

Abstract
An organoiridium–albumin bioconjugate (Ir1‐HSA) was synthesized by reaction of a pendant maleimide ligand with human serum albumin. The phosphorescence of Ir1‐HSA was enhanced significantly compared to parent complex Ir1. The long phosphorescence lifetime and high 1O2 quantum yield of Ir1‐HSA are highly favorable properties for photodynamic therapy. Ir1‐HSA mainly accumulated in the nucleus of living cancer cells and showed remarkable photocytotoxicity against a range of cancer cell lines and tumor spheroids (light IC50; 0.8–5 μm, photo‐cytotoxicity index PI=40–60), while remaining non‐toxic to normal cells and normal cell spheroids, even after photo‐irradiation. This nucleus‐targeting organoiridium‐albumin is a strong candidate photosensitizer for anticancer photodynamic therapy.
Keywords: albumin, organoiridium, photodynamic therapy, photosensitizers
Photodynamic therapy (PDT) is a non‐invasive cancer therapy,1 which uses photosensitizers and light to convert cellular triplet oxygen (3O2) into highly reactive and cell‐damaging singlet oxygen (1O2).2 Examples of clinical photosensitizers include hematoporphyrin derivatives (Photofrin) and aminolevulinic acid (ALA, a porphyrin precursor).3 In recent years, metal complexes with high luminescence have emerged as promising photo‐theranostic candidates owing to their superior photochemical and photophysical properties.4 An octahedral tris‐N,N‐chelated RuII complex (TLD1433) recently entered clinical trials for bladder PDT,4a and WST 11 (TOOKAD Soluble), a square‐planar PdII bacteriochlorophyll derivative, has been approved for vascular‐targeted PDT.4b
Human serum albumin (HSA) conjugates can be used to delivery anticancer drugs. HSA is abundant in blood serum (ca. 0.6 mm), rich in histidine, and contains a free thiol residue at cysteine‐34.5 Moreover, HSA functions as a physiological antioxidant,6 and binds a wide range of biologically and clinically important molecules.7 The effectiveness of HSA‐coupled anticancer drugs has been established clinically for doxorubicin (INNO206; aldoxorubicin),8 and HSA‐based nanoparticle‐encapsulated paclitaxel (Abraxane).9 Recently, HSA‐functionalized metal complexes (for example, Pt, Ru, Os) have been developed for cancer therapy, illustrating that HSA plays a key role in augmenting anticancer activity.10
Herein we have conjugated a maleimide‐functionalized octahedral organo‐iridium(III) complex (Ir1, Figure 1 a) to HSA, giving rise to a large enhancement in the phosphorescence of Ir1‐HSA compared to Ir1. Ir1‐HSA is notably nontoxic in the dark, but exhibits potent photo‐cytotoxicity with significant selectivity for cancer cells and cancer cell spheroids over normal cells. Ir1‐HSA appears to be the first example of an HSA‐functionalized iridium conjugate which allows targeting of cell nuclei, as well as being an efficient photosensitizer for PDT.
Figure 1.
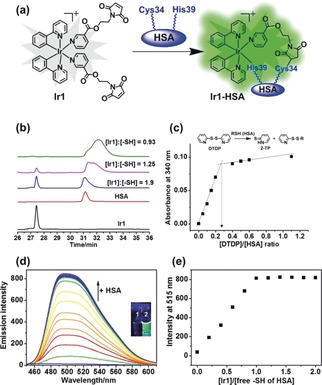
a) The conjugation of Ir1 to HSA. b) Reaction of Ir1 (30 μm) with various concentrations of HSA (0, 60, 90, and 120 μm, concentrations of Cys34 free thiol group were 0, 16.2, 24.3, 32.4 μm, respectively) studied by RP‐HPLC (UV detection at 280 nm). c) Variation of absorbance at 340 nm at various HSA:2,2′‐DTDP ratios ([HSA]=80 μm; [DTDP]=0–110 μm) and determination of the thiol content of HSA. d) Emission spectra of Ir1 (4 μm) in the presence of increasing concentrations of HSA (0–30 μm, [Cys34 free thiol] 0–8.1 μm), in PBS (pH 7.4), reaction time: 20 min at each concentration, λ ex=405 nm; Inset: photos of Ir1 (1) and Ir1‐HSA (2) under UVA irradiation. e) Dependence of the phosphorescence intensity of Ir1 at 515 nm on the concentration of free thiol of HSA, as ratio [Ir1]/[free thiol].
