

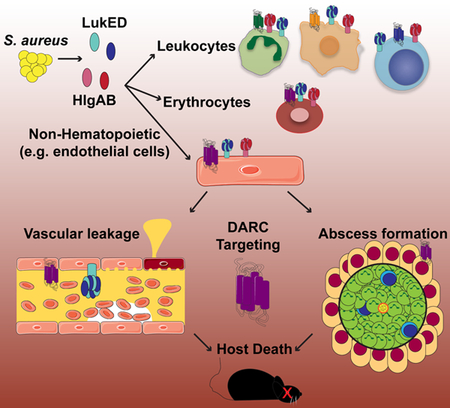
SUMMARY
The pathogenesis of Staphylococcus aureus is thought to depend on the production of pore-forming leukocidins that kill leukocytes and lyse erythrocytes. Two leukocidins, Leukocidin ED (LukED) and γ-Hemolysin AB (HlgAB), are necessary and sufficient to kill mice upon infection and toxin challenge. We demonstrate that LukED and HlgAB cause vascular congestion and derangements in vascular fluid distribution that rapidly cause death in mice. The Duffy antigen receptor for chemokines (DARC) on endothelial cells, rather than leukocytes or erythrocytes, is the critical target for lethality. Consistent with this, LukED and HlgAB injure primary human endothelial cells in a DARC-dependent manner and mice with DARC-deficient endothelial cells are resistant to toxin-mediated lethality. During bloodstream infection in mice, DARC targeting by S. aureus causes increased tissue damage, organ dysfunction, and host death. The potential for S. aureus leukocidins to manipulate vascular integrity highlights the importance of these virulence factors.
Keywords: Staphylococcus aureus, leukocidin, LukED, HlgAB, DARC, ACKR1, endothelial cells
Graphical Abstract
eTOC blurb
Staphylococcus aureus produces a series of leukocidins that contribute to infection. Lubkin et al. show that two leukocidins, LukED and HlgAB, are lethal in vivo by targeting the vasculature via the Duffy antigen receptor for chemokines (DARC). This work highlights the importance of endothelial cell targeting in S. aureus pathogenesis.
INTRODUCTION
S. aureus is a multifaceted pathogen, capable of causing a wide range of diseases, from relatively minor skin and soft tissue infections to invasive, life threatening infections including bacteremia and sepsis (David and Daum, 2010). S. aureus antibiotic resistance continues to increase, as evidenced by the large number of methicillin resistant S. aureus (MRSA) and community acquired MRSA (CA-MRSA) infections in the USA and around the world (Mediavilla et al., 2012), resulting in restricted antimicrobial treatment options. Moreover, invasive S. aureus infections are associated with high mortality rates, even with appropriate antibiotic treatment (Ammerlaan et al., 2009). These realities emphasize the need to better understand the complex pathogenesis of S. aureus infection, especially in fulminant forms of disease, and to develop novel therapeutic modalities.
S. aureus secretes a wide array of virulence factors, one class of which are the bicomponent leukocidins (Spaan et al., 2017). Bacterial isolates causing human infections can produce up to five of these proteins: Leukocidin ED (LukED), γ-Hemolysin AB (HlgAB), γ-Hemolysin CB (HlgCB), Panton-Valentine Leukocidin (PVL) and Leukocidin AB (LukAB). Upon recognition of proteinaceous receptors on host cells, the leukocidins form β-barrel pores in the cell membrane, resulting in the lysis of leukocytes and red blood cells. Though the leukocidins have been studied extensively, we still do not know the precise mechanism by which these toxins facilitate pathology and mortality during severe invasive staphylococcal infection.
RESULTS
Leukocidins cause vascular dysfunction
Intravenous infection of mice with isogenic S. aureus mutants reveals that LukED and HlgAB potentiate lethality additively (Figure 1A, Figure S1A–C). In this acute model, neither LukED nor HlgAB deficiency alleviates S. aureus burdens in the tissues examined (Figure S1D), suggesting that the toxins directly mediate lethality, instead of indirectly by increasing bacterial burden.
Figure 1: LukED and HlgAB cause lethal vascular collapse.

(A) Survival curve of Swiss-Webster mice infected intravenously with 2–3 × 107 CFU of the indicated S. aureus Newman strains (n=7–8 mice per group). See also Figure S1A–D. (B, C) Survival curve of Swiss-Webster mice injected intravenously with purified toxin (n=4 mice per group). See also Figure S1F, G. (D) Quantification of LukD levels in kidneys and hearts of C57BL/6J mice 96 hours post infection (hpi) with 2.5 × 107 CFU of the WT (n=14) or ΔlukED (n=3) S. aureus Newman by ELISA. See also Figure S1E. Representative photographs (E) and H&E staining (F) of ears from Swiss-Webster mice challenged with toxin. Scale bar indicates 100 μM. See also Figure S1H. (G) Hematocrit and (H) temperature change in toxin challenged Swiss Webster mice (n = 5–12 mice per group). (I) Intravital imaging of livers of Swiss Webster mice before and after toxin challenge. Evans blue in red is vascular fluid; Hoechst in blue is nuclei. Scale bars are 80 μM, arrows indicate vessels depleted of fluid. See also Video S1. (J) Evans blue content in organs of toxin challenged Swiss Webster mice without perfusion (n = 7–12 mice per group). See also Figure S1I. Data shown are pooled from (A, B, C, D, G, H, J) or representative of (E, F, I) at least two independent experiments. Where relevant, means ± SEM are shown. Sa = S. aureus. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 (A, B, C, Mantle-Cox test; G, H, J, one-way analysis of the variance (ANOVA) with Tukey’s correction).
To dissect the mechanism of leukocidin induced lethality, we first used an intravenous toxin challenge model. Intravenous challenge with LukED or HlgAB causes rapid death in mice (minimum lethal dose of 0.25 mg/kg for LukED and 0.12 mg/kg for HlgAB Figure 1B, Figure S1F, G). The toxin concentrations needed to exert lethality in vivo are in line with the toxin levels detected in tissues during lethal S. aureus infection, which we found to be on average 2 μg of LukD per gram of kidney tissue and 0.4 μg of LukD per gram of heart tissue (Figure 1D). Of note, there was a direct relationship between the bacterial burden present in tissues and toxin levels (Figure S1E).
We found that death during LukED and HlgAB challenge depends on the ability of these toxins to form pores in host cells, as administration of toxin mutants incapable of pore formation, but capable of receptor binding (Reyes-Robles et al., 2016) is not lethal (Figure 1C). Moreover, this lethal phenotype is specific, as PVL, a leukocidin family member with high homology to LukED and HlgAB but that lacks compatible receptors in mice (Spaan et al., 2017), is not lethal (Figure 1B).
To test if the observed lethality was caused by the leukocidal actions of the toxins, we tested the susceptibility of mice depleted of monocytes, macrophages, or neutrophils. As the leukocidins activate the NLRP3 inflammasome (Holzinger et al., 2012; Mariathasan et al., 2006; Melehani et al., 2015), and systemic inflammasome activation can lead to a similar rapid lethality phenotype (von Moltke et al., 2012), we also tested mice deficient in inflammasome activation (Caspase1/11 and MyD88). To our surprise, all the tested mice were susceptible to toxin challenge (Table S1) leading us to consider non-leukocidal mechanisms of lethality.
We observed that as mice approach endpoint criteria, their extremities become red (Figure 1E). Congruent with this, histology of ears and other organs (Figure 1F, Figure S1H) revealed generalized vascular congestion, suggesting hemoconcentration. We measured the hematocrit of toxin challenged mice and found that indeed it was elevated, confirming that hemoconcentration is caused by toxin challenge (Figure 1G). This was accompanied by a decrease in body temperature (Figure 1H), suggestive of death by a shock-like phenotype. Of note, all these phenotypes were dependent on the pore-forming activity of the toxins (Figure 1G, H).
To observe the effects of toxin on the vasculature in real time, we performed intravital confocal imaging of the liver. We found that upon toxin challenge, the vascular fluid of the liver, labeled with the albumin binding dye Evans blue, rapidly disappeared (Figure 1I, Video S1). The fluid began emptying out of the liver vasculature within a few minutes, before the mice showed signs of approaching endpoint criteria. This suggests that the disappearance of fluid is a cause of impending lethality, rather than an effect. Challenge with mutant toxins unable to form pores had no effect on vascular fluid in the liver. Using the Miles assay (Radu and Chernoff, 2013) we verified the depletion of vascular fluid from both livers and lungs of challenged mice (Figure 1J). Further, we found that vascular fluid leaked out of vessels into the extremities, as seen in the ears and hind limb soft tissue of the mice (Figure 1J, Figure S1I). Altogether, the redistribution of vascular fluid, hemoconcentration, and shock-like death point towards the vasculature as an important target of LukED and HlgAB during intravenous challenge.
Leukocidins target endothelial DARC in vivo
LukED and HlgAB target partially overlapping lists of host receptors. LukED targets CCR5 (Alonzo III et al., 2013), HlgAB targets CCR2 (Spaan et al., 2014), and both toxins target CXCR1, CXCR2 (Reyes-Robles et al., 2013; Spaan et al., 2014), and DARC (Spaan et al., 2015). To determine which of these receptors potentiate the observed toxin-mediated lethality, we challenged mice lacking each of the receptors (Table S1). Surprisingly, only the Darc−/− mice were protected from toxin-induced lethality (Figure 2A). Further, upon toxin challenge, the Darc−/− mice did not exhibit increased hematocrit or derangements in Evans blue distribution (Figure 2B, C).
Figure 2: LukED and HlgAB target endothelial DARC to cause lethality during challenge.

(A) Survival curve of toxin challenged C57BL/6J WT and Darc−/− mice (n = 4–5 mice per group). See also Table S1. (B) Hematocrit of toxin challenged mice (n=3 mice per group). (C) Evans blue content in lungs (left) and ears (right) of toxin challenged WT and Darc−/− mice (n = 3–11 mice per group). (D) Survival curves of toxin challenged bone marrow chimeric mice (n = 4–6 mice per group). See also Figure S2A–D. (E) DARC staining of endothelial cells from the skin of Darcfl/fl and Darcfl/fl Tie2 Cre mice. See also Figure S2F–H. (F) Survival curve of toxin challenged Darcfl/fl and Darcfl/fl Tie2 Cre mice (n = 4–5 mice per group). KO=Darc−/−, bone marrow chimeras annotated as donor → recipient. Data shown are pooled from (A, B, C, D, F), or representative of (E) at least two independent experiments. Where relevant, means ± SEM are shown. ** p<0.01, *** p<0.001.
DARC (also known as atypical chemokine receptor 1 or ACKR1) is a promiscuous receptor that is thought to play a role in regulating chemokine levels and functions in the blood, but the precise nature of this role is unclear (Dawson et al., 2000; Luo et al., 2000). DARC is found on erythrocytes, where it serves as the receptor for several Plasmodium parasites (Horuk et al., 1993; Miller et al., 1975), and the LukED and HlgAB leukocidins (Spaan et al., 2015). DARC is also found on the plasma membrane of nonhematopoietic cells, including epithelial and endothelial cells, particularly in venules (Chaudhuri et al., 1997; Hadley et al., 1994; Peiper et al., 1995; Thiriot et al., 2017). To determine whether leukocidin-induced lethality is mediated by targeting DARC on erythrocytes or nonhematopoietic cells, we generated bone marrow chimeras between WT and Darc−/− mice. Erythrocytes from mice 11 weeks post bone marrow transfer were >90% of donor origin (Figure S2A–D), indicating that bone marrow chimerism was successfully achieved and thus enabling the distinction between hematopoietic and non-hematopoietic DARC. Toxin challenge of these chimeric mice revealed that lethality required DARC on nonhematopoietic cells, and is independent of DARC on erythrocytes (Figure 2D).
Due to the vascular involvement of this phenotype (Figure 1) and the cell type specificity of DARC (Chaudhuri et al., 1997; Hadley et al., 1994; Peiper et al., 1995; Thiriot et al., 2017), we reasoned that endothelial cells were likely the nonhematopoietic cells that the toxins were targeting. To directly test this idea, we generated endothelial specific DARC knockout mice by producing Darcfl/fl mice, and crossing them with Tie2 Cre mice, which express Cre on endothelial cells (Koni et al., 2001). Darcfl/fl Tie2 Cre+ mice, as expected, lacked DARC on their endothelial cells (Figure S2F–H, Figure 2E), and were protected from lethality during LukED and HlgAB challenge (Figure 2F). Taken together, these data demonstrate that lethality during leukocidin challenge involves targeting of DARC on the endothelium.
Leukocidins target human endothelial cells
We next asked if primary human endothelial cells are susceptible to leukocidins. Since DARC is not found on endothelial cells in culture (Lacorre et al., 2004), we transfected primary human pulmonary microvascular endothelial cells (HPMECs) with a plasmid expressing human GFP-DARC. Transfection of DARC rendered HPMECs susceptible to leukocidin-induced membrane damage, which was dependent on pore-formation (Figure 3A).
Figure 3: LukED and HlgAB target primary human endothelial cells in vitro.

(A) Propidium Iodide (PI) uptake by HPMECs transfected with the indicated plasmid after exposure to 0.3 μM of toxin (10 μg/ml) for 3 hours. (B) Immunoblot for DARC and (C) qPCR for DARC transcript in HPMEC after exposure to human whole blood +/− cyclohexamide. (D) PI uptake by HPMEC cells after treatment with human whole blood and exposure to 0.6 μM of toxin (20 μg/ml). (E) Images of and (F) PI uptake by HPMEC cells after transfection with siRNA, treatment with human whole blood, and exposure to 0.6 μM of toxin. Data are pooled from (A, C, D, F) or representative of (B, E) at least three independent experiments. Where relevant, means ± SEM are shown. * p<0.05, ** p<0.01, *** p<0.001 (A, D, F, ANOVA with Tukey’s correction; C, two-tailed student’s t-test).
In the mammalian host, endothelial cells are exposed to blood. We reasoned that exposing cultured cells to whole blood in vitro may restore them to a more in vivo-like state, resulting in increased DARC levels. Indeed, we found that human whole blood increased the expression and production of DARC (Figure 3B, C). The observed increase in DARC levels required new protein synthesis, as it was inhibited by cycloheximide (Figure 3B).
Using this model, we observed that HPMECs are indeed susceptible to leukocidin-mediated membrane damage (Figure 3D). Importantly, the susceptibility of HPMECs upon whole blood exposure is caused by increased DARC expression, and not some other effect of blood exposure, since the cells are protected following DARC depletion with siRNA (Figure 3E, F). Thus, leukocidins can directly target primary human endothelial cells that express DARC.
Leukocidins target DARC during S. aureus infection
We next determined the role of DARC during lethal bloodstream infection with live S. aureus. Compared to wildtype (WT) mice, Darc−/− mice were significantly protected from infection with methicillin sensitive S. aureus (MSSA, strain Newman Figure 4A) and MRSA strains (Figure 4B), including a representative strain of the CAMRSA epidemic clone USA300 (Figure 4C). Darc−/− mice were also protected from the temperature decrease observed upon infection (Figure S4A). Of note, we observed comparable bacterial burdens in the organs surveyed between WT and Darc−/− mice (Figure S4B), suggesting that improved survival is due to loss of toxin activity rather than reduced bacterial burdens in this acute model.
Figure 4: LukED and HlgAB target nonhematopoietic DARC to cause lethality during infection.

Survival curves of C57BL/6J (WT) and Darc−/− mice infected with 2–3.5 × 107 CFU of the indicated strains of S. aureus Newman (A), 1–2.7 × 107 CFU of S. aureus USA500 BK2395 (B), and 0.9–1.3 × 107 CFU of S. aureus LAC (C) (n = 18–24 mice per group). See also Figure S4A, B. (D) Survival curve of bone marrow chimeric mice infected with 1–2 × 107 CFU WT S. aureus Newman (n = 8–9 mice per group). See also Figure S2E. (E) Scoring of kidney pathology from 72–96 hpi of WT and Darc−/− mice infected with 2–3.5 × 107 CFU of S. aureus Newman. (F) H&E staining of hearts and (G) Serum levels of analytes in mice at 96 hpi with 2.5 × 107 CFU of S. aureus Newman. Scale bar in (F) is 1 mm, 100 μm in the magnified images. See also Figure S4C–F. “Baseline” in (G) is the average values for WT and DARC KO mice mock infected (mock infected WT and DARC KO mice had similar values of all analytes measured, See also Table S2). BUN = Blood Urea Nitrogen, ALP = Alkaline Phosphatase. Sa = S. aureus, KO = Darc−/−. Data are pooled from (A-E, G) or representative of (F) at least two independent experiments. Where relevant, means ± SEM are shown. * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001 (A, B, C, D Mantel-Cox test, E, two-tailed linear-by-linear association test, G, one-tailed student’s t-test).
Next, we asked whether the requirement for DARC during S. aureus bloodstream infection is nonhematopoietic using bone marrow chimeric mice. We first confirmed that survival during intravenous infection of bone marrow chimeric mice proceeded as expected in our model (Figure S2E). As with the toxin challenge model, DARC-dependent lethality during infection was mediated through nonhematopoietic cells (Figure 4D), supporting the model that S. aureus, via LukED and HlgAB, targets endothelial DARC to promote lethal bloodstream infection in mice.
DARC potentiates S. aureus pathology
To identify the consequences of leukocidin targeting of DARC during infection we analyzed the gross pathology of infected organs. We found that kidneys from WT mice had more abscesses than from Darc−/− mice (Figure 4E). Histological analysis of infected hearts revealed more numerous and larger abscesses in organs harvested from WT animals than from Darc−/− mice (Figure 4F, Figure S4C–F). Thus, both kidneys and hearts exhibit increased pathology in the presence of DARC, despite having comparable bacterial burdens (Figure S4B). To determine the impact of this pathology on organ function we analyzed serum from WT and Darc−/− mice infected with S. aureus for markers of organ failure. We found that creatinine, blood urea nitrogen, and phosphate (but not calcium) were increased in a DARC dependent manner, consistent with decreased kidney function (Figure 4G, Table S2). Alkaline phosphatase was also increased, but no DARC dependent changes in other associated molecules with liver damage were observed, suggesting damage of a different tissue(s), possibly bone or muscle. Altogether, these data demonstrate that S. aureus mediates DARC-dependent tissue damage, which leads to organ failure and subsequent death during acute bloodstream infection.
DISCUSSION
We demonstrate here that the S. aureus leukocidins LukED and HlgAB target DARC on nonhematopoietic cells, instead of leukocytes or erythrocytes, to cause lethality in mice. Through pore formation, they damage vascular integrity by injuring endothelial cells. After toxin challenge, vascular fluid leaks out of the vessels in the extremities and is depleted from the vital organs, leading to rapid, shock-like death. In infection, the leukocidins lead to death by crippling the function of vital organs which S. aureus has tropism for, by targeting DARC. Thus, we identify endothelial cells as a target for an important family of toxins that have been previously considered to exert their activity primarily as leukotoxins and hemolysins for over 100 years.
The organ specific nature of vascular leakage during toxin challenge is highly unusual. Interestingly, the organ with the most striking degree of vascular leakage is the skin, and this is also the organ with the highest level of venular DARC (Thiriot et al., 2017). Thus, differences in vascular levels of DARC could explain why vascular fluid accumulates in the extremities and is depleted from the internal organs. Of course, hemodynamic differences, or differences in vascular structure could be involved as well.
Another open question is how the toxin pores lead to increased vascular permeability. The obvious mechanism is by inducing cell death. However, we do not have evidence of endothelial cell death in vivo. An alternative possibility is that the toxins exert a subtler effect on endothelial cells by altering signaling pathways in response to pore-mediated changes in ion fluxes. Studies evaluating the effect of leukocidins on tight junctions (Claudin-5) and adherens junctions (VE-Cadherin) using the blood exposed primary human cell model did not reveal any notable changes in protein levels or localization in the cells (Figure S3A, B). Similarly, we were unable to detect any striking changes in endothelial cell staining (CD31) or tight junctions (ZO-1) in vivo in ears of toxin challenged mice (Figure S3C, D). Thus, additional studies are needed to unravel how targeting of DARC on endothelial cells leads to the lethal phenotypes described herein.
Many bacteria secrete toxins that kill or otherwise disrupt the function of endothelial cells (Lubkin and Torres, 2017). Our study expands this list to include the S. aureus leukocidins LukED and HlgAB, which target DARC on endothelial cells with potentially lethal consequences. An important question raised by our work is the relevance of DARC in human S. aureus infection. Many polymorphisms of DARC exist in the human population, due to the selective pressure by Plasmodium parasites (Langhi and Bordin, 2006). This gives rise to the Duffy blood group system, including the Duffy null blood group. By far the most common Duffy null polymorphism is the erythroid silent, which is seen in people of African descent (Howes et al., 2011). This allele abolishes DARC expression in the erythroid lineage specifically, maintaining DARC in other cell types including endothelial cells (Peiper et al., 1995). Though these individuals are protected from leukocidin mediated hemolysis (Spaan et al., 2015), they would not be protected from vascular targeting by LukED or HlgAB.
Given our findings in mice, it is possible that in some cases the morbidity and mortality associated with clinical S. aureus infections are exacerbated by leukocidin-induced damage to the vasculature in highly infected organs. Indeed, vascular damage caused by S. aureus leukocidins could play a role in the rapid disease course experienced by some patients (Adem et al., 2005; Kravitz et al., 2005). Our findings could also explain why community-associated isolates, which mostly cause skin and soft tissue infections but are highly cytotoxic, can be so lethal when they become invasive. This is in contrast to many hospital-associated strains, which are associated with lethality as they tend to infect immunocompromised patients, but exhibit lower cytotoxicity in the laboratory environment (Rose et al., 2015). Perhaps strains that are well adapted to causing invasive disease need to modulate their cytotoxic activity so as to not kill their host.
What is the benefit to S. aureus to target endothelial cells? Perhaps in less acute infections, given that the vast majority of S. aureus is extravascular (Cheng et al., 2009), the leukocidins are an important strategy for attracting nutrient-rich vascular fluid to the site of infection. This could also explain how the leukocidins facilitate abscess formation (Figure 4E, F), by increasing local edema and inflammation. Another possibility is that S. aureus evolved the ability to target DARC purely to access iron by lysing erythrocytes (Mazmanian et al., 2003; Spaan et al., 2015), and endothelial targeting is a bystander effect. Regardless, this work adds to our understanding of the leukocidins, which until now have been mainly studied as killers of leukocytes and erythrocytes. The more we study these toxins the more we learn about these truly multifaceted tools of S. aureus pathogenesis.
STAR METHODS:
CONTACT FOR REAGENT AND RESOURCE SHARING:
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Victor J. Torres (Victor.Torres@nyulangone.org).
EXPERIEMENTAL MODEL AND SUBJECT DETAILS:
Ethics statement.
All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of New York University and were performed according to guidelines from the National Institutes of Health (NIH), the Animal Welfare Act, and US Federal Law.
The use of 5 ml blood from healthy human volunteers for induction of DARC in vitro was approved by the Research Ethics Board at St. Michael’s Hospital, Toronto, Canada (REB#17–184). Under the terms of this protocol, no identifying information or personal health information was obtained or kept in order to protect the confidentiality of the volunteers.
S. aureus strains and culture.
The S. aureus strain Newman, a methicillin-sensitive strain and the isogenic ΔlukED, ΔhlgACB::tet, or ΔlukEDΔhlgACB::tet strains, or the representative MRSA USA500 strain BK2395 and its isogenic ΔlukED strain or CA-MRSA USA300 LAC were used. Strains were routinely grown on tryptic soy agar plates (TSA) overnight at 37°C. For infection experiments, a single colony was inoculated into tryptic soy broth (TSB) and grown overnight at 37°C shaking at 180 rpm at 45 degrees. Subsequently, the bacteria were subcultured 1:100 into TSB, washed and normalized based on Optical Density (OD) 600 to 5×108 CFU/ml, then diluted 1:2 to make 2.5×108 CFU/ml or 1:5 to make 1×108 CFU/ml, and 100 μl was used for infections.
Mice.
Swiss-Webster mice were purchased from Envigo. C57BL/6J Darc−/− mice were provided by Dr. G. Scott Worthen (UPenn), re-derived and bred in-house at NYU School of Medicine. C57BL/6J controls were purchased from The Jackson Laboratory and bred in-house. Ccr5−/− mice were purchased from Taconic and bred in-house. Cxcr1−/−, Cxcr2−/−, Ccr2−/− and Myd88−/− mice were purchased from Jackson Laboratory and bred in-house. Experiments with Caspase1/11−/− mice were performed by author E.C., on animals housed at the University of California, Berkeley. Mice were maintained under specific pathogen-free conditions and used age-matched at 4–6 weeks of age for Swiss-Webster mice, and 7–11 weeks of age for C57BL/6J and all C57BL/6J background KO mice (the mice have similar weights at these respective ages). Swiss-Webster experiments were performed with all female mice, and C57BL/6J experiments were performed with female and male (sex-matched) mice. Mice were randomly mixed within their genotypes and sex and then assigned to groups.
Generation of Darcfl/fl mice.
ACKR1tm1a(EUCOMM)Hmgu ES cells were obtained from the International Mouse Phenotyping Consortium, expanded in the presence of G418, injected into blastocysts and subsequently implanted into pseudopregnant females by staff at the NYU Rodent Genetic Engineering Laboratory. The resulting chimeric mice were bred to WT C57BL/6 females to achieve germline transmission; animals carrying the targeted allele were then bred to FLPo mice, which express a flippase to remove the FRT flanked Neomycin resistance and LacZ cassette present in the targeted locus (see schematic in Fig. S4a). Mice carrying the conditional allele free of the Neo/LacZ cassette were then crossed to C57BL/6 mice to confirm germline transmission of the conditional allele and to ensure loss of the gene encoding the FLPo recombinase. To achieve endothelial specific deletion of Darc, the Darcfl/+ mice were crossed with Tie2 Cre male mice. Mice were genotyped for Darc using primers: 1694 and 1697 to distinguish between the wildtype and floxed alleles; and 1694 and 1873 primers to amplify the null allele resulting from Cre-mediated recombination; and 1695 and 1697 primers, enabling amplification of the Neomycin resistance gene. Mice were genotyped for flippase and Tie2 Cre according to the protocols available from The Jackson Laboratory website.
Absence of DARC on endothelial cells in Darcfl/fl Tie2 Cre mice was confirmed by staining of endothelial cells from mouse skin and analyzing these cells by flow cytometry, as previously described (Benck et al., 2016; Thiriot et al., 2017). Briefly, skin was shaved and harvested, and fat was scraped off with a scalpel. Skin was minced and then digested with 2.5 mg/ml Collagenase D, 50 μg/ml DNAse I, and 1× protease inhibitor for 30 minutes at 37°C on a rotisserie. 1–2*105 filtered cells were then stained in a mix of TruStain fcX and the following Abs: CD31 APC, CD45 PerCP Cy5.5, TER-119 A700, CD11b A700. Cells were fixed, permeabilized, and then stained for DARC using a PE labelled Ab or Sheep IgG Isotype control and analyzed by flow cytometry (CytoFLEX S). Cells were stained for DARC both with and without fixation/permeabilization, and the results were similar.
METHOD DETAILS:
Toxin purification from S. aureus.
Leukocidins were purified from S. aureus culture supernatants as described previously (Reyes-Robles et al., 2016). Briefly, culture filtrates were harvested from 5 hour subcultures of S. aureus Newman ΔΔΔΔ strains harboring plasmids with the toxin sequences with His tags grown in tryptic soy broth (TSB) supplemented with chloramphenicol grown at 37°C with 180 rpm shaking. The cultures were centrifuged, filter-sterilized and the His-tagged proteins were purified on nickel-nitrilotriacetic acid (Ni-NTA) agarose resin (Qiagen) columns. Protein concentrations were quantified by measuring the absorbance at 280 nm using an ND-1000 Spectrophotometer (NanoDrop), then multiplying by the Molecular Weight and dividing by the Extinction Coefficient, calculated using the Northwestern University Peptide Properties Calculator (http://biotools.nubic.northwestern.edu/proteincalc.html). All holotoxin concentrations are represented per subunit. Purified leukocidins were tested for LPS with ToxinSensor Chromogenic LAL Endotoxin Assay Kit (GenScript) and found to contain less than 0.006 EU/ml in the maximum amount used for injections.
Murine in vivo toxin challenge experiments.
Mice were challenged with leukocidins as described previously (Reyes-Robles et al., 2016). Mice were anesthetized by inhalation of isoflurane gas (2%) and the toxins were administered systemically at the indicated doses via retro-orbital injection in 100 μl PBS. Time to acute intoxication was recorded after sacrifice of mice displaying signs of morbidity, including ruffled fur, hunched posture, paralysis, inability to walk, inability to consume food or water, or marked difficulty breathing. Photographs were taken with an Olympus digital camera.
In vivo cell depletion studies.
Swiss-Webster mice were injected with 300 μg of either anti-Ly6G to deplete neutrophils, anti-Ly6C to deplete monocytes, anti-GR1 to deplete neutrophils and monocytes, Clodronate Liposomes (Liposoma) to deplete macrophages and monocytes, or isotype controls. Antibodies were purchased from Bio X Cell. Antibodies were injected intraperitoneally 48 hours before toxin challenge as described previously (Alonzo III et al., 2012). Spleens from control animals were isolated, stained with anti-GR1-PE, anti-Ly6G FITC, anti-Ly6C APC, and anti-CD11b-PE-Cy7 antibodies, and analyzed by flow cytometry (LSRII, BD) to confirm depletion of the correct cell type(s).
Hematocrit.
Mice were challenged with toxin as described above, and then blood was obtained by submandibular bleeding at 4–5 minutes post challenge with LukED, and 7–8 minutes post challenge with HlgAB. We chose these time points so that we would be able to reproducibly bleed the mice (at time points after this, we were not able to harvest blood from the mice because there was not enough fluid left in the vasculature). The blood was anti-coagulated using heparin tubes. Hematocrit was then measured using a HemaVet 950 (Drew Scientific) according to standard manufacturer’s instructions.
Temperature.
The temperature of toxin challenged mice was measured 10 minutes after challenge with room temperature toxins using a Small Rodent Infrared Thermometer (Braintree Scientific), taking the measurement on the chest of the mouse, right over the position of the heart. For infections, temperature was measured right before anesthesia of the day of infection, and on subsequent days post infection as shown.
Miles assay.
We performed the Miles assay to assess endothelial barrier disruption in vivo (Radu and Chernoff, 2013). We injected mice anesthetized by inhalation of isoflurane gas (2%) retro-orbitally with 100 l of 1% Evans blue dye in phosphate buffered saline (PBS), and 30 min later injected the toxins as described above. After 30 minutes (or when mice reached endpoint) the mice were sacrificed and the organs were harvested and weighed. In some experiments, where indicated, mice were perfused for 3 minutes with 30 ml PBS through the left ventricle of the heart using a Masterflex L/S Economy Drive and an Easy-Load II Head (Cole-Palmer) set to speed 0.5 before harvest of tissues. In the case of the liver, the left half of the median lobe of liver was taken. Hind limb soft tissue was harvested by cutting the muscle and fat off the bones of both hind limbs. Evans blue was extracted from the tissues by incubating with 1 ml N,N-dimethyl formamide (Fisher) at 37 °C for 24 hours, followed by centrifugation at 9,391 g for 10 min. 100 μl of N,N-dimethyl formamide was then transferred to a 96 well flat-bottom plate in duplicate and OD600 was measured using an EnVision 2103 Multi-label Reader (PerkinElmer), duplicate measurements were averaged. For quantification, an Evans blue standard was diluted in N,N-dimethyl formamide that had been incubated with the respective organs from mice not injected with Evans blue or toxin. For quantifying samples from ears, we placed the N,N-dimethyl formamide in glass tubes and measured the OD600 using a Genesys 20 spectrophotometer (Thermo Scientific), and took at least three readings of each tube and averaged them. The Evans blue content was then divided by the weight of the organs to give the g/gram tissue.
Intravital imaging.
Intravital imaging was performed via a protocol adapted from Girgis et al (Girgis et al., 2014). Mice were anesthetized with ketamine, xylazine, and acepromazine injected intramuscularly and were kept warm on a heating pad during surgery. Livers of anesthetized mice were exposed by cutting through the skin and peritoneum just below the rib cage and coaxing out a lobe of the liver. Mice were then inverted onto a pre-warmed aluminum stage insert with a 2.5 cm window fitted with a 45×50 mm glass coverslip (Fisher Scientific). The liver was stabilized with gauze soaked in PBS to limit movement during imaging and to keep the liver moist. Mice were injected retro-orbitally with 250 μg of Hoechst 33342 to visualize nuclei and 100 μl 1% Evans blue in PBS to visualize vascular fluid. Mice were then transferred immediately to a heated chamber that maintains the microscope, objectives, mice, and stage at 37°C during imaging. Images were acquired on a Leica SP2 AOBS inverted confocal microscope (20× HC PL APO 0.70 air objective) with 405 nm and 488 nm excitation sources and detected using tunable filters. z stacks of a series of x-y planes were collected every 30 seconds with a step size of 2–4 μm and a total thickness of up to 20 μm. Images were collected using Leica LCS software. For image analysis, ImageJ64 (http://imagej.nih.gov/ij) was used to convert three-dimensional stacks into time series and create maximum projections of the z stacks. All images used to create time-lapse series were treated uniformly with a 0.5-pixel median filter and a 1-pixel Gaussian filter.
Generation of Bone Marrow Chimeras.
8- to 11-week-old recipient mice were lethally irradiated with 1,100 cGy split-dosed irradiation. The BM from donor mice was harvested from both tibiae and femora. After lysis of erythrocytes, at least 2 × 106 cells were intravenously injected into recipient irradiated mice. Experiments were performed at least 11 weeks after irradiation, and chimerism was confirmed in every mouse by staining erythrocytes with anti-DARC PE and evaluated via flow cytometric analysis (CytoFLEX S). Chimerism was also analyzed by taking whole blood from the mice 11 weeks post irradiation and testing for hemolysis with toxin. After 30 minutes of incubation with 10 μg/ml LukED or HlgAB at 37°C, supernatants were collected and the OD at 405 nm was measured on an EnVision plate reader (Perkin Elmer). Percent hemolysis was calculated as compared to full lysis by triton.
Murine in vivo infections.
Mice were anesthetized intraperitoneally with 300–350 μl Avertin based on weight (2,2,2-tribromoethanol dissolved in tert-Amyl alcohol and diluted to a final concentration of 2.5% v/v in 0.9% sterile saline), followed by systemic (retro-orbital) injection of 2–3.5 × 107 colony-forming units (CFU) of MSSA (strain Newman) and isogenic mutants, or 1–2 × 107 CFU for infection of chimeric mice. Experiments with MRSA (USA500) used 1–2.7 × 107 CFU, and with CA-MRSA (USA 300) used 0.9–1.3 × 107 CFU. Mice were monitored for signs of morbidity and mortality including loss of <20% of initial weight, after which they were sacrificed and the time to acute morbidity was recorded.
To measure bacterial burdens, levels of serum analytes, and toxin concentrations in infected mice, animals were sacrificed at a time point when ~90% of the WT infected mice were still alive. For Swiss-Webster mice this was 96 hours post infection. For C57BL/6J background mice, this was typically 96 hours post infection for males and 72 hours post infection for females. Organs were harvested and homogenized in 1 ml of 1× PBS, followed by tenfold serial dilutions in 1× PBS, and the dilutions were plated on TSA plates. The plates were incubated overnight at 37°C to quantify bacterial burden. In experiments measuring LukD concentrations, after removing 100 μl for CFU enumeration, 100 μl 10× RIPA buffer and 10 μl 100× HALT protease inhibitor cocktail were added to the homogenized organs. Homogenates were then incubated for 1 hour on ice, centrifuged for 15 min at 20,817 g at 4°C, and supernatants were frozen at −80°C until they were used.
When serum was collected, it was harvested by sub-mandibular bleeding into serum collection tubes (BD) right before sacrificing the mice, and processed as per the manufacturer’s instructions. Whole blood lysates were collected into EDTA coated tubes (BD), and appropriate amounts of 10× RIPA and 100× HALT protease inhibitor cocktail were added and they were processed as described above for organ lysates.
Gross pathology was assessed by scoring whole kidneys as they were harvested using this scale: 0 = no abscesses visible, 1 = 1 or a few abscesses, 2 = several abscesses throughout the kidney tissue or one area that is overwhelmed with abscesses, 3 = kidney tissue is overwhelmed with abscesses. Each mouse was assigned the score of the more severe kidney.
ELISA for LukD concentrations.
Monoclonal antibody to LukD SM1B225.001 was generated by Janssen Biotech Inc. as follows: Balb/c and C3H mice were injected intraperitoneally with recombinant LukD mixed with Complete or Incomplete Freund’s adjuvant 3 times during 6 weeks, and then injected subcutaneously at the base of the tail with LukD and an agonistic anti-mouse CD40 mAb. B cells were enriched from splenocytes by MACS and co-incubated with FO mouse melanoma cells in a polyethylene glycol (PEG)-4000 solution to generate mAb secreting hybridoma cell lines. Hybridomas were screened for specific binding to LukD. The immunoglobulin heavy chain and light chain genes were cloned by RT-PCR, cDNA synthesis, and ligation into plasmids. Plasmids were sequenced, and antibodies were produced by transfection into Expi293F cells using Gibco’s Expifectamine transfection kit.
Immulon 2H flat-bottomed plates were coated with the monoclonal antibody SM1B225.001 in carbonate bicarbonate buffer overnight. After blocking with Blotto, lysates of organ homogenates and whole blood were added to the plate to be captured. LukD was then detected using a polyclonal rabbit anti-LukD serum (Alonzo III et al., 2012) and goat-anti rabbit HRP antibody. The ELISA was developed with 1-step Ultra TMB-ELISA and stopped with 1N Sulfuric acid. OD at 450 nm was then measured on an EnVision plate reader (Perkin Elmer). Concentrations were calculated by comparison with a standard curve of purified LukD in RIPA buffer.
Serum Analysis.
Serum pooled from males at 96 hours post infection was analyzed. Females were excluded because at this timepoint most of the WT females had died. Clinical pathology parameters were evaluated using an Advia 1800 Chemistry Analyzer following standard procedures. Serum samples were evaluated for the following analytes: AST (aspartate aminotransferase; U/L), ALT (alanine aminotransferase; U/L), ALP (alkaline phosphatase; U/L), GGT (gamma-glutamyl transferase; U/L), TBILI (total bilirubin; mg/dL), BUN (blood urea nitrogen; mg/dL), CREAT (creatinine; mg/dL), NA (sodium; mmol/L), K (potassium; mmol/L), Cl (chloride; mmol/L), Ca (calcium; mg/dL), P (phosphorus; mg/dL), TP (total protein; g/dL), ALB (albumin; g/dL), GLU (glucose; mg/dL), CHOL (cholesterol; mg/dL), TRIG (triglycerides; mg/dL), GLOB (globulins; g/dL), A/G (albumin/globulin ratio), DBILI (direct bilirubin; mg/dL), and IBILI (indirect bilirubin; mg/dL).
Histology:
Toxin challenge:
Organs from toxin challenged mice were fixed in 4% paraformaldehyde overnight, washed twice in PBS and then cryopreserved in 15% sucrose for three hours followed by 30% sucrose overnight. Organs were embedded in NEG 50, flash frozen in 2-methylbutane chilled with liquid nitrogen, and cryosectioned at 10 μm. The hematoxylin and eosin (H&E) stained sections were scanned at 40× magnification on a Leica SCN400 scanner for analysis.
Pathology of infected organs:
Mice were infected as above. Organs were harvested into PBS and then transferred into 10% Neutral Buffered Formalin (VWR) for at least 48 hours and transferred to Janssen BioTherapeutics for tissue processing. Tissues were processed into formalin fixed paraffin embedded (FFPE) tissue blocks on an ASP6025 Vacuum Tissue processer (Leica) using standard procedures. Tissues were sectioned after processing and collected onto positively charged glass slides for H&E staining.
In vitro endothelial cell transfections and toxin exposure.
Primary human pulmonary microvascular endothelial cells (HPMEC) were grown on gelatin-coated glass coverslips in a 12-well plate until 70% confluent and transfected with a plasmid containing GFP-tagged human DARC (variant 1) or GFP alone using X-tremeGENE HP DNA according to the manufacturer’s instructions. The final concentration of the plasmid DNA was 2 μg/ml. 24 hours later, cells were exposed to 0.3 μM toxin (this corresponds to 10 μg/ml) in complete EGM-2 media for 3 hours, PI and Nucblue were added to the media. The PI positive cells and DAPI images were recorded and analyzed by ImageXpress Micro XLS & MetaXpress 6 (Molecular DEVICES).
Induction of DARC expression using whole blood.
HPMEC were grown to confluency in 12-well plates and incubated with complete EGM-2 media or with human whole blood from healthy volunteers that was anti-coagulated with heparin with or without cycloheximide (CHX) 100 μg/ml at 37°C, 5% CO2 for 24 hours. Then the media or blood was aspirated and the cells were washed supplemented with PBS with Ca2+ and Mg2+ five times; cells were checked visually to confirm complete removal of blood cells. DARC protein expression was detected by Western blot using anti-DARC antibody.
Measurement of DARC mRNA by qPCR.
HPMEC cells were grown and treated with human whole blood as above. RNA was isolated using the QIAGEN RNA mini kit; cDNA synthesis was carried out using the High-capacity cDNA Reverse Transcription kit according to the manufacturer’s instructions. For each sample, RNA was reverse-transcribed using a T-Gradient Thermoblock (Biometra). qPCR was conducted using power SYBR Green PCR master mix. cDNA was denatured at 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds then 6 0°C for 1 minute, qPCR was performed with the ABI prism 7900 HT (Applied Biosystems), and the data were analyzed with SDS software v 2.1 (Applied Biosystems). Relative gene expression was compared using the comparative CT method. The primers for DARC and 18S rRNA are described in the Key result table. A fixed amount of cellular cDNA was added to each reaction so that 18S rRNA could be used as a reference.
Exposure of HPMEC cells to toxin in vitro after blood exposure.
HPMEC cells were grown and treated with human whole blood as above. The cells were incubated with 0.6 μM of HlgAB (this corresponds to 20 μg/ml) in serum-free EGM-2 media for 5 hours (except where shorter timepoints are indicated), PI and Nucblue were added to the media. The PI positive cells and DAPI images were recorded and analyzed by ImageXpress Micro XLS & MetaXpress 6 (Molecular DEVICES).
Knockdown of DARC by siRNA.
The HPMEC cells were transfected with DARC siRNA (Chen et al., 2015) and non-targeting siRNA control using Lipofectamine RNAiMAX reagent according to the manufacturer’s instructions. After 24 hours, the cells were treated with human whole blood as above. Then the cells were incubated with 0.6 μM of HlgAB in serum-free EGM-2 media for 5 hours. The PI positive cells and DAPI images were recorded and analyzed by ImageXpress Micro XLS & MetaXpress 6 (Molecular DEVICES) and representative pictures of the monolayers were taken by phase contrast microscope.
Immunoblotting for junctional proteins.
Cells were collected and lysed cells in lysis buffer containing 62.5 mM Tris-HCL (pH 6.8), 2% SDS, 10% glycerol, and 10mM DTT. Lysates were resolved on 10% polyacrylamide gel and transferred onto a nitrocellulose membrane and blocked for 1 hour in 5% BSA in TBS + 0.1% Tween-20. Primary antibodies were diluted 1:1000 (1 μg/mL) in 0.5% BSA in TBST and incubated at 4°C overnight on a rocker. Primary antibodies were mouse anti- β-actin, rabbit anti-Claudin 5, goat anti-VE-Cadherin. After washing several times in TBST, membranes were incubated with HRP-conjugated secondary antibodies (1 μg/mL) for 1 hour at room temperature followed by detection by enhanced chemiluminescence and imaging on a ChemiDoc Imaging System (BioRad, Canada). Protein abundance was quantified by densitometry (ImageJ) after normalization to loading control.
Immunofluorescence.
HPMECs were fixed/permeabilized in 100% iced cold methanol at −20°C for 10 min followed by washing 3× with PBS. Cells were blocked with 5% milk in PBS for 30 min at room temperature followed by immunodetection with rabbit anti-Claudin 5 and mouse anti-Ve-Cadherin diluted 1:100 (10 μg/mL) in PBS. After a 1-hour incubation at room temperature, cells were washed 3× in PBS and treated with species-specific, chromophore-conjugated secondary antibodies diluted 1:1000. Cells were then washed and mounted on slides with DAPI-containing (1:5000) Dako Fluorescence Mounting Medium. Slides were imaged using an Olympus Upright DP72 microscope with settings kept constant between conditions.
Immunofluorescence and Confocal Microscopy of ears:
Whole mount ears were prepared as previously described (Yamazaki et al., 2018). Briefly, whole ears were fixed overnight at 4°C in paraformaldehyde (PFA), lysine, and periodate (PLP) buffer. Whole ears were dissected to separate the base and periphery and hair, fat, and connective tissue was removed before staining. Whole ears were stained in 1× phosphate-buffered saline (PBS) containing 0.2% trition-x, 2% fetal calf serum, 2% goat-serum, 0.5% Fc block. Primary antibodies were diluted in staining buffer overnight at 4°C. The primary antibodies were: CD31-APC and Zo-1-AF647. Following 3 washes, the samples were mounted on slides in mounting media. The inner surface of the ear skin was in contact with the coverslip. Images were acquired using a Zeiss LSM 880 confocal microscope (Carl Zeiss) with the Zen Black software. Z-stacks were acquired using an 20× dry objective with a 0.6 zoom. The imaging data were processed and analyzed using Imaris software version 9.0.1.
QUANTIFICATION AND STATISTICAL ANALYSIS:
Statistical details (“n” numbers, tests used, definition of the error bars) are described in the figure legends, and specific “n” numbers and “n” number definitions are given in Table S3. Analyses of flow cytometric data were performed using FlowJo. Statistical significance was determined using Prism 7.0 b, with two-way ANOVA with Tukey’s post hoc test for multiple comparisons, log-rank (Mantel–Cox) test, or unpaired one- or two-tailed Student’s t-test with SEM as indicated. Statistical significance of kidney pathology data was determined using the linear-by-linear association test with the function lbl_test in the coinpackage in R, using ordered outcome.
Supplementary Material
: Exact “n” numbers and “n” number definitions.
Intravital imaging of the livers of female Swiss Webster mice after intravenous challenge with the indicated toxins. Doses: 20 μg PVL, 6 μg HlgAB, 6 μg HlgABmut, 15 μg LukED, 20 μg LukEDmut. Evans blue in red is labeling the vascular fluid; Hoechst in blue is labeling the nuclei.
HIGHLIGHTS:
S. aureus leukocidins target DARC on endothelial cells to cause vascular dysfunction
Targeting DARC leads to organ damage during S. aureus bloodstream infection
Primary human endothelial cells are susceptible to S. aureus leukocidins
Leukocidins target host leukocytes, red blood cells and endothelial cells
ACKNOWLEDGEMENTS:
We thank members of the Torres laboratory for critical insight into this project and helpful comments on the manuscript. We thank Junjie Mei for performing pilot experiments with DARC KO mice, Nathan Majewski for technical oversight of the identification of the anti-LukD mAb, and Debby Preston for the serum chemistry analyses. We also thank Luisa Cimmino and the Iannis Aifantis Lab for the use of their Hemavet and Russel Vance for experiments with MyD88 and Caspase 1/11 KO mice. We thank Alan Feder for help with statistical analysis. We are grateful to Dr. Peter Hare for his thoughtful comments on the manuscript.
This work was supported in part by the US National Institutes of Health under award numbers GM007308, AI007180 and AI124606 to A.L., AI007180 to M.K., DK111139 to P.M., AI121244 to K.C. and V.J.T, and AI105129 to V.J.T, and by the Natural Sciences and Engineering Research Council of Canada under award number RGPIN-2015–05802 to W.L.L. V.J.T. and K.C. are Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Diseases. W.L.L. is supported by a Canada Research Chair in Mechanisms of Endothelial Permeability.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
J.A., P.B., D.W., and A.S.L. are employees of Janssen Research & Development LLC. V.J.T. is an inventor on patents and patent applications filed by New York University, which are currently under the commercial license to Janssen Biotech Inc.
REFERENCES
- Adem PV, Montgomery CP, Husain AN, Koogler TK, Arangelovich V, Humilier M, Boyle-Vavra S, and Daum RS (2005). Staphylococcus aureus sepsis and the Waterhouse-Friderichsen syndrome in children. N Engl J Med 353, 1245–1251. [DOI] [PubMed] [Google Scholar]
- Alonzo F III, Benson MA, Chen J, Novick RP, Shopsin B, and Torres VJ (2012). Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol Microbiol 83, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzo F III, Kozhaya L, Rawlings SA, Reyes-Robles T, DuMont AL, Myszka DG, Landau NR, Unutmaz D, and Torres VJ (2013). CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerlaan H, Seifert H, Harbarth S, Brun-Buisson C, Torres A, Antonelli M, Kluytmans J, and Bonten M (2009). Adequacy of antimicrobial treatment and outcome of Staphylococcus aureus bacteremia in 9 Western European countries. Clin Infect Dis 49, 997–1005. [DOI] [PubMed] [Google Scholar]
- Benck CJ, Martinov T, Fife BT, and Chatterjea D (2016). Isolation of Infiltrating Leukocytes from Mouse Skin Using Enzymatic Digest and Gradient Separation. J Vis Exp, e53638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Roth AJ, and Horswill AR (2010). Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5, e10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A, Nielsen S, Elkjaer ML, Zbrzezna V, Fang F, and Pogo AO (1997). Detection of Duffy antigen in the plasma membranes and caveolae of vascular endothelial and epithelial cells of nonerythroid organs. Blood 89, 701–712. [PubMed] [Google Scholar]
- Chen Y, Liao N, Lu F, Peng H, and Gao J (2015). The role of Duffy antigen receptor for chemokines in keloids. Gene 570, 44–49. [DOI] [PubMed] [Google Scholar]
- Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, and Missiakas DM (2009). Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J 23, 3393–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David MZ, and Daum RS (2010). Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23, 616–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TC, Lentsch AB, Wang Z, Cowhig JE, Rot A, Maeda N, and Peiper SC (2000). Exaggerated response to endotoxin in mice lacking the Duffy antigen/receptor for chemokines (DARC). Blood 96, 1681–1684. [PubMed] [Google Scholar]
- DuMont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, and Torres VJ (2011). Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol 79, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMont AL, Yoong P, Day CJ, Alonzo F 3rd, McDonald WH, Jennings MP, and Torres VJ (2013). Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A 110, 10794–10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie ES, and Lorenz LL (1952). Staphylococcal coagulase; mode of action and antigenicity. J Gen Microbiol 6, 95–107. [DOI] [PubMed] [Google Scholar]
- Girgis NM, Gundra UM, Ward LN, Cabrera M, Frevert U, and Loke P (2014). Ly6C(high) monocytes become alternatively activated macrophages in schistosome granulomas with help from CD4+ cells. PLoS Pathog 10, e1004080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley TJ, Lu ZH, Wasniowska K, Martin AW, Peiper SC, Hesselgesser J, and Horuk R (1994). Postcapillary venule endothelial cells in kidney express a multispecific chemokine receptor that is structurally and functionally identical to the erythroid isoform, which is the Duffy blood group antigen. J Clin Invest 94, 985–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, Broglie PM, Marketon K, Austermann J, Vogl T, et al. (2012). Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol 92, 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, and Miller LH (1993). A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science 261, 1182–1184. [DOI] [PubMed] [Google Scholar]
- Hothorn T, Hornik K, van de Wiel MA and Zeileis A (2006). A Lego System for Conditional Inference. The American Statistician 60, 257–263. [Google Scholar]
- Howes RE, Patil AP, Piel FB, Nyangiri OA, Kabaria CW, Gething PW, Zimmerman PA, Barnadas C, Beall CM, Gebremedhin A, et al. (2011). The global distribution of the Duffy blood group. Nat Commun 2, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, and Flavell RA (2001). Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz GR, Dries DJ, Peterson ML, and Schlievert PM (2005). Purpura fulminans due to Staphylococcus aureus. Clin Infect Dis 40, 941–947. [DOI] [PubMed] [Google Scholar]
- Lacorre DA, Baekkevold ES, Garrido I, Brandtzaeg P, Haraldsen G, Amalric F, and Girard JP (2004). Plasticity of endothelial cells: rapid dedifferentiation of freshly isolated high endothelial venule endothelial cells outside the lymphoid tissue microenvironment. Blood 103, 4164–4172. [DOI] [PubMed] [Google Scholar]
- Langhi DM Jr., and Bordin JO (2006). Duffy blood group and malaria. Hematology 11, 389–398. [DOI] [PubMed] [Google Scholar]
- Lubkin A, and Torres VJ (2017). Bacteria and endothelial cells: a toxic relationship. Curr Opin Microbiol 35, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Chaudhuri A, Zbrzezna V, He Y, and Pogo AO (2000). Deletion of the murine Duffy gene (Dfy) reveals that the Duffy receptor is functionally redundant. Mol Cell Biol 20, 3097–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, and Dixit VM (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, and Schneewind O (2003). Passage of heme-iron across the envelope of Staphylococcus aureus. Science 299, 906–909. [DOI] [PubMed] [Google Scholar]
- Mediavilla JR, Chen L, Mathema B, and Kreiswirth BN (2012). Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA MRSA). Curr Opin Microbiol 15, 588–595. [DOI] [PubMed] [Google Scholar]
- Melehani JH, James DB, DuMont AL, Torres VJ, and Duncan JA (2015). Staphylococcus aureus Leukocidin A/B (LukAB) Kills Human Monocytes via Host NLRP3 and ASC when Extracellular, but Not Intracellular. PLoS Pathog 11, e1004970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LH, Mason SJ, Dvorak JA, McGinniss MH, and Rothman IK (1975). Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science 189, 561–563. [DOI] [PubMed] [Google Scholar]
- Peiper SC, Wang ZX, Neote K, Martin AW, Showell HJ, Conklyn MJ, Ogborne K, Hadley TJ, Lu ZH, Hesselgesser J, et al. (1995). The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy negative individuals who lack the erythrocyte receptor. J Exp Med 181, 1311–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu M, and Chernoff J (2013). An in vivo assay to test blood vessel permeability. J Vis Exp, e50062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Robles T, Alonzo F 3rd, Kozhaya L, Lacy DB, Unutmaz D, and Torres VJ (2013). Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 14, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Robles T, Lubkin A, Alonzo F 3rd, Lacy DB, and Torres VJ (2016). Exploiting dominant-negative toxins to combat Staphylococcus aureus pathogenesis. EMBO Rep 17, 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RB, de Lencastre A, Eisner W, Severina EP, Shopsin B, Kreiswirth BN, and Tomasz A (1998). Molecular epidemiology of methicillin-resistant Staphylococcus aureus in 12 New York hospitals. MRSA Collaborative Study Group. J Infect Dis 178, 164–171. [DOI] [PubMed] [Google Scholar]
- Rose HR, Holzman RS, Altman DR, Smyth DS, Wasserman GA, Kafer JM, Wible M, Mendes RE, Torres VJ, and Shopsin B (2015). Cytotoxic Virulence Predicts Mortality in Nosocomial Pneumonia Due to Methicillin-Resistant Staphylococcus aureus. J Infect Dis 211, 1862–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan AN, Reyes-Robles T, Badiou C, Cochet S, Boguslawski KM, Yoong P, Day CJ, de Haas CJ, van Kessel KP, Vandenesch F, et al. (2015). Staphylococcus aureus Targets the Duffy Antigen Receptor for Chemokines (DARC) to Lyse Erythrocytes. Cell Host Microbe 18, 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan AN, van Strijp JAG, and Torres VJ (2017). Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan AN, Vrieling M, Wallet P, Badiou C, Reyes-Robles T, Ohneck EA, Benito Y, de Haas CJ, Day CJ, Jennings MP, et al. (2014). The staphylococcal toxins gamma-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat Commun 5, 5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiriot A, Perdomo C, Cheng G, Novitzky-Basso I, McArdle S, Kishimoto JK, Barreiro O, Mazo I, Triboulet R, Ley K, et al. (2017). Differential DARC/ACKR1 expression distinguishes venular from non-venular endothelial cells in murine tissues. BMC Biol 15, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, et al. (2012). Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Li W, Yang L, Li P, Cao H, Motegi SI, Udey MC, Bernhard E, Nakamura T, and Mukouyama YS (2018). Whole-Mount Adult Ear Skin Imaging Reveals Defective Neuro-Vascular Branching Morphogenesis in Obese and Type 2 Diabetic Mouse Models. Sci Rep 8, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
: Exact “n” numbers and “n” number definitions.
Intravital imaging of the livers of female Swiss Webster mice after intravenous challenge with the indicated toxins. Doses: 20 μg PVL, 6 μg HlgAB, 6 μg HlgABmut, 15 μg LukED, 20 μg LukEDmut. Evans blue in red is labeling the vascular fluid; Hoechst in blue is labeling the nuclei.