar]
- 7e. Grillot G. F., Gormley W. T., J. Am. Chem. Soc. 1945, 67, 1968. [Google Scholar]
- 8.
- 8a. Vasilopoulos A., Zultanski S. L., Stahl S. S., J. Am. Chem. Soc. 2017, 139, 7705; [DOI] [PubMed] [Google Scholar]
- 8b. Zhang W., Chen P., Liu G., J. Am. Chem. Soc. 2017, 139, 7709, and references therein. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Malekafzali A., Malinovska K., Patureau F. W., New J. Chem. 2017, 41, 6981; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Heitz C., Jones A. W., Oezkaya B. S., Bub C. L., Louillat-Habermeyer M.-L., Wagner V., Patureau F. W., Chem. Eur. J. 2016, 22, 17980; [DOI] [PubMed] [Google Scholar]
- 9c. Louillat M.-L., Biafora A., Legros F., Patureau F. W., Angew. Chem. Int. Ed. 2014, 53, 3505; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3573. [Google Scholar]
- 10.In order to distinguish the kinetic impact of the O−D versus C−D bonds of phenol, [D1]-1 b (OD) was also tested versus natural-abundance phenol, which gave a KIE=k H/k D of ca. 1 (see the Supporting Information). This suggests that C−H cleavage has greater kinetic relevance (KIE=1.7) than O−H cleavage. See also:
- 10a. Op't Holt B. T., Vance M. A., Mirica L. M., Heppner D. E., Stack T. D. P., Solomon E. I., J. Am. Chem. Soc. 2009, 131, 6421; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Hoffmann A., Citek C., Binder S., Goos A., Rübhausen M., Troeppner O., Ivanovic-Burmazovic I., Wasinger E. C., Stack T. D. P., S, Herres-Pawlis , Angew. Chem. Int. Ed. 2013, 52, 5398; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5508; [Google Scholar]
- 10c. Askari M. S., Esguerra K. V. N., Lumb J.-P., Ottenwaelder X., Inorg. Chem. 2015, 54, 8665, and references therein. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Lee I., Chem. Soc. Rev. 1995, 24, 223; [Google Scholar]
- 11b. Poirier R. A., Wang Y., Westaway K. C., J. Am. Chem. Soc. 1994, 116, 2526. [Google Scholar]
- 12.
- 12a. Yi H., Zhang G., Wang H., Huang Z., Wang J., Singh A. K., Lei A., Chem. Rev. 2017, 117, 9016; [DOI] [PubMed] [Google Scholar]
- 12b. Liu Y., Zhou Z., Jia L., Chen Y., J. Org. Chem. 2017, 82, 9198; [DOI] [PubMed] [Google Scholar]
- 12c. Uma Maheswari C., Kumar G. S., Reddy K. R., Curr. Org. Chem. 2016, 20, 512; [Google Scholar]
- 12d. Zhou S.-L., Guo L.-N., Duan X.-H., Eur. J. Org. Chem. 2014, 8094; [Google Scholar]
- 12e. Studer A., Curran D. P., Nat. Chem. 2014, 6, 765; [DOI] [PubMed] [Google Scholar]
- 12f. Liu C., Liu D., Lei A., Acc. Chem. Res. 2014, 47, 3459; [DOI] [PubMed] [Google Scholar]
- 12g. Wang J., Liu C., Yuan J., Lei A., Angew. Chem. Int. Ed. 2013, 52, 2256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2312. See also: [Google Scholar]
- 12h. Scott M., Sud A., Boess E., Klussmann M., J. Org. Chem. 2014, 79, 12033; [DOI] [PubMed] [Google Scholar]
- 12i. Boess E., Wolf L. M., Malakar S., Salamone M., Bietti M., Thiel W., Klussmann M., ACS Catal. 2016, 6, 3253. [Google Scholar]
- 13.
- 13a. Fukuzumi S., Shimoosako K., Suenobu T., Watanabe Y., J. Am. Chem. Soc. 2003, 125, 9074; [DOI] [PubMed] [Google Scholar]
- 13b. Baciocchi E., Bietti M., Gerini M. F., Lanzalunga O., J. Org. Chem. 2005, 70, 5144. [DOI] [PubMed] [Google Scholar]
- 14.O−H bond-dissociation energies (BDEs) of phenols: Lucarini M., Pedrielli P., Pedulli G. F., J. Org. Chem. 1996, 61, 9259. [Google Scholar]
- 15.α-C−H BDE of dimethylaniline: Dombrowski G. W., Dinnocenzo J. P., Farid S., Goodman J. L., Gould I. R., J. Org. Chem. 1999, 64, 427. [Google Scholar]
- 16. Jamsheena V., Mahesha C. K., Joy M. N., Lankalapalli R. S., Org. Lett. 2017, 19, 6614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary