

Abstract
N 6-methyladenosine (m6A) is the most prevalent post-transcriptional modification in eukaryotes, and plays a pivotal role in various biological processes, such as splicing, RNA degradation and RNA–protein interaction. We report here a prediction framework WHISTLE for transcriptome-wide m6A RNA-methylation site prediction. When tested on six independent datasets, our approach, which integrated 35 additional genomic features besides the conventional sequence features, achieved a major improvement in the accuracy of m6A site prediction (average AUC: 0.948 and 0.880 under the full transcript or mature messenger RNA models, respectively) compared to the state-of-the-art computational approaches MethyRNA (AUC: 0.790 and 0.732) and SRAMP (AUC: 0.761 and 0.706). It also out-performed the existing epitranscriptome databases MeT-DB (AUC: 0.798 and 0.744) and RMBase (AUC: 0.786 and 0.736), which were built upon hundreds of epitranscriptome high-throughput sequencing samples. To probe the putative biological processes impacted by changes in an individual m6A site, a network-based approach was implemented according to the ‘guilt-by-association’ principle by integrating RNA methylation profiles, gene expression profiles and protein–protein interaction data. Finally, the WHISTLE web server was built to facilitate the query of our high-accuracy map of the human m6A epitranscriptome, and the server is freely available at: www.xjtlu.edu.cn/biologicalsciences/whistle and http://whistle-epitranscriptome.com.
INTRODUCTION
Large scale analysis has revealed the abundance of RNA modifications in the human epitranscriptome (1). With the recent advances in the exploration of RNA epigenetics, more than 150 types of RNA modifications have been identified (2). Among them, the most prevalent non-cap modification marker present on eukaryotic messenger RNA (mRNA) and long non-coding RNA, N6-methyladenosine (m6A), (3) has emerged as an abundant and dynamically regulated modification (4). m6A was detected within poly-A RNA for the first time in 1974 (5), and has since been characterized in various eukaryotic species. In the past five decades, various studies have demonstrated the biological significance of m6A RNA methylation, which includes roles in the circadian clock (6), regulation of mRNA translation (7), heat shock response (8), microRNA (miRNA) processing (9), DNA damage response (10), RNA–protein interaction (11) and regulation of RNA stability (12). Consequently, the accurate identification of m6A locations is critical for the study and understanding of the downstream effects of RNA modification in biology.
To identify the precise location of m6A sites on mRNA, the first whole-transcriptome m6A profiling technique m6A-seq (or MeRIP-seq) was introduced in 2012 (13,14), in which the m6A containing RNA fragments is immunoprecipitated, purified and then subjected to further analysis. This technique applies high-throughput sequencing to the IP sample enriched with m6A-containing mRNA fragments. In contrast to the input control samples, it typically results in the detection of m6A containing peaks with around 100 nt resolution using MACS, the exomePeak R/Bioconductor Package and other peak callers (15,16). The precise location of the m6A sites may be further narrowed down to base-resolution by searching for the m6A motif RRACH within the peaks detected with m6A signal. Most existing epitranscriptome databases, such as, MeT-DB and RMBase, rely on this very simple strategy (17,18). A major limitation of this method is that it cannot differentiate between a randomly-occurring RRACH motif and a real m6A-containing motif located nearby, i.e. all the RRACH motifs located within an m6A peak will be reported as holding an m6A site, including the chance occurrences, resulting in false positive predictions. Since m6A-seq is currently the most widely used approach for profiling the transcriptome-wide m6A, and a very large number of the m6A-seq samples have been acquired in different studies, the m6A site information extracted from the m6A-seq using the motif search strategy essentially dominates the existing epitranscriptome databases. For this reason, it is not surprising that both MeT-DB (426 544 sites) and RMBase (477 452 sites) report a very large number of transcriptome m6A sites, many of which may be false positive due to a chance RRACH motif located close to a real m6A site (or within an m6A peak).
Besides the m6A-seq technique, single-based resolution techniques such as the miCLIP (19) and m6A-CLIP (20) were also developed. However, these experiments are usually more laborious to perform but still offer limited coverage of the m6A epitranscriptome, since the reported RNA-methylation sites are still restricted to the transcripts more readily expressed under a specific cell/tissue condition. Although base-resolution profiling techniques have not been very widely applied in biological studies due to their expense and difficulties, they provide the ground truth of m6A site information that is necessary for computational prediction. To date, a large number of RNA-methylation site prediction methods and web servers have been developed based on the information extracted from base-resolution techniques since 2015, including the pseudo nucleotide composition based approach iRNA-Methyl by Chen et al. (21) and physical-chemical properties-based approach pRNAm-PC (22). Subsequently, Zhou et al. employed SRAMP, a random forest machine learning framework, to predict mammalian m6A sites using sequence features (23). Many other site predictors have been developed for m6A and other RNA modification, such as MethyRNA (24), RNAMethPre (25), RAM-NPPS (26), Target M6A (27), AthMethPre (28), iRNA-PseColl (29), M6APred-EL (30), iMethyl-STTNC (31), iRNA-PseDNC (32), etc. (33–39). These methods have been recently reviewed (40). These site predictors usually take the transcript sequence as the input and report a number of possible m6A sites as the output, making them very convenient to use. However, to our knowledge, they are exclusively based on the sequence-derived information—even when the secondary structure or other high level features (41) are used, the information is still directly extracted from sequence without considering other potentially useful genomic features, such as, conservation, transcript type and gene annotation. Although the sequence information probably plays a central role, other genomic features may also be helpful in the prediction of m6A sites and thus should be incorporated in the analysis. Additionally, although potentially feasible, none of these approaches have been applied transcriptome-wide to reconstruct the entire m6A epitranscriptome, thus limiting their usage in large-scale or high-throughput analysis.
In this project, we proposed a prediction framework, WHISTLE, which stands for whole-transcriptome m6A site prediction from multiple genomic features. The framework extracted a comprehensive set of domain knowledge based on various genomic features, and integrated them with conventional sequence-derived features for reconstructing a high-accuracy map of the m6A epitranscriptome. The ‘guilt-by-association’ principle was then applied to further annotate the functional relevance of each individual RNA-methylation site by integrating gene expression profiles, RNA methylation profiles and PPI networks.
MATERIALS AND METHODS
Training and testing data for m6A site prediction
The data used for training and benchmarking in m6A site prediction includes six single-base resolution m6A experiment obtained from five cell types (see Table 1). The base-resolution m6A sites in each experiment were downloaded directly from Gene Expression Omnibus (GEO). The two samples (MOLM13 mi-CLIP sample and the A549 m6A-CLIP) reported based on the human genome assembly hg18 were lifted using UCSC liftOver tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). A total of 20 516 and 17 383 m6A sites out of the original 23 480 and 19 683 sites were lifted to hg19, respectively. Both samples have very large number of (>17000) positive sites that can be used for training and testing after liftOver, and the majority (four out of six) base-resolution samples are based on hg19 and thus do not require extra processing step.
Table 1.
Base-resolution dataset used in m6A site prediction
In the beginning of the performance evaluation procedure, dataset 6 of the base-resolution data (Table 1) was used as the independent testing data, while the other five datasets were used as the training data. The positive training data (m6A sites) was determined as the m6A sites under RRACH consensus motifs that have been reproduced in at least two of the five training datasets. The negative training data (non-m6A sites) was randomly selected from the non-positive RRACH adenosines on the full transcripts containing the positive sites (see Figure 1). Initially, the number of randomly selected negative sites was ten times the number of positive sites. Later, the positive-negative ratio was balanced by randomly splitting the negative samples into 10 random subsets. Consequently, 10 training datasets, each with 1:1 positive-to-negative ratio, were constructed using different negative samples. The negative data was also generated similarly on testing data (Dataset 6), i.e. the negative data were randomly selected non-positive m6A sites from the m6A containing transcripts. The ratio of positive testing data to negative testing data was also kept as 1:10. The testing performances from the 10 independent sessions were averaged.
Figure 1.

Generation of positive and negative data. The transcriptome m6A sites under RRACH consensus motifs that have been reproduced in at least two of the five training datasets were used as positive m6A sites. The negative training data (non-m6A sites) was randomly selected from the non-positive RRACH adenosines on the full transcripts containing the positive sites.
To exclude randomness of testing dataset from the m6A site prediction evaluation, we also applied dataset level leave-one out validation over the six base-resolution datasets. In each round of the dataset level validation, one of the six base-resolution datasets was used as the independent testing data, while the remaining five datasets were used as the training data. The same rules of the training and testing data generation were applied as previously described in each individual test. As the training and testing data were all extracted from different independent experiments, there should be no overfitting problem.
Features for m6A site prediction
Sequence-derived features
The sequence-based information around the RRACH motif was encoded using the same method of m6Apred (44) and MethyRNA (24), which have been shown to be quite effective and achieved good performance in human and yeast m6A site prediction. The sequence feature encodes the nucleotides sequence by three distinct structural chemical properties: ring structures, functional groups and hydrogen bonds. Specifically, adenine and guanine have two ring structures, while cytosine and uracil have only one ring; adenine and cytosine contain the amino group, while guanine and uracil contain the keto group; adenine and uracil can form two hydrogen bonds during hybridization, whereas guanine and cytosine can form three hydrogen bonds. Based on the three structural chemical properties defined above, the i-th nucleotide from sequence S can be encoded by a vector 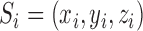 :
:
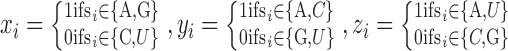 |
(1) |
Therefore, the A, C, G, U can be encoded as a vector of three features (1,1,1), (0,1,0), (1,0,0) and (0,0,1), respectively. Additionally, a feature of the cumulative nucleotide frequency is calculated for each nucleotide position in the sequence. The density of the i-th nucleotide 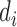 is defined as the sum of all the instances of the i-th nucleotide before the
is defined as the sum of all the instances of the i-th nucleotide before the 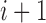 position. The nucleotide frequency
position. The nucleotide frequency 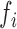 is defined by the following formula:
is defined by the following formula: 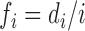 . Using the sequence ‘AUGGACACU’ as an example, the cumulative frequency for adenine is 1.00 (1/1), 0.40 (2/5) and 0.43(3/7) at the first, fifth and seven position, respectively; while the frequency for uracil is 0.50 (1/2) and 0.11 (1/9) at the second and ninth respective position.
. Using the sequence ‘AUGGACACU’ as an example, the cumulative frequency for adenine is 1.00 (1/1), 0.40 (2/5) and 0.43(3/7) at the first, fifth and seven position, respectively; while the frequency for uracil is 0.50 (1/2) and 0.11 (1/9) at the second and ninth respective position.
Genome-derived features
Most existing RNA modification site prediction algorithms use exclusively sequence-based features; however, such features alone may not fully capture the attributes of RNA modification topology. Hence, we generated 35 additional genomic features that may contribute to the prediction. Genomic Features 1–13 are dummy variable features indicating whether the adenosine sites shall fall within the transcript regions that satisfy certain topological properties. All the features in this category are generated by the GenomicFeatures R/Bioconductor package (45) using the transcript annotations hg19 TxDb package. To remove the ambiguity caused by transcript isoforms, only the primary (longest) transcripts of each gene were kept for the extraction of the transcript sub-regions. Genomic Features 14–16 are real valued features defining the relative position of the transcript regions (3′UTR, 5′UTR and whole transcript), i.e. the distance from the adenine to the 5′ end divided by the width of the region. The values are also set to zero for sites that do not belong to the region. Genomic features 17–19 represent the length of the transcript region containing the modification site. The values are also set to zero for sites that not belong to the region. Features 20–22 capture the distance from the adenine sites to the 5′end or 3′end of the splicing junctions. Additionally, the distance to the nearest neighboring m6A sites in the training data is generated to measure the clustering effect of the m6A RNA modification sites. Features 23–26 represent the evolutionary conservation score of the adenosine sites and its flanking regions; two metrics of nucleotide conservation, Phast-Cons score (46) and the fitness consequence scores are used to measure the conservation level of the underlying nucleotide sequence. Features 27 and 28 represent the RNA secondary structures around the adenine site, the RNA secondary structures are predicted using RNAfold from the Vienna RNA package (47). Finally, features 29–35 are the properties of the genes or transcripts containing the m6A sites, such as being the miRNA target genes or housekeeping genes. The annotation of miRNA target sites are from miRanda (48) and TargetScan (49). Supplementary Table S1 contains more details about the genomic features we considered in the prediction.
Machine learning approach used for m6A site prediction
The Support Vector Machine (SVM) is one of the most widely used machine learning algorithms in computational biology. It was previously used for mammalian miRNA target prediction (50), protein kinase-specific phosphorylation sites prediction (51) and mammalian m6A modification sites prediction (24,25). In this project, we used an R language interface of LIBSVM (52) to construct the SVM-based m6A site predictors. Following previous approaches (21,22), the radial basis function was chosen as the kernel function, and the other parameters were set at the default. Random Forest is another popular machine learning algorithm applied in biology data, and one of the earliest mammalian m6A site predictor SRAMP was developed based on the Random Forest approach (23). In this project, we also use Random Forest from the R package randomforest (53) to compare the predictive performance using SVM.
Performance evaluation of m6A site prediction
For both the SVM and random forest classifiers, a 5-fold cross-validation was employed on the training datasets for model selection purpose, and the final performance of the predictor was measured on the independent testing dataset. The receiver operating characteristic curve (sensitivity against 1-specificity) was used to measure the prediction performance under different decision thresholds, and the area under the curve (AUC) was calculated as the main performance evaluation metric.
When evaluating the accuracy of m6A site information stored in existing epitranscriptome m6A site databases MeT-DB Version 2 and RMBase Version 2, the reliability was determined by the number of experiments that support the existence of a specific m6A site, based on which the AUC can be calculated. In addition, the sensitivity (Sn), specificity (Sp) and Matthews correlation coefficient (MCC) were calculated to measure the performance of predictor:
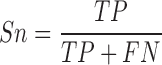 |
(2) |
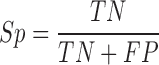 |
(3) |
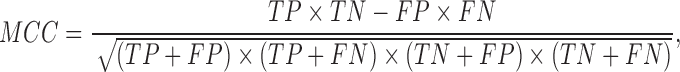 |
(4) |
where, TP, TN, FP and FN represent true positive, true negative, false positive and false negative, respectively. When different methods were compared under AUC, they always use the same positive and negative gold standard dataset, and AUCs were always calculated in the same way. The AUCs of different methods reported in our manuscript are therefore strictly comparable.
Estimate the posterior probability of RNA methylation
The existing machine learning approaches usually report the probability of an m6A motif to be an actual methylation site under the assumption of equal prior probability, i.e. the prior probability of an m6A motif being an m6A site is 0.5. However, it is known in practice that the number of m6A sites is a lot smaller than the number of m6A motifs, so the number of RNA-methylation sites under a specific experimental condition is likely to be significantly over-estimated. To address this bias, a posterior probability of RNA methylation under a specific condition is calculated with: 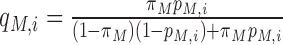 , where,
, where, 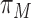 is the prior probability that a transcriptome RRACH motif embraces a true m6A site under a specific model, which is calculated empirically from the 6 base-resolution datasets (see Table 1) as the average number of m6A sites under a condition divided by the number of occurrences of transcriptome RRACH motifs that are supported by at least one m6A record in MeTDB for the mature mRNA model, or RMBase for the full transcript model. These is also the search space of our predicted m6A epitranscriptome.
is the prior probability that a transcriptome RRACH motif embraces a true m6A site under a specific model, which is calculated empirically from the 6 base-resolution datasets (see Table 1) as the average number of m6A sites under a condition divided by the number of occurrences of transcriptome RRACH motifs that are supported by at least one m6A record in MeTDB for the mature mRNA model, or RMBase for the full transcript model. These is also the search space of our predicted m6A epitranscriptome. 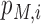 is the predicted probability (or likelihood) of the i-th site being a real m6A site under a specific model M, and
is the predicted probability (or likelihood) of the i-th site being a real m6A site under a specific model M, and 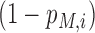 is the probability of the opposite being true.
is the probability of the opposite being true. 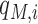 is a posteriori probability of the i-th site being a real m6A site under a specific condition. The posterior probability
is a posteriori probability of the i-th site being a real m6A site under a specific condition. The posterior probability 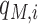 is also reported in the WHISTLE database along with the probability
is also reported in the WHISTLE database along with the probability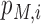 .
.
RESULTS AND DISCUSSION
m6A site prediction
The predictors on the full-transcript data were established first in which the true m6A site and negative sites may be located in both exonic and intronic regions. Because experimental procedures, especially the polyA selection step, may induce bias toward mRNA, we also consider a mature mRNA model, under which, the goal is to predict only exonic m6A sites, and thus only the exonic regions are considered.
We show in Supplementary Table S3 that, although the genome-derived features alone are already very effective for predicting m6A sites, the best performance is achieved when the sequence features and genomic features are combined. Consequently, our m6A site predictor was established based on both the genome-derived features and sequence-derived features.
Feature selection was performed to identify the most effective genomic features for m6A site prediction. Here, datasets 1–5 were used as the training data, while dataset 6 was used as the independent testing data. The relative importance of each genome-derived feature in the prediction was firstly assessed with the Perturb method (54) using the R caret package. Next, the N most important features were retained in the prediction analysis, and the prediction performance was evaluated using a 5-fold cross-validation. As shown in Supplementary Figure S1A, the predictor performance under the full transcript model stops increasing after including the top 14 most important genomic features. The top three most critical genomic features under this model are long exon, miRNA target and conservation score. To achieve the most robust performance and to avoid potential overfitting, only the top 14 genomic features were used in the full transcript model for m6A site prediction purpose in later analysis. Similarly, the top 19 genome-derived features with the highest importance were selected for the mature mRNA model (see Supplementary Figure S1B). The distance to known m6A sites became the most important predictive feature, which demonstrated the clustering effect of m6A modification, followed by long exon and conservation under the mature mRNA model.
The performance of the proposed m6A predictors was then evaluated using independent datasets and compared with competing approaches (Table 2). By combining additional genome-derived features, the performance of our approach was substantially higher in all the tested conditions than MethyRNA and SRAMP, which rely only on information extracted from sequences. WHISTLE achieved AUCs of 0.948 and 0.880 under the full transcript and mature mRNA modes, respectively, representing a major improvement compared to MethyRNA (0.790 and 0.732) and SRAMP (0.761 and 0.706).
Table 2.
Performance evaluation of m6A site prediction methods
| Performance on independent dataset (AUC) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Model | Method | A549 | CD8T | Hela | HEK293 (sysy) | HEK293 (abacm) | MOLM13 | Average AUC |
| Full Transcript | WHISTLE | 0.965 | 0.930 | 0.953 | 0.936 | 0.968 | 0.933 | 0.948 |
| MethyRNA* | 0.807 | 0.800 | 0.741 | 0.848 | 0.778 | 0.765 | 0.790 | |
| SRAMP | 0.856 | 0.841 | 0.762 | 0.883 | 0.838 | 0.759 | 0.761# | |
| Mature mRNA | WHISTLE | 0.903 | 0.904 | 0.894 | 0.936 | 0.818 | 0.823 | 0.880 |
| MethyRNA | 0.751 | 0.734 | 0.676 | 0.848 | 0.698 | 0.686 | 0.732 | |
| SRAMP | 0.814 | 0.796 | 0.702 | 0.869 | 0.796 | 0.710 | 0.706# | |
Note: *The MethyRNA approach uses sequence-derived features with SVM (24), which we reproduced faithfully with the same training data of WHISTLE for comparison.
#The SRAMP method was originally trained on A549, CD8T, HEK293 (sysy) and HEK293 (abacm). To avoid overfitting, only Hela and MOLM13 were considered when evaluating its average performance.
Only the m6A sites not previously used as training data were considered during performance evaluation, so the training sites and testing sites have no overlap. Please see Supplementary Table S4 for the results when all sites from the independent testing samples were considered.
A predicted map of human m6A epitranscriptome
With the extensive study of RNA epigenetics, especially the accumulation of large number of m6A-seq datasets, the transcriptome-wide distribution of m6A sites have been summarized and made available from bioinformatics databases, such as MeT-DB (55) and RMBase (56). MeT-DB is the first transcriptome m6A database that provides condition-specific distribution of m6A RNA methylation in human and mouse initially, and later in other species as well; while RMBase is a more comprehensive RNA modification database, supporting more species and more RNA modification types. However, as these two databases overwhelmingly rely on m6A-seq data, and implemented a data processing pipeline that could not differentiate between true and randomly-occurring m6A motif located in close proximity within an m6A peak, the information they provide may not be accurate and should be re-assessed.
The reliability of a specific m6A site in epitranscriptome databases has been measured by the number of experiments that support the record. This metric will be used when evaluating the accuracy of the two databases. Interestingly, as shown in Table 3, when comparing the two epitranscriptome databases, the exomePeak-based MeT-DB database is slightly more accurate than primarily the MACS-based RMBase database. However, even with hundreds of high-throughput sequencing datasets accumulated, existing epitranscriptome databases are still far less accurate than what we may achieve with machine learning approaches (see Table 2).
Table 3.
Performance evaluation for bioinformatics databases
| Group truth dataset used (AUC) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mode | Method | A549 | CD8T | Hela | HEK293 (sysy) | HEK293 (abacm) | MOLM13 | Average AUC |
| Full Transcript | RMBase | 0.825 | 0.788 | 0.832 | 0.837 | 0.701 | 0.733 | 0.786 |
| MetDB | 0.835 | 0.802 | 0.843 | 0.848 | 0.719 | 0.744 | 0.798 | |
| Mature mRNA | RMBase | 0.768 | 0.716 | 0.790 | 0.752 | 0.707 | 0.682 | 0.736 |
| MetDB | 0.775 | 0.730 | 0.795 | 0.762 | 0.716 | 0.683 | 0.744 | |
Note: To ensure the results are comparable to Table 2, only the unique sites not previously reported in the training data of predictors were considered. Please see Supplementary Table S4 for the results when all sites from the independent testing samples were considered.
We thus performed a whole transcriptome prediction of m6A RNA-methylation sites in human to generate a map of human m6A epitranscriptome using our proposed WHISTLE approach. Our predicted map is of substantially higher accuracy (average AUC of 0.948 and 0.880) compared with existing epitranscriptome databases MeT-DB (average AUC of 0.798 and 0.744) and RMBase (average AUC of 0.786 and 0.736) when evaluated on independent base-resolution datasets under both full transcript and mature mRNA mode, respectively. Additionally, we calculated a posterior probability of RNA-methylation site under a specific experimental condition. This provided a more empirical evaluation of the methylation status by taking into consideration the prior probability of an m6A motif being an m6A site, which is estimated from the base-resolution datasets.
Besides CLIP-based approaches, we also tested the accuracy of the proposed method on a high resolution m6A-seq dataset (57). Although still antibody-based, this m6A-seq dataset was generated from an improved protocol and achieved near base resolution (58). As shown in Table 4, when antibody-based m6A-seq technique is used as the ground truth, WHISTLE still substantially outperformed competing approaches under both the full transcript and mature mRNA models.
Table 4.
Performance assessment using high resolution m6A-seq data
| AUC under full transcript model | AUC under mature mRNA model | |
|---|---|---|
| WHISTLE | 0.980 | 0.904 |
| MethyRNA | 0.904 | 0.826 |
| SRAMP | 0.825 | 0.783 |
| RMBase | 0.774 | 0.758 |
| MeTDB | 0.775 | 0.767 |
Note: The high confidence consensus m6A sites detected in more than two of the total six high resolution m6A-seq experiments (57) were considered. Similar as before, MethyRNA and WHISTLE used the same m6A datasets for training. Only the unique sites not previously reported in the training data were considered here. Please see Supplementary Table S5 for the results when all sites from the independent testing samples were considered.
Website interface
An online database has been built to host the predicted human m6A epitranscriptome. The individual RNA-methylation sites were then functionally annotated with gene expression data, RNA methylation data and protein–protein interaction data according to the ‘guilt-by-association’ principle (detailed in the Supplementary File S2). As is shown in Figure 2, The website supports queries that may be a methylation site, a gene or a specific biological function under the Gene Ontology framework (59). It also supports the download of the original base-resolution datasets (Table 1) used for site prediction and the entire predicted epitranscriptome map with the functional annotations for large-scale analysis.
Figure 2.

WHISTLE website. The WHISTLE website hosts a functionally annotated high-accuracy predicted map of the human m6A epitranscriptome. The WHISTLE website supports direct query of RNA-methylation sites with respect to a specific GO function or gene. The m6A RNA-methylation sites were predicted from m6A-CLIP data, miCLIP data, sequence features and genome-derived features. And then, the most dynamic RNA-methylation sites were annotated under the Gene Ontology framework using the guilt-by-association principle by integrating gene expression, RNA methylation and protein–protein interaction data. Please Supplementary Figure S2 for the complete data processing pipeline of WHISTLE.
CONCLUSIONS
Along with recent advances in RNA epigenetics, especially, the development of new techniques for profiling the RNA methylome (60,61), computationally deciphering the epitranscriptome from various omic data presents a major challenge to the bioinformatics community. In the past few years, sequence-derived features have been widely used for the prediction of RNA modification sites in human (24), mouse (24), other mammals (23,25), yeast (30,62) and other species; and a few major bioinformatics databases, including MeT-DB (18), RMBase (17), m6AVar (63), MODOMICS (64) and RNAMDB (65) have been built. These databases address various aspects of the RNA modifications including transcriptome-wide distribution, mechanism pathway, relevance to miRNA and RNA-binding proteins, functional variants, etc., and have greatly benefited researchers in this field.
Here, we constructed a functionally annotated high-accuracy predicted map of human m6A epitranscriptome and named it WHISTLE. The most stringent validation strategy was implemented, in which the performance of WHISTLE was assessed on six independent datasets (Tables 2 and 3) and on dataset generated from a different technique (Table 4). By integrating 35 genome-derived features with the conventional sequence-derived features, WHISTLE achieved a substantial improvement in accuracy, under both the full transcript model and the mature mRNA model, compared with existing machine learning-based m6A predictors and the latest epitranscriptome databases.
It is worth noting that, the prediction performance achieved on the full transcript model (AUC: 0.948) may be significantly over-estimated due to the library preparation (polyA selection) of the miCLIP and m6A-CLIP samples used, because they cannot effectively capture the intronic m6A sites. The performance achieved on the mature mRNA model (AUC: 0.880) is probably a more realistic estimate.
A web server WHISTLE was built to enable the direct query of predicted RNA-methylation sites, their putative functions and their potential association to other methylation sites or genes, which provides the requisite data for the further epitranscriptome studies in human.
Our work has provided a computational scheme to study the m6A epitranscriptome based on multi-omics datasets using machine learning and network-based method. In the future, it can be easily expanded to the study of other RNA modifications, such as m1A (66) and Pseudouridine (67), as well as in other species, such as mouse and yeast.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: J.M., R.R., Z.L. and J.P.M. conceived the idea and designed the research; Z.W. constructed the genomic features considered in m6A site prediction and processed the raw data; K.C. performed the m6A site prediction; Q.Z. and X.W. performed the network-based functional annotation of individual m6A sites; K.C. built the website; K.C., Q.Z. and W.Z. drafted the manuscript. All authors read, critically revised and approved the final manuscript. We thank Zoya Farooq at University of Liverpool for her assistance in website building.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31671373]; Jiangsu University Natural Science Program [16KJB180027]; XJTLU Key Programme Special Fund [KSF-T-01]; Jiangsu Six Talent Peak Program [XYDXX-118].
Conflict of interest statement. None declared.
REFERENCES
- 1. Roundtree I.A., Evans M.E., Pan T., He C.. Dynamic RNA modifications in gene expression regulation. Cell. 2017; 169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boccaletto P., Machnicka M.A., Purta E., Piatkowski P., Baginski B., Wirecki T.K., de Crecy-Lagard V., Ross R., Limbach P.A., Kotter A. et al.. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018; 46:D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meyer K.D., Jaffrey S.R.. Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol. 2017; 33:319–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niu Y., Zhao X., Wu Y.S., Li M.M., Wang X.J., Yang Y.G.. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013; 11:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desrosiers R., Friderici K., Rottman F.. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. U.S.A. 1974; 71:3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fustin J.M., Doi M., Yamaguchi Y., Hida H., Nishimura S., Yoshida M., Isagawa T., Morioka M.S., Kakeya H., Manabe I. et al.. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013; 155:793–806. [DOI] [PubMed] [Google Scholar]
- 7. Meyer K.D., Jaffrey S.R.. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014; 15:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou J., Wan J., Gao X., Zhang X., Jaffrey S.R., Qian S.B.. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015; 526:591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alarcon C.R., Lee H., Goodarzi H., Halberg N., Tavazoie S.F.. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015; 519:482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xiang Y., Laurent B., Hsu C.H., Nachtergaele S., Lu Z., Sheng W., Xu C., Chen H., Ouyang J., Wang S. et al.. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017; 543:573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T.. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015; 518:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. et al.. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014; 505:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. et al.. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012; 485:201–206. [DOI] [PubMed] [Google Scholar]
- 14. Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R.. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012; 149:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meng J., Lu Z., Liu H., Zhang L., Zhang S., Chen Y., Rao M.K., Huang Y.. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods. 2014; 69:274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dominissini D., Moshitch-Moshkovitz S., Salmon-Divon M., Amariglio N., Rechavi G.. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013; 8:176–189. [DOI] [PubMed] [Google Scholar]
- 17. Xuan J.-J., Sun W.-J., Lin P.-H., Zhou K.-R., Liu S., Zheng L.-L., Qu L.-H., Yang J.-H.. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018; 46:D327–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu H., Wang H., Wei Z., Zhang S., Hua G., Zhang S.-W., Zhang L., Gao S.-J., Meng J., Chen X. et al.. MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018; 46:D281–D287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Linder B., Grozhik A.V., Olarerin-George A.O., Meydan C., Mason C.E., Jaffrey S.R.. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods. 2015; 12:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ke S., Alemu E.A., Mertens C., Gantman E.C., Fak J.J., Mele A., Haripal B., Zucker-Scharff I., Moore M.J., Park C.Y. et al.. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015; 29:2037–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen W., Feng P., Ding H., Lin H., Chou K.-C.. iRNA-Methyl: identifying N 6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015; 490:26–33. [DOI] [PubMed] [Google Scholar]
- 22. Liu Z., Xiao X., Yu D.-J., Jia J., Qiu W.-R., Chou K.-C.. pRNAm-PC: predicting N 6-methyladenosine sites in RNA sequences via physical-chemical properties. Anal. Biochem. 2016; 497:60–67. [DOI] [PubMed] [Google Scholar]
- 23. Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q.. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016; 44:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen W., Tang H., Lin H.. MethyRNA: a web server for identification of N(6)-methyladenosine sites. J. Biomol. Struct. Dyn. 2017; 35:683–687. [DOI] [PubMed] [Google Scholar]
- 25. Xiang S., Liu K., Yan Z., Zhang Y., Sun Z.. RNAMethPre: a web server for the prediction and query of mRNA m6A sites. PLoS One. 2016; 11:e0162707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xing P., Su R., Guo F., Wei L.. Identifying N(6)-methyladenosine sites using multi-interval nucleotide pair position specificity and support vector machine. Sci. Rep. 2017; 7:46757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li G.Q., Liu Z., Shen H.B., Yu D.J.. TargetM6A: identifying N6-methyladenosine sites from RNA sequences via position-specific nucleotide propensities and a support vector machine. IEEE Trans. Nanobioscience. 2016; 15:674–682. [DOI] [PubMed] [Google Scholar]
- 28. Xiang S., Yan Z., Liu K., Zhang Y., Sun Z.. AthMethPre: a web server for the prediction and query of mRNA m6A sites in Arabidopsis thaliana. Mol. Biosyst. 2016; 12:3333–3337. [DOI] [PubMed] [Google Scholar]
- 29. Feng P., Ding H., Yang H., Chen W., Lin H., Chou K.-C.. iRNA-PseColl: identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Mol. Ther. Nucleic Acids. 2017; 7:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei L., Chen H., Su R.. M6APred-EL: a sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Mol. Ther. Nucleic Acids. 2018; 12:635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akbar S., Hayat M.. iMethyl-STTNC: identification of N6-methyladenosine sites by extending the Idea of SAAC into Chou's PseAAC to formulate RNA sequences. J. Theor. Biol. 2018; 455:205–211. [DOI] [PubMed] [Google Scholar]
- 32. Chen W., Ding H., Zhou X., Lin H., Chou K.C.. iRNA(m6A)-PseDNC: identifying N6-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018; 561–562:59–65. [DOI] [PubMed] [Google Scholar]
- 33. Kuksa P.P., Leung Y.Y., Vandivier L.E., Anderson Z., Gregory B.D., Wang L.-S.. Lusser A. In Silico Identification of RNA Modifications from High- Throughput Sequencing Data Using HAMR. RNA Methylation: Methods and Protocols. 2017; 1562:NY: Springer; 211–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen W., Xing P., Zou Q.. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble support vector machines. Sci. Rep. 2017; 7:40242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feng P., Ding H., Chen W., Lin H.. Identifying RNA 5-methylcytosine sites via pseudo nucleotide compositions. Mol. Biosyst. 2016; 12:3307–3311. [DOI] [PubMed] [Google Scholar]
- 36. Chen W., Feng P., Tang H., Ding H., Lin H.. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics. 2016; 107:255–258. [DOI] [PubMed] [Google Scholar]
- 37. Chen W., Feng P., Ding H., Lin H.. Identifying N6-methyladenosine sites in the Arabidopsis thaliana transcriptome. Mol. Genet. Genomics. 2016; 291:2225–2229. [DOI] [PubMed] [Google Scholar]
- 38. Zhao Z., Peng H., Lan C., Zheng Y., Fang L., Li J.. Imbalance learning for the prediction of N(6)-Methylation sites in mRNAs. BMC Genomics. 2018; 19:574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang H., Lv H., Ding H., Chen W., Lin H.. iRNA-2OM: a sequence-based predictor for identifying 2′-O-Methylation sites in homo sapiens. J. Comput. Biol. 2018; 25:1266–1277. [DOI] [PubMed] [Google Scholar]
- 40. Chen X., Sun Y.-Z., Liu H., Zhang L., Li J.-Q., Meng J.. RNA methylation and diseases: experimental results, databases, web servers and computational models. Brief. Bioinform. 2017; bbx142. [DOI] [PubMed] [Google Scholar]
- 41. Wei L., Su R., Wang B., Li X., Zou Q., Gao X.. Integration of deep feature representations and handcrafted features to improve the prediction of N 6 -methyladenosine sites. Neurocomputing. 2018; 324:3–9. [Google Scholar]
- 42. Vu L.P., Pickering B.F., Cheng Y., Zaccara S., Nguyen D., Minuesa G., Chou T., Chow A., Saletore Y., MacKay M. et al.. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017; 23:1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ke S., Pandya-Jones A., Saito Y., Fak J.J., Vagbo C.B., Geula S., Hanna J.H., Black D.L., Darnell J.E. Jr, Darnell R.B.. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017; 31:990–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roadmap Epigenomics C., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al.. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gruber A.R., Bernhart S.H., Lorenz R.. Picardi E. The ViennaRNA web services. RNA Bioinformatics. 2015; NY: Springer; 307–326. [DOI] [PubMed] [Google Scholar]
- 48. Betel D., Koppal A., Agius P., Sander C., Leslie C.. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010; 11:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Agarwal V., Bell G.W., Nam J.W., Bartel D.P.. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015; 4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu H., Yue D., Chen Y., Gao S.J., Huang Y.. Improving performance of mammalian microRNA target prediction. BMC Bioinformatics. 2010; 11:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wong Y.H., Lee T.Y., Liang H.K., Huang C.M., Wang T.Y., Yang Y.H., Chu C.H., Huang H.D., Ko M.T., Hwang J.K.. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007; 35:W588–W594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chang C.-C., Lin C.-J.. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011; 2:1–27. [Google Scholar]
- 53. Liaw A., Wiener M.. Classification and regression by randomForest. R News. 2002; 2:18–22. [Google Scholar]
- 54. Gevrey M., Dimopoulos I., Lek S.. Review and comparison of methods to study the contribution of variables in artificial neural network models. Ecol. Modell. 2003; 160:249–264. [Google Scholar]
- 55. Liu H., Wang H., Wei Z., Zhang S., Hua G., Zhang S.W., Zhang L., Gao S.J., Meng J., Chen X. et al.. MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018; 46:D281–D287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xuan J.J., Sun W.J., Lin P.H., Zhou K.R., Liu S., Zheng L.L., Qu L.H., Yang J.H.. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018; 46:D327–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schwartz S., Mumbach M.R., Jovanovic M., Wang T., Maciag K., Bushkin G.G., Mertins P., Ter-Ovanesyan D., Habib N., Cacchiarelli D. et al.. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014; 8:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schwartz S., Agarwala Sudeep D., Mumbach Maxwell R., Jovanovic M., Mertins P., Shishkin A., Tabach Y., Mikkelsen Tarjei S., Satija R., Ruvkun G. et al.. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013; 155:1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Consortium, G.O. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2016; 45:D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li X., Xiong X., Yi C.. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods. 2017; 14:23–31. [DOI] [PubMed] [Google Scholar]
- 61. Method of the year 2016: epitranscriptome analysis. Nat Methods. 2017; 14:1. [Google Scholar]
- 62. Chen W., Tran H., Liang Z., Lin H., Zhang L.. Identification and analysis of the N(6)-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Rep. 2015; 5:13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zheng Y., Nie P., Peng D., He Z., Liu M., Xie Y., Miao Y., Zuo Z., Ren J.. m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 2017; 46:D139–D145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Boccaletto P., Machnicka M.A., Purta E., Piątkowski P., Bagiński B., Wirecki T.K., de Crécy-Lagard V., Ross R., Limbach P.A., Kotter A. et al.. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2017; 46:D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cantara W.A., Crain P.F., Rozenski J., McCloskey J.A., Harris K.A., Zhang X., Vendeix F.A., Fabris D., Agris P.F.. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011; 39:D195–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dominissini D., Nachtergaele S., Moshitch-Moshkovitz S., Peer E., Kol N., Ben-Haim M.S., Dai Q., Segni Di, Salmon-Divon A., M., Clark W.C. et al.. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016; 530:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.