The octahedral organo‐iridium(III) complex Ir1, containing two chelated phenylpyridine ligands and two monodentate pyridines functionalized with a maleimide substituent, was synthesized and fully characterized as described in the Supporting Information. It was highly stable in phosphate‐buffered saline (PBS) solution for 12 h in the dark and photostable after 1 h irradiation with blue light (465 nm; Supporting Information, Figure S1).
To investigate the reactivity of the C=C bond of the pendant maleimides of Ir1, the complex was reacted with cysteine (Cys) in a molar ratio of 2[Cys] to 1[Ir1] in [D6]DMSO/D2O (2/1 v/v) at 298 K for 30 min. 1H NMR peaks for the vinyl protons of the maleimide groups at 6.62 ppm disappeared upon the addition of Cys, and new peaks appeared between 2.9 ppm and 3.9 ppm assignable to conjugated Cys (Supporting Information, Figure S2). The intensities of the peaks indicated that Cys reacted with each of the two pendant maleimides of Ir1. The conjugate was further characterised by ESI‐MS (Supporting Information, Figures S3, S4).
To investigate whether the free Cys34 thiol of HSA can similarly react with the C=C in maleimide, Ir1 (30 μm) was incubated with various amounts of HSA (0–120 μm) for 1 h and the products were separated by RP‐HPLC. The peak for Ir1 gradually disappeared with increasing amounts of HSA, with complete reaction observed at 120 μm HSA (Figure 1 b). This molar ratio of [HSA]:[Ir1] of 4:1, is consistent with the free thiol content of the HSA used, determined via reaction with 2,2′‐dithiodipyridine (2,2′‐DTDP) using a slightly modified previously reported approach.11 This gave a thiol content of 0.27±0.1 mol SH per mol HSA (Figure 1 c). Hence, the concentration of free −SH groups from 120 μm HSA was 32.4±1.2 μm, which reacted with 30 μm Ir1, suggesting formation of a 1:1 Ir1:HSA adduct. As Cys34 is in a crevice, it is likely that the second maleimide group is not accessible to a second HSA. RP‐HPLC studies on the time‐dependent binding showed that the reaction was relatively rapid with a half‐life of ca. 20 min (Supporting Information, Figure S5).
Ir1 (4 μm) itself was only weakly emissive in aqueous solution (λ ex=405 nm), whereas Ir1‐HSA showed strong phosphorescence under the same conditions (inset graph Figure 1 d). The phosphorescence of Ir1 gradually increased with increasing concentrations of HSA (Figure 1 d), plateauing at a mol ratio of ca. 1.0 Ir1:HSA(free‐SH), with a phosphorescence enhancement of ca. 53‐fold (Figure 1 e).
To remove free thiol groups from HSA (by oxidation to a disulfide), a 10‐fold molar excess of cystine was added to a 100 μm HSA solution for 24 h at 277 K. Then this HSA‐Cys34‐S‐S‐Cys product was reacted with Ir1 for 30 min. The observed phosphorescence was much weaker compared to the conjugate formed by reaction of HSA‐Cys34 and Ir1 (Figure 2 a), clearly suggesting that the free thiol of Cys34 is the binding site for Ir1.
Figure 2.
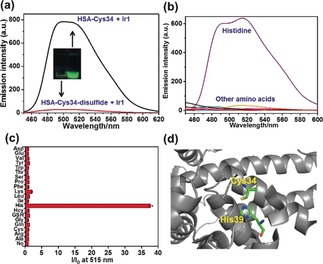
a) Emission intensity of HSA (100 μm) after reaction with 10 mol equiv cystine (giving disulfide formation at Cys34) in PBS (pH 7.4) for 24 h at 277 K, followed by treatment with Ir1 for 30 min; Inset: Images of the reaction mixture compared to Ir1‐HSA under UVA irradiation, showing the decrease in phosphorescence. b) Emission intensity of Ir1 (4 μm) in the presence of various amino acids (100 μm) in PBS solution. c) Emission intensity at 515 nm of Ir1 in the presence (I) vs. the absence (I 0) of amino acids according to (b). d) Structure of HSA (PDB:5IJF); Cys34 and His39 are labeled.
HSA is a large protein (66.5 kDa) with a single‐chain of 585 amino acid residues.5, 6, 7 To provide an indication of which residues of HSA might be involved in the luminescence enhancement of Ir1, the interaction of Ir1 with various amino acids was studied by luminescence analysis (Figure 2 b,c). Interaction of Ir1 with histidine resulted in an emission enhancement of about 37‐fold. In contrast, no significant luminescence enhancement was observed with the other amino acids, including Cys (Figure 2 b,c). Although Ir1 binds to Cys34, other factors appear to be responsible for the enhancement of phosphorescence of Ir1‐HSA. These include interactions with His residues, a strong candidate being nearby His39 (Figure 2 d).
IrIII complexes with weakly bound ligands are known to bind strongly to amino acids/proteins through ligand substitution reactions, especially to histidine/histidine rich proteins, and their use in protein staining has been reported.12 Here it is evident that histidine can switch on the phosphorescence of Ir1. We recorded the ESI‐MS of a mixture of Ir1 and His; two peaks at m/z 656.2 and 903.2 assignable to His‐bound IrIII species were detected (Supporting Information, Figure S6). These results suggest that one sterically‐hindered monodentate maleimide ligand is released after binding of Ir1 to Cys34, being displaced by His39.
As Ir1‐HSA emitted strong phosphorescence, we evaluated Ir1‐HSA as a potential photosensitizer for PDT. Firstly, Ir1 (0.4 mm) was dissolved in 20 mL MeOH:H2O (1:2 v/v), and HSA (0.4 mm) was added. The reaction mixture was stirred for 1 h, followed by Ir1‐HSA purification by dialysis (10 kDa cut‐off filter) to remove unbound Ir1. The Ir1‐HSA conjugate showed a new IR band at 1043 cm−1, characteristic of a new C−S stretch (Supporting Information, Figure S7).
Next the localization of Ir1‐HSA in living A549 lung cancer cells was investigated using confocal microscopy. The cells were incubated with Ir1‐HSA ([Ir]=5 μm) for various times. Real‐time imaging showed that Ir1‐HSA mainly accumulated in the cytoplasm within the first 30 min and then migrated to the nucleus upon further incubation (60 min–120 min; Figure 3). In contrast, the green luminescence of Ir1 alone was observed throughout the cells, both in cytoplasm and nucleus (Supporting Information, Figure S8). This subcellular localization of Ir1 and Ir1‐HSA in A549 cells was also confirmed by ICP‐MS (Supporting Information, Figure S9), demonstrating that iridium localized both in the cytoplasm and nucleus of cells treated with Ir1, whereas it mainly accumulated in the nucleus of Ir1‐HSA treated cells. We further confirmed that the luminescence of Ir1‐HSA was perfectly co‐localized with the blue fluorescence of Hoechst 33 258 (a nucleus dye), with a co‐localization efficiency of 0.82 in live A549 cells (Supporting Information, Figure S10).
Figure 3.
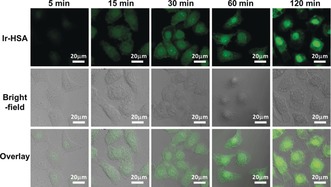
Confocal microscopy images of living A549 cells incubated with Ir1‐HSA ([Ir]=5 μm) in real time (5, 15, 40, 60, 120 min); λ ex=405 nm, λ em=550±20 nm.
To address the question as to whether HSA co‐migrated with Ir, we used an immunofluorescence approach to determine the intracellular localization of HSA. As expected, HSA was not detected in Ir1‐treated A549 cells (Figure 4). However, Ir1‐HSA exposure led to the accumulation of HSA in the cytoplasm and the nuclear membrane. When the cells were treated with HSA, the red luminescence was observed as well. This suggests that, although HSA facilitates delivery of the iridium complex to the nucleus, it does not itself penetrate past the nuclear membrane, and is probably released from Ir1‐HSA before the IrIII complex migrates into the nucleus.
Figure 4.
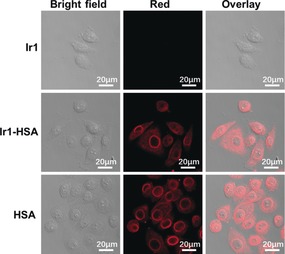
Immunofluorescence staining of HSA in cells exposed to Ir1, HSA, Ir1‐HSA ([Ir]=5 μm, 2 h), respectively. λ ex=563 nm; λ em=580–630 nm.
The phosphorescence quantum yield of Ir1 was very low (0.001) and its phosphorescence lifetime only 182.7 ns in methanol/PBS (1:1, v/v; Figure 5 a; Supporting Information, Table S1) at 298 K. Compared to Ir1, the quantum yield for Ir1‐HSA increased 36× (to 0.036) and its emission lifetime to 871.8 ns. The long phosphorescence lifetime of Ir1‐HSA makes it ideal for 1O2 generation. We used electron paramagnetic resonance (EPR) spectroscopy with 2,2,6,6‐tetramethylpiperidine (TEMP) as a spin trap to detect 1O2 generation by Ir1 and Ir1‐HSA under 465 nm irradiation. As illustrated in Figure 5 b, a characteristic 1:1:1 triplet assignable to 2,2,6,6‐tetramethylpiperidine‐1‐oxyl was observed upon irradiation (20 min). Notably the intensity of the EPR signal generated by Ir1‐HSA was significantly stronger than for Ir1. The 1O2 quantum yield13 (Φ(1O2)) of Ir1‐HSA was 0.83, much higher than that for Ir1 (0.06) upon 465 nm light irradiation (Supporting Information, Table S1).
Figure 5.
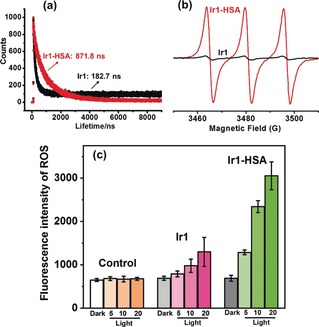
a) Phosphorescence lifetimes of Ir1 and Ir1‐HSA in methanol/PBS (1:1 v/v) at 298 K; b) EPR spectra of Ir1 and Ir1‐HSA with TEMP after 20 min light irradiation (465 nm, 5.76 J cm−2). c) Fluorescence intensity of ROS in A549 cells treated with Ir1 or Ir1‐HSA ([Ir]=5 μm) and ROS probe in the dark or upon light irradiation (5, 10, 20 min). The excitation wavelength of the ROS probe was 520 nm and the fluorescence was measured at 590–625 nm.
The long excited‐state lifetime and very high 1O2 generation quantum yield make Ir1‐HSA a potential PDT agent. To explore this, the dark‐ and photo‐antiproliferative activity of Ir1 and Ir1‐HSA was determined against human cancer cells (lung: A549; hepatoma: Hep‐G2; cisplatin resistant lung: A549R) and normal human cells (lung: MRC‐5; liver: LO2). Cells in the light plate were incubated with Ir1 or Ir1‐HSA for 2 h in the dark, washed with PBS, followed by 20 min irradiation using blue LEDs (465 nm, 5.76 J cm−2), while the dark plate was kept in the dark. Then all cells were allowed to recover over 46 h. No cell death was observed for untreated cells exposed to light (Supporting Information, Figure S11). Ir1 was relatively nontoxic toward A549 cells both in the dark (89.6 μm) and light (53.3 μm) (Table 1). In contrast, Ir1‐HSA was non‐toxic towards A549 cells in the dark (62.3 μm), but became highly cytotoxic upon irradiation (1.1 μm) with a high photocytotoxicity index (PI, PI=dark IC50/light IC50) of 56.6 (Supporting Information, Figure S11). Similar photodynamic efficiency for Ir1‐HSA was also observed for Hep‐G2 and A549R cells. Notably, under the same experimental conditions, both Ir1 and Ir1‐HSA were non‐toxic toward normal cells (MRC‐5 and LO2) (Table 1).
Table 1.
IC50 (μm) values for Ir1 and Ir1‐HSA against 2D and 3D (spheroids) cancer and normal cell lines.
| Cell lines[a] | Ir1 | Ir1‐HSA | ||
|---|---|---|---|---|
| Dark | Light | Dark | Light | |
| A549 | 89.6±3.7 | 53.3±4.5 | 62.3±2.6 | 1.1±0.3 |
| Hep‐G2 | 83.5±3.7 | 54.8±2.6 | 85.6±3.2 | 2.2±0.3 |
| A549R | 75.6±4.1 | 56.2±1.5 | 84.9±5.9 | 2.3±0.2 |
| MRC‐5 | 90.6±1.7 | 76.9±1.6 | 96.4±6.1 | 78.7±2.3 |
| LO2 | 89.3±2.3 | 76.5±0.9 | 88.6±3.0 | 66.4±4.5 |
| A549 spheroid | >100 | >100 | 65.6±5.9 | 4.8±0.2 |
| MRC‐5 spheroid | >100 | >100 | >100 | >100 |
[a] Cells were incubated with the compounds for 2 h in the dark, washed, fresh medium added, followed by incubation in the dark or irradiation at 465 nm (20 min, 5.76 J cm−2), and a further 46 h incubation. IC50 values for Ir1 and Ir1‐HSA are based on Ir concentration. Under the same experimental conditions, 5‐aminolevulinic acid, a clinical PDT agent and cisplatin gave IC50>100 μm both in the dark and upon light irradiation. The IC50 values (concentrations which caused 50 % of cell death) were determined as duplicates of triplicates in three independent sets of experiments. For each data point, the average and standard deviation are reported. For 3D toxicity assays, 8 spheroids were selected for each condition studied.
We further investigated the photocytoxicity in 3D multicellular spheroids (MCSs) with semidiameters of about 400 μm. The cytotoxicities of the complexes toward MCSs were determined by measurement of ATP concentrations using the CellTiter‐Glo 3D Cell Viability Assay (Promega). As shown in Table 1, both Ir1 and Ir1‐HSA were non‐toxic towards A549 cancer spheroids and normal cell spheroids in the dark (IC50>50 μm). However, Ir1‐HSA showed strong phototoxic effects on A549 cancer spheroids upon light irradiation, with an IC50 value of 4.8 μm. We also studied the effect of Ir1‐HSA on the kinetics of 3D MCSs regrowth. After treatment with Ir1‐HSA ([Ir]=5 μm) in the dark, the diameters of 3D MCSs increased slightly after 48 h. However, the MCSs treated with Ir1‐HSA followed by 20 min light irradiation decreased in size over time (Supporting Information, Figure S12).
A reactive oxygen species (ROS) detection assay kit was used to determine whether Ir1‐HSA produced ROS within the cells upon irradiation. Cells treated with the red ROS probe and Ir1 or Ir1‐HSA in the dark showed no evident fluorescence and there was only a very weak fluorescence in the cells treated with Ir1. In contrast, a strong red fluorescence was shown within the cells pre‐treated with Ir1‐HSA following light irradiation (Figure 5 c), suggesting that Ir1‐HSA generated ROS efficiently in cancer cells upon light irradiation.
In summary, we have reported the first example of an organo‐iridium complex‐HSA bioconjugate as a nucleus‐targeted vehicle for anticancer photodynamic therapy. The phosphorescence of Ir1 was greatly enhanced by conjugation to HSA. Interestingly, Ir1‐HSA accumulated mostly in the nucleus of living cancer cells. In contrast to other well‐studied cyclometalated iridium complexes, which mainly located in the cytoplasm, Ir1‐HSA appears to be the first reported nucleus‐targeting photosensitizer. There are only a few reports of the transport of albumin to the nucleus: in response to oxidative stress,14 and via permeabilization of cells with digitonin.15 In our work, it appears that albumin plays an important role in the transport and delivery of Ir1 to the cell nucleus.
Importantly, Ir1‐HSA exhibited a long phosphorescence lifetime and remarkably high 1O2 generation quantum yield along with high photostability, which are essential for an efficient photosensitizer. Ir1‐HSA exhibited excellent photocytotoxicity against a range of cancer cell lines and multicellular spheroids with a high photo‐cytotoxicity index while remaining dormant in normal cells/spheroids, even after photo‐irradiation. All these properties confirm that Ir1‐HSA could be an efficient photosensitizer with novel nucleus‐targeting properties for potential clinical PDT applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the EPSRC (grants EP/G006792 and EP/M027503/1 for P.J.S.), National Natural Science Foundation of China (NSFC, 21701113), the Science and Technology Foundation of Shenzhen (JCYJ20170302144346218) and the Natural Science Foundation of SZU (2018036) for P.Z., Newton Fund (NF160307 for H.H., NF151429 for S.B.) and the Wellcome Trust (209173/Z/17/Z for C.I.) for support. We thank Dr. Lijiang Song for excellent assistance with mass spectrometry; Dr. Ben Breeze with EPR experiments; and Dr. Ivan Prokes with NMR spectroscopy.
P. Zhang, H. Huang, S. Banerjee, G. J. Clarkson, C. Ge, C. Imberti, P. J. Sadler, Angew. Chem. Int. Ed. 2019, 58, 2350.
Contributor Information
Dr. Huaiyi Huang, Email: huanghy87@mail.sysu.edu.cn.
Prof. Peter J. Sadler, Email: P.J.Sadler@warwick.ac.uk.
References
- 1.
- 1a. Heinemann F., Karges J., Gasser G., Acc. Chem. Res. 2017, 50, 2727; [DOI] [PubMed] [Google Scholar]
- 1b. Knoll J. D., Turro C., Coord. Chem. Rev. 2015, 282, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Liu J., Zhang C., Rees T. W., Ke L., Ji L., Chao H., Coord. Chem. Rev. 2018, 363, 17; [Google Scholar]
- 2b. Chen H., Tian J., He W., Guo Z., J. Am. Chem. Soc. 2015, 137, 1539. [DOI] [PubMed] [Google Scholar]
- 3. Dolmans D. E., Fukumura D., Jain R. K., Nat. Rev. Cancer 2003, 3, 380. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Monro S., Colon K. L., Yin H., J. Roque III , Konda P., Gujar S., Thummel R. P., Lilge L., Cameron C. G., McFarland S. A., Chem. Rev. 2018, 10.1021/acs.chemrev.8b00211; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Gill I. S., Azzouzi A. R., Emberton M., Coleman J. A., Coeytaux E., Scherz A., Scardino P. T., J. Urol. 2018, 200, 786; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Zhang P., Chiu C. K. C., Huang H., Lam Y. P. Y., Habtemariam A., Malcomson T., Paterson M. J., Clarkson G. J., O'Connor P. B., Chao H., Sadler P. J., Angew. Chem. Int. Ed. 2017, 56, 14898; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15094; [Google Scholar]
- 4d. Yam V. W., Angew. Chem. Int. Ed. 2015, 54, 8304; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8422; [Google Scholar]
- 4e. Lo K. K., Acc. Chem. Res. 2015, 48, 2985; [DOI] [PubMed] [Google Scholar]
- 4f. Banerjee S., Chakravarty A. R., Acc. Chem. Res. 2015, 48, 2075; [DOI] [PubMed] [Google Scholar]
- 4g. Nam J. S., Kang M. G., Kang J., Park S. Y., Lee S. J. C., Kim H. T., Seo J. K., Kwon O. H., Lim M. H., Rhee H. W., Kwon T. H., J. Am. Chem. Soc. 2016, 138, 10968. [DOI] [PubMed] [Google Scholar]
- 5. He X. M., Carter D. C., Nature 1992, 358, 209. [DOI] [PubMed] [Google Scholar]
- 6. Pirisino R., Simplicio P. Di, Ignesti G., Bianchi G., Barbera P., Pharmacol. Res. Commun. 1988, 20, 545. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Malik N. A., Otiko G., Sadler P. J., J. Inorg. Biochem. 1980, 12, 317; [DOI] [PubMed] [Google Scholar]
- 7b. Roberts J. R., Xiao J., Schliesman B., Parsons D. J., Shaw C. F., Inorg. Chem. 1996, 35, 424; [DOI] [PubMed] [Google Scholar]
- 7c. Ross S. A., Carr C. A., Briet J. W., Lowe G., Anti-Cancer Drug Des. 2000, 15, 431; [PubMed] [Google Scholar]
- 7d. Espósito B. P., Najjar R., Coord. Chem. Rev. 2002, 232, 137. [Google Scholar]
- 8. Kratz F., Curr. Bioact. Compd. 2011, 7, 33. [Google Scholar]
- 9. Miele E., Spinelli G. P., Miele E., Tomao F., Tomao S., Int. J. Nanomed. 2009, 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Mayr J., Heffeter P., Groza D., Galvez L., Koellensperger G., Roller A., Alte B., Haider M., Berger W., Kowol C. R., Keppler B. K., Chem. Sci. 2017, 8, 2241; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Zheng Y. R., Suntharalingam K., Johnstone T. C., Yoo H., Lin W., Brooks J. G., Lippard S. J., J. Am. Chem. Soc. 2014, 136, 8790; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Hanif M., Moon S., Sullivan M. P., Movassaghi S., Kubanik M., Goldstone D. C., Söhnel T., Jamieson S. M. S., Hartinger C. G., J. Inorg. Biochem. 2016, 165, 100; [DOI] [PubMed] [Google Scholar]
- 10d. Chakrabortty S., Agrawalla B. K., Stumper A., Vegi N. M., Fischer S., Reichardt C., Kögler M., Dietzek B., Feuring-Buske M., Buske C., Rau S., Weil T., J. Am. Chem. Soc. 2017, 139, 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stewart A. J., Blindauer C. A., Berezenko S., Sleep D., Tooth D., Sadler P. J., FEBS J. 2005, 272, 353. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Ma X., Jia J., Cao R., Wang X., Fei H., J. Am. Chem. Soc. 2014, 136, 17734; [DOI] [PubMed] [Google Scholar]
- 12b. Ma D. L., Wong W. L., Chung W. H., Chan F. Y., So P. K., Lai T. S., Zhou Z. Y., Leung Y. C., Wong K. Y., Angew. Chem. Int. Ed. 2008, 47, 3735; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3795. [Google Scholar]
- 13. Mari C., Pierroz V., Rubbiani R., Patra M., Hess J., Spingler B., Oehninger L., Schur J., Ott I., Salassa L., Ferrari S., Gasser G., Chem. Eur. J. 2014, 20, 14421. [DOI] [PubMed] [Google Scholar]
- 14. Weber T. J., Negash S., Smallwood H. S., Ramos K. S., Thrall B. D., Squier T. C., Biochemistry 2004, 43, 7443. [DOI] [PubMed] [Google Scholar]
- 15. Mo Y., Barnett M. E., Takemoto D., Davidson H., Kompella U. B., Mol. Vision 2007, 13, 746. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary