


Abstract
Post-transcriptional regulation of gene expression occurs by multiple mechanisms, including subcellular localization of mRNA and alteration of the poly(A) tail length. These mechanisms play crucial roles in the dynamics of cell polarization and embryonic development. Furthermore, mRNAs are emerging therapeutics and chemical alterations to increase their translational efficiency are highly sought after. We show that yeast poly(A) polymerase can be used to install multiple azido-modified adenosine nucleotides to luciferase and eGFP-mRNAs. These mRNAs can be efficiently reacted in a bioorthogonal click reaction with fluorescent reporters without degradation and without sequence alterations in their coding or untranslated regions. Importantly, the modifications in the poly(A) tail impact positively on the translational efficiency of reporter-mRNAs in vitro and in cells. Therefore, covalent fluorescent labeling at the poly(A) tail presents a new way to increase the amount of reporter protein from exogenous mRNA and to label genetically unaltered and translationally active mRNAs.
INTRODUCTION
The key function of mRNAs is translation into proteins and multiple mechanisms act on the mRNA level to regulate gene expression. Among them, asymmetric localization of mRNA plays a fundamental role in large polarized cells and early development (1); hence simple-to-use tools for investigating these processes without interfering with other functions of mRNA are required. In neurons, targeting of mRNAs to dendrites and axons is relevant for intracellular signaling, development and synaptic plasticity. Imaging of mRNAs in neurons and brain tissue has enhanced our understanding of mRNA dynamics, in particular if achieved on the single-molecule level (2). Single-molecule fluorescence in situ hybridization (smFISH) guarantees sensitive detection via multiple fluorophore-labeled probes that are hybridized to a specific RNA, enabling even the detection of a single mRNA molecule (3). However, this approach works best in fixed cells where unbound probes can be removed or more intricate turn-on systems like FIT-probes have to be synthesized (4,5). For tracking mRNA in living cells fluorescently labeled phosphodiester oligodeoxynucleotides (ODNs), which are efficiently taken up by the cell and selectively hybridized to the poly(A) tail were developed (6) and further used to study movement of mRNA in the cell nucleus using photobleaching techniques (7,8). To eliminate fluorescence signal from non-hybridized probe, highly specific and sensitive molecular beacons (MBs) are an interesting and simple-to-use tool for imaging endogenous mRNA (9–11). Live-cell imaging using MBs can be performed with different delivery methods including the use of optimized MBs for the target to prevent unspecific signals (12–14).
In living cells, the most widely used RNA labeling approach is tagging with green fluorescent protein (GFP) via the MS2 system (consisting of the coat protein from bacteriophage MS2 binding to a RNA stem-loop) or alternative RNA-protein pairs from bacteriophages (1). Applications from yeast to mice underscore the importance of this strategy that relies completely on genetically encodable parts (15). Despite the success of the MS2 system, a remaining limitation is the size of the tag that is appended to the mRNA of interest. Typically, 24 MS2 stem loops are appended to the 3′ untranslated region (3′-UTR) of the target RNA and bind 48 molecules of MS2 coat protein (MCP) each fused to GFP. The resulting ribonucleoprotein (RNP) tag exceeds the size of the RNA of interest. Moreover, the MS2 stem loops are recalcitrant to degradation by exoribonuclease Xrn1 when bound to the MCP-GFP fusion protein, which can lead to accumulation of labeled leftover tag after the mRNA decay of the ORF (16), unless an engineered MS2-MCP system with reduced binding affinity is used (17).
A third approach is based on microinjection of labeled mRNA. This approach is particularly useful if genetic alterations are difficult to achieve such as in primary neurons, or if little alteration of the mRNA of interest is desired. Herein, mRNA with a 5′-cap is produced by in vitro transcription in the presence of a fluorophore-labeled UTP, in addition to the four canonical NTPs. The modified UTP is statistically incorporated guaranteeing multiple fluorescence labeling. Such mRNAs were successfully used to visualize mRNA localization in rat neurons (18,19) and in Drosophila (20). Importantly, in this approach, the sequence of the mRNA remains unaltered.
So far, a variety of strategies for the covalent linkage of reporters to RNA has been developed, mostly focusing on cotranscriptional or posttranscriptional enzymatic labeling approaches (21,22). The cotranscriptional approach still requires improvements in cell permeability and salvage pathway compatibility as well as the possibility to apply bioorthogonal click reactions. RNA-modifying enzymes, independent of the broad application of methyltransferases, could be more advantageous (23,24), however the RNA sequence is extended with a tag bearing only one fluorophore.
Labeling mRNAs without interfering with their biological functions is an intricate problem, because functionality is not restricted to the coding region, but the UTRs also contain miRNA and protein binding sites as regulatory elements. In fact, chimeric mRNAs with 3′-UTRs from localized mRNAs were repeatedly shown to be transported and locally translated (25,26). This illustrates that any changes in the sequence, including the UTRs bear the risk to alter the properties of the RNA of interest. Therefore, in addition to approaches relying on fluorescent labeling by extending the sequence (e.g. MS2, aptamers, tRNA-modifying enzymes) (23,24,27–29), methods that do not alter the sequence are required and covalent labeling with small fluorophores has advantages. The body-labeled mRNA is a promising approach, however, multiple fluorophores in the coding region likely interfere with translation by the ribosome. In most cases body-labeled mRNAs are exclusively used to report on mRNA localization but not combined with a translational readout. Strikingly, reports where the protein product of body-labeled mRNAs was tested relied on immunohistochemistry rather than the encoded reporter-protein itself (GFP), suggesting that the amount of protein product was low and translation hampered (18). As an alternative, mRNA can be labeled site-specifically at the 5′-cap (30–33). However, to date only one or two fluorophores can be attached to this relatively small hallmark of eukaryotic mRNAs. Furthermore, since the 5′-cap is involved in numerous processes, small modifications can interfere with eIF4E binding and also with translation (34–36). Therefore, although different concepts for mRNA labeling with their unique strengths have been successfully used for mRNA imaging, the ideal way to label mRNA in living cells—i.e. high fluorescence (e.g. with multiple labels) but without addition of large tags or tags that interfere with the functions of mRNA (most notably translation)—has not been found.
Furthermore, mRNAs are emerging therapeutics in protein replacement therapies and tumor vaccination. Tailoring the pharmacokinetics of mRNAs therapeutics has become an important topic, in particular ways to increase the translational efficiency of exogenous mRNA are highly sought after. Increasing the fraction of correctly capped mRNA as well as additional modified nucleotides throughout the entire transcript (e.g. Ψ and m6A) are routinely used to reduce immunogenicity and increase translation (37,38). Many modified 5′ caps have been synthesized and tested to improve stability and translation, yielding up to 3-fold increased translation compared to the canonical cap (39). Modifications in other regions of the mRNA are being discussed as potential sites to increase the translation but remain underexplored.
We reasoned that the poly(A) tail, which is another hallmark of eukaryotic mRNAs, might be a valuable alternative to (i) conveniently label mRNA, (ii) introduce multiple labels and (iii) to affect translation. The poly(A) tail is a non-coding and repetitive element (typically 150–250 nucleotides long in mammals) (40) generated in a template-independent manner by poly(A) polymerases (PAP). This feature provides the opportunity to install multiple fluorophores. It is non-coding, suggesting that the passage of the ribosome will not be affected. Even the UTRs would remain unaffected by modifying the poly(A) tail and thus the functions of the 3′-UTR as a binding platform for miRNAs and RNA-binding proteins would be maintained. However, the poly(A) tail is involved in circularization and interactions with the 5′-cap which are required for translation initiation, so the impact on translation efficiency cannot be anticipated.
Recently, poly(A) polymerases from different organisms were used for terminal and internal labeling of synthetic and natural RNA via copper-catalyzed azide-alkyne cycloaddition (CuAAC) or copper-free strain-promoted azide-alkyne cycloaddition (SPAAC) (Supplementary Scheme S1) (41). Using optimized conditions, it was possible to site-specifically incorporate a single modified nucleotide of choice (A, C, G, U) containing an azide at the 2′ position site-specifically. We wanted to explore the ability of poly(A) polymerases to incorporate multiple 2′-azido-modified adenosine nucleotides at the 3′-end of mRNA, which could subsequently be labeled by SPAAC. An unnatural poly(A) tail would provide a platform both for multiple labeling of mRNA and for investigating the translation of poly(A) tail-modified mRNA in living cells (Figure 1).
Figure 1.
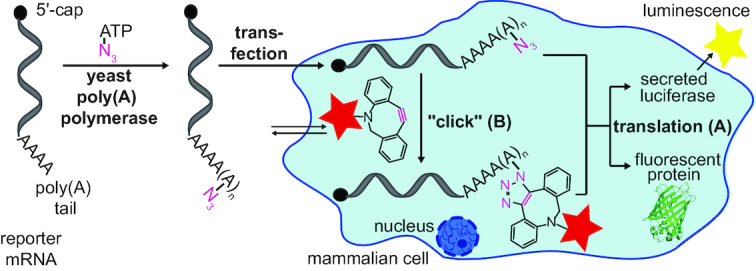
Concept of poly(A) tail labeling for translation and localization analyses of reporter mRNAs. Azido-modified adenosine nucleotides are enzymatically attached at the poly(A) tail of in vitro transcribed reporter mRNAs (luciferase or eGFP) using yeast poly(A) polymerase. (A) Translation analysis. (B) Intracellular labeling.
MATERIALS AND METHODS
3′-End modification analysis of short RNA
The enzymatic addition of 2′-N3-2′-dATP (1 mM) or 3′-N3-2′,3′-ddATP (1 mM) to the 3′-end of RNA (10 μM) was performed in the presence of PAP (600 U) in 1 × PAP reaction buffer for 20 min at 37°C followed by isopropanol precipitation overnight and washing with 70% ethanol. The click reaction was performed with 3′ azido-modified RNA (5 μM) and DBCO-biotin (200 μM) for 45 min at 37°C followed by isopropanol precipitation overnight and washing with 70% ethanol. Then, the modified RNA was analyzed by 15% denat. PAGE. For UHPLC–MS and HPLC analysis, the modified RNA was digested with nuclease P1 (0.33 U) for 4 h at 37°C and then with FastAP (1 U) for 1 h at 37°C. Enzymes were precipitated with HClO4 (200 mM) by incubation for 10 min at room temperature.
Enzymatic synthesis of modified mRNAs at the 5′ cap and the poly(A) tail
For the production of the eGFP-mRNAs the respective eGFP pMRNAxp vector was used (34). For the production of Gaussia luciferase (GLuc) or the Cypridina luciferase (CLuc) mRNAs, the Gaussia or Cypridina luciferase gene, respectively, was cloned into the pMRNAxp mRNAExpressTM vector using BamHI and EcoRI restriction sites. The respective DNA template was amplified in 1 × HF puffer using plasmid (70 ng), dNTP mix (0.5 mM), forward primer (0.5 μM), reverse primer (0.5 μM) and Phusion High-Fidelity DNA Polymerase (1 U). Then, in vitro T7 transcription was performed in 1 × transcription buffer using DNA template (100 ng), A/C/UTP mix (0.5 mM), GTP (0.1 mM), cap analog (1 mM), RiboLock RNase Inhibitor (30 U), T7 RNA polymerase (50 U) and pyrophosphatase (0.1 U) for 3 h at 37°C. Remaining DNA template was digested by incubation with 2 U DNase I for 1 h at 37°C and then, mRNAs were purified using the RNA Clean & Concentrator™-5 Kit (Zymo Research). The enzymatic addition of 2′-N3-2′-dATP (1 mM) to the poly(A) tail of ARCA-capped mRNA (∼3–4 μg) was performed in the presence of PAP (600 U) in 1 × PAP reaction buffer for 20 min at 37°C followed by isopropanol precipitation overnight and washing with 70% ethanol. The click reaction was performed with azido-modified mRNA (500 ng) and DBCO-SRB (50 μM) for 45 min at 37°C followed by isopropanol precipitation overnight and washing with 70% ethanol. Then, the modified mRNA was analyzed by 7.5% denat. PAGE.
Cell culture and toxicity assays
For isolation of total RNA, HeLa cells were cultured in MEM Earle's media (Merck) supplemented with l-glutamine (2 mM), non-essential amino acids (1%), penicillin and streptomycin (1%), and fetal calf serum (FCS, 10%) under standard conditions (5% CO2, 37°C). One day before transfection, 3 × 104 cells were seeded in media (100 μl) in a 96-well plate. Cells were transfected using Metafectene® Pro (0.3 μl) (Biontex) in MEM Earle's media (4.7 μl) and modified mRNA (0.1 μg) in MEM Earle's media (5 μl) for 6 h at 37°C in a total volume of 100 μl per well. Then, media with transfection reagent was removed and the cells were incubated overnight in serum free DMEM media without phenol red (Merck). The cell cytotoxicity was measured using the LDH Cytotoxicity Detection Kit (Takara Bio Inc.) 24 h after transfection according to the manufacturer's protocol. The cell viability was measured using the Vybrant® MTT Cell Proliferation Assay Kit (Thermo Fisher Scientific) 24 h after transfection according to the manufacturer's protocol. A volume of 100 μl 0.04 N HCl in isopropanol was used instead of the SDS–HCl solution to dissolve the formazan. The cell proliferation was measured using the BrdU Cell Proliferation Assay Kit (Merck Millipore) 24 h after transfection according to the manufacturer's protocol.
Cell culture and total RNA isolation or Western Blot analysis
For isolation of total RNA and western blot analysis, HeLa cells were cultured as described above. One day before transfection, 5 × 105 cells were seeded in media (2 ml) in a 6-well plate. Cells were transfected using Metafectene® Pro (6 μl) in MEM Earle's media (94 μl) and modified mRNA (2 μg) in MEM Earle's media (100 μl) for 6 h at 37°C in a total volume of 2 ml per well. Then, media with transfection reagent was removed and the cells were incubated overnight in media. For co-transfection with two different mRNAs, 1 μg of each mRNA was used. For total RNA isolation, 24 h after transfection, cells were incubated with lysis buffer (1 ml). The total RNA was extracted using phenol-chloroform (4:1 and 2:1) followed by isopropanol precipitation overnight. For western blots, cell supernatant was 10× concentrated by lyophilisation, dissolved in 1× PBS and stored at –20°C. Cells were lysed using the protocol of the manufacturer for CelLytic™ M (Sigma Aldrich). Cell lysate was stored at –80°C.
Reverse transcription and quantitative real-time PCR (RT-qPCR)
Isolated total RNA (1 μg) was incubated with DNase I (2 U) in 1 × DNase reaction buffer in a total volume of 10 μl for 30 min at 37°C to degrade residual DNA. The enzymes were then inactivated by addition of EDTA (5 mM) and incubation for 2 min at 65°C. For reverse transcription, half of the digested reaction mixture was incubated in 1 × RT buffer, dNTPs (0.5 mM) with reverse primers for GLuc, CLuc or eGFP (5 μM) and Maxima H Minus Reverse Transcriptase (25 U) for 10 min at 25°C followed by 30 min at 50°C and finally 5 min at 85°C. Using the other half of the digested reaction mixture, the reverse transcription was performed with the reverse primer for ß-actin, respectively. The qPCR was performed in a total volume of 20 μl containing cDNA mixture (2 μl, 1:5 in ddH2O), forward primer (0.5 μM), reverse primer (0.5 μM) and 1 × iTaq Universal SYBR® Green Supermix (Bio-Rad). The following PCR program was conducted: (i) initial denaturation (95°C for 3 min), (ii) denaturation (95°C for 5 s), (iii) elongation (57°C for 30 s), (iv) plate read, (v) 39 × cycle (ii–iv), (vi) melt curve (60°C—95°C, 0.5°C/4 s), (vii) plate read. Data analysis was performed with the CFX Manager 3.1 (Bio-Rad).
Western blots
To determine the protein concentration of cell lysate or cell supernatant, the Bradford assay was performed using BSA calibration standards (80–0 μg/ml) and a dilution of cell lysate or cell supernatant (1:50). Samples (50 μl) were incubated (15 min, rt, exclusion of light) with 1× Roti®-Quant (Roth) staining solution (200 μl) and then, the extinction at 595 nm was determined.
Proteins were separated via tris-glycine-PAGE (10% protein gel, 120 V, 1.5 h, rt, for protein detection from cell lysate 50 μg protein and for protein detection from 10× concentrated cell supernatant 100 μg protein was loaded) and then the proteins were transferred onto nitrocellulose membrane Roti®-NC (Roth) in semi-dry transfer buffer with 80 mA for 75 min at rt. To control protein transfer efficiency, a Ponceau S stain was performed. The membrane was cut into suitable pieces for subsequent antibody treatment and washed with 1× PBS + 0.01% Tween (PBST). Blocking of the membrane was performed in blocking buffer for 1 h at rt, followed by incubation with the respective primary antibodies overnight at 4°C and three times washing with PBST for 5 min at rt. The membrane pieces were incubated with HRP conjugating secondary antibodies for 1 h at rt and washed three times with PBST afterwards. For chemiluminescence detection the EZ-ECL Chemiluminescence detection kit (Biological Industries) was used and results were analyzed with a Chemo Star Advanced Fluorescence & ECL Imager (Intas). Bands were quantified with ImageJ.
Cell culture and sample collection for luminescence or in-cell labeling
For microscopy and luminescence experiments, HeLa cells were cultured as described above. One day before transfection, 2 × 105 cells were seeded in media (1 ml) in a 12-well plate. Cells were transfected using Metafectene® Pro (3 μl) in MEM Earle's media (47 μl) and modified mRNA (1 μg) in MEM Earle's media (50 μl) for 6 h at 37°C in a total volume of 1 ml per well. Then, media with transfection reagent was removed and the cells were incubated overnight in media. For co-transfection with mRNAs, 500 ng of each mRNA were used. For subsequent luminescence measurements, an aliquot of 100 μl of cell media was taken 24 h after transfection and stored at –20°C. For click reactions, cells were incubated with DBCO-SRB (3 μM) for 1 h at 37°C directly after transfection followed by one washing step with media and incubation with media for 1 h to release excess fluorophore. Media was changed again and cells were incubated overnight to release remaining fluorophore.
Luminescence measurements
Luminescence measurements were performed using the Gaussia-Juice Luciferase Assay Kit (pjk) or the Pierce®Cypridina Luciferase Flash Assay Kit (Thermo Scientific). The luciferase activity was determined by adding 5 μl of the supernatant (for Gaussia luminescence a 1:20 dilution in 1 × PBS [pH 7.5] was used) to a 96-well plate followed by injection of 50 μl freshly prepared reaction mixture with an acquisition time of 3 s (duration of signal acquisition).
Fluorescence In Situ Hybridization (FISH) and fluorescence microscopy
One day after transfection, the cells—grown and treated on glass coverslips—were fixed and hybridized with Stellaris eGFP probe with Quasar 670 dye (BioCat) using the protocol for adherent cells from Stellaris® RNA FISH (Biosearch Technologies). Images were taken in a channel for eGFP fluorescence (λexc. = 488 nm, λem. = 510 nm), a channel for SRB fluorescence (λexc. = 568 nm, λem. = 585 nm), a channel for DAPI fluorescence (λexc. = 358 nm, λem. = 461 nm), a channel for Quasar 670 fluorescence (λexc. = 647 nm, λem. = 670 nm) and in the differential interference correlation (DIC) channel.
Colocalization analysis
An object based colocalization analysis was performed using Fiji of the ImageJ software. To identify the respective region of interests, the ‘Analyze Particles’ plugin has been used with a size filter of 4–1000 pixels to exclude large and unspecific areas. The images were previously processed using a median filter (radius: 3 pixels) and the ‘Subtract Background’ function (sliding parabolic, radius: 20 pixels). To binarize the images, a manual threshold was set. After creating the region of interests, it was examined whether a signal was detectable within each region of interest for both channels. For each condition the same settings were used.
Cell culture and live-cell imaging
For time-course microscopy experiments, HeLa cells were culture as described above. One day before transfection, 1.2 × 105 cells were seeded in media (1 mL) in a cell imaging coverglass chamber (Eppendorf). Cells were transfected using Metafectene® Pro (1.8 μl) in MEM Earle's media (48.2 μl) and modified mRNA (0.6 μg) in MEM Earl's media (50 μl) for 6 h at 37°C in a total volume of 1 ml per chamber. Then, media with transfection reagent was removed and cells were incubated in media. Images were taken in a channel for eGFP fluorescence (λexc. = 488 nm, λem. = 510 nm), a channel for SRB fluorescence (λexc. = 568 nm, λem. = 585 nm) and in the differential interference correlation (DIC) channel at different time points (2.5, 5.5, 7.5, 23 and 31 h) after start of transfection.
RESULTS
Polyadenylation of test RNAs with yeast poly(A) polymerase and azido-functionalized ATP
Building on recent work from Winz et al. (41), we first evaluated the enzymatic addition of azido-modified ATPs using yeast poly(A) polymerase (PAP) to test short RNAs (30 nt) as model system for our subsequent studies of the poly(A) tail modification and labeling of reporter mRNAs (Figure 2). ATPs with azido-functionalities at either the C-2′ position (2′-N3-2′-dATP) or the C-3′ position (3′-N3-2′,3′-ddATP) were employed as substrate for yeast PAP (Supplementary Figure S1). While the former nucleotide can yield RNA with multiple 2′-azido-modified deoxyadenosine nucleotide units at the 3′-end, the latter only permits addition of a single 3′-azido-dideoxyadenosine nucleotide, which cannot be further elongated due to the lacking hydroxyl group at the 3′ position. Following the enzymatic introduction of azido-groups, the bioorthogonal strain-promoted azide-alkyne cycloaddition (SPAAC) (32,34) was performed using dibenzocyclooctyne-PEG4-biotin (DBCO-biotin, Supplementary Figure S2) to obtain the corresponding click product (Figure 2A).
Figure 2.

Enzymatic addition of modified adenosines to the 3′-end of short test RNA. (A) Scheme illustrating 3′-end modification of test RNA with 2′-N3-2′-dATP or 3′-N3-2′,3′-ddATP using yeast PAP followed by click reaction with DBCO-biotin. Using 2′-N3-2′-dATP, multiple 2′-N3-2′-deoxyadenosine nucleotides were added at the poly(A) tail and then reacted quantitatively to the corresponding click product. Using 3′-N3-2′,3′-ddATP, a single 3′-N3′-2′,3′-dideoxyadenosine nucleotide is added and reacted during click reaction. (B, C) MS analysis of adenylated and clicked RNA after digestion and dephosphorylation to ribonucleosides: 2′-click-2′-dA 2 (calculated mass of [C49H64N13O11S]+ = 1042.4563, found: 1042.4565) or 3′-click-2′,3′-ddA 4 (calculated mass of [C49H64N13O10S]+ = 1026.4614, found: 1026.4616), respectively. (D) HPLC analysis of adenylated test RNA before and after click reaction. Samples were prepared as in B, C. (E) PAGE analysis of intact modified test RNAs (15% PAA gel, 12 W, 3 h, rt). SYBR™ Gold staining (Invitrogen) is shown.
After enzymatic digestion and dephosphorylation of these differently adenylated and clicked RNAs, LC–MS analysis confirmed the successful enzymatic modification and labeling of short test RNA using both types of N3-modified ATPs (Figure 2B and C). The corresponding masses [M+H]+ and [M+Na]+ for the four modified adenosines 1–4 were identified (Figure 2B and C and Supplementary Figures S3–S5). Importantly, HPLC measurements of the differently adenylated and clicked RNAs after degradation showed complete conversion of the azide to the triazole heterocycle and thus, quantitative click reaction (Figure 2D).
To determine how many 2′-N3-2′-dAs were on average added to the 3′-end of the test RNA, PAGE analysis was conducted before and after the click reaction with DBCO-biotin. Figure 2E shows that yeast PAP appended 2–6 (ø≈ 3) 2′-N3-2-dAs (lane 4) or 0–1 3′-N3-2′,3′-ddAs (lane 2), respectively. After the click reaction we observed a biotin-induced shift of the RNA bands (lanes 1 and 3), indicating again efficient conversion in the click reaction (Figure 2E). These results show that enzymatic addition of multiple azido-modified adenosine nucleotides is possible and that the azido groups can be efficiently converted in the SPAAC reaction, suggesting that multiple reporter groups can be easily installed. With respect to the natural substrate ATP, however, the in vitro incorporation of azido-modified adenosines is compromised—in accordance with the published data (41,42).
Fluorescence labeling of reporter mRNAs with azido-modified poly(A) tail
With the reaction conditions for successful multiple addition of 2′-N3-2′-dA nucleotides to the 3′-end of short test RNA in hand, we investigated whether genuine reporter mRNAs can also be labeled at their poly(A) tail. To this end, we cloned the genes for the secreted Gaussia luciferase (GLuc), Cypridina luciferase (CLuc) or eGFP, respectively, into the pMRNAxp mRNAExpressTM plasmid. We then produced and purified reporter mRNAs containing a 5′-cap, 5′- and 3′-UTRs as well as a genetically encoded poly(A) tail by in vitro transcription using T7 RNA polymerase as previously described (34). For the 5′-cap, the anti-reverse cap analog (ARCA, 3′-O-Me-m7G(5′)ppp(5′)G, Supplementary Figure S6), was used, as it can only be incorporated in the correct orientation (43) and is therefore used for production of therapeutic mRNAs. An ApppG-capped mRNA served as negative control, because it is not translated but stabilizes mRNA relative to a triphosphate and similar to a regular 5′ cap (Supplementary Figure S6). These reporter mRNAs were then enzymatically modified using yeast PAP followed by SPAAC using the strained alkyne-fluorophore-conjugate dibenzocyclooctyne-PEG4-5/6-sulforhodamine B (DBCO-SRB) (Figure 3A and Supplementary Figure S2). Thus, four reporter mRNAs with different modifications, i.e. ARCA- and ApppG-capped mRNAs with normal poly(A) tail and two ARCA-capped mRNAs with modified poly(A) tails—one with azido-modifications (ARCA-N3) and the other one with attached SRB-conjugates (ARCA-click)—were produced. Analysis by gel electrophoresis and in-gel fluorescence revealed a bright fluorescent band upon irradiation of the SRB fluorophore at λexc. = 532 nm at the expected length (GLuc: ∼ 800 nt, eGFP: ∼ 1200 nt), indicating successful and efficient labeling (Figure 3B and C and Supplementary Figure S7). Controls without click reaction but with the azido-modified poly(A) tail did not show a fluorescent band at λem. = 575 nm. All constructs were detected at the expected length in SYBR Gold staining, independent of the labeling reaction (Figure 3B and C), confirming that neither of the two modification steps causes degradation, even in the case of long reporter-mRNAs (Figure 3B and C). To elucidate whether the mRNAs were labeled by multiple fluorophores, we prepared reporter-mRNAs containing a single fluorophore as control, using 3′-N3-2′,3′-ddATP followed by SPAAC. In comparison to the mRNAs labeled via 2′-N3-2′-dA in the poly(A) tail, these control reporter-mRNAs showed only a very faint fluorescent band (Supplementary Figure S8). We therefore conclude that we were able to enzymatically append multiple azido-modifications also to long reporter-mRNAs and to efficiently install multiple fluorescent labels in their poly(A) tail, in line with our results obtained for small test-RNAs.
Figure 3.
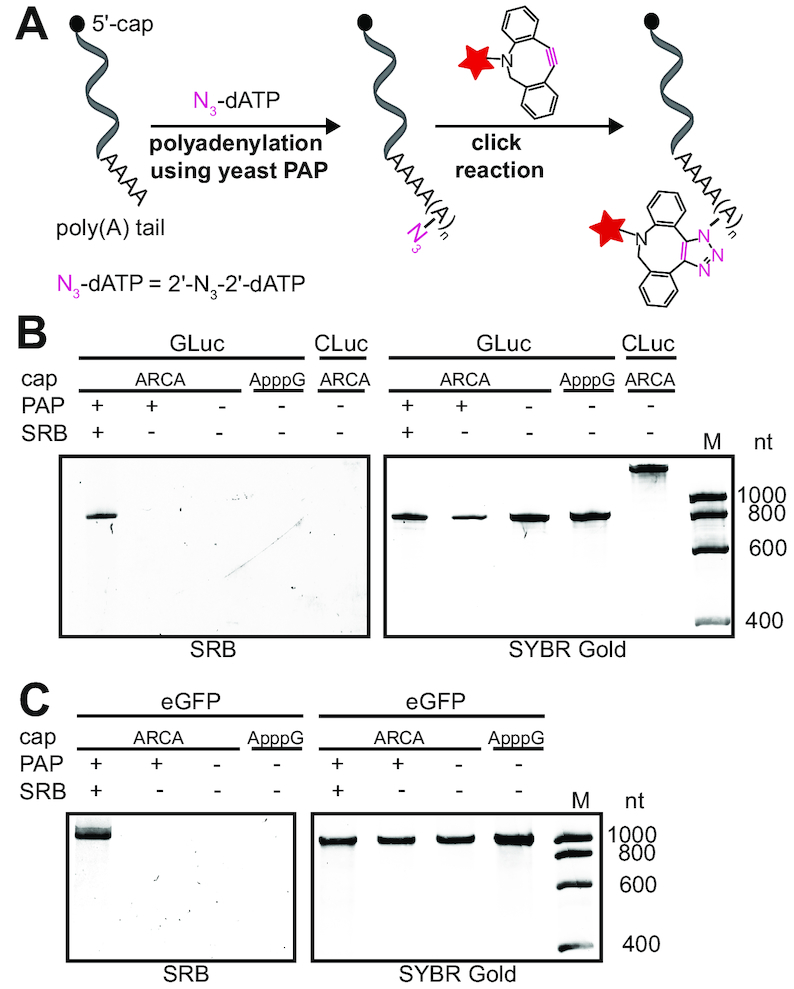
In vitro fluorescence labeling of poly(A) tail-modified reporter mRNAs. (A) Concept for poly(A) tail modification of mRNAs with 2′-N3-2′-dATP using yeast PAP followed by click reaction using DBCO-SRB. (B, C) Luciferase (GLuc, CLuc) and eGFP mRNAs were analyzed by PAGE (7.5% PAA gel, 10 W, 1 h, rt) and visualized using SRB fluorescence (λex = 532 nm) and SYBR™ Gold (Invitrogen) staining.
Toxicity of poly(A) tail-modified reporter mRNAs
Next, we wanted to investigate whether mRNA with modified adenosine nucleotides would cause toxic effects in cells. To this end, HeLa cells were transfected with the respective mRNAs and toxicity was measured based on the lactate dehydrogenase (LDH) cytotoxicity assay, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay and the bromodeoxyuridine (BrdU) cell proliferation assay. Cells transfected with ARCA-capped eGFP-mRNAs with or without clicked poly(A) tail showed a similar fraction of dead cells (41% versus 42%), which was in the same range as cells transfected with the respective DNA, i.e. peGFP-C1 plasmid (38%, Supplementary Figure S9A). As expected, the fraction of dead cells was slightly increased for all transfected compared to non-transfected cell samples (26%, Supplementary Figure S9A). Similarly, the viability of cells was only marginally reduced upon transfection with differently modified mRNAs or peGFP-C1 plasmid (96–97% compared to untransfected cells, Supplementary Figure S9B). The proliferation of transfected cells was slightly reduced (∼90%). Here, cells transfected with mRNA were slightly more affected (∼90%) than cells transfected with peGFP-C1 plasmid (97%), albeit within the margin of experimental error. There was no difference in proliferation between cells transfected with unmodified or modified mRNAs, respectively (Supplementary Figure S9C). In summary, these data show that the modifications to the poly(A) tails of the mRNA do not induce toxicity.
Poly(A) tail-modified luciferase mRNA exhibit enhanced translation
To assess the effect of modifications at the poly(A) tail on translation, we prepared a set of differently modified reporter mRNAs and transfected HeLa cells with different combinations thereof (Figure 4A). First, we transfected HeLa cells with GLuc-mRNAs bearing different modifications at the poly(A) tail (ARCA-N3, ARCA-click) and analyzed the amount of protein produced on western blots (Figure 4B). Again, ARCA-capped mRNA served as positive control and ApppG-capped mRNA as negative control. Western blot analysis showed that GLuc protein was significantly increased when the poly(A) tail was modified (Figure 4B). This effect was more pronounced when the poly(A) tail was clicked with DBCO-SRB (ARCA-click) compared to azido-modified poly(A) tail (ARCA-N3).
Figure 4.
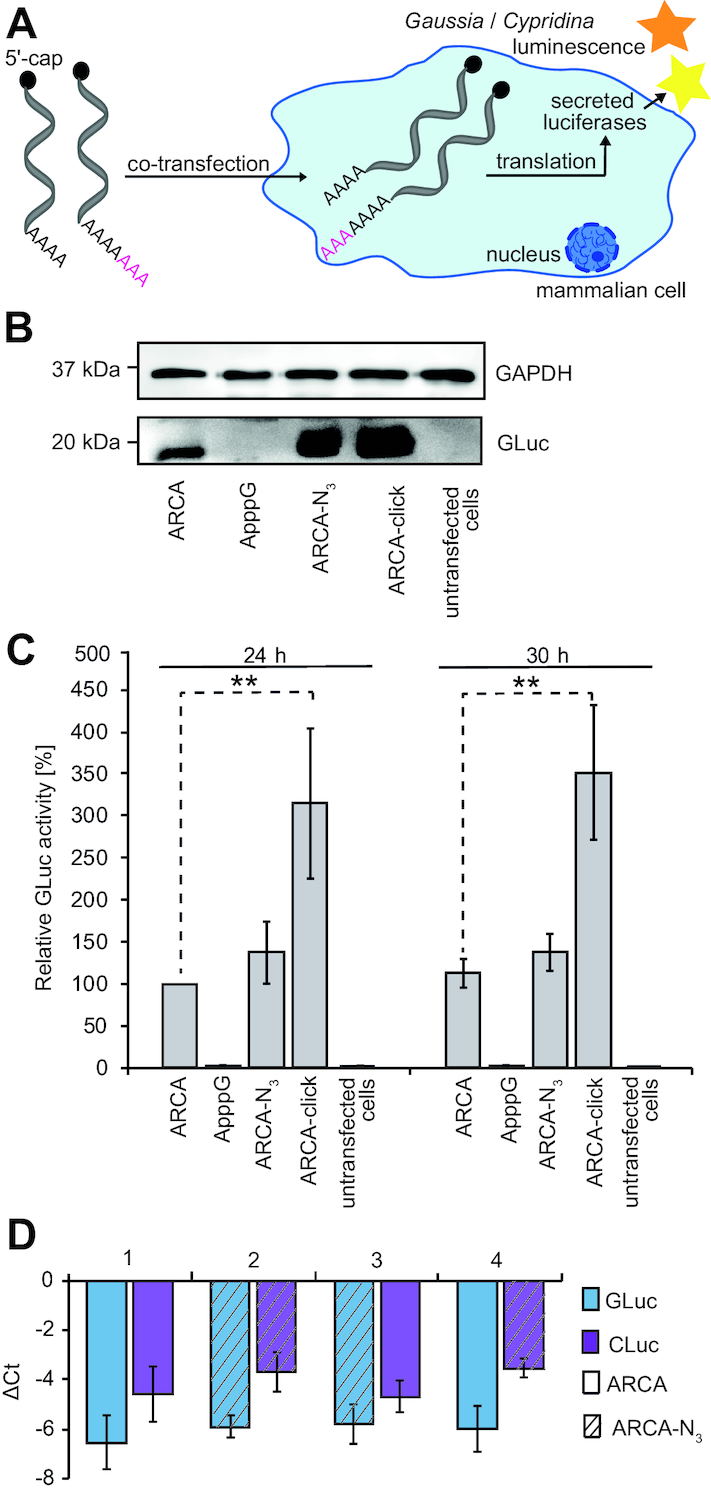
Stability and translational efficiency of poly(A) tail-modified luciferase mRNAs. (A) Scheme illustrating translation assay in mammalian cells of poly(A)-modified luciferase mRNAs followed by luminescence analysis of secreted luciferases. (B) Western blot analysis of GLuc protein in the supernatant of cells transfected with indicated GLuc-mRNA constructs. With the cell lysate of corresponding samples, GAPDH protein level from the respective cell lysate is shown as control. (C) Translational efficiency of differently modified GLuc-mRNAs relative to co-transfected CLuc-mRNA. The GLuc luminescence was referenced to the luminescence from ARCA-capped CLuc-mRNA and then normalized to ARCA-GLuc mRNA after 24 h (≙ 100%). Data and error bars show average and standard deviation of three independent experiments measured in duplicate. Statistical comparisons performed by two-tailed t-test with P < 0.01 (**) and P < 0.001 (***), statistical comparisons to ApppG and untransfected cells are not shown). (D) Comparison of GLuc- and CLuc-mRNA levels by qPCR at 24 h after transfection for different combinations of modified poly(A) tails, as indicated in the legend. The GLuc or CLuc mRNA level was quantified by qPCR and compared to the ß-actin-mRNA level by generating ΔCt = Ct(target)-Ct(reference). Data and error bars show averages and standard deviations of two independent experiments with two technical replicates measured in duplicate. Abbreviations: Ct: cycle threshold, target: GLuc- or CLuc-mRNA, reference: ß-actin-mRNA, ApppG / ARCA: capped 5′-end, N3: azido-modified poly(A) tail, click: clicked poly(A) tail).
To rule out that differences in transfection might be responsible for this effect, we co-transfected HeLa cells with GLuc- and CLuc-mRNAs with differently modified poly(A) tails and determined the relative luciferase activities from the supernatant (Figure 4C). All measurements were normalized to the luciferase activity obtained with ARCA-capped GLuc-mRNA after 24 h (≙100%, Figure 4C). Again, GLuc-mRNA with an azido-modified poly(A) tail (ARCA-N3, Figure 4C) showed increased translational activity both after 24 and 30 h (∼140% or 150%). The measured GLuc activity was further increased when the GLuc-mRNA with the azido-modified poly(A) tail was clicked to DBCO-SRB (ARCA-click), yielding >300% of translational activity relative to the regular ARCA-capped GLuc-mRNA (ARCA, Figure 4C). The negative control with ApppG-capped GLuc-mRNA gave negligible luciferase activity (4%). These data confirm that poly(A) tail-modified GLuc-mRNA used in this study are more efficiently translated as observed in western blots and show that differences in transfection are not responsible for the effect. To rule out that the positive effect on translation was caused by higher purity of the clicked mRNA (as previously described in Ref (44)), we carried out additional precipitation and column purifications for all mRNAs (Supplementary Figure S10). Also with these additional purification steps, the translational efficiency of mRNA with poly(A) tail modification and click (ARCA-click) remained at >240% (ARCA GLuc-mRNA ≙ 100%).
To elucidate whether the observed increase in GLuc activity is a result of different mRNA levels in the cell (e.g. as a result of different stability or relative transfection efficiency), we analyzed and quantified the mRNA levels of differently poly(A) tail-modified mRNAs by qPCR. First, we confirmed that the modified GLuc-mRNA was detectable 24 h after transfection at high levels relative to endogenous ß-actin-mRNA (Supplementary Figure S11A, Supplementary Table S3). To decipher the effect of modifications on the poly(A) tail, we then co-transfected HeLa cells with all combinations of azido-modified GLuc- and CLuc-mRNAs used in this study and measured their abundance after 24 h relative to ß-actin-mRNA (Figure 4D). These data revealed similar levels of GLuc-mRNA in all experiments, suggesting that azido-modifications in the poly(A) tail do not interfere with degradation (1–4 in Figure 4D, Supplementary Table S4). CLuc-mRNAs were in all cases slightly less abundant than GLuc-mRNA and the azido-modified CLuc-mRNAs were less abundant than CLuc-mRNAs with unmodified poly(A) tails, suggesting a decrease in stability in the case of CLuc-mRNA (1–4 in Figure 4D and Supplementary Table S4). These data indicate that the azido-modifications have no stabilizing effect and do not increase the mRNA levels (be it by increased stability or increased transfection).
Taken together, the results above show that (i) installing azido-modifications at the poly(A) tail is efficient for multiple labeling of mRNAs, (ii) the resulting mRNAs are not more abundant or stable, but (iii) are more efficiently translated than the ARCA-capped mRNA without poly(A) modifications.
Translation of poly(A) tail-modified eGFP mRNAs
Encouraged by the efficient translation of poly(A) tail-modified luciferase-mRNAs, we next tested eGFP-mRNA produced in a similar manner to analyze whether the resulting eGFP can be imaged in HeLa cells using confocal microscopy (Figure 5A). First, we validated by qPCR that differently modified eGFP-mRNAs were present in HeLa cells 24 h after transfection at significant levels (Supplementary Figure S11B, Supplementary Table S3)—similar to the results obtained with luciferase mRNAs. Next, we transfected HeLa cells with differently modified eGFP-mRNAs (ARCA, ApppG, ARCA-N3 and ARCA-click) and analyzed the amount of eGFP-protein as a function of poly(A) modifications on western blot (Figure 5B). In line with the results obtained for luciferase-mRNAs shown above, quantification revealed that ARCA-N3-eGFP-mRNA yielded significantly more eGFP (150 ± 10%) compared to regular ARCA-mRNA and that even more protein was produced from ARCA-click-eGFP-mRNA (320 ± 30%). GAPDH levels were identical in all samples, indicating that transfection with modified eGFP mRNAs does not impair translation in general (Figure 5B).
Figure 5.
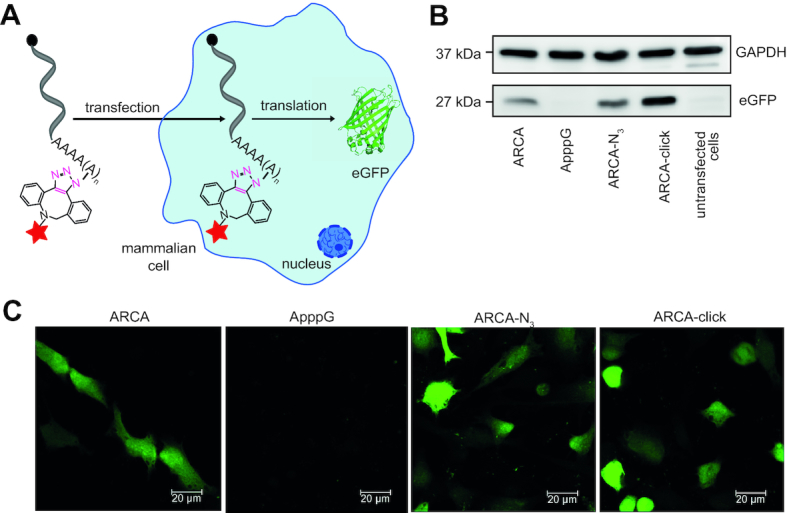
Poly(A) tail-modified eGFP-mRNA is a translationally active mRNA. (A) Scheme illustrating transfection of HeLa cells with poly(A) tail-modified mRNAs (ARCA-N3, ARCA-click) or with unmodified control (ARCA, ApppG) and following translation into protein. (B) The eGFP and GAPDH protein level of different cell samples was analyzed on western blots. (C) Confocal microscopy shows eGFP fluorescence after transfection with indicated modified mRNAs. 24 h after transfection, cells were fixed for imaging. Scale bars are 20 μm.
Furthermore, we imaged HeLa cells transfected with different eGFP-mRNAs on a confocal microscope. Cell samples transfected with eGFP-mRNAs (except for the negative control ApppG) developed green fluorescence indicating efficient translation (Figure 5C). The images corroborate that the modified poly(A) tails in ARCA-N3 and ARCA-click mRNAs positively impact on translation and thus confirm the results from western blots and from the luciferase-mRNAs.
Imaging and in-cell labeling of poly(A) tail-modified eGFP mRNAs
We showed above that the SPAAC reaction can be used for efficient fluorescent labeling of mRNAs with multiple fluorescent SRB-dyes in vitro (Figure 3). Using HeLa cells, we now wanted to test, whether this covalent labeling with fluorescent dyes will enable detection of mRNA localization after transfection. We transfected HeLa cells with ARCA-click-eGFP-mRNAs and imaged them after fixation and permeabilization (Figure 6, right panel). In the SRB channel, small punctated signals were clearly visible in the perinuclear and cytosolic region, indicating that multiple labeling at the poly(A) tail enables imaging of mRNAs after transfection (Figure 6B, right panel; Supplementary Figures S12 and S13). To ensure that the observed fluorescent signals originate indeed from labeled mRNAs and not SRB-fluorophore alone (e.g. after degradation of mRNA or removal of the fluorophore from the mRNAs), we performed smFISH using Stellaris RNA FISH probes with Quasar670 fluorophores. Images from smFISH showed the same punctate staining that was colocalized with the SRB-signal (Supplementary Figure S14A and B), indicating that ARCA-click mRNAs can be easily detected in cells by confocal imaging. Labeling mRNA in the poly(A) tail with multiple fluorophores therefore presents a valuable new strategy to label mRNA with multiple small organic dyes and obtain the sensitivity required for detection in cells without altering the encoded sequence (Figure 6B, right panel). Furthermore, the labeled mRNAs are actively translated (Figures 4 and 5). This could also be confirmed by live-cell images at different timepoints after transfection with ARCA-click-eGFP-mRNA. Again, the SRB-signal shows the evenly distributed punctated pattern over the entire time (Supplementary Figures S15 and S16).
Figure 6.
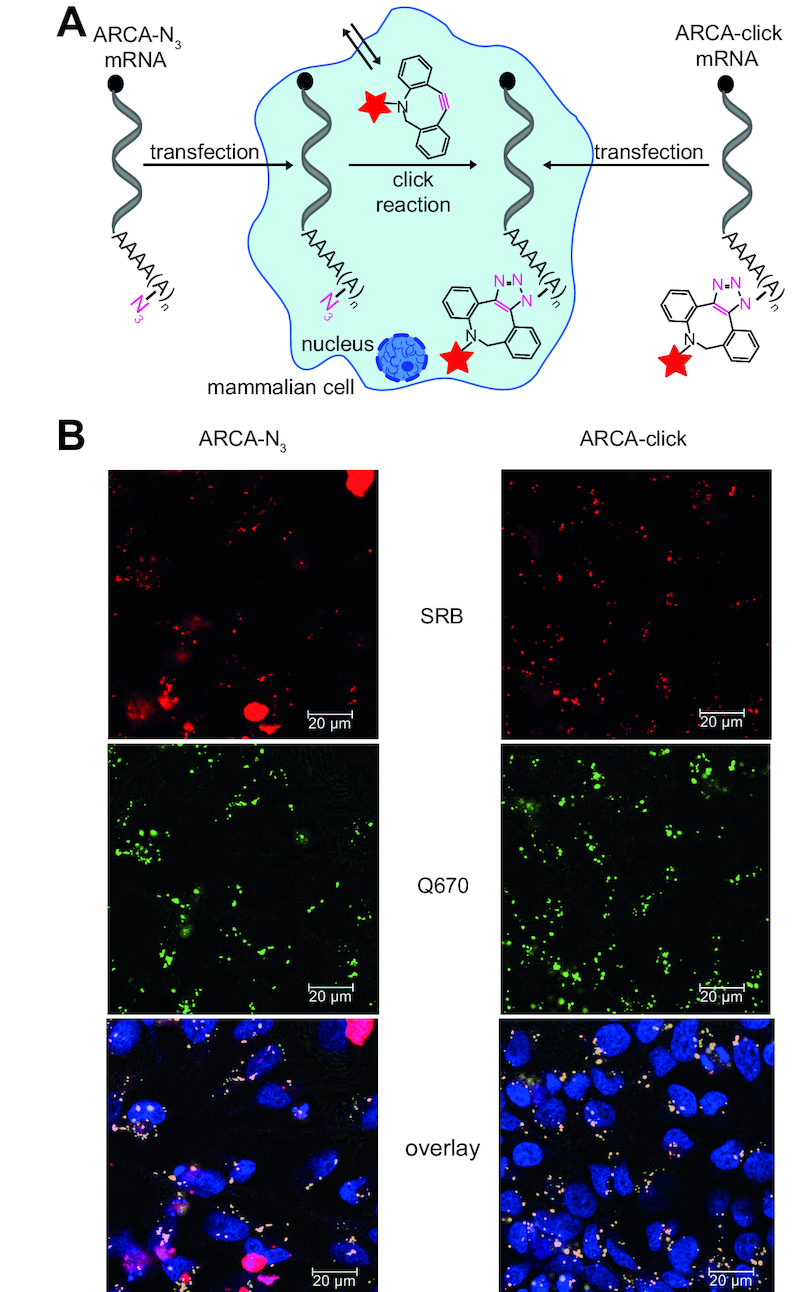
eGFP-mRNA with azido-modified poly(A) tail can be labeled in living cells. (A) Scheme illustrating in-cell click reaction in mammalian cells transfected with ARCA-N3-eGFP-mRNA. (B) Confocal microscopy of HeLa cells transfected with eGFP-mRNA with azido modifications at the poly(A) tail (ARCA-N3) or with in vitro clicked poly(A) tail (ARCA-click). Sample with ARCA-N3-eGFP-mRNA was then subjected to DBCO-SRB treatment to achieve bioorthogonal fluorescence labeling in living cells, followed by washing steps. After incubation overnight, all cell samples were fixed and hybridized using Stellaris eGFP probe with Quasar 670 dye. SRB and Q670 fluorescence show mRNA labeling. Same cell samples were used as for translational analysis (Figure 5). Scale bars are 20 μm.
Since the SPAAC reaction is bioorthogonal—i.e. non-toxic and not interfering with cellular components—it can in principle be performed in living cells. Covalent labeling in living cells presents an important step towards complete intracellular labeling, but has been particularly difficult for mRNAs that are intracellular and less abundant than the respective proteins or ribosomal RNAs. We figured that our poly(A) labeling approach might overcome current limitations of intracellular mRNA labeling reactions because it offers multiple azido-groups for reaction (Figure 6A, left panel). We transfected HeLa cells with ARCA-N3-eGFP-mRNAs and then incubated them with cell-permeable and non-toxic DBCO-SRB (34) for 1 h (Figure 6A, left panel). Excess fluorophore was removed overnight, cells were fixed and prepared for imaging, including smFISH for eGFP-mRNA (using Stellaris FISH Quasar 670). Confocal images of cells transfected with ARCA-N3-eGFP-mRNA showed a bright red SRB fluorescence signal over the background fluorescence that consisted of many small punctated signals but also big red dots (Figure 6B, left panel). The small punctated signal show a similar distribution to the exogenous labeled mRNA (ARCA-click-eGFP-mRNA) after transfection (Figure 6B, right panel; Supplementary Figures S12 and S13), indicating that the intracellular click reaction was successful. Moreover, this pattern colocalized with smFISH for the eGFP-mRNA sequence (Supplementary Figure S14C and D). Importantly, cells transfected with eGFP-mRNAs without azido-modifications (i.e. ARCA, ApppG) and treated with SRB-DBCO under the same conditions did not show the pattern of many small dots (Supplementary Figure S17). However, the occasional large big red dots were observed in all cell samples (i.e. ARCA-N3 or untransfected cells) treated with DBCO-SRB (Figure 6B left panel, Supplementary Figures S12 and S17). These large red dots can be clearly distinguished from the punctated mRNA stainings. Closer inspection showed that the big dots spanned the entire z-axis of a cell (Supplementary Figure S18), suggesting that they originated from precipitate or background reaction of DBCO-SRB (45,46) or that—despite washing—the dye molecules were not completely removed. Importantly, the big red dots did not overlap with the Q670 signal from smFISH, confirming that their origin is not associated with mRNA.
Taken together, these data show that our approach can be used to efficiently label mRNA at the poly(A) tail in vitro and in cells. Labeling mRNAs in vitro with multiple fluorophores makes them amenable to imaging their localization after transfection into cells in a regular confocal microscope without changing their sequence and while maintaining translation. This approach will prove useful for investigating mRNA localization in primary cells and model organisms like zebrafish, where labeled mRNAs can be easily injected. Importantly, the alterations to the mRNA are minimal compared to other approaches that are widely used to date (MS2, aptamers, etc). The ability to perform the click reaction in cells opens the door to image different time points by clicking different fluorophores but also to analyze mRNA-protein complexes by isolation via the poly(A) tail clicked to biotin.
DISCUSSION
In this work, we showed that yeast poly(A) polymerase can be readily used to append multiple azido-modified adenosine nucleotides to different mRNAs using commercially available 2′-N3-2′-dATP. Quantification revealed that on average three modified adenosines are installed. Importantly, the azido-modified adenosines were completely converted in a SPAAC reaction using different commercially available DBCO-conjugates. Therefore, our approach can be universally used to efficiently label both short and long RNAs with multiple tags, such as biotin or fluorophores.
The multiple poly(A) tail labeling with SRB significantly increased translation of different reporter-mRNAs in vitro and in cells, as confirmed by independent methods. Therefore, our approach has enormous potential in the field of therapeutic mRNAs, where ways to change the pharmacokinetic properties of mRNAs are needed. In particular, ways to increase translation of a therapeutically relevant mRNA are highly sought after (47). Current approaches have focused on the mRNA cap and yielded the now generally used ARCA cap, increasing translation ∼2-fold as well as additional modifications in the triphosphate bridge of the cap that further enhanced translation ∼3-fold (39). In addition, internal modifications (Ψ and m6A) are generally used to reduce immunogenicity of mRNA, but some of them (m6A) also positively impact on translation (37,38). Our approach suggests that modifications at the poly(A) tail might be a valuable alternative strategy. We observed a 3-fold increase in translation efficiency for mRNAs that were clicked to multiple SRB-dyes in their poly(A) tail. This effect is in the range of recent reports for cap modifications and bears potential to give even higher effects if combined with those.
Currently, we can only speculate about the mechanism by which labeling the poly(A) tail increases the translational efficiency, but we could rule out effects on transfection efficiency and stabilization at the mRNA level. Since the increase in translation efficiency is higher after the click reaction with DBCO-SRB (3-fold compared to 1.3-fold) it is reasonable to assume that the fluorophore itself can promote formation of the translationally active complex. Normally, the poly(A) tail interacts with poly(A) binding proteins (PABPs) and consequently with the eIF4F cap-binding complex (comprising eIF4E, eIF4A and eIF4G) in a circularized configuration facilitating mRNA translation (48,49). Furthermore, the poly(A) tail directly interacts with the eukaryotic deadenlyases PAN2-PAN3 and the poly(A) ribonuclease (PARN). It serves as the main point of attack for deadenlyation-dependent decay and is involved in controlling the highly complex processes of poly(A) tail length regulation, gene expression and mRNA stability (reviewed in (49–53)). It is most likely that the fluorophores we introduced at the poly(A) tail facilitate interaction with PABP. Evidence in favor of this hypothesis comes from the observation that related fluorescent dyes (rhodamine B) were reported to interact with specific proteins, including methylthioadenosine phosphorylase, BSA or lysozyme (54–56).
Key advantages of multiple poly(A) tail labeling are that the mRNAs remain completely unaltered in their sequence—both in the coding and in the untranslated regions—and that small fluorescent dyes are covalently attached. The combination of these features sets the method apart from most labeling approaches for mRNA to date that have been applied to cells. Approaches for mRNA labeling to date require either extremely large tags, such as the 24 stem loops in combination with 48 MCP-GFP proteins typically used in the MS2 system (28), or smaller extensions, such as specific RNAs engineered with a 8 tandem MB target repeat sequence (57) or aptamers, that however still alter the native mRNA sequence (24,27,29,58,59). Since the 3′-UTR of mRNAs contains multiple miRNA and protein binding sites and is heavily involved in localization, alternative adenylation, and other regulatory processes, changes made to the sequence may interfere with these processes. Our work shows that even small alterations in the poly(A) tail that do not change the sequence at all have effects on translation, illustrating that care must be taken when interpreting the results obtained from approaches where RNA was modified more drastically. For smaller tags, attachment of multiple fluorophores has not been described to date to the best of our knowledge. However, significant turn-on effects and very good sensitivities could be achieved (27,58,59). SPAAC reactions with turn-on effects are still rare (60), but potentially alternative bioorthogonal reactions, in particular the tetrazine-ligation with a turn-on dye (61) could be useful to further boost the sensitivity of our approach in the future.
In the long run, our approach aims at investigating the behavior of minimally altered mRNAs in cells and developing organisms. This includes investigation of its subcellular localization but also analysis of interacting RNA-binding proteins. Here, the azido-groups could be used to conveniently isolate poly(A) tail-modified genetically unaltered mRNAs via biotin/streptavidin with their protein binding partners. The enzymatic addition of the modified poly(A) tail to the mRNA of interest is performed in vitro, the click reaction can be performed in vitro or in living cells. In any case, the mRNA of interest then has to be introduced into the cell. Our study shows that transfection of mammalian cells with mRNAs is reliable and efficient, in line with previous work (34). The subcellular localization of mRNA after transfection was perinuclear and cytoplasmic—independent of the poly(A) tail modification, as confirmed by smFISH. However, the signal obtained from the labeled mRNA colocalized with smFISH for the eGFP sequence does not show the typical fine smFISH pattern of a single RNA detection, but the dots may represent multiple mRNAs within endosomes. Since the mRNA is actively translated it has to be (at least partially) available to the ribosomes. Previous studies showed that part of the mRNA delivered via lipofection localizes in cytoplasmic vesicles, most likely due to compartmentalization in the endosomal system (62). Assuming that this is correct and knowing that such mRNA likely does not contribute to translation, there is potential to further increase the translation of exogenous mRNAs after lipofection, e.g. by RNA modifications (62). Importantly, mRNA can also be injected into developing organisms and this is a standard procedure for one cell stage zebrafish embryos (63–65) or Drosophila (66). We anticipate that research in this field will directly benefit from our easy to use method for mRNA labeling. Finally, the observation that facile and covalent modification of mRNAs with fluorophores at the poly(A) tail leads to a significant increase in translational efficiency raises hope to use this approach as a novel strategy to increase translation, e.g. of mRNAs for developmental studies in model organisms or even for therapeutic mRNAs.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr W. Dörner and S. Wulff for UHPLC–MS assistance, Dr K. Chandrasekaran for helpful discussions and experimental assistance as well as K. Arnke and N. Püllen for experimental support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [RE2796/6-1 and RE2796/2-1 to A.R., EXC1003 to A.R.]; Fonds der Chemischen Industrie [doctoral fellowship to L.A., Dozentenpreis to A.R.]. Funding for open access charge: Deutsche Forschungsgemeinschaft [RE2796/2-1 and RE2796/6-1].
Conflict of interest statement. None declared.
REFERENCES
- 1. Buxbaum A.R., Haimovich G., Singer R.H.. In the right place at the right time: visualizing and understanding mRNA localization. Nat. Rev. Mol. Cell Biol. 2015; 16:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buxbaum A.R., Yoon Y.J., Singer R.H., Park H.Y.. Single-molecule insights into mRNA dynamics in neurons. Trends Cell Biol. 2015; 25:468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pichon X., Bastide A., Safieddine A., Chouaib R., Samacoits A., Basyuk E., Peter M., Mueller F., Bertrand E.. Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J. Cell Biol. 2016; 214:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hövelmann F., Gaspar I., Ephrussi A., Seitz O.. Brightness enhanced DNA FIT-probes for wash-free RNA imaging in tissue. J. Am. Chem. Soc. 2013; 135:19025–19032. [DOI] [PubMed] [Google Scholar]
- 5. Hövelmann F., Seitz O.. DNA stains as surrogate nucleobases in fluorogenic hybridization probes. Acc. Res. Chem. 2016; 49:714–723. [DOI] [PubMed] [Google Scholar]
- 6. Politz J.C., Taneja K.L., Singer R.H.. Characterization of hybridization between synthetic oligodeoxynucleotides and RNA in living cells. Nucleic Acids Res. 1995; 23:4946–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Politz J.C., Browne E.S., Wolf D.E., Pederson T.. Intranuclear diffusion and hybridization state of oligonucleotides measured by fluorescence correlation spectroscopy in living cells. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:6043–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molenaar C., Abdulle A., Gena A., Tanke H.J., Dirks R.W.. Poly(A)+ RNAs roam the cell nucleus and pass through speckle domains in transcriptionally active and inactive cells. J. Cell Biol. 2004; 165:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tyagi S., Kramer F.R.. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 1996; 14:303–308. [DOI] [PubMed] [Google Scholar]
- 10. Mhlanga M.M., Vargas D.Y., Fung C.W., Kramer F.R., Tyagi S.. tRNA-linked molecular beacons for imaging mRNAs in the cytoplasm of living cells. Nucleic Acids Res. 2005; 33:1902–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Medley C.D., Drake T.J., Tomasini J.M., Rogers R.J., Tan W.. Simultaneous monitoring of the expression of multiple genes inside of single breast carcinoma cells. Anal. Chem. 2005; 77:4713–4718. [DOI] [PubMed] [Google Scholar]
- 12. Santangelo P.J., Nix B., Tsourkas A., Bao G.. Dual FRET molecular beacons for mRNA detection in living cells. Nucleic Acids Res. 2004; 32:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen A.K., Behlke M.A., Tsourkas A.. Efficient cytosolic delivery of molecular beacon conjugates and flow cytometric analysis of target RNA. Nucleic Acids Res. 2008; 36:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nitin N., Santangelo P.J., Kim G., Nie S., Bao G.. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res. 2004; 32:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rath A.K., Rentmeister A.. Genetically encoded tools for RNA imaging in living cells. Curr. Opin. Biotechnol. 2015; 31:42–49. [DOI] [PubMed] [Google Scholar]
- 16. Garcia J.F., Parker R.. MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2-MCP system. RNA. 2015; 21:1393–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tutucci E., Vera M., Biswas J., Garcia J., Parker R., Singer R.H.. An improved MS2 system for accurate reporting of the mRNA life cycle. Nat. Methods. 2018; 15:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao Y., Tatavarty V., Korza G., Levin M.K., Carson J.H.. Multiplexed dendritic targeting of alpha calcium calmodulin-dependent protein kinase II, neurogranin, and activity-regulated cytoskeleton-associated protein RNAs by the A2 pathway. Mol. Biol. Cell. 2008; 19:2311–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tübing F., Vendra G., Mikl M., Macchi P., Thomas S., Kiebler M.A.. Dendritically localized transcripts are sorted into distinct ribonucleoprotein particles that display fast directional motility along dendrites of hippocampal neurons. J. Neurosci. 2010; 30:4160–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilkie G.S., Davis I.. Drosophila wingless and pair-rule transcripts localize apically by dynein-mediated transport of RNA particles. Cell. 2001; 105:209–219. [DOI] [PubMed] [Google Scholar]
- 21. Holstein J.M., Rentmeister A.. Current covalent modification methods for detecting RNA in fixed and living cells. Methods. 2016; 98:18–25. [DOI] [PubMed] [Google Scholar]
- 22. Anhäuser L., Rentmeister A.. Enzyme-mediated tagging of RNA. Curr. Opin. Biotechnol. 2017; 48:69–76. [DOI] [PubMed] [Google Scholar]
- 23. Li F., Dong J., Hu X., Gong W., Li J., Shen J., Tian H., Wang J.. A covalent approach for site-specific RNA labeling in Mammalian cells. Angew. Chem. Int. Ed. Engl. 2015; 54:4597–4602. [DOI] [PubMed] [Google Scholar]
- 24. Alexander S.C., Busby K.N., Cole C.M., Zhou C.Y., Devaraj N.K.. Site-specific covalent labeling of RNA by enzymatic transglycosylation. J. Am. Chem. Soc. 2015; 137:12756–12759. [DOI] [PubMed] [Google Scholar]
- 25. Aakalu G., Smith W.B., Nguyen N., Jiang C., Schuman E.M.. Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron. 2001; 30:489–502. [DOI] [PubMed] [Google Scholar]
- 26. Shan J., Munro T.P., Barbarese E., Carson J.H., Smith R.. A molecular mechanism for mRNA trafficking in neuronal dendrites. J. Neurosci. 2003; 23:8859–8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dolgosheina E.V., Jeng S.C., Panchapakesan S.S., Cojocaru R., Chen P.S., Wilson P.D., Hawkins N., Wiggins P.A., Unrau P.J.. RNA mango aptamer-fluorophore: a bright, high-affinity complex for RNA labeling and tracking. ACS Chem. Biol. 2014; 9:2412–2420. [DOI] [PubMed] [Google Scholar]
- 28. Lionnet T., Czaplinski K., Darzacq X., Shav-Tal Y., Wells A.L., Chao J.A., Park H.Y., de Turris V., Lopez-Jones M., Singer R.H.. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat. Methods. 2011; 8:165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paige J.S., Wu K.Y., Jaffrey S.R.. RNA mimics of green fluorescent protein. Science. 2011; 333:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holstein J.M., Muttach F., Schiefelbein S.H.H., Rentmeister A.. Dual 5′ cap labeling based on regioselective RNA methyltransferases and bioorthogonal reactions. Chem. - Eur. J. 2016; 23:6165–6173. [DOI] [PubMed] [Google Scholar]
- 31. Holstein J.M., Stummer D., Rentmeister A.. Enzymatic modification of 5′-capped RNA with a 4-vinylbenzyl group provides a platform for photoclick and inverse electron-demand Diels-Alder reaction. Chem. Sci. 2015; 6:1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holstein J.M., Schulz D., Rentmeister A.. Bioorthogonal site-specific labeling of the 5′-cap structure in eukaryotic mRNAs. Chem. Commun. 2014; 50:4478–4481. [DOI] [PubMed] [Google Scholar]
- 33. Schulz D., Holstein J.M., Rentmeister A.. A chemo-enzymatic approach for site-specific modification of the RNA cap. Angew. Chem., Int. Ed. 2013; 52:7874–7878. [DOI] [PubMed] [Google Scholar]
- 34. Holstein J.M., Anhäuser L., Rentmeister A.. Modifying the 5′-cap for click reactions of eukaryotic mRNA and to tune translation efficiency in living cells. Angew. Chem. Int. Ed. 2016; 55:10899–10903. [DOI] [PubMed] [Google Scholar]
- 35. Muttach F., Mäsing F., Studer A., Rentmeister A.. New AdoMet analogues as tools for enzymatic transfer of photo-cross-linkers and capturing RNA-protein interactions. Chem. - Eur. J. 2017; 23:5988–5993. [DOI] [PubMed] [Google Scholar]
- 36. Mamot A., Sikorski P., Warminski M., Kowalska J., Jemielity J.. Azido-functionalized 5′-cap analogs for preparation of translationally active mRNAs suitable for fluorescent labeling in living cells. Angew. Chem. Int. Ed. 2017; 56:15628–15632. [DOI] [PubMed] [Google Scholar]
- 37. Karikó K., Muramatsu H., Welsh F.A., Ludwig J., Kato H., Akira S., Weissman D.. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008; 16:1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao B.S., Roundtree I.A., He C.. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017; 18:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wojtczak B.A., Sikorski P.J., Fac-Dabrowska K., Nowicka A., Warminski M., Kubacka D., Nowak E., Nowotny M., Kowalska J., Jemielity J.. 5′-Phosphorothiolate dinucleotide cap analogues: reagents for messenger RNA modification and potent small-molecular inhibitors of decapping enzymes. J. Am. Chem. Soc. 2018; 140:5987–5999. [DOI] [PubMed] [Google Scholar]
- 40. Tian B., Hu J., Zhang H., Lutz C.S.. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005; 33:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Winz M.L., Samanta A., Benzinger D., Jäschke A.. Site-specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-free strain-promoted click chemistry. Nucleic Acids Res. 2012; 40:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin G., Keller W.. Tailing and 3′-end labeling of RNA with yeast poly(A) polymerase and various nucleotides. RNA. 1998; 4:226–230. [PMC free article] [PubMed] [Google Scholar]
- 43. Stepinski J., Waddell C., Stolarski R., Darzynkiewicz E., Rhoads R.E.. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3′-O-methyl)GpppG and 7-methyl (3′-deoxy)GpppG. RNA. 2001; 7:1486–1495. [PMC free article] [PubMed] [Google Scholar]
- 44. Karikó K., Muramatsu H., Ludwig J., Weissman D.. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011; 39:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beatty K.E., Fisk J.D., Smart B.P., Lu Y.Y., Szychowski J., Hangauer M.J., Baskin J.M., Bertozzi C.R., Tirrell D.A.. Live-cell imaging of cellular proteins by a strain-promoted azide-alkyne cycloaddition. ChemBioChem. 2010; 11:2092–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim E.J., Kang D.W., Leucke H.F., Bond M.R., Ghosh S., Love D.C., Ahn J.S., Kang D.O., Hanover J.A.. Optimizing the selectivity of DIFO-based reagents for intracellular bioorthogonal applications. Carbohydr. Res. 2013; 377:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuhn A.N., Beißert T., Simon P., Vallazza B., Buck J., Davies B.P., Tureci O., Sahin U.. mRNA as a versatile tool for exogenous protein expression. Curr. Gene Ther. 2012; 12:347–361. [DOI] [PubMed] [Google Scholar]
- 48. Jackson R.J., Hellen C.U., Pestova T.V.. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010; 11:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weill L., Belloc E., Bava F.A., Méndez R.. Translational control by changes in poly(A) tail length: recycling mRNAs. Nat. Struct. Mol. Biol. 2012; 19:577–585. [DOI] [PubMed] [Google Scholar]
- 50. Parker R., Song H.. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 2004; 11:121–127. [DOI] [PubMed] [Google Scholar]
- 51. Garneau N.L., Wilusz J., Wilusz C.J.. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007; 8:113–126. [DOI] [PubMed] [Google Scholar]
- 52. Goldstrohm A.C., Wickens M.. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008; 9:337–344. [DOI] [PubMed] [Google Scholar]
- 53. Eckmann C.R., Rammelt C., Wahle E.. Control of poly(A) tail length. Wiley Interdiscip. Rev.: RNA. 2011; 2:348–361. [DOI] [PubMed] [Google Scholar]
- 54. Milian S., Satish L., Kesh S., Chaudhary Y.S., Sahoo H.. Interaction of lysozyme with rhodamine B: a combined analysis of spectroscopic & molecular docking. J. Photochem. Photobiol. B. 2016; 162:248–257. [DOI] [PubMed] [Google Scholar]
- 55. Cai H.-H., Zhong X., Yang P.-H., Wei W., Chen J., Cai J.. Probing site-selective binding of rhodamine B to bovine serum albumin. Colloids Surf. A. 2010; 372:35–40. [Google Scholar]
- 56. Bartasun P., Cieśliński H., Bujacz A., Wierzbicka-Woś A., Kur J.. A study on the interaction of rhodamine B with methylthioadenosine phosphorylase protein sourced from an antarctic soil metagenomic library. PLoS One. 2013; 8:e55697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen M., Ma Z., Wu X., Mao S., Yang Y., Tan J., Krueger C.J., Chen A.K.. A molecular beacon-based approach for live-cell imaging of RNA transcripts with minimal target engineering at the single-molecule level. Sci. Rep. 2017; 7:1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sunbul M., Jäschke A.. Contact-mediated quenching for RNA imaging in bacteria with a fluorophore-binding aptamer. Angew. Chem. Int. Ed. 2013; 52:13401–13404. [DOI] [PubMed] [Google Scholar]
- 59. Arora A., Sunbul M., Jäschke A.. Dual-colour imaging of RNAs using quencher- and fluorophore-binding aptamers. Nucleic Acids Res. 2015; 43:e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Friscourt F., Fahrni C.J., Boons G.J.. A fluorogenic probe for the catalyst-free detection of azide-tagged molecules. J. Am. Chem. Soc. 2012; 134:18809–18815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Muttach F., Muthmann N., Reichert D., Anhäuser L., Rentmeister A.. A benzylic linker promotes methyltransferase catalyzed norbornene transfer for rapid bioorthogonal tetrazine ligation. Chem. Sci. 2017; 8:7947–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kirschman J.L., Bhosle S., Vanover D., Blanchard E.L., Loomis K.H., Zurla C., Murray K., Lam B.C., Santangelo P.J.. Characterizing exogenous mRNA delivery, trafficking, cytoplasmic release and RNA-protein correlations at the level of single cells. Nucleic Acids Res. 2017; 45:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gilmour D., Jessen J.R., Lin S.. Manipulating Gene Expression in the Zebrafish. Zebrafish - A Practical Approach. 2002; 261:Oxford: University Press; 121–143. [Google Scholar]
- 64. Hruscha A., Krawitz P., Rechenberg A., Heinrich V., Hecht J., Haass C., Schmid B.. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development. 2013; 140:4982–4987. [DOI] [PubMed] [Google Scholar]
- 65. Chandrasekaran K.S., Rentmeister A.. Clicking a fish: click chemistry of different biomolecules in Danio rerio. Biochemistry. 2018; 58:24–30. [DOI] [PubMed] [Google Scholar]
- 66. Cha B.-J., Koppetsch B.S., Theurkauf W.E.. In vivo analysis of Drosophila bicoid mRNA localization reveals a novel microtubule-dependent axis specification pathway. Cell. 2001; 106:35–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.