

Abstract
We report a visible‐light‐mediated organocatalytic strategy for the enantioselective acyl radical conjugate addition to enals, leading to valuable 1,4‐dicarbonyl compounds. The process capitalizes upon the excited‐state reactivity of 4‐acyl‐1,4‐dihydropyridines that, upon visible‐light absorption, can trigger the generation of acyl radicals. By means of a chiral amine catalyst, iminium ion activation of enals ensures a stereoselective radical trap. We also demonstrate how the combination of this acylation process with a second catalyst‐controlled bond‐forming event allows to selectively access the full matrix of all possible stereoisomers of the resulting 2,3‐substituted 1,4‐dicarbonyl products.
Keywords: acyl radicals, organocatalysis, photochemistry, stereodivergence, Stetter reaction
Chiral 1,4‐dicarbonyl compounds are versatile synthetic intermediates1 and important structural elements found in a wide variety of natural products and pharmaceutical agents.2 However, their direct and stereocontrolled preparation is difficult. While effective methods that rely on chiral auxiliaries were recently reported,3 catalytic asymmetric variants to access enantioenriched 1,4‐dicarbonyl compounds are rare.4 This is mainly because the stereoselective union of two carbonyl units in a 1,4 relation generally requires a polarity inversion of one of the carbonyl substrates (Figure 1 a, path i). The Stetter reaction5 is the prototypical example of such umpolung 6 reactivity: an electrophilic aldehyde is converted into a nucleophilic acyl anion equivalent (e.g. the Breslow intermediate) that can attack α,β‐unsaturated carbonyl compounds to afford the target 1,4‐dicarbonyl products. Despite the tremendous potential of this approach, the literature contains only a few examples of enantioselective intermolecular catalytic Stetter reactions.7 In a strategically similar approach, acyl silanes served as acyl anion precursors to develop a catalytic asymmetric acylation of α,β‐unsaturated amides.8
Figure 1.
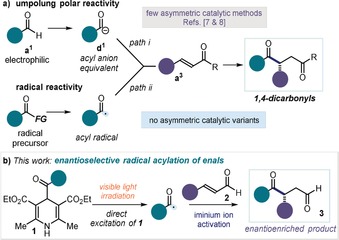
a) Intermolecular strategies to access 1,4‐dicarbonyl compounds from two carbonyl subunits: i) polar approach based on umpolung reactivity and ii) acyl radical conjugate addition to α,β‐unsaturated carbonyl compounds; FG: functional group. b) Proposed photochemical stereocontrolled iminium ion‐mediated conjugate addition of acyl radicals, generated upon direct excitation of 4‐acyl‐1,4‐dihydropyridines 1, to enals 2.
An alternative strategic disconnection to directly access 1,4‐dicarbonyls relies on radical manifolds (Figure 1 a, path ii). Specifically, the Giese‐type addition of acyl radical intermediates9 to α,β‐unsaturated carbonyl compounds offers a valuable alternative to the use of acyl anion equivalents.10 However, an enantioselective catalytic version of this radical approach has not yet been achieved, mainly because the high reactivity of acyl radicals hampers their effective stereocontrolled trap. Herein, we report a photochemical organocatalytic protocol11 that addresses this deficit in enantioselective synthesis (Figure 1 b). We show that the activation of enals 2 by means of iminium ion formation12 triggers the enantioselective interception of photochemically generated acyl radicals to afford enantioenriched 1,4‐dicarbonyls 3.
The choice of a suitable acyl radical precursor was guided by our recent findings that 4‐alkyl‐1,4‐dihydropyridines, upon visible‐light excitation and at ambient temperature, directly afford C(sp 3)‐centered radicals.13 In analogy to this photochemical pattern, we surmised that the excited‐state reactivity of structurally related 4‐acyl‐1,4‐dihydropyridines (acyl‐DHPs, 1) could generate the target acyl radical under mild conditions (Figure 1 b). To test our plan's feasibility, we used the benzoyl derivative 1 a (Bz‐DHP) as the model substrate. 1 a can be readily synthesized as a stable crystalline yellow solid from commercially available phenylglyoxal. UV‐vis spectroscopic analysis established that 1 a can absorb in the visible frequency region (Figure 2 a). The ability of 1 a to trigger the formation of benzoyl radicals upon simple photoexcitation at 460 nm was corroborated by EPR studies, conducted at 77 K. The EPR spectrum showed an isotropic X‐band absorption with g factor=2.0008 (Figure 2 b), which is consistent with literature on the characterization of benzoyl radicals.14 In addition, irradiating a CH3CN solution of 1 a with a single high‐power visible‐light‐emitting diode (LED, λ max=460 nm, irradiance of 30 mW cm−2) and in the presence of the radical scavenger 2,2,6,6‐tetramethyl‐1‐piperidinyloxy (TEMPO, 1 equiv) led to the formation of the benzoyl‐TEMPO adduct in 45 % yield (details in Section H of the Supporting Information). Overall, these experiments indicate that the simple photoexcitation of 1 a can trigger the formation of benzoyl radicals.
Figure 2.
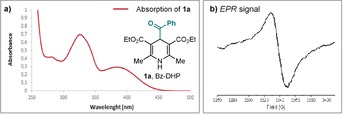
a) Absorption spectrum of 1 a in CH3CN (0.15 mm). b) EPR spectrum of the benzoyl radical generated from 1 a at 77 K after 170 min of light irradiation at 460 nm (30 mW cm−2).
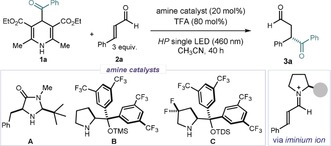
With a suitable acyl radical precursor in hand, we then focused on developing the photochemical asymmetric iminium ion‐mediated radical addition. We selected cinnamaldehyde 2 a and Bz‐DHP 1 a as model substrates (Table 1). The experiments were conducted in CH3CN using a blue (HP) LED (λ max=460 nm)15 with an irradiance at 30 mW cm−2, as controlled by an external power supply (full details of the illumination set‐up are reported in the Supporting Information, Figure S1). A blank reaction conducted at ambient temperature in the absence of any chiral amine catalyst delivered the 1,4‐dicarbonyl product 3 a in 15 % yield after only 4 hours (entry 1). This result highlights the intrinsic challenge of developing an enantioselective variant, which requires the chiral catalyst to override a fast racemic background reaction. To mitigate the uncatalyzed path, we performed further experiments under cryogenic conditions (−15 °C). We used chiral secondary amine catalysts with an established profile in promoting asymmetric iminium‐ion‐mediated processes. The imidazolidinone catalyst A 16a afforded product 3 a in high yield but low stereocontrol (entry 2), while the diarylprolinol silylether B 16b inferred a slightly higher enantiomeric excess, but at the expense of reactivity (entry 3). Interestingly, the yield of product 3 a correlated positively with the electrophilicity of the iminium ions (catalyst A forms a more reactive iminium ion than B upon condensation with 2 a).17 This observation prompted us to use the gem‐difluorinated diarylprolinol silylether catalyst C, which we previously designed for the photo‐activation of iminium ions.15a We reasoned that the incorporation of electron‐withdrawing fluorine atoms would facilitate the stereoselective acyl radical trap by providing a chiral iminium ion with an enhanced electrophilicity. Pleasingly, product 3 a was formed in high yield and good enantioselectivity under catalysis by C (74 % ee, entry 4). Lowering the irradiance to 20 mW cm−2, thus modulating the amount of acyl radicals generated, further improved the system's efficiency, providing optimal conditions (entry 5, 3 a formed in 86 % yield and 76 % ee). Interestingly, the reaction maintained the same efficiency under green light irradiation (LED with λ max=525 nm), but required an unpractically long time (92 vs. 40 hours, entry 6). Finally, the reactivity was completely inhibited in the absence of light, demonstrating the photochemical nature of the process (entry 7).
Table 1.
Optimization studies.[a]
| Entry | Catalyst | T [°C] | λ | mW/cm2 | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|
| 1[d] | none | 25 | 460 | 30 | 15 | – |
| 2 | A | −15 | 460 | 30 | 85 | −14 |
| 3 | B | −15 | 460 | 30 | 43 | 27 |
| 4 | C | −15 | 460 | 30 | 84 | 74 |
| 5 | C | −15 | 460 | 20 | 96 (88) | 76 |
| 6[e] | C | −15 | 525 | 30 | 74 | 76 |
| 7 | C | −15 | none | 0 | 0 |
[a] Reactions performed on a 0.1 mmol scale for 40 h using 0.2 mL of solvent under illumination by a single high‐power (HP) LED. [b] Yield of 3 a determined by 1H NMR analysis of the crude mixture using trichloroethylene as the internal standard; yields of the isolated 3 a are reported in brackets. [c] Enantiomeric excess determined by UPC2 analysis on a chiral stationary phase. [d] Reaction time: 4 h. [e] Reaction time: 92 h. TFA: trifluoroacetic acid; TMS: trimethylsilyl; TDS: thexyl‐dimethylsilyl.
Adopting the optimized conditions described in Table 1, entry 5, we then investigated the generality of the photo‐organocatalytic asymmetric acyl radical conjugate addition (Figure 3). We first evaluated the reactivity of differently substituted acyl radicals, photochemically generated from the precursor acyl‐DHPs 1, towards the addition to cinnamaldehyde. For aromatic moieties in 1, different substitution patterns were tolerated well, regardless of their electronic and steric properties, affording the corresponding 1,4‐dicarbonyl products 3 a–h in high yields and moderate to good stereocontrol. Heteroaryl frameworks can also be included in the product, as shown for the furanyl‐ and thienyl‐substituted adducts 3 i and 3 j, respectively. Finally, alkyl substituents on the acyl group could be readily introduced (products 3 k–m), including the sterically demanding adamantyl group (3 k), which is used in medicinal chemistry to improve ADME properties of lead compounds.18 Interestingly, we did not observe, under the reaction conditions, any byproducts arising from competing decarbonylation (C=O loss) of the aliphatic acyl radical.
Figure 3.
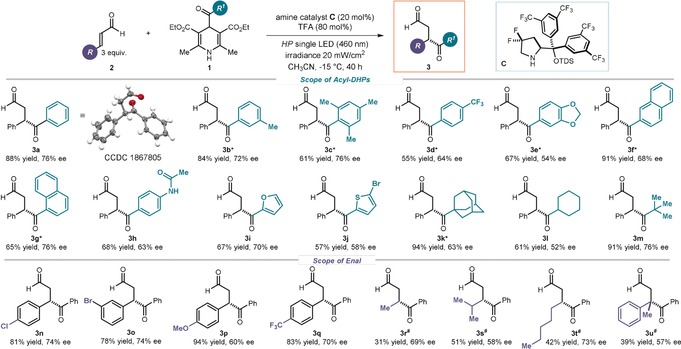
Survey of the acyl DHPs 1 and enals 2 that can participate in the acyl radical conjugate addition. Reactions performed on a 0.1 mmol scale using 3 equiv of enal 2 in 0.2 mL of CH3CN under illumination at 460 nm with an irradiance at 20 mW cm−2. Yields and enantiomeric excesses of the isolated products 3 are indicated below each entry (average of two runs per substrate). *Irradiance: 30 mW cm−2. #Using 2 equiv of enal 2.
Experiments to probe the scope of the cinnamaldehyde component 2 revealed that a range of substituents are tolerated on the aryl ring (products 3 n–q). Importantly, aliphatic enals with short, encumbered, or long fragments at the β position also reacted smoothly, affording the corresponding 1,4‐dicarbonyl products 3 r–t. Finally, the acyl radical conjugate addition to β,β′‐disubstituted (E)‐3‐phenylbut‐2‐enal enabled us to enantioselectively forge a quaternary carbon stereocenter (3 u).
Crystals from compound 3 a were suitable for X‐ray crystallographic analysis,19 which established the stereochemical course of the radical process.
To demonstrate the synthetic utility of the method, we prepared the acylated product 3 a on a synthetically useful scale (1 mmol scale, 74 % yield and 70 % ee). 3 a served as an intermediate for the preparation of the biologically active (S)‐(+)‐4‐(4‐(2‐methoxyphenyl)piperazin‐1‐yl)‐1,2‐diphenylbutan‐1‐one (4), a serotonin 5HT1A receptor antagonist.20 4 was synthesized in a single step by reductive amination of 3 a without eroding the stereochemical integrity of the progenitor (Scheme 1).
Scheme 1.

Synthesis of serotonin 5HT1A receptor antagonist 4; STAB=sodium triacetoxyborohydride.
We recognized that the structure of the β‐acylated products 3 could provide the opportunity to stereoselectively access 2,3‐disubstituted 1,4‐dicarbonyls, which are valuable chiral motifs in natural products and drug scaffolds.2 Since the aldehyde‐containing product 3 bears the chemical handle required for enamine activation, we envisioned the possibility of combining the iminium‐ion‐mediated acyl radical addition process with a second bond‐forming event controlled by a different organic catalyst (aldehyde α‐functionalization). This sequential iminium ion‐enamine activation approach would lead to the target 2,3‐disubstituted 1,4‐dicarbonyl adduct 5 (Scheme 2). Ideally, the identification of two distinct chiral catalysts that specifically trigger the two mechanistically orthogonal steps of the process could enable selective access to any product enantiomer or diastereomer of 5 by judicious catalyst selection.21 In implementing this stereodivergent plan, we capitalized on the electron‐poor nature of the difluorinated catalyst C, which makes it very suitable for iminium ion activation. In contrast, these electronic properties greatly hamper the condensation of C with the adduct 3 to generate the enamine intermediate. We hypothesized that the addition of the more electron‐rich amine catalyst B would therefore exclusively assume the control of the enamine‐mediated step. Switching the enantiomer of the specific enamine catalyst B would therefore ensure a selective access to both diastereoisomeric 2,3‐difunctionalized products 5 with high enantioselectivity.
Scheme 2.
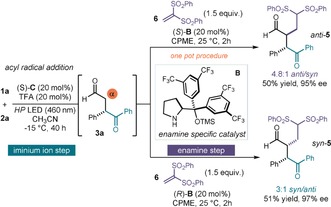
One‐pot stereodivergent synthesis of 2,3‐difunctionalized 1,4‐dicarbonyl compounds via cycle‐specific iminium ion/enamine catalysis; cyclopentyl methyl ether (CPME).
This plan was tested by performing the photochemical β‐acylation of cinnamaldehyde 2 a and Bz‐DHP 1 a catalyzed by the fluorinated catalyst (S)‐C. After completion of the radical addition step, the aminocatalyst (S)‐B (20 mol %) was added along with 1,1‐bis(phenylsulfonyl)ethylene 6 as a reactive Michael acceptor22 and cyclopentylmethylether (CPME) as the solvent (Scheme 2). This one‐pot procedure granted access to the 2,3‐disubsitituted product 5 with high enantioselectivity (95 % ee) and good anti diastereoselectivity (4.8:1 anti/syn). In consonance with our design plan, using the other enantiomer of the enamine catalyst B while retaining the iminium specific catalyst C isomer resulted in complete reversal of diastereocontrol to provide the syn adduct 5 without loss in reaction efficiency or enantioselectivity (51 yield, 97 % ee, 3:1 syn/anti).
In summary, we have demonstrated that easily accessible 4‐acyl‐1,4‐dihydropyridines can generate acyl radicals upon irradiation with visible light. The mild reaction conditions of this photochemical radical‐generating strategy were used to develop the first reported example of enantioselective catalytic acyl radical conjugate addition. This iminium‐ion‐mediated process affords valuable enantioenriched acyclic 1,4‐dicarbonyl compounds and can be used for the stereoselective synthesis of a biologically relevant molecule. We also demonstrated that, by combining this acylation process with a second catalyst‐controlled bond‐forming event, it is possible to selectively access 2,3‐substituted 1,4‐dicarbonyl products using a one‐pot procedure, and that both stereoisomers can become available by judicious catalyst selection. Efforts are ongoing to expand the synthetic potential of this asymmetric acyl radical addition strategy and fully elucidate the reaction mechanism.15
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank MINECO (CTQ2016‐75520‐P) and the European Research Council (ERC 681840—CATA‐LUX) for financial support. A.V.‐P. thanks the CONACyT (Consejo Nacional de Ciencia y Tecnología, Mexico‐Ref. 237346) for a postdoctoral fellowship. We thank Dr. Suva Paria for preliminary investigations and Professor Maurizio Fagnoni (University of Pavia) for useful discussions.
G. Goti, B. Bieszczad, A. Vega-Peñaloza, P. Melchiorre, Angew. Chem. Int. Ed. 2019, 58, 1213.
Contributor Information
Dr. Giulio Goti, http://www.iciq.org/research/research_group/prof‐paolo‐melchiorre/.
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Paal C., Ber. Dtsch. Chem. Ges. 1884, 17, 2756; [Google Scholar]
- 1b. Knorr L., Ber. Dtsch. Chem. Ges. 1884, 17, 2863. [Google Scholar]
- 2.
- 2a. Whittaker M., Floyd C. D., Brown P., Gearing A. J. H., Chem. Rev. 1999, 99, 2735; [DOI] [PubMed] [Google Scholar]
- 2b. DeMartino M. P., Chen K., Baran P. S., J. Am. Chem. Soc. 2008, 130, 11546 and references therein; [DOI] [PubMed] [Google Scholar]
- 2c. Gavai A. V., et al., ACS Med. Chem. Lett. 2015, 6, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Kaldre D., Klose I., Maulide N., Science 2018, 361, 664; [DOI] [PubMed] [Google Scholar]
- 3b. Robinson E. E., Thomson R. J., J. Am. Chem. Soc. 2018, 140, 1956; [DOI] [PubMed] [Google Scholar]
- 3c. Baran P. S., DeMartino M. P., Angew. Chem. Int. Ed. 2006, 45, 7083; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7241; [Google Scholar]
- 3d. Kise N., Tokioka K., Aoyama Y., Matsumura Y., J. Org. Chem. 1995, 60, 1100. [Google Scholar]
- 4.
- 4a. Huang S., Kötzner L., De C. K., List B., J. Am. Chem. Soc. 2015, 137, 3446; [DOI] [PubMed] [Google Scholar]
- 4b. Jang H.-Y., Hong J.-B., MacMilllan D. W. C., J. Am. Chem. Soc. 2007, 129, 7004. [DOI] [PubMed] [Google Scholar]
- 5. Stetter H., Schreckenberg M., Angew. Chem. Int. Ed. Engl. 1973, 12, 81; [Google Scholar]; Angew. Chem. 1973, 85, 89. [Google Scholar]
- 6. Seebach D., Angew. Chem. Int. Ed. Engl. 1979, 18, 239; [Google Scholar]; Angew. Chem. 1979, 91, 259. [Google Scholar]
- 7.
- 7a. Enders D., Han J., Henseler A., Chem. Commun. 2008, 3989; [DOI] [PubMed] [Google Scholar]
- 7b. Enders D., Han J., Synthesis 2008, 3864; [Google Scholar]
- 7c. Liu Q., Perreault S., Rovis T., J. Am. Chem. Soc. 2008, 130, 14066; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. DiRocco D. A., Rovis T., J. Am. Chem. Soc. 2011, 133, 10402; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Jousseaume T., Wurz N. E., Glorius F., Angew. Chem. Int. Ed. 2011, 50, 1410; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1446; For a review: [Google Scholar]
- 7f. Enders D., Balensiefer T., Acc. Chem. Res. 2004, 37, 534. [DOI] [PubMed] [Google Scholar]
- 8. Nahm M. R., Potnick J. R., White P. S., Johnson J. S., J. Am. Chem. Soc. 2006, 128, 2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For acyl radical properties, reactivity, and classical methodologies for their generation, see: Chatgilialoglu C., Crich D., Komatsu M., Ryu I., Chem. Rev. 1999, 99, 1991. [DOI] [PubMed] [Google Scholar]
- 10.For examples of non-stereocontrolled acyl radical addition to electron-poor olefins, see:
- 10a. Scheffold R., Orlinski R., J. Am. Chem. Soc. 1983, 105, 7200; [Google Scholar]
- 10b. Esposti S., Dondi D., Fagnoni M., Albini A., Angew. Chem. Int. Ed. 2007, 46, 2531; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2583; [Google Scholar]
- 10c. Bergonzini G., Cassani C., Wallentin C.-J., Angew. Chem. Int. Ed. 2015, 54, 14066; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14272; [Google Scholar]
- 10d. Wang G.-Z., Shang R., Cheng W.-M., Fu Y., Org. Lett. 2015, 17, 4830; [DOI] [PubMed] [Google Scholar]
- 10e. Capaldo L., Riccardi R., Ravelli D., Fagnoni M., ACS Catal. 2018, 8, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Silvi M., Melchiorre P., Nature 2018, 554, 41. [DOI] [PubMed] [Google Scholar]
- 12. MacMillan D. W. C., Nature 2008, 455, 304. [DOI] [PubMed] [Google Scholar]
- 13. Buzzetti L., Prieto A., Raha Roy S., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 15039; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15235. [Google Scholar]
- 14. Krusic P. J., Rettig T. A., J. Am. Chem. Soc. 1970, 92, 722. [Google Scholar]
- 15.Irradiance at 460 nm secured the selective excitation of the acyl-DHP substrate 1. The resulting acyl radical is then stereoselectively intercepted by the ground-state electrophilic chiral iminium ion. The excitation of the transiently generated chiral iminium ion cannot be operative under these conditions, since this intermediate cannot absorb wavelengths longer than 430 nm, see:
- 15a. Silvi M., Verrier C., Rey Y. P., Buzzetti L., Melchiorre P., Nat. Chem. 2017, 9, 868; [DOI] [PubMed] [Google Scholar]
- 15b. Verrier C., Alandini N., Pezzetta C., Moliterno M., Buzzetti L., Hepburn H. B., Vega-Peñaloza A., Silvi M., Melchiorre P., ACS Catal. 2018, 8, 1062; [Google Scholar]
- 15c. Mazzarella D., Crisenza G. E. M., Melchiorre P., J. Am. Chem. Soc. 2018, 140, 8439. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Lelais G., MacMillan D. W. C., Aldrichimica Acta 2006, 39, 79; [Google Scholar]
- 16b. Jensen K. L., Dickmeiss G., Jiang H., Albrecht Ł., Jørgensen K. A., Acc. Chem. Res. 2012, 45, 248. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Lakhdar S., Ofial A. R., Mayr H., J. Phys. Org. Chem. 2010, 23, 886; [Google Scholar]
- 17b. Lakhdar S., Ammer J., Mayr H., Angew. Chem. Int. Ed. 2011, 50, 9953; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10127; [Google Scholar]
- 17c. Lakhdar S., Tokuyasu T., Mayr H., Angew. Chem. Int. Ed. 2008, 47, 8723; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8851. [Google Scholar]
- 18. Lamoureux G., Artavia G., Curr. Med. Chem. 2010, 17, 2967. [DOI] [PubMed] [Google Scholar]
- 19.CCDC 1867805 (3 a) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 20. Kohlman T. D., Xu Y.-C., Godfrey A. G., O'Toole J. C., Zhang T. Y., EP 924205-A1-19990623, 1999.
- 21.For the use of cyclic specific catalysts in organocatalytic cascade processes, see:
- 21a. Simmons B., Walji A. M., MacMillan D. W. C., Angew. Chem. Int. Ed. 2009, 48, 4349; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4413; [Google Scholar]
- 21b. Huang Y., Walji A. M., Larsen C. H., MacMillan D. W. C., J. Am. Chem. Soc. 2005, 127, 15051. [DOI] [PubMed] [Google Scholar]
- 22. Sulzer-Mossé S., Alexakis A., Mareda J., Bollot G., Bernardinelli G., Filinchuk Y., Chem. Eur. J. 2009, 15, 3204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary